
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic Ru–Ir/Rh complexes for catalytic allyl alcohol reduction to propylene†
Kanade
Kawaji
,
Mina
Tsujiwaki
,
Ayaka
Kiso
,
Yukina
Kitajo
,
Manami
Kitamura
,
Minako
Nishimura
,
Junya
Horikawa
,
Haruto
Ikushuma
,
Shin
Takemoto
 * and
Hiroyuki
Matsuzaka
* and
Hiroyuki
Matsuzaka
Department of Chemistry, Graduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka City, Osaka 558-8585, Japan. E-mail: stakemoto@omu.ac.jp
First published on 6th August 2024
Abstract
Bimetallic Ru–Ir/Rh complexes with the Ru-based metalloligand cis-(bpy)2Ru(PPh2)2 (RuP2) serve as catalysts for the selective reduction of allyl alcohol to propylene, employing H2 gas or an electrochemical setup. Metal–metal bonded key π-allyl intermediates [(RuP2)M(η3-C3H5)]2+ (M = Ir, Rh) are identified, advancing the understanding of the catalytic processes.
The catalytic reduction of C–O single bonds has gained significant attention in recent years as a crucial step for utilizing biomass as a sustainable feedstock in the chemical industry.1,2 Specifically, the chemoselective deoxygenation of allyl alcohol offers a promising route to bio-based propylene.3 This approach is particularly attractive because allyl alcohol can be readily derived from glycerol,4,5 an abundant byproduct of biodiesel production, and propylene is one of the most important feedstocks in the petrochemical industry.
Traditionally, reducing allylic alcohols to the corresponding olefins involves converting the starting alcohol to an allylic ester, followed by palladium-catalyzed allylic substitution with a hydride donor.6 However, direct catalytic hydrogenation typically reduces the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, leaving the C–O bond intact.7 Notably, Funabiki et al. and Alper et al. have demonstrated that the cobalt hydride complex [CoH(CN)5]3− selectively catalyzes the reduction of allylic alcohols to the corresponding olefins under H2 (1 atm), albeit with low turnover numbers (TONs < 10).8 More recently, Saito et al. developed an efficient Pd/TiO2 photocatalyst that promotes the reduction of allylic alcohols to olefins with methanol as the hydrogen source.3a However, when H2 gas was used instead of methanol, the reduction of the CC bond predominated. Additionally, limited research exists on the electroreduction of allyl alcohol on platinum electrodes in acidic media, where the formation of a mixture of propylene and propane was observed, with the selectivity significantly affected by the presence of adsorbed methanol.9
C bond, leaving the C–O bond intact.7 Notably, Funabiki et al. and Alper et al. have demonstrated that the cobalt hydride complex [CoH(CN)5]3− selectively catalyzes the reduction of allylic alcohols to the corresponding olefins under H2 (1 atm), albeit with low turnover numbers (TONs < 10).8 More recently, Saito et al. developed an efficient Pd/TiO2 photocatalyst that promotes the reduction of allylic alcohols to olefins with methanol as the hydrogen source.3a However, when H2 gas was used instead of methanol, the reduction of the CC bond predominated. Additionally, limited research exists on the electroreduction of allyl alcohol on platinum electrodes in acidic media, where the formation of a mixture of propylene and propane was observed, with the selectivity significantly affected by the presence of adsorbed methanol.9
We previously reported that the dicationic iridium(III) hydride complex [(RuP2)IrH(NCMe)3][BF4]2 ([1][BF4]2) containing the ruthenium(II) phosphido complex, RuP2, as a metalloligand efficiently catalyzes the hydrogenation of internal alkynes to E-alkenes with high chemo- and stereoselectivity.10 Building on this success and recognizing the potential of acidic transition metal hydrides to activate allylic C–OH bonds and form metal π-allyl complexes,11 we present the use of [1][BF4]2 as a catalyst for the chemoselective reduction of allyl alcohol to propylene under 1 atm H2. We also report electrochemical deoxygenation of allyl alcohol to propylene using a RuP2 rhodium complex as a catalyst. The π-allyl complexes [(RuP2)M(η3-C3H5)]2+ (M = Ir, Rh) have been identified as key catalytic intermediates.
