
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrongly reducing helical phenothiazines as recyclable organophotoredox catalysts†
Haru
Ando
a,
Hiroyoshi
Takamura
 a,
Isao
Kadota
*a and
Kenta
Tanaka
*b
a,
Isao
Kadota
*a and
Kenta
Tanaka
*b
aDepartment of Chemistry, Graduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushima-Naka, Kitaku, Okayama 700-8530, Japan. E-mail: kadota-i@okayama-u.ac.jp
bResearch Institute for Interdisciplinary Science, Okayama University, 3-1-1 Tsushima-Naka, Kitaku, Okayama 700-8530, Japan. E-mail: ktanaka@okayama-u.ac.jp
First published on 19th March 2024
Abstract
Recyclable phenothiazine organophotoredox catalysts (PTHS 1–3, E1/2ox* = −2.34 to −2.40 V vs. SCE) have been developed. When the recycling performance was evaluated, PTHS-1 could be recovered at least four times without loss of its catalytic activity. These recyclable organophotoredox catalysts represent a promising tool for sustainable organic synthesis.
Recycling photocatalysts has attracted much attention as a critical factor in emerging chemical technologies in terms of environmental concerns and economic benefits. While recycling heterogeneous photocatalysts such as semiconductors and polymers has been widely developed due to their easy separation and reusability, that of homogeneous photocatalysts such as metal-based polypyridyl complexes of ruthenium and iridium as well as organic dyes has received less attention.1 Although there are some examples of recycling metal-based polypyridyl complexes,2 recycling organophotocatalysts remains much less explored and is usually limited to the use of polymer methods.3 Recently, a nanofiltration process for the recovery of the 4CzIPN organophotocatalyst under continuous-flow conditions has been reported, albeit the recycling performance was not examined.4 Considering that organophotoredox catalysts are cost-effective and of low toxicity, the development of an approach for their recycling is highly desirable in the context of sustainable organic synthesis.
10-Aryl phenothiazines are widely used as photocatalysts for photoredox reactions and atom-transfer-radical-addition polymerizations in organic chemistry (Fig. 1; e.g., PTH-1).5 Due to their low excited-state oxidation potentials (E1/2ox* ≈ −2.10 V vs. SCE), a number of 10-aryl-phenothiazine-catalyzed photoredox reactions that proceed via oxidative quenching cycles have been developed.6 However, the high reactivity of the p-position relative to the nitrogen atom on 10-aryl phenothiazines renders these prone to reacting with electrophiles.7 Thus, several modified 10-aryl phenothiazine catalysts have recently been explored (Fig. 1; PTH 2–4).8 Despite these advances, the development of more stable and sustainable photoredox catalysts remains highly desirable.
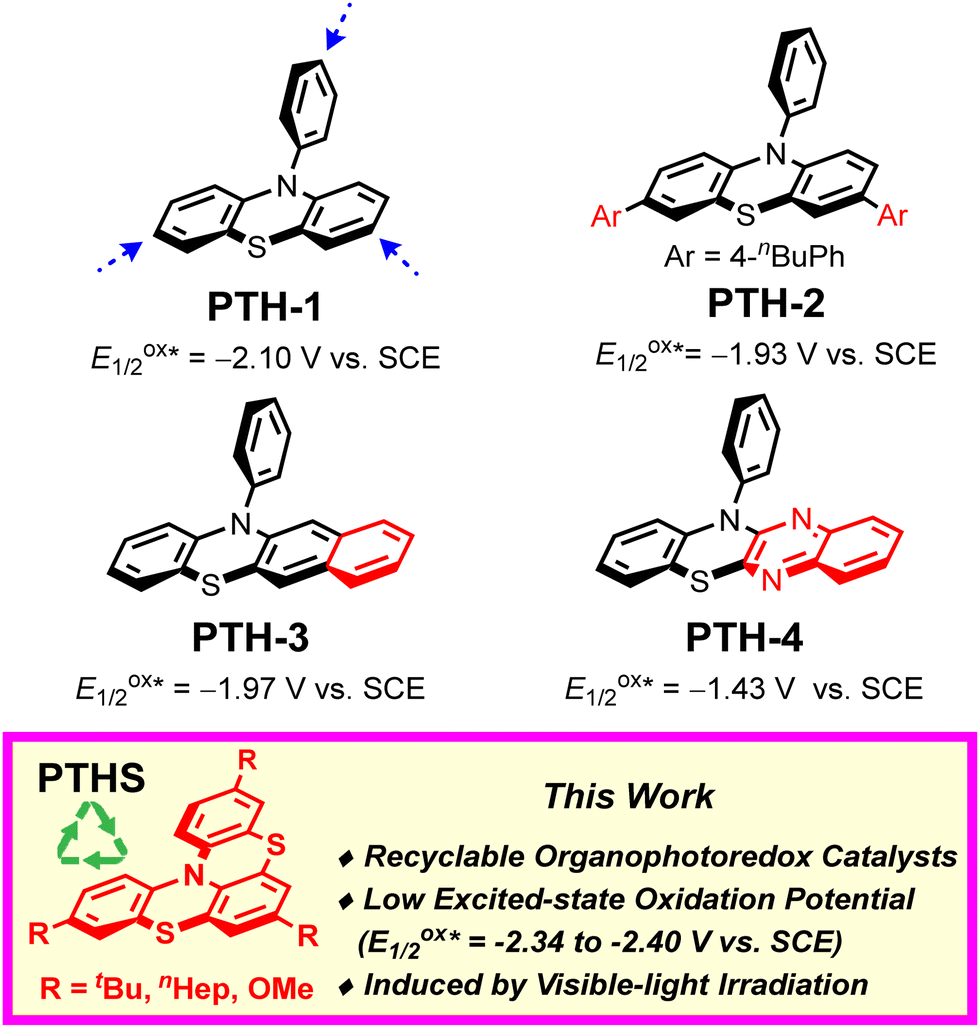 | ||
| Fig. 1 Representative phenothiazine catalysts. | ||
