
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic aerobic α-oxygenation of amides to imides using a highly durable decatungstate tetraphenylphosphonium salt†
Chen
Gu
,
Takafumi
Yatabe
 ,
Kazuya
Yamaguchi
and
Kosuke
Suzuki
*
,
Kazuya
Yamaguchi
and
Kosuke
Suzuki
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ksuzuki@appchem.t.u-tokyo.ac.jp
First published on 10th April 2024
Abstract
Decatungstate is a potent photocatalyst for hydrogen atom transfer (HAT) but faces degradation issues when using a typical tetra-n-butylammonium salt. Herein, we employed tetraphenylphosphonium as a countercation to yield a highly durable and efficient HAT photocatalyst, enabling α-oxygenation of amides to their corresponding imides using O2 as an oxidant.
Hydrogen atom transfer (HAT) reactions, which involve homolytic cleavage of C–H bonds by hydrogen abstractors, are notable methods for the direct C–H functionalization of various organic molecules without prefunctionalization (directing group) and high acidity of C–H bonds in substrates.1,2 Photocatalysts play a crucial role in facilitating HAT as an eco-friendly method.2,3 Decatungstate ([W10O32]4−, W10), a type of metal oxide cluster (namely, polyoxometalate),4 is a widely employed HAT photocatalyst.3,5 Experimental and theoretical studies have elucidated its mechanism as a photocatalyst.6 Upon UV light irradiation, W10 undergoes intramolecular ligand-to-metal charge transfer (LMCT) from O2− to W6+, forming a singlet excited state. This excited state promptly decays to a relaxed state, wO, which exhibits remarkable reactivity and can react with substrates via both HAT and single-electron-transfer (SET) mechanisms. Exploiting the robust HAT capability of W10, various synthetic reactions, such as oxygenation, dehydrogenation, and C–X (X = C, N, S, F, Si) bond formation, have been possible.2,5,7–9 Typically, W10 is employed as a tetra-n-butylammonium (TBA) salt (TBAW10) in photocatalytic organic reactions because of its solubility in organic solvents and availability from chemical reagent suppliers. However, photoexcited W10 undergoes HAT not only with substrates but also with its countercation, TBA, decomposing TBA cations during photocatalytic reactions and causing various issues (though these are rarely mentioned in the literature)—decreased solubility and activity of TBAW10, difficulties in its reusability, and impurity formation in its products.10
To address the issues, we propose using a countercation with a structure that has a higher bond dissociation energy (BDE) than the alkyl group of TBA. Because the BDE of the C–H bond of benzene (113 kcal mol−1) is substantially higher than those of the C–H bonds of alkyl groups (95–100 kcal mol−1),11 aryl groups in cations may provide higher resistance to HAT by W10 photocatalysis while retaining solubility in organic solvents. Specifically, a tetraphenylphosphonium (TPP) cation, which possesses no structures other than aryl groups, is inherently more chemically and thermally stable than quaternary alkylammonium cations. Thus, employing TPP as the countercation of W10 promises considerable durability improvement compared with conventional alkylammonium cations.
Imides are essential structures in natural compounds and pharmaceuticals.12 Conventional methods for imide synthesis involve the acylation of amides with acyl halides,13 aldehydes,14 esters,15 thioesters,16 and acid anhydrides.17 Nevertheless, these methods require stoichiometric amounts of reactants, resulting in a high environmental impact. Therefore, the direct synthesis of imides through α-oxygenation of amides has garnered considerable attention18 because of its reduced number of reaction steps, use of various readily available amide structures, and the potential for using O2 as an oxidant under mild conditions, particularly when combined with photocatalysis. Several photocatalytic systems have been explored for this reaction; however, there are still challenges, such as limited substrate scope, excessive oxidant usage, and prolonged reaction time (Table S1, ESI†).12,18
In this study, we synthesized a highly durable and efficient W10 photocatalyst as a TPP salt (TPPW10, TPP4[W10O32]). Notably, TPPW10 demonstrated efficient photocatalysis for direct imide synthesis via α-oxygenation of several amides using O2 as an oxidant (Fig. 1). By employing TPP as a countercation, cation degradation by W10 during the photocatalytic reaction was circumvented, and TPPW10 exhibited substantial activity for the α-oxygenation compared with typical photocatalysts. Further, we demonstrated that TPPW10 can be readily recovered after the reaction and reused without loss of its high activity.
