
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adaptive water oxidation catalysis on a carboxylate-sulfonate ligand with low onset potential†
Jing
Yang‡
*a,
Shaoqi
Zhan‡
e,
Linqin
Wang
c,
Hao
Yang
d,
Lele
Duan
 c,
Xiaolei
Fan
bf,
Tianqi
Liu
*b and
Licheng
Sun
*cd
c,
Xiaolei
Fan
bf,
Tianqi
Liu
*b and
Licheng
Sun
*cd
aCollege of Health Science and Environmental Engineering, Shenzhen Technology University, Shenzhen 518118, China. E-mail: yangjing2@sztu.edu.cn
bInstitute of Wenzhou, Zhejiang University, 325005, Wenzhou, China. E-mail: tianqil@kth.se
cCenter of Artificial Photosynthesis for Solar Fuels, School of Science, Westlake University, 310024 Hangzhou, China. E-mail: sunlicheng@westlake.edu.cn
dDepartment of Chemistry, School of Engineering Sciences in Chemistry Biotechnology and Health, KTH Royal Institute of Technology, 10044 Stockholm, Sweden
eDepartment of Chemistry – Ångström, Uppsala University, Box 523, 751 20 Uppsala, Sweden
fDepartment of Chemical Engineering, School of Engineering, The University of Manchester, Oxford Road, Manchester M13 9PL, UK
First published on 22nd May 2024
Abstract
A water oxidation catalyst Ru-bcs (bcs = 2,2′-bipyridine-6′-carboxylate-6-sulfonate) with a hybrid ligand was reported. Ru-bcs utilizes the electron-donating properties of carboxylate ligands and the on-demand coordination feature of sulfonate ligands to enable a low onset potential of 1.21 V vs. NHE and a high TOF over 1000 s−1 at pH 7. The adaptive chemistry uncovered in this work provides new perspectives for developing molecular catalysts with high efficiency under low driving forces.
The pursuit of water oxidation catalysts with both high turnover frequency (TOF) and low onset potential is a focal point in artificial photosynthesis.1–4 The oxygen-evolving complex (OEC) in photosystem II (PSII) exemplifies such a catalytic system, exhibiting TOF ranging from 100 to 400 s−1 under solar irradiation.5,6 The driving force for water oxidation in PSII directly originates from the tyrosine/tyrosyl radical pair, which possesses a redox potential of 1.2 eV, corresponding to the 1.2 V anodic potential in electrochemical water oxidation.6 Various negatively charged ligands play pivotal roles in this scenario. Specifically, five negatively charged oxygen atoms in the first coordination sphere stabilize the Mn4Ca cluster, thereby reducing the potential for oxygen evolution. Additionally, protein residues in the second coordination sphere, such as carboxylates, stabilize catalytic intermediates and manage the local proton transfer, thus accelerating catalytic kinetics.7–9
To replicate the performance of the OEC, polypyridine-based anionic ligands were employed in artificial water oxidation catalysts,10,11 leading to the development of several catalysts that demonstrate outstanding performance in terms of either onset potential or TOFs. As illustrated in Fig. S2a (ESI†), incorporation of a sulfonate group into Ru-tpy (Grotjahn-cat, tpy = terpyridine) catalysts has led to significantly enhanced TOF exceeding 2000 s−1 under neutral conditions, albeit with a compromise of a 1.61 V onset potential versus the normal hydrogen electrode (NHE) under pH 7, possibly due to the presence of electron-neutral coordination only in the first coordination sphere.12 Introducing the negatively charged carboxylate and/or pyrrolato group in the first coordination sphere can effectively reduce the onset potential (around 1.35 V vs. NHE under pH 7), while the other dangling