Treatment of [1][BF4]2 with 5 equiv. of allyl alcohol in acetone at 50 °C resulted in the formation of the π-allyl complex [2][BF4]2 as shown in Fig. 1(a). This complex was isolated as a red crystalline solid in 82% yield. The 1H NMR spectrum for [2][BF4]2 revealed distinct signals attributed to the π-allyl protons: central CH (5.81 ppm), syn-CH2 (5.67 and 5.33 ppm), and anti-CH2 (3.45–3.37 ppm). Furthermore, the 31P{1H} NMR spectrum displayed two mutually coupled doublets at 225.9 and 224.8 ppm (2JPP = 28 Hz), indicating a significant downfield shift in comparison to the precursor [1][BF4]2 (−71.9, −80.5 ppm, 2JPP = 99 Hz). These shifts are characteristic of μ-PPh2 ligands bridging metals with a strong interaction.12
 | ||
| Fig. 1 (a) Synthesis of [2][BF4]2. (b) X-ray structure of the cationic part of [2][OTf]2. Ellipsoids are drawn at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ru1–Ir1 2.7776(3), Ir1–C1 2.199(4), Ir1–C2 2.208(4), Ir1–C3 2.142(6), Ir1–C3A 2.189(11), Ir1–P1 2.2218(9), Ir1–P2 2.2271(9), Ru1–P1 2.3508(9), Ru1–P2 2.3384(9), P1–Ir1–P2 109.06(3), P1–Ru1–P2 101.19(3), Ru1–P1–Ir1 74.75(3), Ru1–P2–Ir1 74.90(3). Due to a disorder in the orientation of the π-allyl ligand, the central carbon atom was treated as split atoms (C3 and C3A); only the one with a larger occupancy (C3) is shown. (c) Optimized geometry and selected frontier orbitals of [(RuP2)Ir(η3-allyl)]2+ ([2]2+). | ||
Single-crystal X-ray analysis on the triflate salt [2][OTf]2 confirmed the presence of an Ir(η3-allyl) unit coordinated by a RuP2 ligand, as illustrated in Fig. 1(b). The η3-allyl ligand is oriented perpendicular to the planar RuIrP2 core, causing the two bpy ligands and consequently the two PPh2 moieties to be non-equivalent, as evidenced by the 31P NMR spectrum. The short Ru–Ir distance (2.7776(3) Å), along with the acute Ru–P–Ir bond angles, indicate the presence of a metal–metal bond. Supporting this interpretation, orbital representations derived from density functional theory (DFT) analysis, depicted in Fig. 1(c), further elucidate the nature of this interaction. Specifically, HOMO−14 showcases a Ru–Ir σ-bond with similar contribution from Ru (51%) and Ir (41%), indicating a predominantly covalent character. Other orbitals from HOMO to HOMO−13 reveal various metal centered interactions, including an Ir-centered lone pair (5dz2) in the HOMO and two sets of orbitals indicative of Ru–Ir π/π* (HOMO−1 and HOMO−12) and δ/δ* (HOMO−2 and HOMO−4) interactions. The resulting (σ)2(π)2(π*)2(δ)2(δ*)2(Ir 5dz2)2 electron configuration is consistent with a metal–metal σ-bond between d5 Ru(III) and d7 Ir(II) centers.
