
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation mechanism of phenols by copper(II)–halide complexes†
Lan
Yang
,
Rin
Ito
,
Hideki
Sugimoto
 ,
Yuma
Morimoto
and
Shinobu
Itoh
*
,
Yuma
Morimoto
and
Shinobu
Itoh
*
Department of Molecular Chemistry, Division of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: shinobu@chem.eng.osaka-u.ac.jp
First published on 21st June 2024
Abstract
The mechanism of oxidation of phenols by tetrahedral copper(II)–halide complexes was investigated to demonstrate that phenols with an electron-withdrawing substituent are oxidized via a proton-transfer/electron-transfer (PTET) mechanism, whereas phenols with an electron-donating substituent involve a concerted proton/electron transfer (CPET) mechanism. The importance of the tetrahedral geometry of the metal centre as well as the effects of the halide ligands of the substrates were explored.
The redox reactivity of transition-metal complexes depends largely on the geometry of the metal centre. In the case of copper(II) complexes with similar donor groups, for example, the oxidation ability of copper(II) complexes taking a tetrahedral (Td) geometry may be higher than those having a square planar, square pyramidal, or trigonal bipyramidal structure, since the Td geometry is more suited to the copper(I) oxidation state compared to the others. To explore the redox reactivity of such tetrahedral copper(II) complexes, we have developed a series of copper(II) complexes, [CuII(TMG3tach)X]+ (1X), where TMG3tach is an N3-tridentate ligand consisting of cis,cis-1,3,5-triaminocyclohexane (tach) and N,N,N′,N′-tetramethylguanidino (TMG) substituents, and X is an anionic co-ligand (F−, Cl−, Br−, I−, MeO−, C6F5O−, C6F5S−, or ROO−).1–3 Reactivity studies of the halide complexes demonstrated that they undergo CuII–X bond cleavage, and in the case of X = F−, Cl−, and Br−, they induce C–H bond activation of an external substrate, such as 1,4-cyclohexadiene (CHD) with a weak C–H bond (76.0 ± 1.2 kcal mol−1),4 to give the corresponding copper(I) complex and benzene as the oxidation product.3 Such C–H bond activation reactivity of transition-metal halide complexes has been reported by using high-valent transition-metal halide complexes of NiIII, PdIV, CuIII, and AuIII, where the higher oxidation state of the metal ions induces homolytic cleavage of the metal–halide bond.5–14 In the case of 1X, on the other hand, the metal centre has a normal CuII oxidation state, but not a high-valent metal ion such as CuIII. Thus, we suspected that such reactivity of halide complexes can be attributed to their tetrahedrally distorted geometry, which induces CuII–X bond homolysis to give CuI and X˙, the latter of which formally abstracts a hydrogen atom from the substrate.3 Unfortunately, however, the oxidation reaction of CHD by 1X was too slow to perform a detailed kinetic analysis.
In this study, we further examined the reactivity of 1X (X = F−, Cl−, Br−, or I−) toward phenolic substrates (4-substituted-2,6-di-tert-butylphenol PYH, Scheme 1) in order to shed light on the O–H bond activation mechanism by copper(II)–halide complexes. Phenols are often used as a mechanistic probe for hydrogen atom transfer reactions. To examine the geometric effect on the reactivity of copper(II)–halide complexes, we also employed copper(II)–halide complexes 2X (X = Cl− or Br−) supported by a tripodal tetradentate ligand TMG3tren (1,1,1-tris(2-(N2-(1,1,3,3-tetramethylguanidino))ethyl)amine),15–17 that enforces the trigonal bipyramidal (Tbp) geometry of the metal centre (Fig. 1).
