
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChalcogen bonding interaction between ebselen and nitrite promote N-nitrosation of amines†
Tuhin
Sahana
,
Adwaith K.
Valappil
and
Subrata
Kundu
 *
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram (IISER–TVM), Thiruvananthapuram – 695551, India. E-mail: skundu@iisertvm.ac.in; skundu.chem@gmail.com
First published on 27th June 2024
Abstract
Ebselen (EbSe), a therapeutically significant molecule, is shown to exhibit chalcogen bonding interaction with nitrite anion (ONO−). This report suggests that the σ-holes of EbSe are powerful for offering weak but influential interactions towards biologically relevant ONO−, thereby assisting oxidative transformations like N-nitrosation of aromatic amines.
Ebselen (EbSe) is a lipid-soluble organoselenium compound with remarkable therapeutic potential against a wide array of disease conditions related to the heart, tumour growth, inflammation, and many more.1 The biological activities of EbSe are primarily attributed to its efficiency in extinguishing highly reactive oxidants (Fig. 1A).2 For instance, EbSe exhibits glutathione peroxidase (GPx) activity,3 thereby providing protection for various organisms against reactive oxygen species (ROS). GPx catalyses reduction of hydroperoxide (ROOH) in the presence of glutathione (GSH)/thiol cofactors (Fig. 1B). EbSe is also known to scavenge the incredibly reactive peroxynitrite anion (ONOO−) mostly by transforming it to nitrite anion (ONO−), along with the formation of the corresponding selenoxide species EbSeO through oxygen-atom transfer reactivity of peroxynitrite (Fig. 1C),4 while EbSe catalysed isomerization of ONOO− to nitrate anion (ONO2−) has also been suggested in the literature.5
 | ||
| Fig. 1 (A) and (B) GPx activity of EbSe and a commonly proposed mechanism. (C) EbSe mediated transformations of the peroxynitrite anion (ONOO−) via the corresponding adduct complex EbSe···OONO−. | ||
Understanding the interactions of EbSe with various reactive oxidants is of prime importance in order to utilize ebselen or related derivatives as an efficient mediator of the antioxidant activities in the biological milieu. The widely accepted mechanism for the GPx activity of EbSe indicates an initial nucleophilic attack of thiol GSH leading to the heterolytic cleavage of the Se–N bond in EbSe to produce the corresponding selenenyl sulfide EbSeSG as the intermediate species prior to the formation of the active reductant EbSeH in the presence of thiol (Fig. 1B).3 Alternatively, the ROS like peroxide (ROOH) may directly interact with EbSe to yield selenoxide species EbSeO through a formal oxygen-atom transfer from the ROS, and thus may quench the oxidizing capacity of the ROS. A previous computational study on the reactions of EbSe and ONOO− suggests that an initial coordination of the peroxynitrite anion at the Se-site of EbSe is exothermic and yields a transient adduct complex EbSe⋯OONO− (Fig. 1C),6 which subsequently promotes the cleavage of the O–O bond in EbSe⋯OONO− prior to its transformation to either (EbSeO + ONO−) or (EbSe + ONO2−). It is noteworthy that the initial steps for all the above-mentioned mechanistic postulates involve the Se-site of EbSe as the LUMO features a σ*(Se–N) character with a predominant contribution from the Se-orbital (Fig. 2A).7 Moreover, adduct complexes of EbSe chalcogen bonded to a variety of coordinating solvents (e.g. acetonitrile, tetrahydrofuran, dimethylsulfoxide) and pyridine derivatives have been illustrated earlier.8–10 Notably, a previous study based on high resolution X-ray diffraction data as well as computational analyses on EbSe suggest that the Se-site contains two σ-holes (σ1 and σ2), along the σ*(Se–N) and σ*(Se–C) directions, respectively (Fig. 2B), which are capable of engaging in directional non-covalent Se⋯X (X = O, N) interactions.11,12 Based on the computational studies, these σ-holes have been proposed to recognize physiologically relevant small molecular species such as H2O, H2O2, O2˙−, and ˙OH primarily through Se⋯O interaction. In fact, the previously proposed interactions of the Se-site in EbSe with peroxynitrite (OONO−) or nitrate (ONO2−) perhaps originate from the noncovalent interactions at the Se-based σ1-hole.12