To explore the recyclability of organophotoredox photocatalysts, we have designed and synthesized recyclable phenothiazine organophotoredox catalysts (Fig. 1; PTHS).9 The catalyst design is based on the following considerations: (i) in order to increase the stability of the catalyst and the reducing properties, bulky and electron-donating groups such as the tBu group are introduced at the p-position relative to the nitrogen atom on the phenothiazine catalysts;10 (ii) in order to absorb visible light and increase the stability of the radical intermediate, the phenothiazine catalysts are endowed with a thia-bridged helically shaped structure, which expands the π conjugation compared to 10-phenylphenothiazine (PTH-1).10
We initially synthesized a series of phenothiazine catalysts (PTHS 1–3) that were obtained in short steps and a moderate yield using commercially available starting materials to probe the relationship between their structure and physical properties (Table 1).11,12 Interestingly, we found that these catalysts have low excited-state oxidation potentials (E1/2ox* = −2.34 to −2.40 V vs. SCE) compared to other phenothiazine catalysts such as PTH 1–4. Therefore, PTHS 1–3 were expected to reduce various substrates via oxidative quenching cycles. In addition, the PTHS catalysts exhibit an absorption band in the visible spectrum, which indicates that they can be activated by visible light.
|
|
|||||
|---|---|---|---|---|---|
| Catalyst | E 1/2(C˙+/C*)a (V) | E 1/2(C˙+/C)b (V) | E 0,0 (eV) | Excitation λmax (nm) | Emission λmax (nm) |
| a Excited-state oxidation potentials were estimated on the basis of the ground-state redox potentials and the intersection of the absorption and emission bands. b Determined by cyclic voltammetry in CH2Cl2vs. SCE.1 | |||||
| PTHS-1 | −2.34 | 0.86 | 3.20 | 317 | 449 |
| PTHS-2 | −2.35 | 0.80 | 3.15 | 316 | 460 |
| PTHS-3 | −2.40 | 0.64 | 3.04 | 314 | 475 |

With these promising results in hand, we applied the PTHS catalysts to various types of photoredox reactions. First, we examined the three-component oxytrifluoromethylation of 1,1-diphenylethylene (2; Table 2).13 The reactions proceeded smoothly in the presence of a catalytic amount of PTHS 1–3 (1.0 mol%) to give the desired product (3) in a good yield. In addition, a variety of alkenes can be applied to the reaction.11 Blank experiments in the absence of a catalyst or light confirmed that the reaction requires a PTHS catalyst and irradiation with blue LEDs to proceed.11 Since the PTHS catalysts have low excited-state oxidation potentials, the CF3 radical was smoothly generated from Umemoto's reagent (1; Ep/2 = −0.25 V vs. SCE)14 and then reacted with 2 to give the desired product (3). This organophotocatalytic reaction is significantly more cost-effective and sustainable than previously reported methods based on transition-metal catalysts.13
|
|
||
|---|---|---|
| Entry | Catalyst | Yield (%) |
a All reactions were carried out with 1 (0.105 mmol), 2 (0.1 mmol), and the catalyst (1.0 mol%) in acetone/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). :1, v/v) at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W).
