
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of actin dimerization inducers inspired by actin-depolymerizing macrolides†
Moeka
Itakura
a,
Didik Huswo
Utomo
ab and
Masaki
Kita
 *a
*a
aGraduate School of Bioagricultural Sciences, Nagoya University, Nagoya 464-8601, Japan. E-mail: mkita@agr.nagoya-u.ac.jp
bBioinformatics Research Center, Indonesian Institute of Bioinformatics, Malang, Jawa Timur 65162, Indonesia
First published on 15th April 2024
Abstract
Several natural cytotoxic C2-symmetric bis-lactones, such as swinholide A and rhizopodin, sequester actin dimer from the actin network and potently inhibit actin dynamics. To develop new protein–protein interaction (PPI) modulators, we synthesized structurally simplified actin-binding side-chain dimers of antitumor macrolide aplyronine A. By fixing the two side-chains closer than those of rhizopodin, the C4 linker analog depolymerized filamentous actin more potently than natural aplyronines. Cross-link experiments revealed that actin dimer was formed by treatment with the C4 linker analog. Molecular dynamics simulations showed that this analog significantly changed the interaction and spatial arrangement of the two actins compared to those in rhizopodin to provide a highly distorted and twisted orientation in the complex. Our study may promote the development of PPI-based anticancer and other drug leads related to cytoskeletal dynamics.
Actin filament is an essential component of the cytoskeleton in eukaryotes, and modulation of its protein–protein interactions (PPIs) is a promising method for controlling cytoskeletal dynamics and cellular signaling pathways.1 Several marine cytotoxic macrolides are known to potently depolymerize actin and form a 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex, such as in aplyronine A (ApA) (Fig. 1c),2–4 mycalolides/kabiramides,5 and reidispongiolide A,6 which have similar 11-carbon aliphatic side-chains terminating in an N-methyl enamide group. These side-chains similarly insert into the hydrophobic cleft of actin, sequestering the actin monomer (G-actin) and inhibiting polymerization to filamentous actin (F-actin).7 In addition, ApA is a unique PPI inducer for actin and α,β-tubulin heterodimer to inhibit microtubule assembly against cancer cells (IC50 = 10 pM against the human cervical carcinoma cell line HeLa S3).8 The basis of the molecular interaction between ApA and actin has been established by X-ray crystallography (PDB ID: 1WUA),9 photolabeling experiments,10 and binding kinetics analysis.11 In addition, recent molecular modeling studies have proposed the structure of actin-ApA-tubulin heterotrimeric complexes and their inhibitory effects on microtubule assembly.12