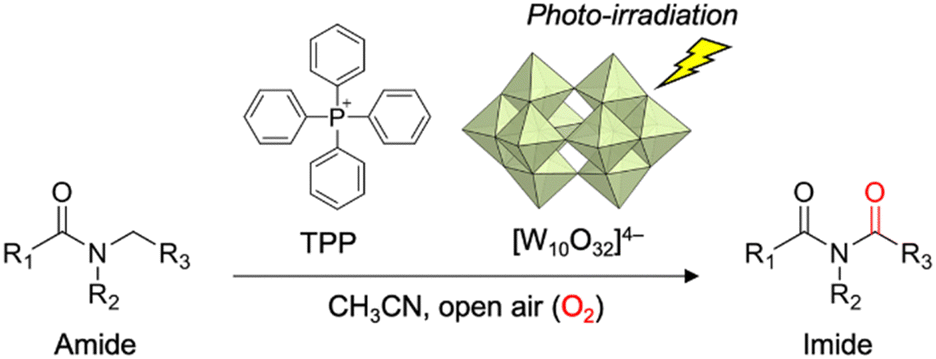 | ||
| Fig. 1 Direct imide synthesis via photocatalytic aerobic α-oxygenation of amides using highly durable and efficient TPPW10. | ||
Synthesis of TPPW10 was conducted via the cation exchange of an in situ formed sodium salt of W10 by reacting with excess amounts of TPP bromide in a hot aqueous solution (see ESI† for details). The obtained TPPW10 was characterized by electrospray ionization-mass spectrometry (ESI-MS), infrared (IR), UV-vis, 1H nuclear magnetic resonance (NMR), and 31P NMR spectroscopies. In the ESI-MS spectrum in acetonitrile (CH3CN), two sets of signals were observed at m/z 2193.4 and 4047.0 that are attributable to [TPP6W10O32]2+ and [TPP5W10O32]+, respectively (Fig. S1, ESI†). The IR spectrum exhibited characteristic absorption bands derived from W–O–W and W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of W10 (Fig. S2, ESI†). The UV-vis spectrum in CH3CN exhibited an absorption band of O-to-W LMCT (λmax = 324 nm) (Fig. S3, ESI†), and it was confirmed that the TPP cation has no absorption in the region above 300 nm (Fig. S4, ESI†). The 1H NMR spectrum exhibited signals ascribed to the aryl groups of TPP, and the 31P NMR spectrum exhibited a single peak attributed to the P atom in TPP (Fig. S5, ESI†). Moreover, single crystals suitable for the X-ray diffraction study were obtained by recrystallization in a mixed solvent of CH3CN and diethyl ether, and the TPPW10 structure was unambiguously determined using single-crystal structural analysis (Fig. S6, ESI†).
O bonds of W10 (Fig. S2, ESI†). The UV-vis spectrum in CH3CN exhibited an absorption band of O-to-W LMCT (λmax = 324 nm) (Fig. S3, ESI†), and it was confirmed that the TPP cation has no absorption in the region above 300 nm (Fig. S4, ESI†). The 1H NMR spectrum exhibited signals ascribed to the aryl groups of TPP, and the 31P NMR spectrum exhibited a single peak attributed to the P atom in TPP (Fig. S5, ESI†). Moreover, single crystals suitable for the X-ray diffraction study were obtained by recrystallization in a mixed solvent of CH3CN and diethyl ether, and the TPPW10 structure was unambiguously determined using single-crystal structural analysis (Fig. S6, ESI†).