|
||
| 1 | PTHS-1 | 79 |
| 2 | PTHS-2 | 72 |
| 3 | PTHS-3 | 80 |

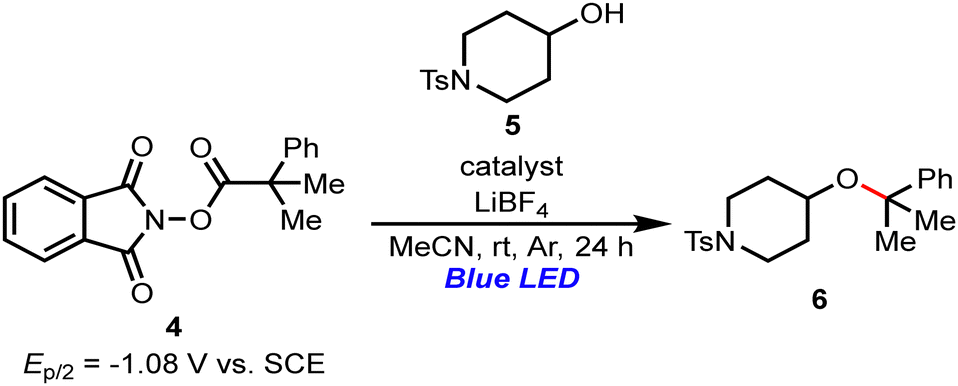
Next, we investigated a visible-light-mediated decarboxylative C(sp3)–O bond formation (Table 3). Nagao and Ohmiya have reported that PTH-3 catalyzes the decarboxylative coupling between aliphatic alcohol 5 and redox-active esters such as 4 (Ep/2 = −1.08 V vs. SCE).8b It thus seems feasible to speculate that the PTHS catalysts might also be applied to the decarboxylative C(sp3)–O bond formation from ester 4 to provide the corresponding ether (6) in a moderate yield.
|
|
||
|---|---|---|
| Entry | Catalyst | Yield (%) |
| a All reactions were carried out with 4 (0.2 mmol), 5 (0.6 mmol), LiBF4 (10 mol%), and the catalyst (10 mol%) in MeCN at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). | ||
| 1 | PTHS-1 | 60 |
| 2 | PTHS-2 | 48 |
| 3 | PTHS-3 | 40 |
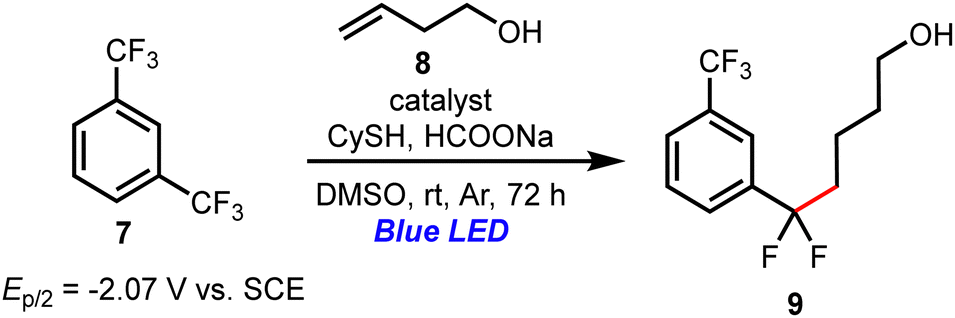
The results of the defluoroalkylation of 1,3-bis(trifluoromethyl)benzene (7) with unactivated alkenes are shown in Table 4.15 When 7 (Ep/2 = −2.07 V vs. SCE) was treated with 3-buten-1-ol (8) in the presence of PTHS-1 and PTHS-2, the reaction proceeded effectively to give 9 in a moderate to good yield (Table 4, entries 1 and 2). In contrast, PTHS-3 furnished a trace amount of the product and unknown byproducts, and 1,3-bis(trifluoromethyl)benzene was not recovered due to its low boiling point (Table 4, entry 3). PTHS-3 has a low oxidation potential compared to PTHS-1 and 2 (PTHS-3: E1/2(C˙+/C) = 0.64 V vs. SCE, PTHS-1: E1/2(C˙+/C) = 0.86 V vs. SCE, PTHS-2: E1/2(C˙+/C) = 0.80 V vs. SCE), suggesting that the oxidation of sodium formate would not proceed efficiently.15 Given that the excited-state oxidation potential of PTH-3 is lower than the reduction potential of 7, the reaction with PTH-3 was also inefficient (Table 4, entry 4).