:1 complex, such as in aplyronine A (ApA) (Fig. 1c),2–4 mycalolides/kabiramides,5 and reidispongiolide A,6 which have similar 11-carbon aliphatic side-chains terminating in an N-methyl enamide group. These side-chains similarly insert into the hydrophobic cleft of actin, sequestering the actin monomer (G-actin) and inhibiting polymerization to filamentous actin (F-actin).7 In addition, ApA is a unique PPI inducer for actin and α,β-tubulin heterodimer to inhibit microtubule assembly against cancer cells (IC50 = 10 pM against the human cervical carcinoma cell line HeLa S3).8 The basis of the molecular interaction between ApA and actin has been established by X-ray crystallography (PDB ID: 1WUA),9 photolabeling experiments,10 and binding kinetics analysis.11 In addition, recent molecular modeling studies have proposed the structure of actin-ApA-tubulin heterotrimeric complexes and their inhibitory effects on microtubule assembly.12
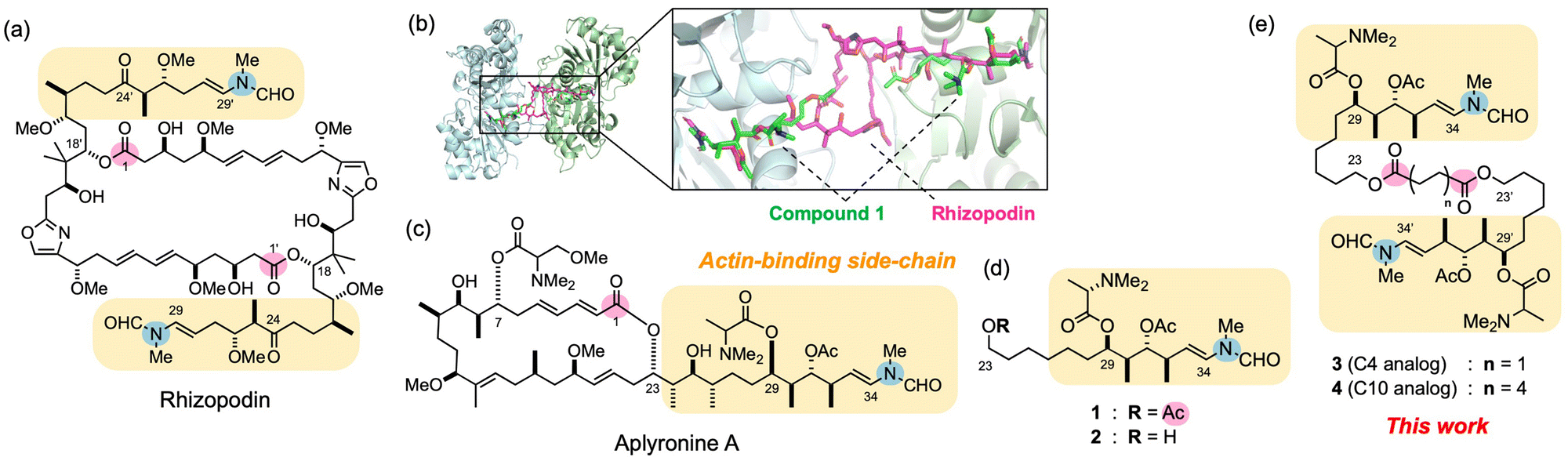 | ||
| Fig. 1 Design of actin-binding side-chain dimers of aplyronine A (ApA) inspired by the bis-lactone rhizopodin. (a) Structures of actin-depolymerizing natural product rhizopodin. Actin-binding sites are highlighted in rounded yellow squares. The lactone carbonyl carbon atoms (C1 and C1′) and terminal enamide nitrogen atoms are highlighted in pink and blue circles, respectively. (b) Binding modes of rhizopodin (magenta) and the ApA side-chain analog 1 (green) on actin. The actin-1 complex obtained by molecular modeling studies19 was superimposed on the actin–rhizopodin complex (left) and viewed from the bottom side (right). (c)–(e) Structures of ApA and its side-chain monomer and dimer analogs in this work. | ||
In addition to monomeric macrolides, several natural bis-lactones sequester the actin dimer from the actin network, and potently inhibit the nuclearization and polymerization of actin.1 For example, swinholide A and bistheonellide A are 44- and 40-membered, C2-symmetric dimeric lactones from the marine sponge Theonella swinhoei.13 All of these natural bis-lactones intervene between two actin molecules, form tertiary complexes with each of its side-chains bound to G-actin, and inhibit polymerization to F-actin.14 In addition, rhizopodin, a 38-membered, C2-symmetric bis-lactone from the myxobacterium Myxococcus stipitatus,15,16 also shows potent cytotoxicity and actin-depolymerizing activity (Fig. 1a). It bears two sets of oxazole rings and C11 side-chains that terminate in N-methyl enamide groups. X-ray crystallographic analysis of the actin–rhizopodin complex17 revealed that each of its side-chains binds to the hydrophobic clefts of subdomains (SD) 1 and 3 on two different actin molecules to form a 2:1 complex (Fig. 1b). Through the C1–C18 and C1′–C18′ bis-lactone structure, including two pairs of rigid oxazole and diene moieties, rhizopodin fixes the spatial arrangement of its side-chain parts, and induces PPI between two actin molecules.