 | ||
| Fig. 2 (A) LUMO of ebselen calculated at the B3PW91/6-311G(2df,p) level of theory. (B) Overview of this work. The arrows indicate the location of σ-holes (σ1 and σ2). | ||
While the interactions of EbSe with various ROS (such as peroxides, peroxynitrite, and hydroxyl radical) were previously assessed computationally,4–6,11 the reactivity of nitrite anion (ONO−) with ebselen (EbSe)/ebsulfur (EbS) is not known to the best of our knowledge. Inspired by our recent work on the generation of reactive intermediates from the reaction of nitrite anion and organosulfur compounds,13 the objective of this work is to probe the interactions/reactions between EbSe/EbS and ONO− using in-depth NMR spectroscopic and computational analyses. This work reveals that the noncovalent interactions in the resultant adduct complex EbSe⋯ONO− amplify the reactivity of the otherwise stable nitrite anion towards aromatic amines (Fig. 2B).
Multinuclear (1H, 13C, 77Se) NMR spectra of an authentic sample of EbSe and a solution consisting of an equimolar mixture of tetra-n-butylammonium nitrite [TBA][ONO] and ebselen (EbSe) in CDCl3 at room temperature were compared for gaining insights into the interaction between EbSe and the ONO− anion. 77Se NMR spectroscopic investigation on a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of EbSe and nitrite anion depicts a chemical shift at δ(77Se) = 942.19 ppm, whereas an authentic sample of EbSe shows δ(77Se) = 961.40 ppm (Fig. 3 and Fig. S1, ESI†). The upfield shift of the 77Se resonance for EbSe originates due to Se⋯O interaction in the EbSe⋯ONO− adduct and this is consistent with the previously reported EbSe complexes chalcogen-bonded to the guest molecules.8 Comparison of the 1H and 13C NMR spectra of the adduct complex EbSe⋯ONO− displays that both the proton and carbon resonances distinctly shift relative to those of an authentic sample of EbSe (Fig. S2 and S3, ESI†). Furthermore, NMR experiments such as 13C DEPT135, 1H–1H COSY, and 1H–13C HMQC on EbSe⋯ONO− in CDCl3 at room temperature allow us to assign the chemical shift of each aromatic proton individually (Fig. S4–S9, ESI†). While most of the proton resonances of EbSe undergo minor to moderate upfield shifts upon the coordination of the nitrite anion, the chemical shift of the H4-proton encounters a distinct downfield shift (Fig. 4 and Fig. S2, ESI†). Thus, these findings reveal that the nitrite anion is H-bonded to the H4–C4 site of EbSe in addition to the above-mentioned Se⋯O interaction.
:1 solution of EbSe and nitrite anion depicts a chemical shift at δ(77Se) = 942.19 ppm, whereas an authentic sample of EbSe shows δ(77Se) = 961.40 ppm (Fig. 3 and Fig. S1, ESI†). The upfield shift of the 77Se resonance for EbSe originates due to Se⋯O interaction in the EbSe⋯ONO− adduct and this is consistent with the previously reported EbSe complexes chalcogen-bonded to the guest molecules.8 Comparison of the 1H and 13C NMR spectra of the adduct complex EbSe⋯ONO− displays that both the proton and carbon resonances distinctly shift relative to those of an authentic sample of EbSe (Fig. S2 and S3, ESI†). Furthermore, NMR experiments such as 13C DEPT135, 1H–1H COSY, and 1H–13C HMQC on EbSe⋯ONO− in CDCl3 at room temperature allow us to assign the chemical shift of each aromatic proton individually (Fig. S4–S9, ESI†). While most of the proton resonances of EbSe undergo minor to moderate upfield shifts upon the coordination of the nitrite anion, the chemical shift of the H4-proton encounters a distinct downfield shift (Fig. 4 and Fig. S2, ESI†). Thus, these findings reveal that the nitrite anion is H-bonded to the H4–C4 site of EbSe in addition to the above-mentioned Se⋯O interaction.
