
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplete amide cis–trans switching synchronized with disulfide bond formation and cleavage in a proline-mimicking system†
Yuhe
Cheng
a,
Tadashi
Hyodo
 b,
Kentaro
Yamaguchi
b,
Tomohiko
Ohwada
*a and
Yuko
Otani
*a
b,
Kentaro
Yamaguchi
b,
Tomohiko
Ohwada
*a and
Yuko
Otani
*a
aGraduate School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-0033, Japan. E-mail: otani@mol.f.u-tokyo.ac.jp; ohwada@mol.f.u-tokyo.ac.jp
bSchool of Pharmaceutical Sciences at Kagawa Campus, Tokushima Bunri University, 1314-1 Shido, Sanuki, Kagawa 769-2193, Japan
First published on 20th May 2024
Abstract
A typical naturally occurring disulfide structure in proteins is an 8-membered disulfide ring formed between two adjacent cysteine (Cys–Cys) residues. Based on this structure, we designed 7- to 9-membered disulfide ring molecules, embedded in the 7-azabicyclo[2.2.1]heptane skeleton, that switch their conformation from exclusively trans-amide to exclusively cis-amide upon redox transformation from dithiol to disulfide, and vice versa. Constrained shape of disulfide rings is rare in nature, and the present molecular structure is expected to be a useful fundamental component for the construction of new conformation-switching systems.
Molecules that undergo a dramatic conformational change in a stimulus-dependent manner, that is molecular switches, have potential applications as conformational switches for peptides/proteins and sensing materials.1–3 Among the potential switching mechanisms, amide trans–cis isomerization is one of the most general structure changes in a single molecule, as exemplified in nature by prolyl amide isomerization in ion channels. Prolyl amide trans–cis isomerization causes dramatic conformational changes linked to the functions, i.e., opening and closing, of ion channels.4,5 On the other hand, redox reactions are ubiquitous in cellular processes. In particular, the formation and cleavage of a disulfide bond between the side-chains of two cysteine residues in a peptide or protein are considered to synchronize with the trans–cis conversion of amides in the main-chain, which would cause conformational switching of the whole molecule.6–10 However, this is not the case (see below).
Naturally occurring disulfide rings in proteins are formed between two thiol groups of two adjacent cysteine (Cys–Cys) residues (1), generating an 8-membered lactam ring (2) called a vicinal disulfide ring (VDR), upon oxidative disulfide bond formation (Fig. 1(a)).6,7,9,11,12 While 1 has a trans-amide structure, the Cys–Cys disulfide (2) has various conformers with respect to the amide trans–cis isomerism as well as disulfide ring conformations, due to ring strains arising from the long C–S (1.8 Å) and S–S (2.0 Å) bonds and acute dihedral angle (∠CSSC: 85°).13,14 Indeed, the Cys–Cys disulfide (2) is known to change its conformation depending on the protein environment. Conformations of vicinal cysteine disulfides can be classified into 20 possible structures based on the combination of plus and minus values of dihedral angles, χ1, χ2, χ3, χ2′, and χ1′ along the successive side-chains (Fig. 1(b)).15 In the cases of model tertiary amides (Fig. 1(c)), the reduced open-dithiol form 3 consists of a mixture of cis and trans amides, whereas the cyclic disulfide 4 formed upon oxidation contains only a cis-amide structure.16