In our continuing studies on the structure–activity relationships (SAR) in actin-depolymerizing macrolides,18 we reported structurally-simplified C29–C34 side-chain analogs of ApA, including 1 which possesses a C23 acetoxy group that mimicks the C1 lactone carbonyl group, the C29 N,N-dimethyl-L-alanine ester, and the C34 N-methyl enamide moiety as minimum functional groups (Fig. 1d).19 Interestingly, the binding mode of 1 to actin, particularly in the C29–C34 part, is highly similar to those of the C24–C29 and C24′–C29′ parts of rhizopodin, as well as that of ApA (Fig. 1b). Therefore, by mimicking its unique PPI-modulating activity, we expected to develop novel ligands that potently inhibit actin-polymerization dynamics. Here we describe the design and development of side-chain dimer analogs of ApA in which two molecules of 1 are linked as in bivalent actin-depolymerizing bis-lactones, and evaluate their biological activities and binding modes with actin.
In the X-ray crystal structure of the actin–rhizopodin complex,17 the distance between C1 and C1′ carbonyl carbons was 10.6 Å. Thus, we designed side-chain dimers 3 and 4 connected at the C23 and C23′ acyloxy groups by using succinate (C4) and sebacate (C10) linkers (Fig. 1e). The distances between the linker ester carbons in 3 and 4 were estimated to be 3.9 and 11.4 Å, respectively, when their alkyl chains had zigzag orientations. We initially expected that 3 would be unable to form a complex with actin dimers as with rhizopodin, whereas 4 could form such a complex and improve the activity of 1.
Condensation of primary alcohol 219 with succinic acid or sebacic acid in the presence of EDC·HCl and DMAP gave C4 analog 3 (26%) and C10 analog 4 (32%), respectively (See ESI† for details). The biological activities of 3 and 4 were compared to those of natural aplyronines and monomeric side-chain analogs (Table 1). In the in vitro F-actin sedimentation assay using ultracentrifugation, the amount of F-actin in the precipitate fraction was reduced by treatment with 3 or 4 in a dose-dependent manner (Fig. S1, ESI†). Notably, the C4 dimer 3 showed more potent activity (EC50 = 1.0 μM against 3 μM actin) than ApA and aplyronine C (ApC, a natural congener of ApA that lacks the C7 ester) (EC50 = 1.3–1.4 μM)8b as well as monomeric analogs 1 and 2.19 It is noteworthy that 3 showed more potent actin-depolymerizing activity than ApA, despite the weaker binding affinity of 1 than that of ApA (KD values of their biotin derivatives were 10.1 and 5.2 μM, respectively, based on the Bio-Layer Interferometry analysis). Meanwhile, the C10 dimer 4 also showed moderate activity (EC50 = 20 μM), but this activity was weaker than those of 1 and 3.
| Compound | Actin-depolymerizing activitya EC50 (μM) for 3 μM actin | Cytotoxicity IC50 (μM) | |
|---|---|---|---|
| HCT116 | HEK293 | ||
| a Values indicate the concentrations required to depolymerize F-actin (3 μM for monomer) to 50% of its control amplitude. Averages of two reproducible runs are shown. b See ref. 18. c Not examined. d See ref. 19. e Determined by the fluorescence intensity of pyrenyl actin. See ref. 8b. | |||
| Aplyronine A | 1.4b [1.3]e | 1.0 × 10−4 | 4.2 × 10−4 |
| Aplyronine C | −c[1.4]e | 2.1 × 10−2 | 2.5 × 10−2 |
| 1 | 8.8d | 20 | >50 |
| 2 | 27d | >50 | >50 |
| 3 (C4) | 1.0 | 2.5 | >50 |
| 4 (C10) | 20 | 1.0 | 32 |
As for cytotoxicity, both 3 and 4 moderately inhibited the proliferation of the human colon cancer cell line HCT116 (IC50 = 2.5 and 1.0 μM, respectively), and were 8 and 20 times more potent than 1. However, even with the more active 4, its cytotoxicity was 10000 and 48 times weaker than those of ApA and ApC, respectively. A similar cytotoxicity profile was observed against human embryonal kidney-derived HEK293 cells. Previous SAR studies using HeLa S3 cells showed that several side-chain analogs of aplyronines and mycalolides exhibited potent actin-depolymerizing activity, but little cytotoxicity (IC50 > 10 μM).7,10,18 In addition, fluorescent aplyronines have been shown to rapidly accumulate in the cytoplasm and disassemble the actin cytoskeleton in cells.20 Thus, despite showing much less cytotoxicity than natural aplyronines, both 3 and 4 with an alkyl diester linker might penetrate cells more easily than 1 and 2, and effectively interfere with actin dynamics in HCT116 cells.