|
|
||
|---|---|---|
| Entry | Catalyst | Yield (%) |
| a All reactions were carried out with 7 (0.1 mmol), 8 (0.3 mmol), cyclohexane thiol (10 mol%), sodium formate (0.3 mmol), and the catalyst (10 mol%) in DMSO at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). | ||
| 1 | PTHS-1 | 83 |
| 2 | PTHS-2 | 47 |
| 3 | PTHS-3 | Trace |
| 4 | PTH-3 | 6 |
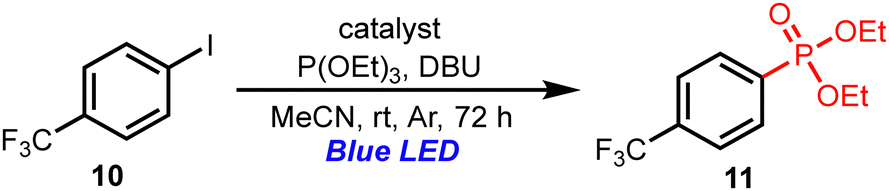
Subsequently, we investigated the photoredox cross-coupling reaction between 4-trifluoromethyliodobenzene (10) and triethylphosphite in the presence of the PTHS catalysts, which afforded aromatic phosphonate 11 in good yield (Table 5).16 Although 10 has a low reduction potential (Ep/2 = −2.16 V vs. SCE),17 the single-electron transfer from the PTHS catalysts to 10 is energetically favorable due to the low excited-state oxidation potentials of the PTHS catalysts (E1/2ox* = −2.34 to −2.40 V vs. SCE). Moreover, various aryl iodides can be suitable for the reactions to give the corresponding products in high yields.11 Accordingly, the PTHS catalysts are suitable for photoredox reactions via oxidation-quenching cycles.
|
|
||
|---|---|---|
| Entry | Catalyst | Yield (%) |
| a All reactions were carried out with 10 (0.1 mmol), triethylphosphite (0.3 mmol), DBU (0.2 mmol), and the catalyst (10 mol%) in MeCN at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). | ||
| 1 | PTHS-1 | 77 |
| 2 | PTHS-2 | 78 |
| 3 | PTHS-3 | 78 |

To examine the stability of the catalysts, we carried out the photochemical sulfonylation of PTH-1 and PTHS-1 (Scheme 1). When PTH-1 was treated with tosyl chloride (TsCl) under irradiation with blue LEDs, monosulfonylated 12 was obtained in 78% yield due to the high reactivity of the p-position relative to the nitrogen atom in 10-aryl phenothiazines.8a In contrast, PTHS-1 was effectively recovered in 95% yield, proving that the presence of tBu groups increases the catalyst stability. Therefore, PTHS-1 is applicable to various photoredox reactions, which cannot be effectively achieved by hitherto reported phenothiazine catalysts.
 | ||
| Scheme 1 Photochemical sulfonylation of phenothiazines. All reactions were carried out with phenothiazines (0.2 mmol) and tosyl chloride (TsCl; 0.2 mmol) in MeCN at room temperature for 24 h under an N2 atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). aPreviously reported product yield.7a | ||
The high stability of PTHS-1 also prompted us to investigate its recycling performance (Table 6). After completion of the cross-coupling reaction of 10 with triethylphosphite, PTHS-1 was collected via extraction with EtOAc and column chromatography, and the ability of recycled PTHS-1 to catalyze the reaction was examined. PTHS-1 could be effectively recovered at least four times without loss of its catalytic activity. In contrast, PTH-1 suffered from an obvious loss of catalytic activity. Thus, PTHS-1 is demonstrably a recyclable organophotocatalyst and suitable for sustainable synthetic methods.
|
|
||||
|---|---|---|---|---|
| Run | 1 | 2 | 3 | 4 |
| a All reactions were carried out with 10 (0.1 mmol), triethylphosphite (0.3 mmol), DBU (0.2 mmol), and the catalyst (10 mol%) in MeCN at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). | ||||
| PTHS-1 yield (%) | 77 | 78 | 75 | 79 |
| PTH-1 yield (%) | 54 | 32 | 23 | 9 |
Finally, when the reaction was performed on a gram scale, the desired product was obtained in 85% yield (1.20 g) with 96% recovery of PTHS-1 (Scheme 2). Thus, PTHS-1 is a highly active catalyst with high recoverability even when used on a gram scale.