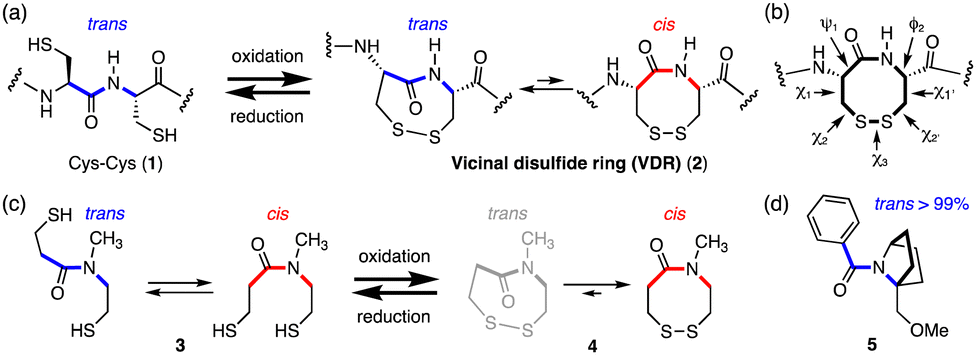 | ||
| Fig. 1 (a) Reversible formation and cleavage of a disulfide bond (2) in neighboring cysteine residues (1). (b) Dihedral angles of the vicinal disulfide ring. (c) The amide trans–cis equilibrium of a model tertiary amide in reduced form (thiol 3) and oxidized form (disulfide 4). (d) Bicyclic amide compound 5. | ||
The most abundant and least strained structure of the vicinal cysteine disulfide ring, observed in the crystal structures of proteins, is the trans-(plus(χ1), minus(χ1′)) left-handed staple structure, that is, trans-(+,−)LHstaple, which has a trans-amide (see Fig. 3(c) box). However, the vicinal disulfide ring conformations in proteins often undergo conformational change in response to nearby ligand binding, and this can generate rare and highly strained conformations such as trans-(+, −)AntiRHHook, which has (χ1, χ2, χ3, χ2′, χ1′) = (+, −, +, +, −) (see Fig. 3(c) box).6,17–19
We have found that the amide in the fused bicyclic pyrrolidine ring (5), that is, a bicyclic proline skeleton bearing a bridgehead substituent, forms exclusively a trans-amide structure, despite being a tertiary amide (Fig. 1(d)).20,21 These results inspired us to consider molecules with a similar bicyclic skeleton, in which the amide isomerism might be switched from exclusively trans to exclusively cis and vice versa, concomitantly with disulfide ring formation and cleavage, respectively.
In this study, we combined a monocyclic proline-mimicking ring system (A series) and a bicyclic proline-mimicking ring system (B series) with a dithiol-disulfide moiety (Fig. 2). The bicyclic ring-fused molecules (B) underwent complete amide cis–trans conversion upon intramolecular disulfide bond formation from the smallest 7-membered to the 9-membered disulfide ring, while the monocyclic ring system (A) gave a trans/cis amide mixture in both the dithiol and disulfide states. We next thoroughly analyzed the conformation and the dynamic isomerization process of the disulfide ring of the B system. We found that the ring takes practically a single conformation strained in shape, and the ring inversion is slow enough to permit detection of the enantiomers with respect to ring structure even at 0 °C, which is unusual in disulfide ring systems. These results suggested that the present bicyclic-ring-fused disulfide ring system might work as a redox-dependent switch.
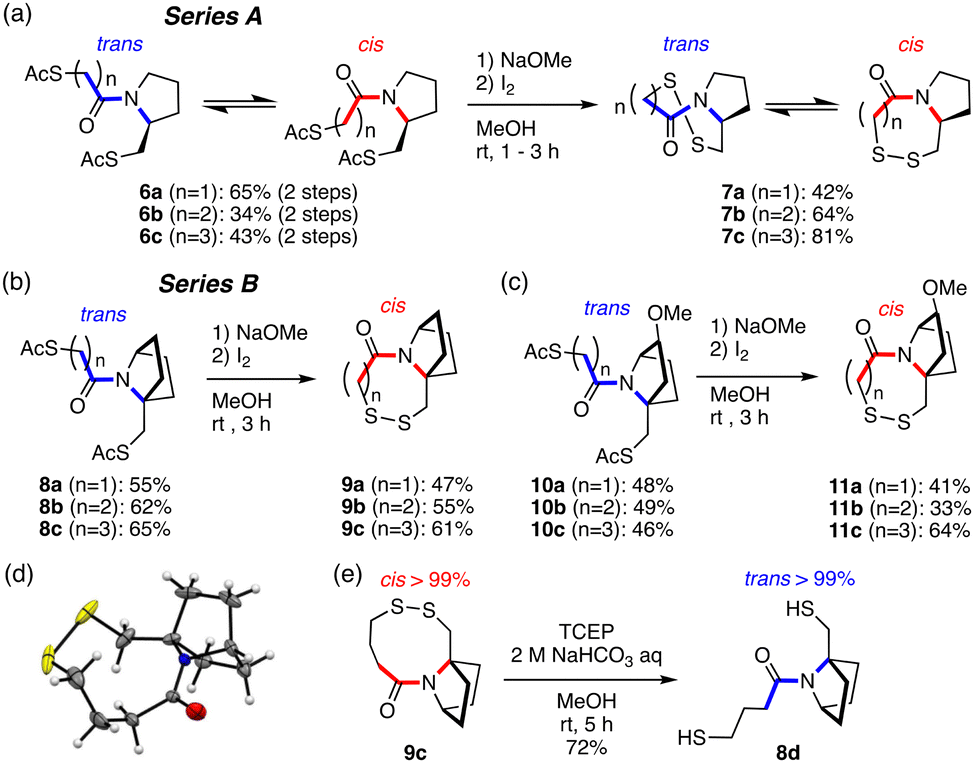 | ||
| Fig. 2 Disulfide ring formation. (a) Series A: monocyclic ring-fused compounds. (b) and (c) Series B: bicyclic ring-fused compounds. (d) Crystal structure of 9c (CCDC no. 2332996).† (e) Reductive cleavage of the disulfide ring of 9c. | ||
To study the redox-induced change of amide cis–trans isomerization, acetyl-protected thiol compounds and disulfide compounds fused with a monocyclic pyrrolidine ring (A: 6a–6c and 7a–7c) or a bicyclic ring (B: 8a–8c, 10a–10c and 9a–9c, 11a–11c) with seven-, eight- and nine-membered rings were synthesized (Fig. 2(a)–(c) and Fig. S1, S2, ESI†). Acetyl groups were deprotected in basic conditions to afford thiol in situ, which was transformed to the disulfide by oxidation with I2. The crystal structure of the 9-membered ring disulfide fused with a bicyclic ring (9c) shows a cis-amide