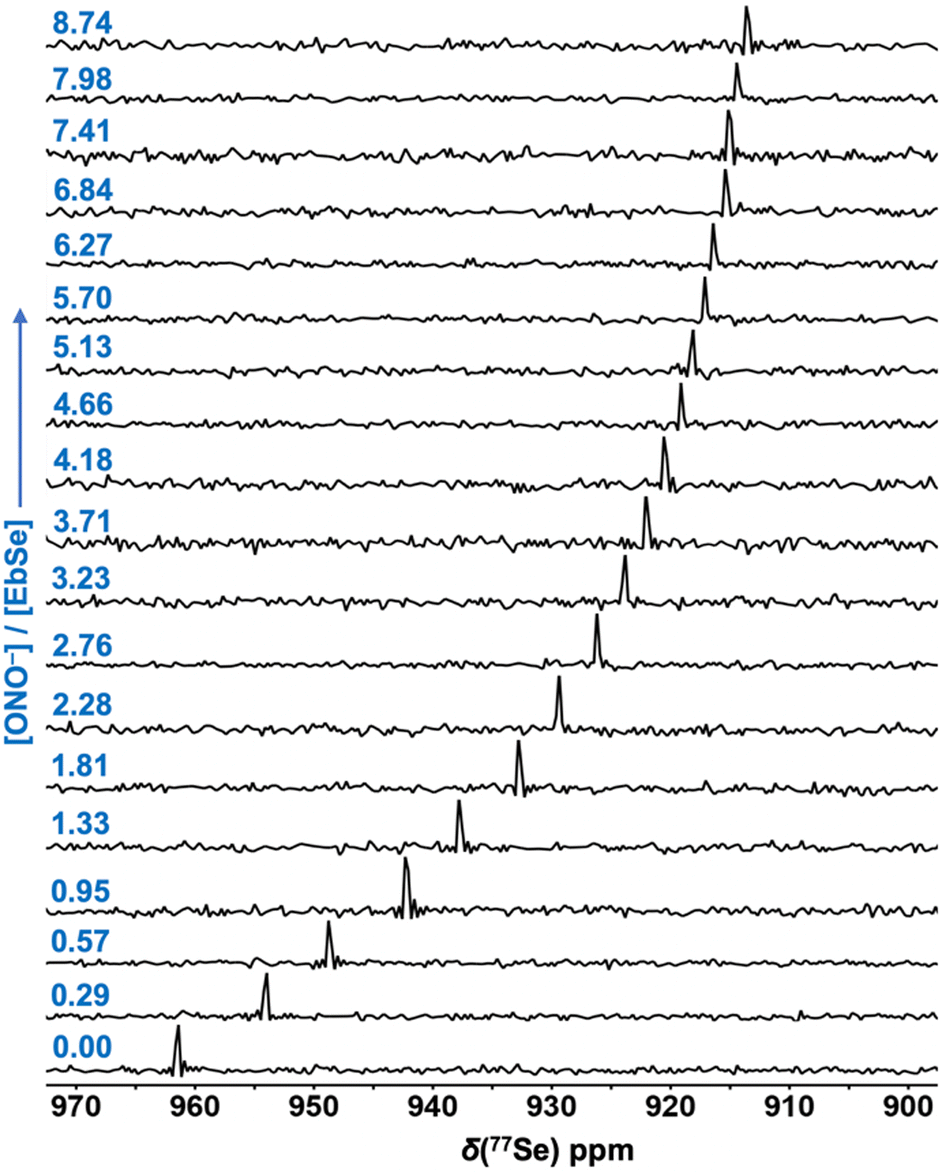 | ||
| Fig. 3 77Se NMR (95 MHz) spectra of EbSe in CDCl3 at room temperature upon increasing the ratio of nitrite anion and EbSe from 0.00 to 8.74 (as shown in the left panel). | ||
 | ||
| Fig. 4 1H NMR spectra of EbSe in CDCl3 at room temperature upon increasing the ratio of nitrite anion and EbSe from 0.00 to 8.74 (as shown in the left panel). The peak marked with * originates from the CDCl3 solvent residual peak. See Fig. 2A for the numbering scheme. | ||
77Se and 1H NMR spectra of EbSe with a systematic variation in the amount of added [TBA][ONO], where [ONO−]/[EbSe] ranged from 0.00 to 8.74, in CDCl3 at room temperature were recorded (Fig. 3 and 4). The increasing amount of nitrite anion equivalent in the solution of EbSe shows a gradual upfield shift of the 77Se resonance δ(77Se) (Fig. 3). A fitting of this shift in δ(77Se) with the 1:1 host–guest interaction model provides an association constant (Ka) of 25.22 M−1 (logKa = 1.40) (Fig. S10, ESI†).14,15 Notably, the association constant for EbSe⋯ONO− is moderately higher as compared to that of the previously reported EbSe⋯HMPA complex (HMPA = hexamethylphosphoramide) and this may be attributed to factors such as the anionic charge on the guest.8 In order to understand the role of the coulombic charge of the guest molecule on ebselen adduct complexes, 77Se and 1H NMR studies were carried out on the solutions of tetra-n-butylammonium nitrate [TBA][ONO2] and EbSe in CDCl3 at room temperature. Both the 77Se and 1H NMR studies suggest very weak association between EbSe and ONO2− anion (Fig. S12 and S13, ESI†). Notably, nitrate is known to be poorly coordinating as compared to nitrite due to more delocalization of the negative charge.16
We turned to investigate the binding of nitrite anion with a sulfur analogue of EbSe, namely 2-(4-methylphenyl)-1,2-benzisothiazol-3(2H)-one (EbS). Unlike the LUMO of EbSe, the LUMO of EbS shows a minor contribution from σ*(S–N) (Fig. S14, ESI†).7 A series of 1H NMR spectra recorded on the samples consisting of EbS and varied equivalents of [TBA][ONO] in CDCl3 at room temperature show negligible shifts of the proton resonances (Fig. S15, ESI†). Thus, this suggests that EbS is not an effective host for nitrite anions, as it is not capable of offering a strong S···O interaction with the weak σ-hole of a relatively lighter chalcogen site.