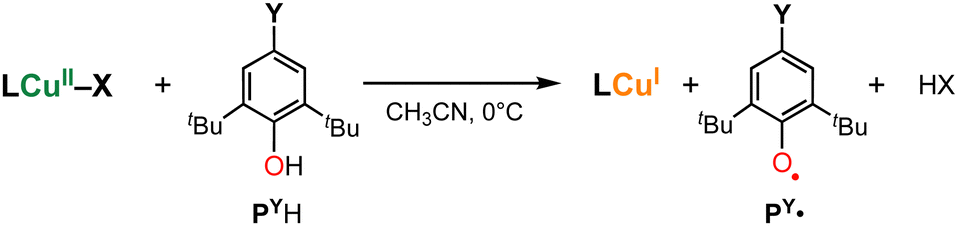 | ||
| Scheme 1 Reaction of copper(II)–halide complexes and 4-substituted-2,6-di-tert-butylphenol (PYH). | ||
 | ||
| Fig. 1 ChemDraw structures of 1X and 2X. | ||
Fig. 2(a) shows the spectral changes observed upon the addition of PtBuH (Y = tert-butyl, 12.5 mM) to 1Br (0.25 mM) in CH3CN at 0 °C under an N2 atmosphere as a typical example, where the absorption bands at 410 and 560 nm due to 1Br gradually decrease with a concomitant increase in the absorption bands at 380, 400 and 626 nm, obeying first-order kinetics (see the first-order plot shown in the inset to Fig. 2(a)). The absorption bands of the post-reaction solution at 380, 400 and 626 nm are identical to those of the 2,4,6-tri-tert-butylphenoxyl radical (PtBu˙), and its quantitative formation based on 1Br was confirmed using the reported ε values of PtBu˙.18 The formation of the phenoxyl radical PtBu˙ and the copper(I) complex was further confirmed by the EPR spectrum shown in Fig. S1 (ESI†), where only an EPR signal ascribable to PtBu˙ was observed at g = 2.0041,19 but the EPR signals due to the copper(II) complex 1Br completely disappeared. The fate of the generated HBr will be discussed below.
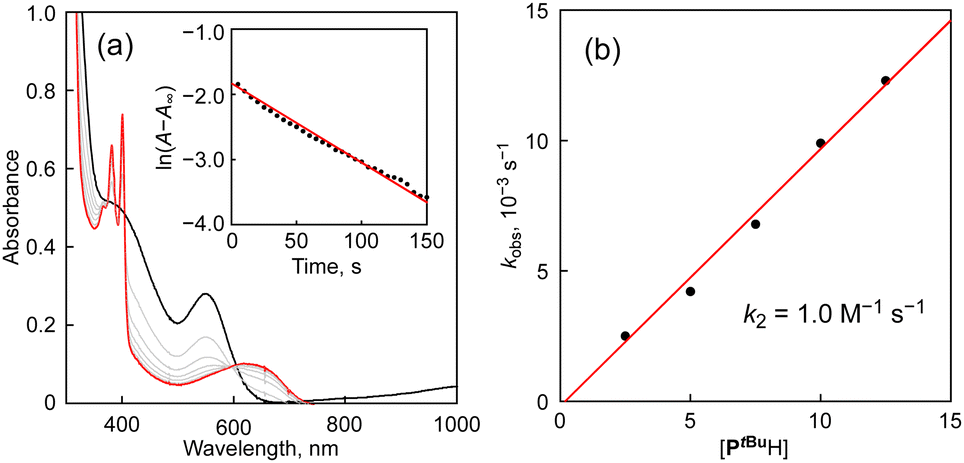 | ||
| Fig. 2 (a) UV-vis spectral changes for the reaction of 1Br (0.25 mM) with 2,4,6-tri-tert-butylphenol (PtBuH, 12.5 mM) in CH3CN at 0 °C. Inset: A pseudo-first-order plot based on the absorption change at 410 nm. (b) Plot of kobsvs. substrate concentration. | ||
The pseudo-first-order rate constant (kobs) obtained from the plot of ln(A − A∞) against the reaction time (inset of Fig. 2(a)) showed linear correlation with the concentration of PtBuH, as shown in Fig. 2(b), from which the second-order rate constant (k2) was determined to be 1.0 M−1 s−1 from the slope.
Importantly, both 2Cl and 2Br taking a Tbp geometry did not show such reactivity toward PtBuH under the same reaction conditions, clearly demonstrating that the Td geometry of the metal centre is crucial to inducing O–H bond activation reactivity.