structure (Fig. 2(d)), which is consistent with the solution conformation (see below). The amide structure was analyzed by means of 1H-NMR and 2D-NMR (COSY, TOCSY, NOESY, and HSQC) techniques. For analysis of thiol compounds, acetyl-protected thiol compounds were utilized as equivalents to thiol compounds as they are more stable under air conditions. It was found that pyrrolidine-fused compounds 6a–6c (A series) form a mixture of trans and cis amide structures (the average trans–cis ratio is around 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :34), with trans-amide being the major conformer (Table 1 and Fig. S3, ESI†). After oxidation, the cis-amide conformation is preferred in disulfides 7a–7c. While the 7-membered ring disulfide (7a) forms only the cis-amide conformation, the ratio of the cis isomer decreases (7b: cis:trans = 90:10; 7c: cis:trans = 85:15) as the ring size becomes larger (7 → 8 → 9-membered) (Table 1 and Fig. S4, ESI†). This is probably because the pyrrolidine ring strain is released in the 9-membered disulfide ring, allowing the amide bond to rotate to give trans-amide, which would be equivalent to the trans and cis amide mixture of the natural 8-membered vicinal disulfide ring system (VDR) 2 (Fig. 1(a)).
:34), with trans-amide being the major conformer (Table 1 and Fig. S3, ESI†). After oxidation, the cis-amide conformation is preferred in disulfides 7a–7c. While the 7-membered ring disulfide (7a) forms only the cis-amide conformation, the ratio of the cis isomer decreases (7b: cis:trans = 90:10; 7c: cis:trans = 85:15) as the ring size becomes larger (7 → 8 → 9-membered) (Table 1 and Fig. S4, ESI†). This is probably because the pyrrolidine ring strain is released in the 9-membered disulfide ring, allowing the amide bond to rotate to give trans-amide, which would be equivalent to the trans and cis amide mixture of the natural 8-membered vicinal disulfide ring system (VDR) 2 (Fig. 1(a)).
| Solvent |
cis:transa (%) |
Disulfide ring size | Solvent |
cis:trans (%) |
||
|---|---|---|---|---|---|---|
| a Measured at 297.3 K. b Measured at 297.3 K and 273.2 K. | ||||||
| 6a | CD3OD | 34:66 |
7a | 7 | CD3OD, CDCl3 | >99:<1a |
| 6b | CD3OD | 35:65 |
7b | 8 | CD3OD | 90:10b |
| 6c | CD3OD | 34:66 |
7c | 9 | CD3OD | 85:15b |
| 8a | CD3OD, CDCl3 | <1:>99 |
9a | 7 | CDCl3 | >99:<1a |
| 8b | CD3OD, CDCl3 | <1:>99 |
9b | 8 | CDCl3 | >99:<1b |
| 8c | CD3OD, CDCl3 | <1:>99 |
9c | 9 | CDCl3 | >99:<1b |
| 10a | CDCl3 | <1:>99 |
11a | 7 | CD3OD, CDCl3 | >99:<1a |
| 10b | CDCl3 | <1:>99 |
11b | 8 | CD3OD, CDCl3 | >99:<1b |
| 10c | CDCl3 | <1:>99 |
11c | 9 | CDCl3 | >99:<1b |
In contrast, the bicyclic ring-fused compounds (B series) showed complete selectivity. The reduced forms, that is acetyl-protected dithiol compounds (8a–8c and 10a–10c), form only the trans-amide structure in CD3OD and in CDCl3 (Fig. S5, ESI†). Upon disulfide formation, the corresponding disulfides (9a–9c and 11a–11c) form only the cis-amide structure in CD3OD and in CDCl3 (Table 1 and Fig. S6, ESI†), irrespective of the ring size of the disulfide ring and the substituent on the bicyclic ring (11a–11c). The complete trans/cis selectivity of the amide is due at least in part to the fact that the disulfide ring and the azabicyclic ring share a bond (N1–C8), constraining the dihedral angle (C2–N1–C8–C7) (Fig. 3(a)).