Unfortunately, several attempts to grow crystals of the adduct complex EbSe⋯ONO− failed due to the limited choice of suitable non-coordinating solvents. Hence, two possible isomers of EbSe⋯ONO− (syn/anti) and EbSe⋯ONO2− were calculated at the B3PW91/6-311G(2df,p) level of density functional theory (DFT) (Fig. 5 and S16, S17, ESI†).7 The syn- and anti-isomers are nearly isoenergetic as the relative energy differs marginally by 2.18 kcal mol−1. The DFT optimized structure of the syn-isomer of EbSe⋯ONO− depicts a Se···O2 interatomic distance of 2.211 Å along with an elongated Se–N1 bond (2.069 Å in syn-EbSe⋯ONO−) in comparison to the Se–N1 bond length of 1.875 Å in free EbSe (Fig. 5A and Table S2, ESI†). Similarly, the anti-isomer also shows comparable metrical parameters (Se···O2 2.168 Å and Se–N1 2.043 Å). It is noteworthy that these Se···O interatomic distances in both the syn- and anti-isomers of EbSe⋯ONO− are significantly shorter in comparison to the sum of the van der Waals radii Se + O 3.42 Å (Fig. S16, ESI†), thereby suggesting noncovalent interactions between the Se-site and nitrite anion. Moreover, these Se···O distances are comparable to the Se···N distances ranging from 2.304(1) to 2.6166(15) Å obtained from the X-ray crystal structures of the adducts of EbSe with various N-bases.9 In contrast to the notable Se–N1 bond elongation EbSe⋯ONO−, changes in other bond lengths of EbSe upon the coordination of the nitrite anion are negligible. Moreover, the nearly linear N1–Se···O2 angle 169.11° and 170.17° for the syn- and anti-isomers indicates the directional nature of the chalcogen bonding interaction of the nitrite anion with the σ-hole along the more polar σ*(Se–N) direction,12 thereby resulting in additional H-bonding interaction with the H4-site and consistent with the 1H NMR studies (Fig. 3 and 4). Interaction of the nitrite anion with EbSe also results in a marginal elongation of the O2–N2 bond by 0.032 (/0.048) Å and contraction of the N2–O3 bond by 0.039 (/0.047) Å as observed for the syn (/anti) isomers of EbSe⋯ONO−. The geometry optimized structure of EbSe⋯ONO2− shows a Se···O2 interatomic distance of 2.295 Å, which is longer than that in EbSe⋯ONO−. Consequently, the Se–N1 distance of 1.990 Å in EbSe⋯ONO2− is shorter relative to that in EbSe⋯ONO− (Fig. 5 and Fig. S17, ESI†). Thus, these findings from the computational and NMR studies correlate well and unambiguously demonstrate that chalcogen bonding interaction in EbSe⋯ONO− is stronger than in EbSe⋯ONO2−.
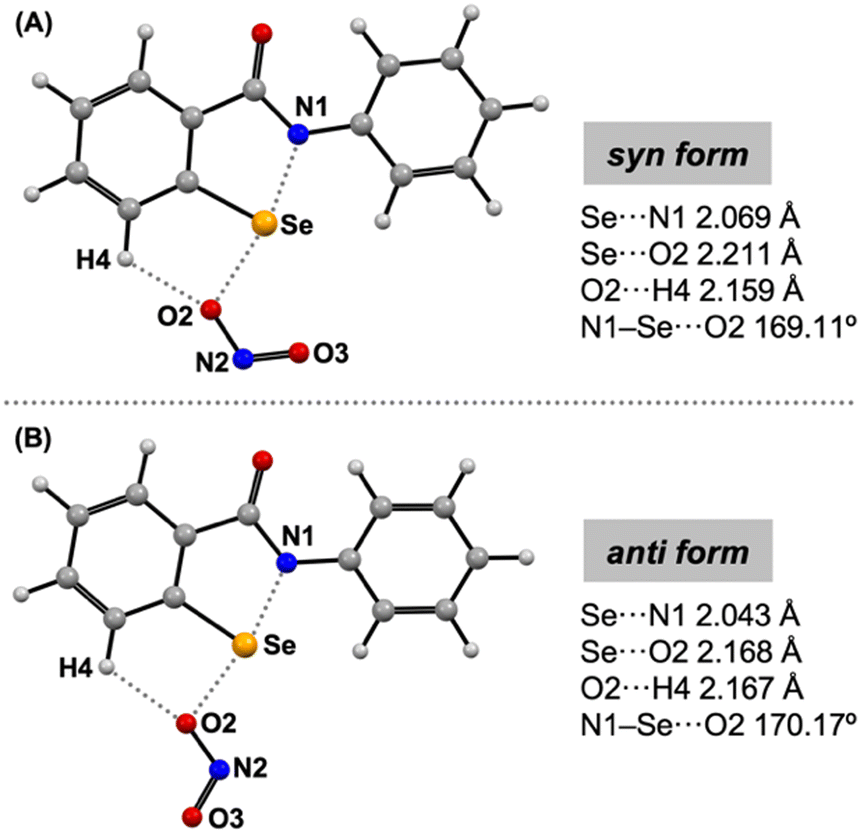 | ||
Fig. 5 DFT optimized (B3PW91/6-311G(2df,p)) structures of EbSe···ONO− in syn-form (A) and anti-form (B) with respect to the Se···O–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety. O moiety. | ||
Aiming to understand the implications of such chalcogen bonding interaction on the reactivity profile of the nitrite anion, N-nitrosation of amines in the presence and absence of EbSe was investigated. An equimolar reaction of [TBA][ONO] and di-p-tolylamine (Tol2NH) in the presence of EbSe (1.0 equiv.) in dichloromethane at room temperature provides the corresponding N-nitrosamine Tol2NNO in 61% yield (Table 1, Fig. S18–S20, ESI†). Notably, a control reaction of [TBA][ONO] and Tol2NH under the analogous reaction conditions in the absence of EbSe yields a trace (7%) amount of Tol2NNO. Interestingly, the above-mentioned reaction in the presence of EbSe in coordinating solvents like acetonitrile or tetrahydrofuran does not provide Tol2NNO. Thus, these observations suggest that (a) the activation of nitrite through the chalcogen bonding interaction with EbSe is critical for N-nitrosation of amine; (b) coordinating solvents compete for the chalcogen bonding interactions, thereby hindering the activation of the nitrite anion and inhibiting N-nitrosation. Similarly, inhibition by a product like N-nitrosated amine cannot be ruled out. A reaction of [TBA][ONO] and Tol2NH (1:1) in the presence of EbSe (10 mol%) in dichloromethane gives 52% yield of Tol2NNO (considering nitrite as the limiting reagent). A comparison of the yields of Tol2NNO upon systematic variation of either nitrite anion or amine equivalents keeping EbSe loading fixed demonstrates that (a) the yield of the reaction drops in the presence of higher equivalent of amine; (b) higher equivalent of the nitrite anion enhances the efficiency of N-nitrosation. While arylamines (such as di-p-tolylamine and N-methylphenyl amine) efficiently undergo N-nitrosation in the presence of nitrite anion and EbSe (Fig. S21, ESI†), aliphatic amine (e.g. diisopropylamine) provides a trace yield under the analogous reaction conditions. This may be attributed to the higher basicity of the aliphatic amines, thereby competing against the nitrite anion in interacting with EbSe. Thus, these findings indicate that the proposed in situ generated EbSe···ONO− adduct complex is capable of catalysing N-nitrosation reaction, while competitive binding of the amine substrate may impede the reaction outcome. This activation of the nitrite anion at the Se-site of EbSe and subsequent N-nitrosation are somewhat reminiscent of the nitrosating reactivity of the nitrite towards nucleophiles in the presence of Lewis acidic metal sites.17 Gutmann–Beckett analysis to evaluate the Lewis acidic nature of EbSe, however, does not suggest any considerable Lewis acidic nature of EbSe (Fig. S22, ESI†).18
| Entry | Reaction | Yield of N-nitrosamine (%) |
|---|---|---|
| 1 | [TBA][NO2] + (Tol)2NH (Control) | 7 |
| 2 | [TBA][NO2] + (Tol)2NH + EbSe | 61 |
| 3 | [TBA][NO2] + (Tol)2NH + EbS | 37 |
| 4 | [TBA][NO2] + (Tol)2NH + EbSe (10 mol%) | 52 |
| 5 | 2 [TBA][NO2] + (Tol)2NH + EbSe (10 mol%) | 100 |
| 6 | [TBA][NO2] + 2 (Tol)2NH + EbSe (10 mol%) | 48 |
| 7 | [TBA][NO2] + (Tol)2NH + EbS (10 mol%) | 28 |
| 8 | [TBA][NO2] + PhNHMe + EbSe | 91 |
| 9 | [TBA][NO2] + iPr2NH + EbSe | Trace |
In conclusion, this work provides spectroscopic illustrations of various weak interactions between two physiologically significant molecular entities, namely nitrite anion (ONO−) and ebselen EbSe, while such experimental demonstration of EbSe interaction was previously limited to various stable guests like coordinating solvents and N-bases.8–10 Though weak in nature, the Se-centered σ-hole interaction in EbSe···ONO− is shown to be effective in promoting aromatic amine oxidation leading to N-nitrosamine, a potential carcinogen.19 Of broader significance, this work underscores that ebselen may be capable of activating biologically relevant oxidants and mediate oxidative modifications of the amino acids in the absence of a redox cofactors like thiol.