Then, the reactions of 1Br and a series of phenol derivatives PYH (Y = OMe, Et, H, CHO, or COMe) were examined under the same reaction conditions (in CH3CN at 0 °C) to gain an insight into the mechanism of the phenol oxidation reaction. The kinetic analysis data are given in Fig. S2–S6 (ESI†). In all cases except PCOMeH, the reaction obeyed first-order kinetics in the presence of an excess amount of PYH (a pseudo-first-order reaction condition) and plots of the observed first-order rate constants (kobs) against the substrate concentration exhibited linear correlation, from which the second-order rate constants (k2) were determined from the slopes. For PCOMeH, the second-order rate constant (k2) was determined in the presence of a stoichiometric amount of the substrate (under a second-order reaction condition, Fig. S6, ESI†). In the case of POMeH as the substrate, the quantitative formation of the phenoxyl radical product POMe˙ was also confirmed by the appearance of its characteristic absorption bands at 387, 406 and 542 nm (Fig. S3, ESI†).20 On the other hand, the final organic product of the reactions with other phenols PYH (Y = Et, H, CHO, or COMe) were confirmed to be 3,3′,5,5′-tetra-tert-butyl-[1,1′-bi(cyclohexylidene)]-2,2′,5,5′-tetraene-4,4′-dione by MALDI-TOF mass spectra, as shown in Fig. S7 (ESI†), which was formed by the C–C coupling reaction of the generated phenoxyl radical species PY˙ and the subsequent elimination of Y2 from the dimeric intermediate (Scheme S1, ESI†).10
Fig. 3 shows a Hammett plot of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k2 against σp. As can clearly be seen, the reaction rate (k2) increases linearly as the electron-withdrawing (EW) ability of the p-substituent Y increases (increasing the σp value) in going from Y = Et to COMe with a Hammett ρ value of 4.0. In the cases of Y = OMe or tBu, however, the data points deviated from the linear line, where the reaction rates were larger than those expected from the linear correlation. Such a phenomenon was also observed in the reactions of phenol derivatives with copper(III)-superoxide (CuIII–OO˙) and nickel(III)–fluoride (NiIII–F) complexes supported by 2,6-diamidepyridine ligands.10,21 The authors suggested a change of reaction mechanism across the series of phenol derivatives. Namely, the reactions where the phenols have an electron-withdrawing (EW) substituent involve a PTET (proton transfer following electron transfer) mechanism, whereas the oxidation of phenols with an electron-donating (ED) substituent includes a hydrogen atom transfer (HAT) or concerted proton/electron transfer (CPET) mechanism. Kinetic deuterium isotope effects (KIEs) were determined to be 1.6 and 1.7 for the oxidation of POMeH(D) and PtBuH(D), respectively (Fig. S8 and S9, ESI†). Such a small KIE value was reported in the oxidation of PHH by an NiIII–Cl complex supported by a 2,6-diamidepyridine ligand, for which the CPET mechanism was proposed.9 On the other hand, no kinetic deuterium isotope effect was observed (KIE = 1.0) in the oxidation of PCOMeH(D) with an EW-substituent by 1Br (Fig. S10, ESI†).
k2 against σp. As can clearly be seen, the reaction rate (k2) increases linearly as the electron-withdrawing (EW) ability of the p-substituent Y increases (increasing the σp value) in going from Y = Et to COMe with a Hammett ρ value of 4.0. In the cases of Y = OMe or tBu, however, the data points deviated from the linear line, where the reaction rates were larger than those expected from the linear correlation. Such a phenomenon was also observed in the reactions of phenol derivatives with copper(III)-superoxide (CuIII–OO˙) and nickel(III)–fluoride (NiIII–F) complexes supported by 2,6-diamidepyridine ligands.10,21 The authors suggested a change of reaction mechanism across the series of phenol derivatives. Namely, the reactions where the phenols have an electron-withdrawing (EW) substituent involve a PTET (proton transfer following electron transfer) mechanism, whereas the oxidation of phenols with an electron-donating (ED) substituent includes a hydrogen atom transfer (HAT) or concerted proton/electron transfer (CPET) mechanism. Kinetic deuterium isotope effects (KIEs) were determined to be 1.6 and 1.7 for the oxidation of POMeH(D) and PtBuH(D), respectively (Fig. S8 and S9, ESI†). Such a small KIE value was reported in the oxidation of PHH by an NiIII–Cl complex supported by a 2,6-diamidepyridine ligand, for which the CPET mechanism was proposed.9 On the other hand, no kinetic deuterium isotope effect was observed (KIE = 1.0) in the oxidation of PCOMeH(D) with an EW-substituent by 1Br (Fig. S10, ESI†).