The selective activation of allyl alcohol C–O bond by [1][BF4]2 prompted an investigation into the complex's potential as a catalyst for hydrogenolysis of allylic C–OH bonds. The findings from our initial screening, using cinnamyl alcohol as the test substrate, are summarized in Table 1. In the presence of 1 mol% of [1][BF4]2, the alcohol was reduced with H2 (1 atm) in acetone at 50 °C, yielding trans-β-methylstyrene (A; 76%) and n-propylbenzene (B; 6%) as outlined in entry 1. The presence of methanesulfonic acid (MsOH) as a co-catalyst (1 mol%) notably increased the yield of A to 95% (entry 2). Conversely, the addition of triethylamine, a basic additive, completely inhibited the catalytic activity (entry 3).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additivea | Yieldb (%) | |
| A | B | |||
| a All additives were 1 mol% relative to cinnamyl alcohol. b Determined by 1H NMR. c BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl. d dppe = Ph2PCH2CH2PPh2. e dcype = Cy2PCH2CH2PCy2 (Cy = cyclohexyl). f dppf = 1,1′-bis(diphenylphosphino)ferrocene. | ||||
| 1 | [1][BF4]2 | None | 76 | 6 |
| 2 | [1][BF4]2 | MsOH | 95 | 5 |
| 3 | [1][BF4]2 | Et3N | 0 | 0 |
| 4 | [Ir(cod)((S)-BINAP)][BF4]c | MsOH | 0 | 0 |
| 5 | [Ir(cod)(dppe)][BF4]d | MsOH | 3 | 0 |
| 6 | [Ir(cod)(dcype)][BF4]e | MsOH | 3 | 2 |
| 7 | [Ir(cod)(dcype)][BF4] | MsOH + HBF4 | 3 | 2 |
| 8 | [Ir(cod)(dppf)][BF4]f | MsOH + HBF4 | 12 | 0 |
| 9 | [Ru(bpy)2(PHPh2)2][BF4]2 | MsOH | 0 | 0 |
| 10 | None | MsOH | 0 | 0 |
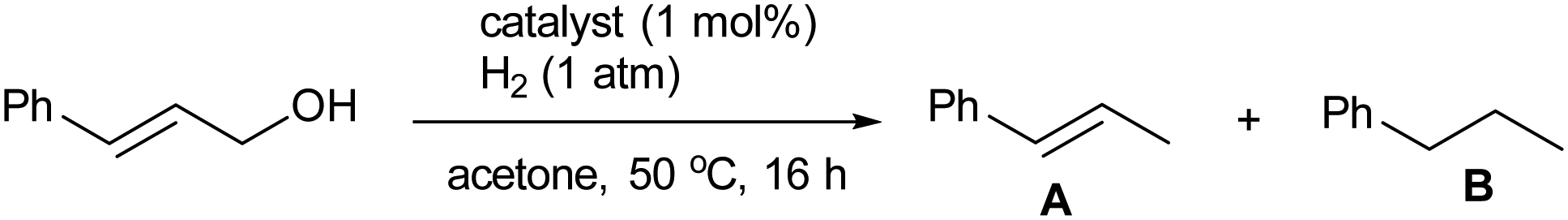
Exploration of mononuclear iridium complexes of the type [Ir(cod)(diphosphine)]+ (entries 4–7) yielded minimal amount of A (0–12%), despite the potential in situ formation of a dicationic Ir(III) hydride species analogous to [1][BF4]2 in the presence of HBF4 (entries 7 and 8). For entry 8, we have separately confirmed the formation of an Ir(III) hydride complex [IrH(cod)(MeCN)(dppf)][BF4]2 (see ESI†). Similarly, the mononuclear ruthenium complex [Ru(bpy)2(PHPh)2][BF4]2 (entry 9) and MsOH alone (entry 10) showed no catalytic activity. These results highlight the exceptional catalytic performance of [1][BF4]2, likely attributable to the synergistic interaction between its iridium and ruthenium components.
Utilizing the catalyst mixture of [1][BF4]2 and MsOH (1 mol% each) enabled the selective deoxygenation of parent allyl alcohol with H2 gas (1 atm), yielding propylene with an impressive 98% efficiency, as illustrated in Fig. 2. To further explore the potential of this catalytic system, we conducted the reduction of neat allyl alcohol, aiming to achieve higher propylene TONs. Remarkably, at 80 °C using 5 mL (73 mmol) of allyl alcohol with 1.0 μmol of [1][BF4]2 and 0.10 mmol of MsOH, we observed a propylene TON of 2850 (relative to [1][BF4]2), as depicted in Fig. 3. Notably, only trace amounts of propane were detected, emphasizing the selectivity of this reaction under the specified conditions.