carboxylate could competitively coordinate with the catalytic center at high oxidation states, leading to the blockage of active sites. Therefore, the Ru-tda and Ru-t5a catalysts require an activation process to generate catalytically active Ru-aqua species under high potential.13,14 Catalyst Ru-tpa bearing a dangling phosphate with a lower pKa value (Fig. S2d, ESI†) can increase the TOFmax value to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 under neutral condition.15,16 However, the stronger coordinating ability of phosphonate (Fig. S2c, ESI†) necessitates a more complicated activation process and structural evolution for the catalyst to generate catalytically active species Ru-tpaO (tpaO = 3-(hydroxo-[2,2′:6′,2′′-terpyridine]-6,6′′-diyl)bis(phosphonate)). To circumvent this activation process, we previously developed Ru-tds catalysts using sulfonate ligands with the lowest pKa and weaker coordinating ability, effectively addressing the issue of incomplete utilization of catalytic sites.17 Although the catalysts above demonstrate excellent performance regarding TOF or TOFmax, they fall short in achieving a low onset potential compared to Ru-bda of 1.14 V vs. NHE under neutral conditions.18–20
000 s−1 under neutral condition.15,16 However, the stronger coordinating ability of phosphonate (Fig. S2c, ESI†) necessitates a more complicated activation process and structural evolution for the catalyst to generate catalytically active species Ru-tpaO (tpaO = 3-(hydroxo-[2,2′:6′,2′′-terpyridine]-6,6′′-diyl)bis(phosphonate)). To circumvent this activation process, we previously developed Ru-tds catalysts using sulfonate ligands with the lowest pKa and weaker coordinating ability, effectively addressing the issue of incomplete utilization of catalytic sites.17 Although the catalysts above demonstrate excellent performance regarding TOF or TOFmax, they fall short in achieving a low onset potential compared to Ru-bda of 1.14 V vs. NHE under neutral conditions.18–20
Designing a catalyst that can simultaneously achieve both low onset potential and high TOF still poses a timely challenge. The hybrid catalyst Ru-bpc,21 illustrated in Fig. S2b (ESI†), combines the favorable characteristics of Ru-bda18,19,22–24 (low onset potential) and Ru-bpa25,26 (fast proton transfer kinetics), showcasing a low barrier of merely 9.1 kcal mol−1 for O–O formation. Computational results suggest that the phosphonate remains coordinated with ruthenium during water nucleophilic attack, thereby leaving only one proton-accepting site with pKa around 7.1 (Fig. S2d, –PO3H−, ESI†) available to expedite proton transfer. We envisage that through the hybridization of Ru-bda with Ru-bds27 and capitalizing on the adaptive coordination feature of sulfonate,17 a dangling proton-accepting site with a lower pKa value of around 0.7 can be maintained during the catalytic process to enhance the rate of water oxidation.
In this work, an adaptive water oxidation catalyst Ru-bcs (Fig. 1) with a carboxylate-sulfonate hybrid ligand is tailor-designed. The catalyst displays both low onset potential of 1.21 V vs. NHE and high TOF over 1000 s−1 at pH 7. These values represent the lowest onset potential and among the highest rates observed for molecular catalysts with water nucleophilic attack (WNA) mechanism operating under neutral pH conditions (Fig. S3, ESI†). The adaptive chemistry uncovered here provides new perspectives for developing molecular catalysts with high efficiency under low driving forces.