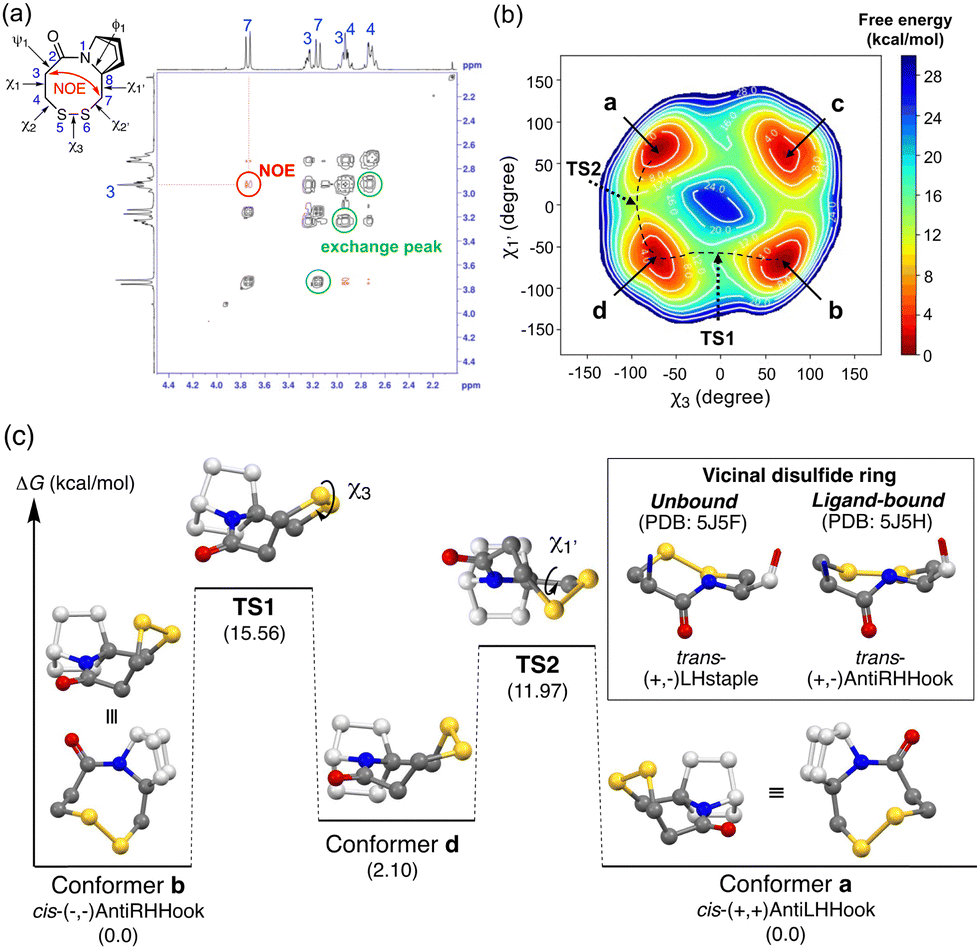 | ||
| Fig. 3 (a) The NOESY spectrum of 9b in CDCl3 at 273.2 K. (b) Free energy landscape obtained by metadynamics simulation of 9b for 20 ns with the OPLS3e force field in CHCl3. (c) Free energy diagram of the disulfide ring isomerization process of 9b calculated at the APFD/6-311+G(2d,p) level (SCRF = IEFPCM, solvent = CHCl3). Hydrogens are omitted for clarity. The carbons of the disulfide ring are shown in gray and other carbons are shown in light gray. Relative energies are shown in parentheses. In a box, side views of the two crystal structures of vicinal disulfide in acetylcholine binding protein in a ligand-unbound state (left) and in the ligand-bound state (right) are also shown. | ||
Furthermore, the disulfide compound 9c was reduced with tris(2-carboxyethyl)phosphine (TCEP) to afford dithiol 8d (Fig. 2(e)). 8d forms exclusively the trans-amide structure, as in 8c (Fig. S7, ESI†). 8d could be readily acetylated to afford 8c (see ESI†). These results strongly suggest that the amide cis–trans isomerism can be controlled reversibly by redox reaction.