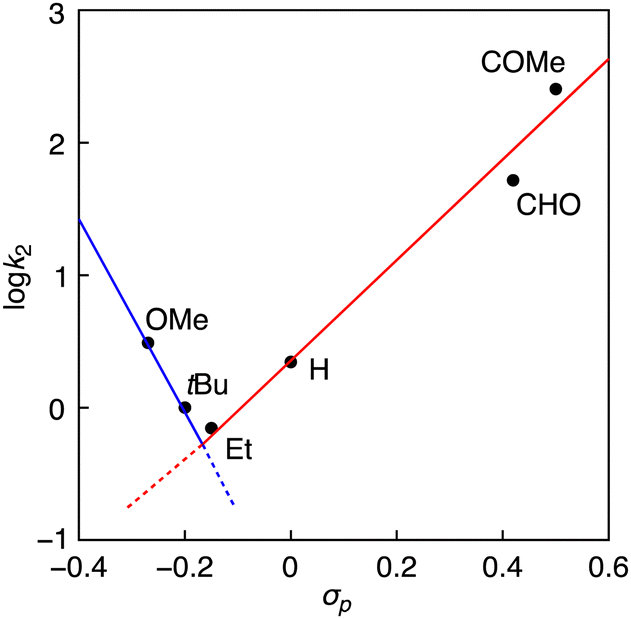 | ||
| Fig. 3 Hammett plot for the reaction of 1Br and PYH. | ||
To explain these kinetic results, we propose the reaction mechanism illustrated in Scheme 2.
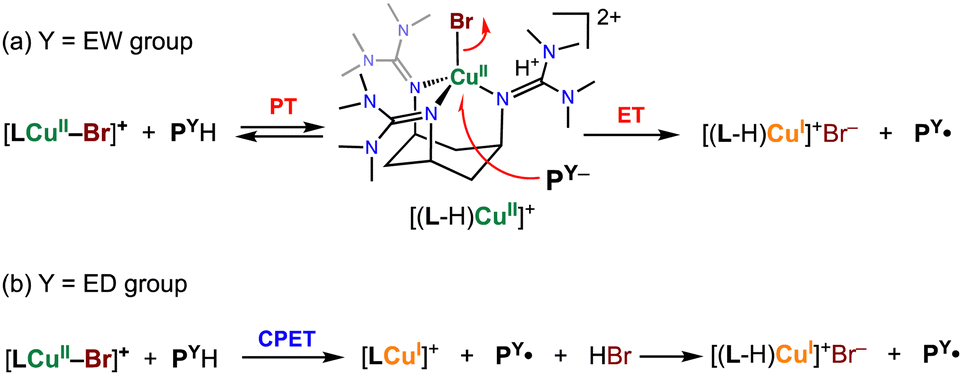 | ||
| Scheme 2 Proposed mechanism for the phenol oxidation reaction by 1X. | ||
For phenol substrates with an EW-substituent, such as PCOMeH, deprotonation of the phenol substrate by one of the TMG substituents of the supporting ligand takes place, causing an acid–base equilibrium (initial PT process in Scheme 2(a)). Then, electron transfer from the generated phenolate to copper(II) ion occurs concomitantly with dissociation of Br−, giving a copper(I) complex and the phenoxyl radical product PCOMe˙. Dissociated Br− forms a guanidinium salt with the protonated TMG substituent (Scheme 2(a)). Thus, the reductive dissociation process of Br− from the CuII–Br centre is rate-limiting. This mechanism is consistent with the fact that no KIE was observed, as mentioned above.