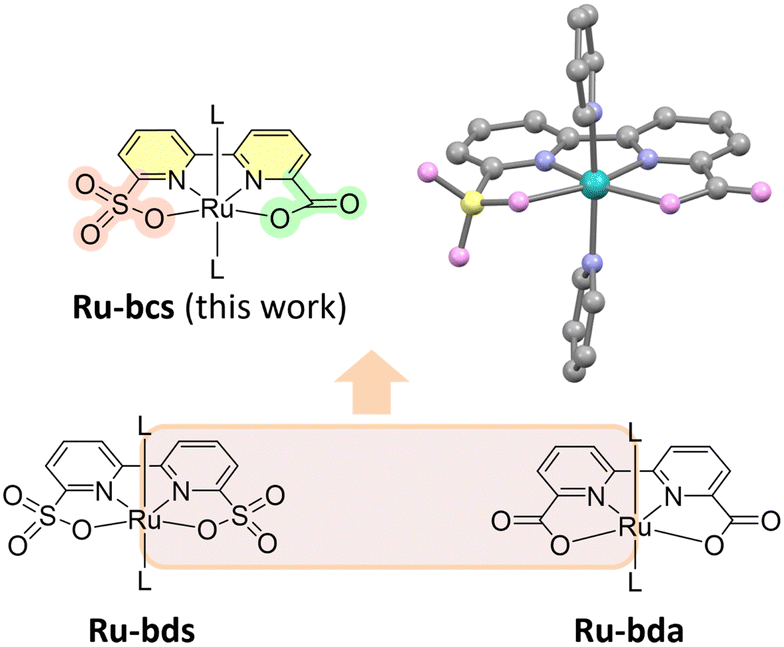 | ||
| Fig. 1 Single crystal structure of Ru-bcs with thermal ellipsoids at 50% probability (hydrogen atoms and solvent molecules are omitted for clarity) and structures of water oxidation catalysts discussed in the paper; L is pyridine-based ligand. bcs = 2,2′-bipyridine-6′-carboxylic-6-sulfonate, bds = 2,2′-bipyridine-6,6′-disulfonate, bda = 2,2′-bipyridine-6,6′-dicarboxylate. | ||
The X-ray crystal structure of Ru-bcs is illustrated in Fig. 1 and Fig. S1 (ESI†), with detailed information provided in the ESI.† We initially accessed the catalytic water oxidation activity using Ce(IV) as a chemical oxidant. Kinetic measurements revealed that oxygen evolution is a first-order reaction with respect to catalyst concentrations. The catalytic performance of Ru-bcs under Ce(IV)-driven water oxidation conditions is less effective (Fig. S4 and S5, TOF = 0.68 s−1, TON = 1013, ESI†) compared to Ru-bda. Under similar catalyst concentrations, Ru-bda with picoline as the axial ligand displays TOFs ranging from 14.7 to 32.8 s−1.19 The low activity may be attributed to the relatively small driving force of Ce(IV) (E0 = 1.6 V vs. NHE)28 compared to the catalytic onset potential of Ru-bcs (around 1.6 V at pH 1). Under acidic conditions, a reversible peak appeared around 0.9 V (Fig. S6, grey, ESI†), corresponding to the single electron process of Ru(II) → Ru(III), which occurs approximately 300 mV lower in potential compared to that of Ru-bds. This redox signal became irreversible once the potential exceeded 1.5 V (blue and red solid lines in Fig. S6, ESI†), suggesting structural evolution of the catalysts at high potential.17 A new reduction signal around 1.1 V emerged in the reverse CV scan, which can be assigned to the reduction of the newly generated Ru-aqua species. Similar phenomenon is also observed under neutral conditions (Fig. 2) and will be discussed in detail later. An increase in current in the range of 1.6–1.8 V provides evidence of catalytic water oxidation.