We next investigated the conformation of the disulfide ring of 9b bearing an 8-membered ring, in which the amide conformation is exclusively cis. 9b shows a single set of 1H-NMR signals in CDCl3 and CD3OD. The NOESY spectrum showed an NOE signal between H(3) and H(7), which is consistent with the cis-amide structure (Fig. 3(a), red cross-peaks). Furthermore, this NOESY spectrum also showed exchange peaks between two geminal hydrogen atoms on the disulfide ring carbons (C3, C4, and C7) (Fig. 3(a), black cross-peaks). These results strongly indicate that two equilibrium conformers of the disulfide ring structure are enantiomers with respect to the amide plane. Metadynamics simulations along the collective coordinate of the χ3 and χ1′ torsion angles suggested four low-energy conformers a–d (Fig. 3(b)). Subsequent DFT-optimization identified a pair of enantiomers with respect to ring inversion, conformer (enantiomer) a and conformer (enantiomer) b, as energy-minimum structures having the same energy (Fig. 3(b) and (c)). The experimental homonuclear 1H–1H vicinal (3J) coupling constants between hydrogen atoms on C3 and C4 matched well with those calculated using the Karplus equation based on the DFT-optimized structures of conformers a/b (experimental: 13.2 Hz and 5.2 Hz in CD3OD, calculated: 12.4, 3.7 Hz, respectively) (Table S1, ESI†). These results are consistent with the view that conformers a and b are mutually exchanging slowly on the NMR time scale. Similarly, two energy minimum structures of MeO-substituted 11b were obtained. Due to the presence of another stereogenic center on the bicyclic structure (C3′ with a methoxy substituent), these two structures are diastereomers, that is, conformer b′, the lowest-energy structure, is similar to conformer b of 9b and conformer a′, the second-lowest energy structure, is similar to conformer a of 9b (Fig. S8, ESI†). Conformer b′ has the disulfide sulfur atoms anti to the methoxy substituent with respect to the amide plane, and conformer a′ has the disulfide sulfur atoms syn to the methoxy substituent. NMR chemical shifts of conformer b′ and conformer a′ were calculated with the gauge-independent atomic orbital (GIAO) method based on the DFT-optimized structures. The calculated chemical shifts of conformer b′ and conformer a′ are in good agreement with the chemical shifts of the major and minor conformers in 1H-NMR, respectively. The calculated ratio of major b′ and minor a′ conformers based on the DFT energy difference was 60:40, which matches well with the experimental ratio of major and minor conformers (74:26) (Fig. S9, ESI†).
The dihedral angles are (χ1, χ2, χ3, χ2′, χ1′) = (+59°, +57°, −73°, −28°, +72°) for conformer a and (−59°, −57°, +73°, +28°, −72°) for conformer b (Table S2, ESI†). Judging from the sign of the dihedral angle of the disulfide ring, conformer a (a′) forms the cis-(+, +)AntiLHHook conformation and conformer b (b′) forms the cis-(−, −)AntiRHHook conformation, which are rare in terms of natural monocyclic 8-membered disulfide ring structures (Fig. 3(c) box). Although the shape of this disulfide ring can be classified as highly strained, the unique bicyclic skeleton stabilizes the putative highly strained disulfide ring structure, and ring inversion is slow enough to permit discrimination of the enantiomers arising from the ring conformation. The values of the SS dihedral angle (Table S3, ESI†) and redox potentials of the disulfide compounds (Fig. S16, ESI†) are also supported for strain-free conformations. Different from previously reported derivatives of vicinal disulfide, the present bicyclic ring-fused disulfide compounds form only the cis-amide conformation, and therefore we can elucidate the rate-determining interconversion barriers between the two ring conformations. The rate-determining isomerization barriers (ΔG‡) between conformer a and b in 9b and 9c and between conformer a′ and b′ in 11b and 11c were estimated by exchange spectroscopy (EXSY) and line-shape analysis (Table 2 and Fig. S10–S15, ESI†).