We employed TPPW10 as a photocatalyst for aerobic α-oxygenation of amides to synthesize their corresponding imides. Initially, 1-methyl-2-pyrrolidone (1a) was used as a model substrate for the reaction (Table 1). With 0.7-mol% TPPW10, 1a was oxygenated and selectively converted to the corresponding imide product, N-methylsuccinimide (2a), in 83% yield by photoirradiation from a Xe lamp (λ > 350 nm) for 4 h under an air atmosphere in CH3CN (Table 1, entry 1; Fig. S7, ESI†). The reaction also proceeded using LED (λ = 365, 405 nm) instead of a Xe lamp (Table 1, entries 3 and 4). When using TBAW10, which is a widely used TBA salt of W10, the yield of 2a was 73% (Table 1, entry 5). The reaction did not proceed when performed in the dark or under an Ar atmosphere (Table 1, entries 6 and 7), indicating that the photoexcitation of W10 and O2 is essential for the reaction. In addition, oxygenation did not proceed when using sodium tungstate (Na2WO4; Table 1, entry 8). Other polyoxometalate photocatalysts, TBA3[α-PW12O40] and TBA4H[γ-PV2W10O40],19 were ineffective for the reaction (Table 1, entries 9 and 10). Notably, TPPW10 showed considerably higher activity than organic molecular HAT photocatalysts, such as benzophenone, xanthone, anthraquinone, 2-chloroanthraquinone, and eosin Y (Table 1, entry 1 vs. entries 11–15). Although TiO2 is a frequently-used heterogeneous inorganic photocatalyst, it showed lower 1a conversion and 2a yield compared to those by TPPW10, and byproduct 3a was generated in almost the same amount as 2a (Table 1, entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Conv. (%) | Yield (%) | |
| 1a | 2a | 3a | ||
| a Reaction conditions: 1a (0.6 mmol), photocatalyst (0.7 mol%), CH3CN (3 mL), open air, and 4-h photoirradiation using Xe lamp (λ > 350 nm). Conversions and yields were determined by gas chromatography analysis using N-methylphthalimide as an internal standard. LED (λ = 365 nm). b LED (λ = 405 nm). | ||||
| 1 | TPPW10 | >99 | 83 | 1 |
| 2 | TPPW10 (reuse) | >99 | 80 | 4 |
| 3a | TPPW10 (LED, 365 nm) | >99 | 70 | 1 |
| 4b | TPPW10 (LED, 405 nm) | >99 | 73 | 1 |
| 5 | TBAW10 | >99 | 73 | 1 |
| 6 | TPPW10 (dark) | <1 | <1 | <1 |
| 7 | TPPW10 (Ar) | <1 | <1 | <1 |
| 8 | Na2WO4 | 2 | <1 | <1 |
| 9 | TBA3[α-PW12O40] | 4 | 2 | <1 |
| 10 | TBA4H[γ-PV2W10O40] | 10 | 2 | 1 |
| 11 | Benzophenone | 43 | 18 | 2 |
| 12 | Xanthone | 18 | 6 | <1 |
| 13 | Anthraquinone | 23 | 9 | 1 |
| 14 | 2-Chloroanthraquinone | 14 | 8 | 2 |
| 15 | Eosin Y | 17 | 8 | 1 |
| 16 | TiO2 (P25) | 32 | 9 | 7 |
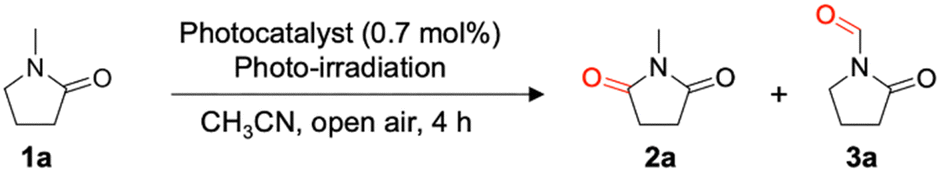
To investigate the structures of TPP cation and W10 anion after the photocatalytic α-oxygenation of 1a under the conditions described in Table 1, TPPW10 was recovered as a powder by adding the concentrated reaction solution to ethyl acetate, a poor solvent for TPPW10, followed by filtration and vacuum drying (Fig. S8, ESI†). The 1H NMR spectrum of the recovered TPPW10 exhibited signals in the range of 7.6–8.0 ppm, attributable to the aryl groups of TPP, and other signals derived from the oxidation of TPP were unobserved (Fig. 2a). The 31P NMR spectrum showed only a single signal at 23.3 ppm, attributable to the P atom in TPP (Fig. 2b). These results confirm that the TPP cation structure was maintained after the photocatalytic reaction. Using IR spectroscopy, we also confirmed that the W10 anion structure was maintained after the photocatalytic reaction (Fig. 2c and Fig. S9, ESI†). Further, the ESI-MS spectrum of the recovered catalyst exhibited signals attributable to TPPW10 (m/z 2194.0 and 4047.5 for [TPP6W10O32]2+ and [TPP5W10O32]+, respectively; Fig. S10, ESI†). Notably, the recovered TPPW10 can be reused for imide synthesis without loss of its high photocatalytic activity; 1a was converted to 2a in 80% yield using the recovered TPPW10 (0.7 mol%) after 4-h photoirradiation (Table 1, entry 2).