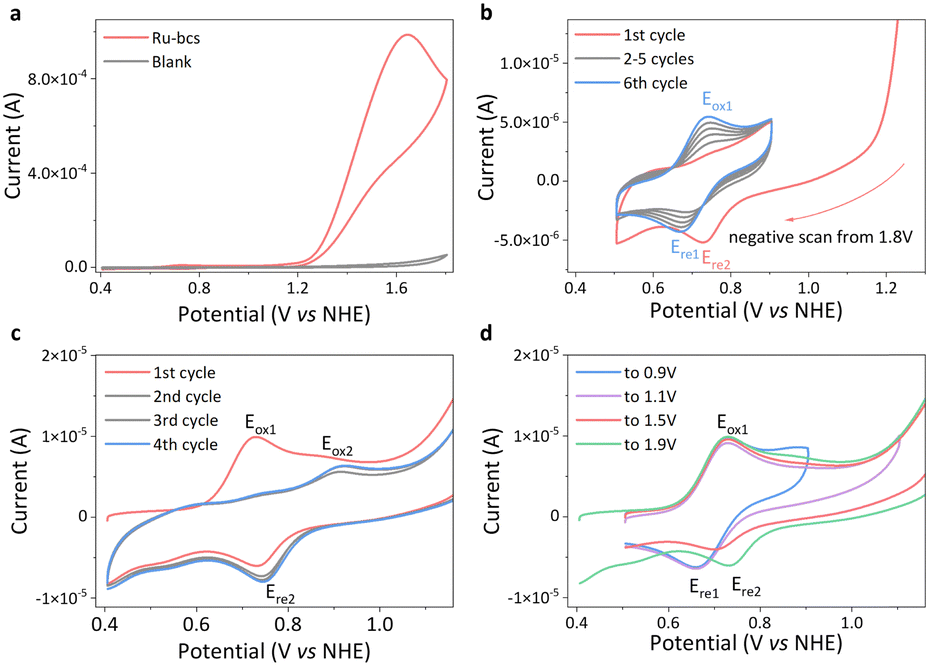 | ||
| Fig. 2 (a) CV of 1 mM Ru-bcs in pH 6.95 phosphate buffer (0.2 M) containing 10% CF3CH2OH at a scanning rate of 100 mV s−1, working electrode: GC. (b) Negative-scan CV from 1.8 V, working electrode: GC. (c) 1st to 4th CV scans of Ru-bcs, and (d) CVs of Ru-bcs at different potential windows, working electrode: GC. | ||
The cyclic voltammetry (CV) curve of Ru-bcs (Fig. 2a) reveals a significant enhancement in catalytic current at pH 7, with an onset potential of 1.21 V vs. NHE, the lowest reported so far for molecular water oxidation catalysts with WNA mechanism under neutral conditions. Specifically, (1) for catalysts that do not require an activation process, Ru-bcs exhibits onset potentials 400 mV and 140 mV lower compared to those of Grotjahn-Cat12 and Ru-tds,17 respectively, albeit with relatively lower TOFs (1000 vs. 2000 s−1). (2) Thanks to its tetra-coordinated equatorial ligand and coordination-adaptive sulfonate group, Ru-bcs displays an onset potential approximately 140 mV lower than that of Ru-tda,13 Ru-tpa,16 and Ru-t5a,14 which typically requires an activation process before application in water oxidation. However, since the above three analogous catalysts utilized the foot-of-the-wave analysis (FOWA) method to determine TOFmax, we are unable to directly compare their catalytic performance. (3) We also conducted a preliminary comparison with the catalysts that require dual-site participation for O–O bond formation. The onset potential of Ru-bcs is similar to that of Ru-bds (1.24 V),27 slightly higher than that of Ru-bda (1.14 V).19 Its advantage lies in the WNA mechanism, which will be discussed later. Catalysts with the WNA mechanism are more likely to be applicable in practical scenarios such as surface immobilization on electrodes.
The generation of oxygen was confirmed by controlled potential electrolysis (CPE) experiment with the faradaic efficiency for water oxidation exceeding 94% (Fig. S7, ESI†). The molecular nature of the catalyst after CPE is also confirmed by CV tests (Fig. S8 and S9, ESI†). At 1.6 V applied potential, the catalytic current density of Ru-bcs reaches a value over 14 mA cm−2 (Fig. 2a), which is comparable to the-state-of-the-art Ru-bds catalyst under identical conditions.27 Consequently, we focused on studying the electrochemical water oxidation by Ru-bcs under neutral conditions in the rest of this investigation. CV and differential pulse voltammetry (DPV) were employed to electrochemically characterize Ru-bcs. In the first CV scan, one oxidation signal is clearly observed at 0.73 V (Eox1, Fig. 2c), associated with the RuIII/II redox couple. The peak current exhibits a linear relationship with the square root of the scan rate (Fig. S10 and S11, ESI†), indicating a diffusion-controlled electrochemical process. Utilizing the Randles–Sevcik equation, we calculated the diffusion coefficient to be 2.50 × 10−6 cm2 s−1. The catalytic currents, which are independent of scan rate between 600 to 1000 mV s−1 (Fig. S12–S14, ESI†), were used to estimate the catalytic TOF using eqn (S1) in the ESI† (Note S1), providing a reliable method to compare the TOF of Ru-bcs with the majority of reported catalysts. The obtained TOF value of 1135 ± 152 s−1 (Ecat = 1.8 V) is comparable to the rate of natural photosynthesis and represents one of the highest activities reported thus far for molecular catalysts with WNA mechanism operating under neutral conditions. The RuIV/III redox couple is too kinetically unfavorable to be distinguished at pH 7. The large irreversible anodic current appearing at the onset potential around 1.21 V indicates electrocatalytic oxygen evolution from water, where RuIV might trigger oxygen formation.