The values obtained by the two methods were in good agreement. The isomerization barriers are 15–16 kcal mol−1, and are similar in magnitude for all compounds, independent of the disulfide ring size.
The magnitude of the isomerization barrier is consistent with the reported value for isomerization between two major conformers of vicinal disulfide.13 These values are higher than the reported values for acyclic benzyl phenyl disulfide 12 (7.7 kcal mol−1).22
Both metadynamics simulations and DFT calculations suggested that ring inversion between conformer a and conformer b proceeds in multiple steps, containing two major transition states; one for disulfide bond rotation, represented by χ3 (from b to d, TS1) and the other for disulfide ring inversion, represented by χ1′ (from d to a, TS2) (Fig. 3(b) and (c)). DFT calculation suggested that the disulfide bond rotation (χ3 rotation: TS1) is the rate-determining step rather than the disulfide ring conformational change (TS2) (Fig. 3(c)). The calculated rotation barrier of the disulfide bond (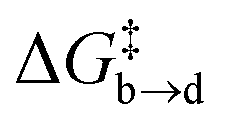 ) in 9b is 15.56 kcal mol−1, which is consistent with the experimentally observed barrier (15.9 kcal mol−1). On the other hand, disulfide ring isomerization proceeds in multiple steps, with multiple intermediates between d and a, including rotation of the bridgehead substituent (χ1′ rotation) and amide nitrogen inversion (change of ϕ1). The highest barrier to ring isomerization is 11.97 kcal mol−1 (a → TS2). These results strongly indicate that disulfide bond rotation is the rate-determining step.
) in 9b is 15.56 kcal mol−1, which is consistent with the experimentally observed barrier (15.9 kcal mol−1). On the other hand, disulfide ring isomerization proceeds in multiple steps, with multiple intermediates between d and a, including rotation of the bridgehead substituent (χ1′ rotation) and amide nitrogen inversion (change of ϕ1). The highest barrier to ring isomerization is 11.97 kcal mol−1 (a → TS2). These results strongly indicate that disulfide bond rotation is the rate-determining step.
Furthermore, the rotation barrier of the disulfide bond, i.e., the energy barrier along TS1, of various compounds was estimated by DFT calculation (Fig. 3(c) and Table S3, ESI†). The magnitudes of the disulfide bond rotation barrier of cyclic disulfides (4, 7b, 9b and 9c) are similar, independent of the ring size, while still being higher than that of acyclic benzyl phenyl disulfide (12). The rotation barrier of the disulfide bond is clearly higher than that of the carbon–carbon bond in the same skeleton: CC_9b, in which the sulfur atoms in 9b are replaced by methylene groups, respectively, showed a lower barrier. These results demonstrate that the disulfide ring imposes strong constraints.
In summary, redox-induced complete amide cis–trans switching was achieved in a single molecule by intramolecular disulfide stapling through fusion of the disulfide to a bicyclic ring system. The obtained bicyclic-fused 8-membered disulfide compounds take essentially a single kind of conformer with respect to the disulfide ring. Ring conversion is slow enough to permit the detection of a pair of enantiomers in the case of 9b and two nearly mirror-image disulfide conformers in the case of 11b. The disulfide ring conformations are highly strained, and can be classified as cis-(+, +)AntiLHHook and cis-(−, −)AntiRHHook, respectively, which are rare conformations in nature. The disulfide ring allowed for a ring structure that was considerably distorted, resulting in a complete switch of the amide. It should be feasible to incorporate these molecular systems into peptides to enable reversible conformational switching.23
This work was supported by JSPS KAKENHI grant numbers JP 23K06022 (YO) and 23H02600 (TO) and by Asahi Glass Foundation (YO) and Astellas Foundation for Research on Metabolic Disorders (YO). DFT calculations were performed using the Research Center for Computational Science, Okazaki, Japan (projects: 23-IMS-C035, 22-IMS-C040). We thank Prof. M. Kanai, Dr K. J. Malawska, and Mr T. Fukuta, Graduate School of Pharmaceutical Sciences, University of Tokyo, for the assistance with cyclic voltammetry measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- J. L. Zhang, J. Q. Zhong, J. D. Lin, W. P. Hu, K. Wu, G. Q. Xu, A. T. S. Wee and W. Chen, Chem. Soc. Rev., 2015, 44, 2998–3022 RSC.