 | ||
| Fig. 2 (a) 1H, (b) 31P nuclear magnetic resonance spectra (CD3CN), and (c) infrared spectra of the fresh and recovered TPPW10 after the photocatalytic aerobic α-oxygenation of 1a. | ||
Although we also attempted to recover TBAW10via the same procedure used for TPPW10 after the photocatalytic reaction of 1a, a sticky sample was formed, and the catalyst was difficult to be recovered by filtration. When we performed a reuse experiment for the photocatalytic reaction of 1a using the sticky catalyst, the yield of 2a became notably lower (45% yield) than the reaction using fresh TBAW10 (73% yield). To analyze the structure of TBA cations after the reaction, we performed the photocatalytic α-oxygenation of 1a using TBAW10 in deuterated acetonitrile (CD3CN) and measured the 1H NMR spectrum of the reaction solution after a 4-h reaction. The 1H NMR spectrum showed that the peaks of the alkyl groups of TBA completely disappeared (Fig. S11, ESI†), indicating TBA degradation.
Next, we investigated the reaction mechanism by performing several experiments. When a radical scavenger, 2,2,6,6-tetramethylpiperidine 1-oxyl, was added to the reaction solution, the conversion of 1a and yield of 2a considerably decreased (Table S2, ESI†), suggesting that the reaction proceeded via radical generation. To identify the oxygen source in the reaction, 18O-isotope labeling experiments were performed (Fig. 3), and the products were analyzed by gas chromatography (GC) and GC-mass spectrometry (GC-MS). When 1a α-oxygenation was performed using TPPW10 photocatalysis under an 18O2 atmosphere, 2a was obtained in 81% yield. The formation of 18O-labeled 2a with an 18O content of 88% was revealed via GC-MS analysis using the ratio of peak intensities at m/z 115 (18O-labeled 2a) and m/z 113 (2a) (Fig. S12, ESI†). In addition, when 1a α-oxygenation was performed in the presence of H218O (59 mg, 5 equivalents with respect to 1a) under an air atmosphere, GC-MS analysis revealed the formation of 18O-labeled 2a with an 18O content of 32%, indicating that H2O also acted as the oxygen source of the reaction (minor path; Fig. S13a, ESI†). Even when the amount of H218O was increased (294 mg, 15 equivalents with respect to 1a), the 18O content remained almost unchanged (Fig. S13b, ESI†). Therefore, the major oxygen source of this reaction is O2 rather than H2O.
 | ||
| Fig. 3 18O labeling experiments for the photocatalytic aerobic α-oxygenation of 1a using TPPW10: (a) under an 18O2 atmosphere and (b) in the presence of H218O (59 mg). Reaction conditions: 1a (0.6 mmol), TPPW10 (0.7 mol%), CH3CN (3 mL), and 4-h photoirradiation using Xe lamp (λ > 350 nm). | ||
Based on the above results, we proposed a plausible reaction mechanism (Fig. 4). A carbon-centered radical was generated through the HAT process on the α-position of 1a by the photoexcited W10. In contrast, it has been reported that SET on 1a by photoexcited W10 does not occur.20 The generated carbon-centered radical on 1a reacted with O2 to form a peroxyl radical (R–OO˙), which further reacted with 1a or the reduced W10 to form hydroperoxide, finally resulting in the formation of 2a (major path A). Another plausible reaction path involves the reaction with H2O (minor path B): the carbon-centered radical through HAT on 1a reacted with the photoexcited W10 through SET to form an iminium cation, which underwent nucleophilic attack by H2O to form hydroxylated 1a and then further oxidized to form 2a.