To explain the high actin-depolymerizing activity of the C4 analog 3, we attempted to observe its actin dimer complex in solution. However, no dimer or oligomer formation was detected by gel permeation HPLC or Native-PAGE analysis due to the weak interaction of the side-chain moiety with actin19 (Fig. 2a, b, and Fig. S2, ESI†). Thus, the actin-3 complex was treated with NHS-diazirine followed by UV365 irradiation to form covalent bonds. SDS-PAGE analysis revealed that a cross-linked actin dimer (86 kDa) was formed only in the presence of 3 (Fig. 2c), which supported the formation of the 2:1 actin-3 complex in solution.
 | ||
| Fig. 2 Observation of the actin dimer complex with 3. (a) Gel permeation HPLC analysis. The monomeric actin–ApA and actin–3 complexes were detected (tR = 20.3 min), but not their dimeric ones (expected tR = 17.8 min). (b) HR-CN (high resolution clear native) PAGE analysis. The actin complex with ApA was detected as a major band at 43 kDa (monomer), but not with 3. (c) Cross-link experiments. Actin (5 μM) was treated with NHS–diazirine in the absence or presence of 3 (25 μM) in G-buffer at rt for 20 h, followed by UV365 irradiation for 1 min. An arrow indicates the cross-linked actin dimer (86 kDa) on SDS-PAGE. | ||
To explain the unique activities of 3 and 4, docking simulations with two actin molecules were performed. The initial models of the actin-3 and actin-4 complexes were constructed from two sets of an actin-1 complex19 superimposed on an actin-rhizopodin complex (Fig. 1b). In the lowest-energy models obtained by docking simulation using rigid models, both 3 and 4 similarly bound to actin at the binding site of 1. We next performed molecular dynamics (MD) simulations to obtain reliable models of the 2:1 actin-side-chain dimer complex. Using YASARA software, we evaluated actin-3, -4, and -rhizopodin complexes at a density of 0.997 g mL−1, 298 K, and pH 7.4. After 100 ns simulations, all three complexes were relatively stable, but the actin-3 complex exhibited the largest fluctuation in terms of the conformational stabilities of the actin and ligands (RMSD = 8.71 and 5.74 Å on average, respectively), followed by the actin-4 complex (6.30 and 4.24 Å) and the actin-rhizopodin complex (3.84 and 2.20 Å) (Fig. S3, ESI†). Similarly, for the actin-3 complex, the binding energy significantly decreased and the radius of gyration increased, whereas these changes were relatively small in the other two complexes.
In the crystal structure of the actin-rhizopodin complex, two actin molecules almost directly face each other (Fig. 1b). This arrangement facilitates PPI at SD1 of one actin and SD3 of the other actin, and vice versa. In fact, two pairs of electrostatic attractions by oppositely charged residues (Lys328 and Asp25) are observed, along with several hydrophobic interactions, such as at Gly23, Asp24, Pro333, and Glu334 (Fig. S4, ESI†). Meanwhile, our MD simulations for 100 ns revealed that these PPIs and interactions with rhizopodin were well maintained (Fig. S5 and S6, ESI†). As for 3 and 4, both of their side-chain moieties interacted with actin and stabilized PPIs between SD1 and SD3 of two actins, similar to rhizopodin (Fig. 3a). The protein–ligand interactions diagram calculated with MOE also showed that the hydrophobic contacts of 3 and 4 on actin were highly conserved with those of the C29–C34 part of ApA8c,9 and 1,19 including Tyr143, Gly146, Thr148, Gly168, Tyr169, Leu346, Leu349, Thr351, Phe352, and Met355 (Fig. S6, ESI†). However, the two actins had a highly distorted and twisted orientation on the actin–3 complex, whereas they aligned parallel and faced frontally on the actin-4 complex. In the actin-3 complex, several residues around the ligand participated in the PPIs, such as G146, L349, S350, and T351, which were not included in the actin–rhizopodin complex (Fig. S7, ESI†).