In the 2nd to 4th cycles of CV scan (Fig. 2c), the intensity of the Eox1 peak decreases, while a new oxidation peak at 0.92 V (Eox2) and a reduction peak (Ere2) at 0.75 V emerge. These peaks likely correspond to the redox behavior of different forms of RuII-aqua species associated with the dynamic behavior of RuII-bcs in the presence of water molecules (Scheme S5, ESI†). We tentatively assign Eox1 to the one-electron oxidation process of species 1 and 2, which contain coordinated negatively charged ligands, due to its relatively low potential compared with Eox2. During O–O bond formation, the sulfonate group may dissociate to accelerate the proton transfer rate, leading to the predominance of species 3 and 4 after turnover, which contain uncoordinated negatively charged ligands, in the subsequent CV cycles. This hypothesis also finds support in subsequent evidence. The Ere1 is the predominant reduction signal under low applied potentials (Fig. 2d, blue and purple). As the applied potential increases, the equilibrium shifts towards species 3 and 4, with Ere2 becoming the primary reduction signal (Fig. 2d, red and green). To rule out the possibility that the Ere2 signal originates from the heterogeneous active species generated by catalyst degradation at high potentials, we conducted negative scan CV tests, as shown in Fig. 2b. The species 3 and 4 generated during the negative sweep CV beginning at 1.8 V, but subsequently disappeared in the following narrow-range repeat CVs. This indicates that Ere2 might arise from diffusible active species rather than degradation species adsorbed on the electrode, such as ruthenium oxide. Meanwhile, the Eox1 signal gradually strengthens, further supporting the increased concentration of species 1 and 2 at the electrical double layer. However, we lack direct evidence to exclude the possibility of diffusible degradation product formation. Density functional theory calculations reveal that the RuIII/II redox couple is at 0.70 V. With the RuII-aqua and RuIII-aqua species (Fig. S15, ESI†), six-coordinated structures are slightly more stable with 0.94 kcal mol−1 and 0.1 kcal mol−1 lower in free energy, respectively, leading to a redox potential of 0.73 V. The oxidation of six-coordinated RuII-aqua to six-coordinated RuIII–OH (6.2 kcal mol−1 lower than the seven-coordinated structure) occurs at 0.81 V at pH 7. Further oxidation yields the seven-coordinated RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, as the calculated pKa of RuIV–OH species is −1.14, with a redox potential of 0.90 V for RuIII–OH/RuIVO. This seven-coordinate RuIVO species is 0.72 kcal mol−1 lower in free energy than its six-coordinated counterpart, indicating the feasibility of sulfonate coordination. Subsequent oxidation of seven-coordinated RuIVO species to RuVO species has a high oxidation potential of 1.53 V, much larger than the experimental onset potential 1.21 V. Therefore, the calculations support the catalytic O–O bond formation initiates at seven-coordinated RuIVO species. The sulfonate coordination/decoordination in the RuIVO may aid the proton transfer during the O–O bond formation step.