On the other hand, for the reaction of phenols with an ED-substituent, like POMeH, such a proton transfer from the phenol hardly occurs due to the higher pKa of the phenolic proton of POMeH, so that concerted proton/electron transfer (CPET) becomes the major pathway (Scheme 2(b)). In this case as well, the generated HBr eventually forms a guanidinium salt with the TMG substituent of the supporting ligand (Scheme 2(b)).
To gain further insight into the reaction mechanism, the effects of the halide ligands X were examined using PCOMeH with EW-substituents as substrates under the same reaction conditions. Fig. S12–S14 (ESI†) show the kinetic analysis data for the reactions of 1F, 1Cl, and 1I, respectively. The k2 values for these reactions were determined in the presence of a stoichiometric amount of the substrate (a second-order reaction condition), since the reactions of PCOMe were too fast under the pseudo-first-order reaction conditions (in the presence of an excess amount of the substrate). Thus, the plot of (A0 − A)/[Cu]0(A − A∞) against time gave a straight line passing through the origin, from which the second-order rate constant (k2) was obtained as the slope of the linear line, as listed in Table 1. Reported Cu–X bond dissociation energy (BDE) values are also included in Table 1 and the plot of logk2 against the BDE of Cu–X is shown in Fig. 4.
| F | Cl | Br | I | |
|---|---|---|---|---|
| k 2/102 (M−1 s−1) | 1.25 | 0.50 | 2.53 | 10.34 |
| BDE (kcal mol−1) of Cu–X4 | 103 | 90 | 80 | 70 |
 | ||
| Fig. 4 Plot of logk2vs. BDECu–X for the reaction of 1X and PCOMeH. | ||
Notably, the logk2 of 1I, 1Br and ICl exhibited very good linear correlation with the reported BDE values4 of Cu–X, where the weaker the Cu–X bond, the faster the reaction rate. This is consistent with the proposed mechanism involving the rate-limiting reductive Cu–X dissociation reaction for the oxidation of PCOMeH.
On the other hand, the logk2 value of 1F is significantly larger than that predicted from the linear line, as shown in Fig. 4. This result clearly indicates that the reaction mechanism of 1F is different from that of the other complexes (1I, 1Br, or ICl). That is, the oxidation of the phenol by 1F may involve a CPET mechanism rather than a PTET mechanism. This may be due to the extremely strong BDE of HF (136 kcal mol−1)4 compared to those of the others (HCl: 103 kcal mol−1, HBr: 88 kcal mol−1, HI: 71 kcal mol−1).4 In fact, we obtained a KIE value of 1.4, similar to those of the CPET reactions mentioned above (KIE = 1.6–1.7, Fig. S11, ESI†). The formation of free HF was confirmed by 19F-NMR for the reaction of 1F with PCOMeH in CH3CN (vide infra), where the fluorine signal of HF was detected at δ = −148 ppm and its yield was estimated as 78% based on the copper(II) complex using OTf− as an internal standard (Fig. S15, ESI†).
In summary, we have demonstrated that tetrahedrally distorted copper(II)–halide complexes 1X supported by a TMG3tach ligand showed oxidation ability toward phenol derivatives, where substrates with an EW-substituent are oxidized via a proton-transfer/electron-transfer (PTET) mechanism and those with an ED-substituent undergo a concerted proton/electron transfer (CPET) mechanism. The importance of the tetrahedral geometry of the metal centre was also demonstrated by comparing the reactivity with that of a copper(II)–halide complex 2X with a trigonal bipyramidal geometry. Moreover, the TMG group is shown to work as a proton acceptor from the phenol substrate in the PTET mechanism, and in the reaction of PCOMe and 1F, the strong BDE of HF (the product) greatly enhanced the reactivity. Further mechanistic studies are being conducted to gain more detailed insights into the reaction mechanism.