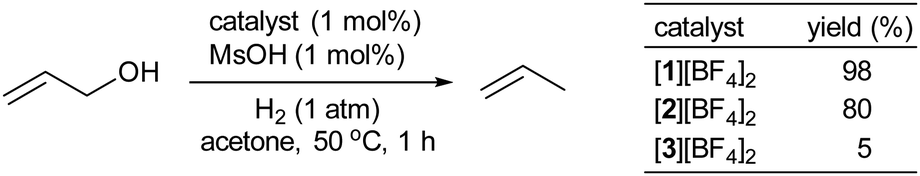 | ||
| Fig. 2 Catalytic hydrogenolysis of allyl alcohol with 1 mol% [1][BF4]2, [2][BF4]2, or [3][BF4]2. | ||
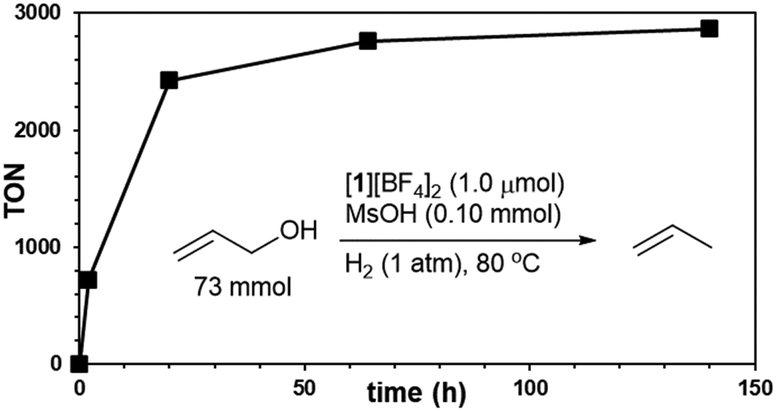 | ||
| Fig. 3 Time course of propylene TON. The reaction was performed in neat allyl alcohol (5.0 mL, 73 mmol) with Ru–Ir catalyst [1][BF4]2 (1.0 μmol) and MsOH (0.10 mmol) at 80 °C in a sealed vessel filled with H2 (1 atm initial pressure, 114 mL inner volume). | ||
To elucidate the mechanisms underlying the hydrogenolysis of allyl alcohol mediated by the [1][BF4]2 + MsOH catalyst, we undertook a series of stoichiometric and catalytic experiments. Initially, a reaction between [1][BF4]2 and allyl alcohol in acetone produced a mixture containing [2][BF4]2 and free MeCN. This mixture was then exposed to H2 gas, leading to the anticipated regeneration of [1][BF4]2 (observed in a 67% NMR yield) along with propylene production. Interestingly, this reaction also yielded a new species, [3][BF4]2, in 19% NMR yield, identified as a product of bpy ligand cyclometallation, as shown in Fig. 4. Complex [3][BF4]2 was separately synthesized through the thermolysis of [2][BF4]2 in the presence of MeCN, with its structure confirmed crystallographically as depicted in Fig. 5. Subsequent testing of the isolated complexes [2][BF4]2 and [3][BF4]2 as catalysts for allyl alcohol hydrogenolysis revealed that while [2][BF4]2 maintained catalytic activity comparable to [1][BF4]2, [3][BF4]2 exhibited significantly lower activity (Fig. 2). This suggests that the conversion of [2][BF4]2 to [3][BF4]2 acts as a catalyst deactivation pathway (Fig. 4). Notably, [3][BF4]2 remained unchanged when exposed to H2 (1 atm) at 50 °C in acetone, reinforcing its role as a deactivation product.