O species, as the calculated pKa of RuIV–OH species is −1.14, with a redox potential of 0.90 V for RuIII–OH/RuIVO. This seven-coordinate RuIVO species is 0.72 kcal mol−1 lower in free energy than its six-coordinated counterpart, indicating the feasibility of sulfonate coordination. Subsequent oxidation of seven-coordinated RuIVO species to RuVO species has a high oxidation potential of 1.53 V, much larger than the experimental onset potential 1.21 V. Therefore, the calculations support the catalytic O–O bond formation initiates at seven-coordinated RuIVO species. The sulfonate coordination/decoordination in the RuIVO may aid the proton transfer during the O–O bond formation step.
The DPVs of Ru-bcs at various pH values were measured to study the proton and electron transfer processes during water activation and O–O bond formation (Fig. S16 and S17, ESI†). A pH-independent oxidation process around 0.86 V appears in the pH range of 1–4, suggesting a pure electron transfer process under acidic conditions. The peak-to-peak separation (ΔEp) at pH 1 of the signal is 56 mV, indicating a 1-electron transfer process of RuIII/II (the solid grey line in Fig. S6, ESI†).29 Above pH 4, the first redox couple becomes pH dependent with a slope of −59 mV per pH, indicating the removal of a proton-coupled with the removal of an electron. For RuIV/III, a one-electron-one-proton PCET process is observed over the whole pH range from 1 to 12 with a slope of −58 mV per pH, relative to the generation of RuIV–OH or RuIV–O. Further oxidation of RuIV is not observed via DPVs. The catalytic mechanism by Ru-bcs is summarized in Scheme 1 based on the electrochemical data mentioned above. It should be noted that the proposed cycle is only applicable under neutral to alkaline conditions. A RuIV–OH with either 6- or 7-coordination configuration could be generated under acidic media (Scheme S4, ESI†). Ce(IV)-driven water oxidation activity is linearly dependent on the catalyst concentrations (Fig. S5, ESI†), suggesting that the RDS should take place on a single-site in accordance with a WNA pathway. To further prove the WNA mechanism, H/D kinetic isotope effects (KIE) on electrochemical water oxidation were investigated by measuring CV curves of Ru-bcs in both H2O and D2O (Fig. S18, ESI†). The method to correct the overpotential in H2O and D2O was described in Note S2 (ESI†). A large difference between these two catalytic curves was observed, and the calculated KIE value is 2.06. The primary KIE is in line with direct O–H bond breaking in the rate-determining step (rds) of the electrochemical water oxidation.
 | ||
| Scheme 1 Proposed catalytic cycle of Ru-bcs at neutral condition. Axial ligands are omitted for clarity. | ||
In conclusion, the incorporation of a hybrid carboxylate-sulfonate ligand into the ruthenium complex has led to the creation of coordination-adaptive spheres: (1) water can coordinate with Ru through ligand exchange at the initial state, enabling Ru-bcs to generate high-valent active species via PCET under neutral conditions. This process is supported by Pourbaix diagram and calculations. (2) The distorted coordination geometry allows sulfonate and aqua ligands to coordinate with Ru simultaneously in the high-valent state (7-coordination). These electron-rich ligands ensure the achievement of a low onset potential. Additionally, compared to dicarboxylate ligands, the less electron-donating sulfonate ligands may destabilize the high-valent Ru–oxo species to enhance its reactivity, enabling it to catalyze O–O bond formation at the Ru(IV) state. (3) During O–O bond formation, the sulfonate may also decoordinate to accelerate proton transfer kinetics. Consequently, both low onset potential (1.21 V vs. NHE) and high TOF (over 1000 s−1) were obtained under neutral conditions. Findings of the work suggest that the introduction of coordination-adaptive ligands to molecular catalysts could be a generic strategy to promote electrocatalytic oxidation reactions, such as water and ammonia oxidation.