 | ||
| Fig. 3 Structures of actin dimer complexes with 3 and 4 obtained by MD simulation for 100 ns. (a) PPI analysis. Interacting residues of the two actin chains are shown as magenta and cyan surface models, respectively. Ligands are highlighted with green stick models. (b) Structures of the ligands on the actin complex. Two molecules of ligand 1 on actin (grey) obtained by the docking simulation using induced-fit models19 are superimposed. The lactone carbonyl carbon atoms and terminal enamide nitrogen atoms are highlighted with pink and blue circles, respectively. The distances between the two ester atoms and the two terminal enamide N atoms of the ligands are shown in Å. | ||
The distance between the two linker ester carbons in the actin-3 complex was only 3.2 Å, which was much smaller than those of rhizopodin and 4 (10.2 and 8.1 Å, respectively) (Fig. 3b). Therefore, the two actins bound to 3 had their ligand-binding sites closer to each other than those of rhizopodin and needed to adopt a highly distorted structure. This effect might enable 3 to sequester the actin dimer from the microfilament network, and to enhance in vitro actin-depolymerizing activity. On the other hand, 4 linked with sebacic acid maintained PPI between two actins at an appropriate distance similar to lysopodin. However, unlike rhizopodin (Fig. S5, ESI†), 4 did not induce PPI at the top site (around Ser239–Glu241), but instead induced a salt bridge between Asp25 and Arg147. These differences might give flexibility to the actin dimers with 4, and decrease activity.
For comparison, MD simulations of phthalate 5 and 4,4′-biphenyldicarboxylate 6 were performed for 50 ns (Fig. S8 and S9, ESI†). Due to rigid and conjugated sp2 carbon linkers, the distances of the terminal ester carbons in 5 and 6 are almost fixed at around 3.1 and 9.7 Å, respectively. As a result, 5 produced a distorted dimeric actin complex similar to 3, while 6 gave a parallel complex similar to 4, along with similar PPIs between SD1 and SD3 of actin (Fig. S10, ESI†). These results suggested that the length of the linkers was highly important for actin-binding side-chain dimers to fix their spatial arrangement and modulate PPIs on the actin dimer.
In summary, we developed structurally simplified side-chain dimers of an antitumor macrolide ApA that regulates actin-polymerization dynamics in a unique fashion. In particular, the C4 linker analog 3 depolymerized F-actin more potently than natural aplyronines by fixing the two actin-binding side-chains closer than those in bis-lactone rhizopodin. Further application of actin-dimerization inducers may promote studies on the modes of action related to cytoskeletal dynamics, as well as the development of PPI-based anticancer and other drug leads.
This work was supported in part by JSPS grants (19H02839, 23H03823 and 24K01635). Support was also provided by the Astellas Foundation for Research on Metabolic Disorders and the Naito Foundation.
Conflicts of interest
There are no conflicts to declare.References
- M. Kita and H. Kigoshi, Nat. Prod. Rep., 2015, 32, 534 RSC.
- (a) K. Yamada, M. Ojika, T. Ishigaki, Y. Yoshida, H. Ekimoto and M. Arakawa, J. Am. Chem. Soc., 1993, 115, 11020 CrossRef CAS; (b) M. Ojika, H. Kigoshi, T. Ishigaki, I. Tsukada, T. Tsuboi, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7441 CrossRef CAS.