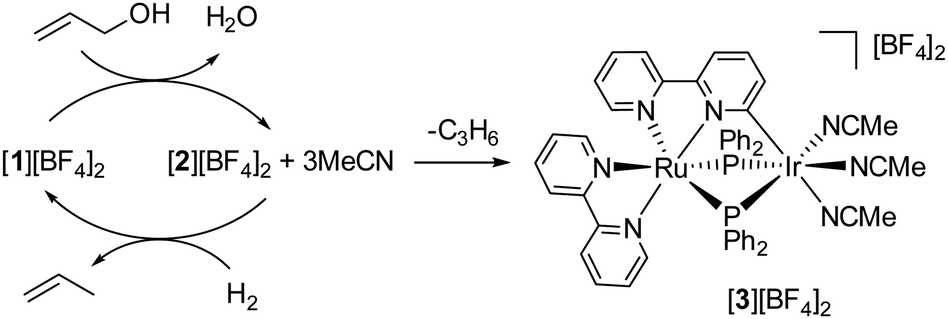 | ||
| Fig. 4 A plausible catalytic cycle and a catalyst deactivation pathway. | ||
 | ||
| Fig. 5 X-ray structure of the cationic part of [3][BF4]2. Ellipsoids are drawn at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ir1–C1 1.993(11), Ir1–P1 2.326(3), Ir1–P2 2.311(3), Ir1–N5 2.118(12), Ir1–N6 2.135(14), Ir1–N7 2.091(11), Ru1–P1 2.328(3), Ru1–P2 2.305(3), Ru1–N1 2.053(8), Ru1–N2 2.138(9), P1–Ir1–P2 76.73(11), P1–Ru1–P2 76.81(11), Ru1–P1–Ir1 93.83(11), Ru1–P2–Ir1 94.89(11). | ||
Further investigations into the impact of MsOH on the catalytic process revealed that it accelerated the conversion of [1][BF4]2 to [2][BF4]2 in the presence of allyl alcohol (Fig. 6a), suggesting proton transfer from Ir–H to the coordinated allyl alcohol,11 where the added acid can act as a proton shuttle. Additionally, MsOH effectively inhibited the thermolysis of [2][BF4]2 to [3][BF4]2 (Fig. 6b), thereby suppressing the catalyst deactivation pathway. These observations highlight the beneficial effects of the added MsOH on the overall catalytic efficiency of the hydrogenolysis reaction, though the molecular grounds underlying these effects warrant further investigations.
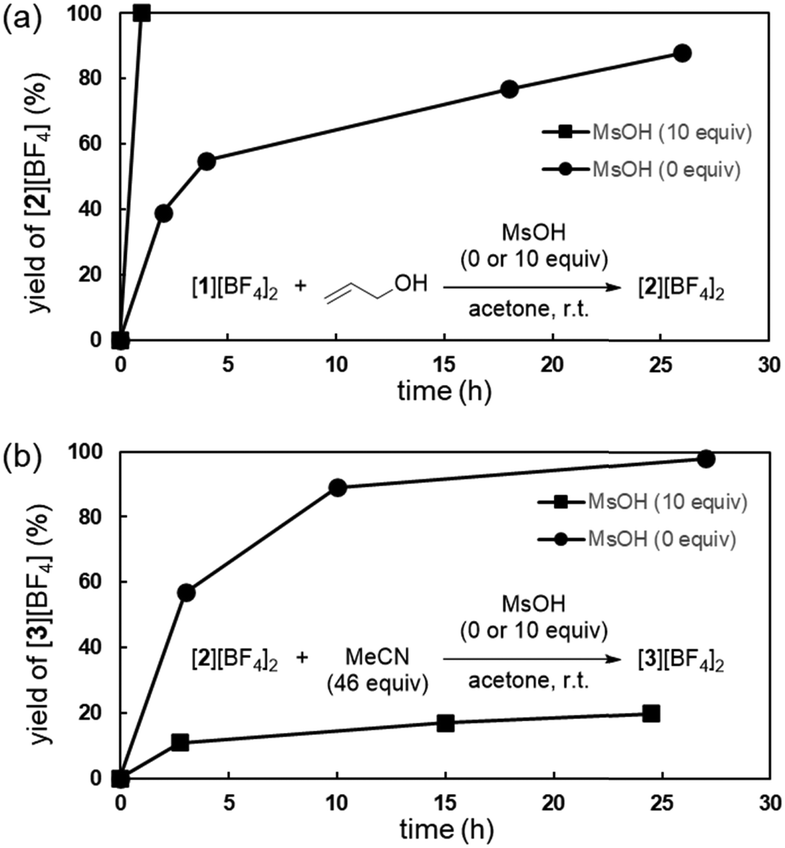 | ||
| Fig. 6 Effect of MsOH (a) on the formation of [2][BF4]2 from [1][BF4]2 and allyl alcohol, and (b) on the formation of [3][BF4]2 by thermolysis of [2][BF4]2 in the presence of MeCN. | ||
In our quest to explore alternative methods for reducing allyl alcohol, we investigated electrochemical techniques using a divided cell setup (Fig. 7a). The cathodic electrolyte consisted of a mixture of allyl alcohol (5 mL) and water (5 mL), a metal catalyst (0.010 mmol), and MsOH (1.0 mmol). Employing the Ru–Ir complex [1][BF4]2 as the catalyst resulted in the formation of H2 as the main product, indicating a strong preference for proton reduction. In contrast, using the Ru–Rh complex [(RuP2)Rh(cod)][OTf] ([4][OTf]; cod = 1,5-cyclooctadiene),10 demonstrated a clear preference for the electroreduction of allyl alcohol, giving propylene with 77% selectivity and 40% faradaic efficiency in 1 h at room temperature (Fig. 7a).