- K. P. Lu, G. Finn, T. H. Lee and L. K. Nicholson, Nat. Chem. Biol., 2007, 3, 619–629 CrossRef CAS PubMed.
- S. C. R. Lummis, D. L. Beene, L. W. Lee, H. A. Lester, R. W. Broadhurst and D. A. Dougherty, Nature, 2005, 438, 248–252 CrossRef CAS PubMed.
- K. K. A. Reddy, M. Jayashree, P. C. V. Govindu and K. H. Gowd, Proteins: Struct., Funct., Bioinf., 2021, 89, 599–613 CrossRef CAS PubMed.
- X. Wang, M. Connor, R. Smith, M. W. Maciejewski, M. E. H. Howden, G. M. Nicholson, M. J. Christie and G. F. King, Nat. Struct. Biol., 2000, 7, 505–513 CrossRef CAS PubMed.
- J. S. Richardson, L. L. Videau, C. J. Williams and D. C. Richardson, J. Mol. Biol., 2017, 429, 1321–1335 CrossRef CAS PubMed.
- A. P. Blum, K. R. Gleitsman, H. A. Lester and D. A. Dougherty, J. Biol. Chem., 2011, 286, 32251–32258 CrossRef CAS PubMed.
- T. Shi, S. M. Spain and D. L. Rabenstein, Angew. Chem., Int. Ed., 2006, 45, 1780–1783 CrossRef CAS PubMed.
- J. S. Richardson, L. L. Videau, C. J. Williams and D. C. Richardson, J. Mol. Biol., 2017, 429, 1321–1335 CrossRef CAS PubMed.
- A. D. De Araujo, V. Herzig, M. J. Windley, S. Dziemborowicz, M. Mobli, G. M. Nicholson, P. F. Alewood and G. F. King, Antioxid. Redox Signaling, 2013, 19, 1976–1980 CrossRef PubMed.
- D. Z. Avizonis, S. Farr-Jones, P. A. Kosen and V. J. Basus, J. Am. Chem. Soc., 1996, 118, 13031–13039 CrossRef CAS.
- C. J. Creighton, C. H. Reynolds, D. H. S. Lee, G. C. Leo and A. B. Reitz, J. Am. Chem. Soc., 2001, 123, 12664–12669 CrossRef CAS PubMed.
- B. Schmidt, L. Ho and P. J. Hogg, Biochemistry, 2006, 45, 7429–7433 CrossRef CAS PubMed.
- E. L. Ruggles, P. B. Deker and R. J. Hondal, Tetrahedron, 2009, 65, 1257–1267 CrossRef CAS PubMed.
- K. Kaczanowska, G. A. Camacho Hernandez, L. Bendiks, L. Kohs, J. M. Cornejo-Bravo, M. Harel, M. G. Finn and P. Taylor, J. Am. Chem. Soc., 2017, 139, 3676–3684 CrossRef CAS PubMed.
- J. J. Bellizzi III, J. Widom, C. Kemp, J.-Y. Lu, A. K. Das, S. L. Hofmann and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4573–4578 CrossRef PubMed.
- A. K. Das, J. J. Bellizzi, S. Tandel, E. Biehl, J. Clardy and S. L. Hofmann, J. Biol. Chem., 2000, 275, 23847–23851 CrossRef CAS PubMed.
- M. Hosoya, Y. Otani, M. Kawahata, K. Yamaguchi and T. Ohwada, J. Am. Chem. Soc., 2010, 132, 14780–14789 CrossRef CAS PubMed.
- Y. Otani, X. Liu, H. Ohno, S. Wang, L. Zhai, A. Su, M. Kawahata, K. Yamaguchi and T. Ohwada, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- R. R. Fraser, G. Boussard, J. K. Saunders, J. B. Lambert and C. E. Mixan, J. Am. Chem. Soc., 1971, 93, 3822–3823 CrossRef CAS.
- A. Brust, C. I. A. Wang, N. L. Daly, J. Kennerly, M. Sadeghi, M. J. Christie, R. J. Lewis, M. Mobli and P. F. Alewood, Angew. Chem., Int. Ed., 2013, 52, 12020–12023 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332996. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01096e |
| This journal is © The Royal Society of Chemistry 2024 |