- (a) H. Kigoshi, M. Ojika, T. Ishigaki, K. Suenaga, T. Mutou, A. Sakakura, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7443 CrossRef CAS; (b) I. Hayakawa, K. Saito, S. Matsumoto, S. Kobayashi, A. Taniguchi, K. Kobayashi, Y. Fujii, T. Kaneko and H. Kigoshi, Org. Biomol. Chem., 2017, 15, 124 RSC; (c) T. R. Pettigrew, R. J. Porter, S. J. Walsh, M. P. Housden, N. Y. S. Lam, J. S. Carroll, J. S. Parker, D. R. Spring and I. Paterson, Chem. Commun., 2020, 56, 1529 RSC; (d) T. Ohyoshi, A. Takano, I. Kikuchi, T. Ogura, M. Namiki, Y. Miyazaki, T. Hirano, S. Konishi, Y. Ebihara, K. Takeno, I. Hayakawa and H. Kigoshi, Org. Biomol. Chem., 2022, 20, 2922 RSC.
- (a) K. Yamada, M. Ojika, H. Kigoshi and K. Suenaga, Nat. Prod. Rep., 2009, 26, 27 RSC; (b) M. Kita and H. Kigoshi, in Handbook of Anticancer Drugs from Marine Origin, ed. S.-K. Kim, Springer, 2015, pp. 701–740 Search PubMed.
- (a) P. Liu and J. S. Panek, J. Am. Chem. Soc., 2000, 122, 1235 CrossRef CAS; (b) J. S. Panek and P. Liu, J. Am. Chem. Soc., 2000, 122, 11090 CrossRef CAS; (c) M. Kita, H. Oka, A. Usui, T. Ishitsuka, Y. Mogi, H. Watanabe, M. Tsunoda and H. Kigoshi, Angew. Chem., Int. Ed., 2015, 54, 14174 CrossRef CAS PubMed; (d) B. V. Pipaliya, D. N. Trofimova, R. L. Grange, M. Aeluri, X. Deng, K. Shah, A. W. Craig, J. S. Allingham and P. A. Evans, J. Am. Chem. Soc., 2021, 143, 6847 CrossRef CAS PubMed.
- (a) J. S. Allingham, A. Zampella, M. V. D'Auria and I. Rayment, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14527 CrossRef CAS PubMed; (b) I. Paterson, K. Ashton, R. Britton, G. Cecere, G. Chouraqui, G. J. Florence and J. Stafford, Angew. Chem., Int. Ed., 2007, 46, 6167 CrossRef PubMed.
- (a) H. Kigoshi, K. Suenaga, T. Mutou, T. Ishigaki, T. Atsumi, H. Ishiwata, A. Sakakura, T. Ogawa, M. Ojika and K. Yamada, J. Org. Chem., 1996, 61, 5326 CrossRef CAS; (b) S.-Y. Saito, S. Watabe, H. Ozaki, H. Kigoshi, K. Yamada, N. Fusetani and H. Karaki, J. Biochem., 1996, 120, 552 CrossRef CAS.
- (a) M. Kita, Y. Hirayama, K. Yoneda, K. Yamagishi, T. Chinen, T. Usui, E. Sumiya, M. Uesugi and H. Kigoshi, J. Am. Chem. Soc., 2013, 135, 18089 CrossRef CAS PubMed; (b) M. Kita, Y. Hirayama, K. Yamagishi, K. Yoneda, R. Fujisawa and H. Kigoshi, J. Am. Chem. Soc., 2012, 134, 20314 CrossRef CAS PubMed; (c) M. Kita, Y. Hirayama, M. Sugiyama and H. Kigoshi, Angew. Chem., Int. Ed., 2011, 50, 9871 CrossRef CAS.
- K. Hirata, S. Muraoka, K. Suenaga, T. Kuroda, K. Kato, H. Tanaka, M. Yamamoto, M. Takata, K. Yamada and H. Kigoshi, J. Mol. Biol., 2006, 356, 945 CrossRef CAS PubMed.