This work was supported by JST-CREST (JPMJCR16P1 to SI), Grant in Aid for Scientific Research (B) (JSPS 23K26669 to SI) and JST SPRING (JPMJSP2138 to YL).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Shimizu, Y. Morimoto, D. Faltermeier, M. Kerscher, S. Paria, T. Abe, H. Sugimoto, N. Fujieda, K. Asano, T. Suzuki, P. Comba and S. Itoh, Inorg. Chem., 2017, 56, 9634–9645 CrossRef CAS PubMed.
- I. Shimizu, Y. Morimoto, G. Velmurugan, T. Gupta, S. Paria, T. Ohta, H. Sugimoto, T. Ogura, P. Comba and S. Itoh, Chem. – Eur. J., 2019, 25, 11157–11165 CrossRef CAS PubMed.
- Y. Lan, Y. Morimoto, I. Shimizu, H. Sugimoto and S. Itoh, Inorg. Chem., 2023, 62, 10539–10547 CrossRef CAS PubMed.
- Y.-R. Luo, Conprehensive Handbook of Chemical Bond Energy, CRC Press, Boca Raton, London, New York, 2007 Search PubMed.
- M. H. Pérez-Temprano, J. M. Racowski, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 4097–4100 CrossRef PubMed.
- S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S.-L. Zheng, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472–6475 CrossRef CAS PubMed.
- S. J. Hwang, B. L. Anderson, D. C. Powers, A. G. Maher, R. G. Hadt and D. G. Nocera, Organometallics, 2015, 34, 4766–4774 CrossRef CAS.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- P. Mondal, P. Pirovano, A. Das, E. R. Farquhar and A. R. McDonald, J. Am. Chem. Soc., 2018, 140, 1834–1841 CrossRef CAS PubMed.
- P. Mondal and A. R. McDonald, Chem. – Eur. J., 2020, 26, 10083–10089 CrossRef CAS PubMed.
- D. Gygi, M. I. Gonzalez, S. J. Hwang, K. T. Xia, Y. Qin, E. J. Johnson, F. Gygi, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 6060–6064 CrossRef CAS PubMed.
- S. K. Kariofillis and A. G. Doyle, Acc. Chem. Res., 2021, 54, 988–1000 CrossRef CAS PubMed.
- J. K. Bower, A. D. Cypcar, B. Henriquez, S. C. E. Stieber and S. Zhang, J. Am. Chem. Soc., 2020, 142, 8514–8521 CrossRef CAS PubMed.
- M. Lovisari, R. Gericke, B. Twamley and A. R. McDonald, Inorg. Chem., 2021, 60, 15610–15616 CrossRef CAS PubMed.
- K. W. Krockert, F. Garg, J. Heck, M. V. Heinz, J. Lange, R. Schmidt, A. Hoffmann and S. Herres-Pawlis, Dalton Trans., 2024, 53, 2973–2990 RSC.
- V. Raab, J. Kipke, O. Burghaus and J. Sundermeyer, Inorg. Chem., 2001, 40, 6964–6971 CrossRef CAS PubMed.
- H. Wittmann, V. Raab, A. Schorm, J. Plackmeyer and J. Sundermeyer, Eur. J. Inorg. Chem., 2001, 1937–1948 CrossRef CAS.
- V. W. Manner, T. F. Markle, J. H. Freudenthal, J. P. Roth and J. M. Mayer, Chem. Commun., 2008, 256–258 RSC.
- D. Ar, A. F. R. Kilpatrick, B. Cula, C. Herwig and C. Limberg, Inorg. Chem., 2021, 60, 13844–13853 CrossRef CAS PubMed.
- J. P. Collman, R. A. Decréau and C. J. Sunderland, Chem. Commun., 2006, 3894–3896 RSC.
- W. D. Bailey, D. Dhar, A. C. Cramblitt and W. B. Tolman, J. Am. Chem. Soc., 2019, 141, 5470–5480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, kinetic analysis data, EPR and MALDI-TOF mass spectra of the post-reaction solutions and 19F-NMR for the detection of HF. See DOI: https://doi.org/10.1039/d4cc02483d |
| This journal is © The Royal Society of Chemistry 2024 |