JY acknowledges the financial support from the National Natural Science Foundation of China (No. 22101186) and Natural Science Foundation of Top Talent of SZTU (No. GDRC202114). LS acknowledges the financial support from the National Key R&D Program of China (2022YFA0911900), the National Natural Science Foundation of China (22088102), and the Starting-up Package of Westlake University. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) (allocations NAISS 2024/5-22 and NAISS 2024/6-26), partially funded by the Swedish Research Council through grant agreement no. 2022-06725.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Liu and L. Sun, Sci. Bull., 2023, 68, 854–856 CrossRef CAS PubMed.
- T. Liu and L. Sun, J. Energy Chem., 2022, 73, 643–645 CrossRef.
- D. W. Shaffer, Y. Xie and J. J. Concepcion, Chem. Soc. Rev., 2017, 46, 6170–6193 RSC.
- Q.-F. Chen, Z.-Y. Cheng, R.-Z. Liao and M.-T. Zhang, J. Am. Chem. Soc., 2021, 143, 19761–19768 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed.
- B. Das, A. Rahaman, A. Shatskiy, O. Verho, M. D. Kärkäs and B. Åkermark, Acc. Chem. Res., 2021, 54, 3326–3337 CrossRef CAS PubMed.
- A. G. Nash, C. J. Breyer, B. D. Vincenzini, G. I. Elliott, J. Niklas, O. G. Poluektov, A. L. Rheingold, D. K. Smith, D. G. Musaev and D. B. Grotjahn, Angew. Chem., Int. Ed., 2021, 60, 1540–1545 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, M. Pipelier, J. Lebreton, D. Dubreuil, J. Benet-Buchholz, X. Sala, A. Tessier and A. Llobet, ACS Catal., 2018, 8, 2039–2048 CrossRef CAS.
- N. Vereshchuk, J. Holub, M. Gil-Sepulcre, J. Benet-Buchholz and A. Llobet, ACS Catal., 2021, 11, 5240–5247 CrossRef CAS.
- N. Vereshchuk, R. Matheu, J. Benet-Buchholz, M. Pipelier, J. Lebreton, D. Dubreuil, A. Tessier, C. Gimbert-Suriñach, M. Z. Ertem and A. Llobet, J. Am. Chem. Soc., 2020, 142, 5068–5077 CrossRef CAS PubMed.
- T. Liu, S. Zhan, N. Shen, L. Wang, Z. Szabó, H. Yang, M. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2023, 145, 11818–11828 CrossRef CAS PubMed.
- L. Duan, A. Fischer, Y. Xu and L. Sun, J. Am. Chem. Soc., 2009, 131, 10397–10399 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, J. Am. Chem. Soc., 2017, 139, 15347–15355 CrossRef CAS PubMed.
- L. Wang, L. Duan, Y. Wang, M. S. Ahlquist and L. Sun, Chem. Commun., 2014, 50, 12947–12950 RSC.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, Inorg. Chem., 2016, 55, 12024–12035 CrossRef CAS PubMed.
- N. Song, J. J. Concepcion, R. A. Binstead, J. A. Rudd, A. K. Vannucci, C. J. Dares, M. K. Coggins and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4935–4940 CrossRef CAS PubMed.
- Y. Xie, D. W. Shaffer, A. Lewandowska-Andralojc, D. J. Szalda and J. J. Concepcion, Angew. Chem., Int. Ed., 2016, 55, 8067–8071 CrossRef CAS PubMed.
- J. M. Kamdar, D. C. Marelius, C. E. Moore, A. L. Rheingold, D. K. Smith and D. B. Grotjahn, ChemCatChem, 2016, 8, 3045–3049 CrossRef CAS.
- J. Yang, L. Wang, S. Zhan, H. Zou, H. Chen, M. S. G. Ahlquist, L. Duan and L. Sun, Nat. Commun., 2021, 12, 373 CrossRef CAS PubMed.
- V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862–1891 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2347388. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02303j |
| ‡ These authors contributed equally: JY and SZ. |
| This journal is © The Royal Society of Chemistry 2024 |