 | ||
| Fig. 7 (a) Electrochemical deoxygenation of allyl alcohol using [4][OTf] in a divided cell. (b) Stoichiometric reactions. (c) Proposed catalytic cycle for the cathodic reaction. | ||
A stoichiometric reaction between [4][OTf] and allyl alcohol, in the presence of MsOH, yielded a red crystalline solid, formulated as [(RuP2)Rh(η3-C3H5)][OTf][OMs]·HOMs ([5][OTf] [OMs]·HOMs) in 46% yield (see ESI†). Further purification of this intermediate through recrystallization from MeOH–Et2O gave [5][OTf]2 (Fig. 7b). The single-crystal X-ray structure of [5][OTf]2 (Fig. 7c) revealed it to be isostructural with its iridium analogue [1][OTf]2. Treatment of [5][OTf]2 with 2 equiv. of Cp2Co in the presence of MsOH and COD generated [4][OTf] (>99% NMR yield) and propylene (65% yield) (Fig. 7b).
Based on these results and given the fact that both [4][OTf] and [5][OTf]2 were inactive for the reduction of allyl alcohol with H2, we propose a catalytic cycle for the reduction of allyl alcohol with H+ and e− as outlined in Fig. 7c. The cycle initiates with the formation of a cationic Rh(I) solvent complex, [(RuP2)Rh(solv)n]+ (solv = allyl alcohol) from [4]+ and allyl alcohol. This intermediate is then transformed into the dicationic π-allyl species [5]2+ upon protonation. Two-electron reduction of [5]2+ followed by the protonolysis of the resulting neutral Rh(I) π-allyl complex completes the catalytic cycle.
In summary, we have demonstrated that the Ir/Rh complexes with the RuP2 metalloligand enable the selective reduction of allyl alcohol to propylene via H2 activation or electrochemical deoxygenation. This provides a promising route for producing industrial chemicals from bio-based renewable resources. The identification of both metal–metal bonded ([2]2+ and [5]2+) and non-metal–metal bonded intermediates ([1]2+ and [4]+) during the reaction highlights the flexible metal-centered reactivity of the RuP2 ligand, suggesting its potential for further applications in various catalytic processes.
We thank JSPS (18H04268, 20H02758, 21K05088, 22K19054) and Masuyakinen basic research foundation for financial support.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2347094–2347096 and 2367600 contain supplementary crystallographic data for this article.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC; (b) M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed; (c) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- (a) P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef PubMed; (b) M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2010, 47, 2946–2948 RSC; (c) J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed; (d) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed; (e) T. V. Stein, T. Weigand, C. Merkens, J. Klankermayer and W. Leitner, ChemCatChem, 2013, 5, 439–441 CrossRef; (f) S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 6296 CrossRef PubMed; (g) N. I. Saper and J. F. Hartwig, J. Am. Chem. Soc., 2017, 137, 17667–17676 CrossRef PubMed; (h) R. Seki, N. Hara, T. Saito and Y. Nakao, J. Am. Chem. Soc., 2021, 143, 6388–6394 CrossRef CAS PubMed; (i) A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershøj, G. M. F. Batista, A. Sommerfeldt, S. Frølich, H. Birkedal and T. Skrydstrup, Nature, 2023, 617, 730–737 CrossRef CAS PubMed; (j) C.-H. Hung, T.-H. Wang, G. P. A. Yap, J. C. Juan and T.-G. Ong, ChemCatChem, 2023, 15, e202300249 CrossRef CAS; (k) Y. Liao, K. Takahashi and K. Nozaki, J. Am. Chem. Soc., 2024, 146, 2419–2425 CrossRef CAS PubMed.