S. K. gratefully acknowledges CRG/2021/001174 from SERB. The authors are thankful for the support from IISER-TVM.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. Noguchi, Arch. Biochem. Biophys., 2016, 595, 109–112 CrossRef CAS PubMed; (b) M. J. Parnham and H. Sies, Biochem. Pharmacol., 2013, 86, 1248–1253 CrossRef CAS PubMed; (c) G. K. Azad and R. S. Tomar, Mol. Biol. Rep., 2014, 41, 4865–4879 CrossRef CAS PubMed.
- T. Schewe, Gen. Pharmacol. Vasc. Syst., 1995, 26, 1153–1169 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1419 CrossRef CAS PubMed.
- H. Masumoto, R. Kissner, W. H. Koppenol and H. Sies, Chem. Res. Toxicol., 1996, 398, 179–182 Search PubMed.
- K. P. Bhabak, A. A. Vernekar, S. R. Jakka, G. Roy and G. Mugesh, Org. Biomol. Chem., 2011, 9, 5193–5200 RSC.
- D. G. Musaev, Y. V. Geletii, C. L. Hill and K. Hirao, J. Am. Chem. Soc., 2003, 125, 3877–3888 CrossRef CAS PubMed.
- See (ESI†) for the computational details.
- A. Daolio, P. Scilabra, M. E. Di Pietro, C. Resnati, K. Rissanen and G. Resnati, New J. Chem., 2020, 44, 20697–20703 RSC.
- T. Fellowes, E. Lee, J. Tran, R. Xu, A. Barber, S. C. Brydon, J. K. Clegg and J. M. White, Cryst. Growth Des., 2023, 23, 7179–7188 CrossRef CAS.
- T. Fellowes, M. A. Sani and J. White, Chem. – Eur. J., 2024, e202400385 CrossRef CAS PubMed.
- S. P. Thomas, K. Satheeshkumar, G. Mugesh and T. N. Gururow, Chem. – Eur. J., 2015, 21, 6793–6800 CrossRef CAS PubMed.
- σ1 hole is stronger than σ2, as the former is aligned along the σ* direction of the more polar Se–N bond (Fig. 2). See ref. 11 for details.
- G. Kolliyedath, T. Sahana, S. M. Johnson and S. Kundu, Angew. Chem., Int. Ed., 2023, 62, e202313187 CrossRef CAS PubMed.
- P. Thordarson, bindfit – supramolecular.org.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- A. Mondal, K. P. Reddy, S. Som, D. Chopra and S. Kundu, Inorg. Chem., 2022, 61, 20337–20345 CrossRef CAS PubMed.
- T. Sahana, A. K. Valappil, A. S. P. R. Amma and S. Kundu, ACS Org. Inorg., 2023, 3, 246–253 CrossRef CAS PubMed.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS.
- (a) E. J. Olajos, Ecotoxicol. Environ. Saf., 1977, 1, 175–196 CrossRef CAS PubMed; (b) W. Lijinsky, Cancer Metastasis Rev., 1987, 6, 301–356 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02137a |
| This journal is © The Royal Society of Chemistry 2024 |