 | ||
| Fig. 4 Proposed reaction mechanism for the photocatalytic aerobic α-oxygenation of 1a to 2a using W10via hydrogen atom transfer (HAT, major path A) and single electron transfer (SET, minor path B). | ||
Finally, we investigated the substrate scope of the TPPW10-catalyzed system (Fig. 5). The reactions of pyrrolidone derivatives with methyl, ethyl, and phenyl groups substituting the N atoms proceeded efficiently to yield their corresponding imides (2a–2c). In this process, cyclic methylene moieties were selectively oxidized. This was applicable even when the ring size of the cyclic amides increased; for example, a corresponding imide (2d) was selectively obtained from 1-methyl-2-piperidone. Not only cyclic tertiary amides but also noncyclic amides can be applied to the reaction; for example, the reaction of N-methyacetamide yielded the corresponding imide 2g. Notably, using the TPPW10-catalyzed system, various secondary cyclic, noncyclic, and benzylic amides can also be converted to their corresponding imides (2f–2j). However, this system was not effective to obtain imide products 2k and 2l owing to dealkylation during the photocatalytic reactions.
 | ||
Fig. 5 Substrate scope for photocatalytic aerobic α-oxygenation of 1a using TPPW10. Reaction conditions: substrate (0.6 mmol), TPPW10 (0.7 mol%), CH3CN (3 mL), open air, and 4-h photoirradiation using Xe lamp (λ > 350 nm). Yields were determined by gas chromatography using N-methylphthalimide as internal standard (n.d. = not detected). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 h, b6 h. 5 h, b6 h. | ||
In conclusion, we synthesized a highly durable and reactive decatungstate tetraphenylphosphonium salt (TPPW10), which efficiently promoted the photocatalytic aerobic α-oxygenation of various tertiary and secondary amides to their corresponding imides. TPPW10 showed significantly higher activity for the photocatalytic aerobic α-oxygenation of imides than previously reported photocatalytic systems. Further, TPPW10 can be easily recovered after the photocatalytic reaction and reused for the photocatalytic reaction without notable loss of its high activity; the structures of both the TPP cation and W10 anion were maintained in the recovered catalyst.
This study was supported in part by JST FOREST (JPMJFR213M), JSPS KAKENHI (22H04971), and the JSPS Core-to-Core program. We would like to thank Dr Li and Mr Hachimura for their contribution in the preliminary experiments.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36 CrossRef CAS PubMed
.
-
(a) L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376 RSC
-
(a) A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1 RSC
-
(a)
M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 CrossRef
-
(a) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701 CrossRef CAS
-
(a) C. Tanielian, Coord. Chem. Rev., 1998, 178, 1165 CrossRef
-
(a) C. Tanielian, K. Duffy and A. Jones, J. Phys. Chem. B, 1997, 101, 4276 CrossRef CAS
- J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef PubMed
-
(a) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92 CrossRef CAS PubMed
-
(a) T. Yamase, N. Takabayashi and M. Kaji, J. Chem. Soc., Dalton Trans., 1984, 793 RSC
-
(a)
Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 CrossRef
- J. Sperry, Synthesis, 2011, 3569 CrossRef CAS
- M. Klinge, H. Cheng, T. M. Zabriskie and J. C. Vederas, J. Chem. Soc., Chem. Commun., 1994, 1379 RSC
- L. Wang, H. Fu, Y. Jiang and Y. Zhao, Chem. – Eur. J., 2008, 14, 10722 CrossRef CAS PubMed
- M. B. Andrus, W. Li and R. F. Keyes, Tetrahedron Lett., 1998, 39, 5465 CrossRef CAS
- F. Wang, H. Liu, H. Fu, Y. Jiang and Y. Zhao, Adv. Synth. Catal., 2009, 351, 246 CrossRef CAS
- D. Davidson and H. Skovronek, J. Am. Chem. Soc., 1958, 80, 376 CrossRef CAS
-
(a) J. C. Gramain, R. Remuson and Y. Troin, J. Chem. Soc., Chem. Commun., 1976, 194 RSC
- C. Li, K. Suzuki, N. Mizuno and K. Yamaguchi, Chem. Commun., 2018, 54, 7127 RSC
- S. Angioni, D. Ravelli, D. Emma, D. Dondi, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2008, 350, 2209 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods and spectroscopic data. CCDC 2336024. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01016g |
| This journal is © The Royal Society of Chemistry 2024 |