- (a) J. Caner, Z. Liu, Y. Takada, A. Kudo, H. Naka and S. Saito, Catal. Sci. Technol., 2014, 4, 4093–4098 RSC; (b) J. R. Dethlefsen, D. Lupp, A. Teshome, L. B. Nielsen and P. Fristrup, ACS Catal., 2015, 5, 3638–3647 CrossRef CAS; (c) Y. Takada, J. Caner, S. Kaliyamoorthy, H. Naka and S. Saito, Chem. – Eur. J., 2017, 23, 18025–18032 CrossRef CAS PubMed.
- (a) E. Arceo, P. Marsden, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3357–3359 RSC; (b) M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed; (c) M. Kim and H. Lee, ACS Sustainable Chem. Eng., 2017, 5, 11371–11376 CrossRef CAS.
- For catalytic synthesis of propylene from hydrogenolysis of glycerol, see: (a) L. Yu, J. Yuan, Q. Zhang, Y.-M. Liu, H.-Y. He, K.-N. Fan and Y. Cao, ChemSusChem, 2014, 7, 743–747 CrossRef CAS PubMed; (b) V. Zacharopoulou, E. S. Vasiliadou and A. A. Lemonidou, Green Chem., 2015, 17, 903–912 RSC; (c) C. J. A. Mota, V. L. C. Gonçalves, J. E. Mellizo, A. M. Rocco, J. C. Fadigas and R. Gambetta, J. Mol. Catal. A, 2016, 422, 158–164 CrossRef CAS; (d) Y. Nakagawa, M. Tamura and K. Tomishige, Res. Chem. Intermed., 2018, 44, 3879–3903 CrossRef CAS.
- J. Tsuji, I. Minami and I. Shimizu, Synthesis, 1986, 623–627 CrossRef CAS.
- J. G. Wadkar and R. V. Chaudhari, J. Mol. Catal., 1983, 22, 103–116 CrossRef CAS.
- (a) T. Funabiki, Y. Yamazaki and K. Tarama, J. Chem. Soc., Chem. Commun., 1978, 63–65 RSC; (b) J.-T. Lee and H. Alper, Tetrahedron Lett., 1992, 33, 4101–4104 Search PubMed.
- (a) G. Horányi, Electrochim. Acta, 1986, 31, 1095–1103 CrossRef; (b) E. Pastor, S. Wasmus, T. Iwasita, M. C. Arévalo, S. González and A. J. Arvia, J. Electroanal. Chem., 1993, 353, 81–100 CrossRef CAS; (c) P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed.
- S. Takemoto, M. Kitamura, S. Saruwatari, A. Isono, Y. Takada, R. Nishimori, M. Tsujiwaki, N. Sakaue and H. Matsuzaka, Dalton Trans., 2019, 48, 1161–1165 RSC.
- F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968–10969 CrossRef CAS PubMed.
- For example, see: (a) P. E. Kreter and D. W. Meek, Inorg. Chem., 1983, 22(2), 319–326 CrossRef CAS; (b) A. E. Findlay, S. Leelasubcharoen, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Dalton Trans., 2010, 39, 9264–9269 RSC; (c) D. O. Downing, P. Zavalij and B. W. Eichhorn, Inorg. Chim. Acta, 2011, 375(1), 329–332 CrossRef CAS; (d) B. Pan, D. A. Evers-McGregor, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2013, 52, 9583–9589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and crystallographic details. CCDC 2347094–2347096 and 2367600. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01711k |
| This journal is © The Royal Society of Chemistry 2024 |