- (a) T. Kuroda, K. Suenaga, A. Sakakura, T. Handa, K. Okamoto and H. Kigoshi, Bioconjugate Chem., 2006, 17, 524 CrossRef CAS; (b) M. Kita, K. Yamagishi, K. Tsuchiya, Y. Seguchi, H. Nakane and H. Kigoshi, Bioorg. Med. Chem., 2017, 25, 6322 CrossRef CAS PubMed; (c) K. Yoneda, Y. Hu, M. Kita and H. Kigoshi, Sci. Rep., 2015, 5, 17853 CrossRef CAS PubMed.
- Y. Hirayama, K. Yamagishi, T. Suzuki, H. Kawagishi, M. Kita and H. Kigoshi, Bioorg. Med. Chem., 2016, 24, 2809 CrossRef CAS PubMed.
- (a) D. H. Utomo and M. Kita, Bull. Chem. Soc. Jpn., 2023, 96, 120 CrossRef CAS; (b) M. Kita, in New Tide of Natural Product Chemistry, ed. H. Ishikawa and H. Takayama, Springer, 2023, pp. 59–75 Search PubMed.
- (a) M. Kobayashi, J. Tanaka, T. Katori and I. Kitagawa, Chem. Pharm. Bull., 1990, 38, 2960 CrossRef CAS PubMed; (b) J. Tanaka, T. Higa, M. Kobayahshi and I. Kitagawa, Chem. Pharm. Bull., 1990, 38, 2967 CrossRef CAS; (c) M. R. Bubb, I. Spector, A. D. Bershadsky and E. D. Korn, J. Biol. Chem., 1995, 270, 3463 CrossRef CAS PubMed; (d) V. A. Klenchin, R. King, J. Tanaka, G. Marriott and I. Rayment, Chem. Biol., 2005, 12, 287 CrossRef CAS PubMed; (e) I. Shin, S. Hong and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14246 CrossRef CAS PubMed.
- S. Saito, S. Watabe, H. Ozaki, M. Kobayashi, T. Suzuki, H. Kobayashi, N. Fusetani and H. Karaki, J. Biochem., 1998, 123, 571 CrossRef CAS PubMed.
- (a) F. Sasse, H. Steinmetz, G. Höfle and H. Reichenbach, J. Antibiot., 1993, 46, 741 CrossRef CAS PubMed; (b) T. M. Gronewold, F. Sasse, H. Lünsdorf and H. Reichenbach, Cell Tissue Res., 1999, 295, 121 CrossRef CAS PubMed.
- (a) R. Jansen, H. Steinmetz, F. Sasse, W.-D. Schubert, G. Hagelüken, S. C. Albrecht and R. Mller, Tetrahedron Lett., 2008, 49, 5796 CrossRef CAS; (b) N. Horstmann and D. Menche, Chem. Commun., 2008, 5173 RSC.
- G. Hagelueken, S. C. Albrecht, H. Steinmetz, R. Jansen, D. W. Heinz, M. Kalesse and W. D. Schubert, Angew. Chem., Int. Ed., 2009, 48, 595 CrossRef CAS PubMed.
- K. Futaki, M. Takahashi, K. Tanabe, A. Fujieda, H. Kigoshi and M. Kita, ACS Omega, 2019, 4, 8598 CrossRef CAS PubMed.
- D. H. Utomo, A. Fujieda, K. Tanaka, M. Takahashi, K. Futaki, K. Tanabe, H. Kigoshi and M. Kita, Chem. Commun., 2021, 57, 10540 RSC.
- M. Kita, K. Yoneda, Y. Hirayama, K. Yamagishi, Y. Saito, Y. Sugiyama, Y. Miwa, O. Ohno, M. Morita, K. Suenaga and H. Kigoshi, ChemBioChem, 2012, 13, 1754 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01304b |
| This journal is © The Royal Society of Chemistry 2024 |