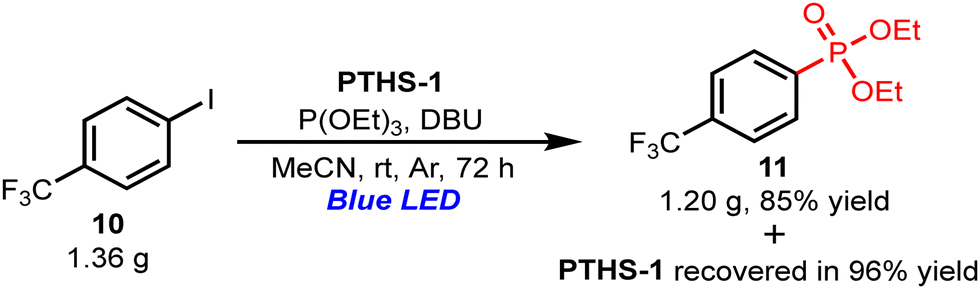 | ||
| Scheme 2 Gram-scale photoredox cross-coupling reaction of 4-trifluoromethylbenzene with triethylphosphite. The reaction was carried out with 10 (5.0 mmol), triethylphosphite (15.0 mmol), DBU (10.0 mmol), and PTHS-1 (10 mol%) in MeCN at room temperature under an Ar atmosphere and blue LED irradiation (λmax = 425 nm, 18 W). | ||
In summary, we have developed strongly reducing and recyclable phenothiazine organophotoredox catalysts (PTHS 1–3) that can be activated by visible light. These catalysts exhibit relatively low excited-state oxidation potentials (E1/2ox* = −2.34 to −2.40 V vs. SCE). The PTHS-1 can be recovered at least four times without loss of its catalytic activity in the cross-coupling reaction of 4-trifluoromethyliodobenzene (Ep/2 = −2.16 V vs. SCE), highlighting its potential for sustainable photocatalysis. These recyclable organophotoredox catalysts thus represent a promising tool for sustainable organic synthesis.
This work was supported by JKA and its promotion funds from KEIRIN RACE, Murata Science Foundation, and Wesco Scientific Promotion Foundation. We appreciate the assistance of the Division of Instrumental Analysis at Okayama University with NMR spectroscopy and high-resolution mass spectrometry.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Abramov, S. Bonardd, R. Pérez-Ruiz and D. D. Díaz, Adv. Synth. Catal., 2022, 364, 2–17 CrossRef CAS; (b) M. Zheng, I. Ghosh, B. Konig and X. Wang, ChemCatChem, 2019, 11, 703–706 CrossRef CAS.
- (a) Z. Wu, X. Liao, L. Yuan, Y. Wang, Y. Zheng, J. Zuo and Y. Pan, Chem. – Eur. J., 2020, 26, 5694–5700 CrossRef CAS PubMed; (b) X. Zhang, Y. Li, X. Hao, K. Jun, R. Zhang and C. Duan, Tetrahedron, 2018, 74, 7358–7363 CrossRef CAS; (c) X. Zhang, Y. Li, X. Hao, X. Jin, K. Jin, R. Zhang and C. Duan, Tetrahedron, 2018, 74, 1742–1748 CrossRef CAS; (d) J. Xia, G. Deng, M. Zhou, W. Liu, P. Xie and J. Li, Synlett, 2012, 2707–2713 CAS.
- (a) J. J. Lessard, G. M. Scheutz, A. B. Korpusik, R. A. Olson, C. A. Figg and B. S. Sumerlin, Polym. Chem., 2021, 12, 2205–2209 RSC; (b) M. Peavy and C. Hobbs, Tetrahedron Lett., 2021, 65, 152759 CrossRef CAS.
- Z. Wen, D. Pintossi, M. Nuño and T. Noël, Nat. Commun., 2022, 13, 6147 CrossRef CAS PubMed.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS.
- Representative report see: O. P. Williams, A. F. Chmiel, M. Mikhael, D. M. Bates, C. S. Yeung and Z. K. Wickens, Angew. Chem., Int. Ed., 2023, 62, e202300178 CrossRef CAS PubMed.
- (a) J. Liu, H. Liu, X. Guo, Z. Wang, X. Wu, J. Li and C. Zhu, Green Chem., 2023, 25, 3847–3851 RSC; (b) A. Jiménez-Almarza, A. López-Magano, R. Mas-Ballesté and J. Alemán, ACS Appl. Mater. Interfaces, 2022, 14, 16258–16268 CrossRef; (c) S. Jana, C. Empel, C. Pei, T. V. Nguyen and R. M. Koenigs, Adv. Synth. Catal., 2020, 362, 5721–5727 CrossRef CAS; (d) C. Liu, Y. Shen and K. Yuan, Org. Biomol. Chem., 2019, 17, 5009–5013 RSC; (e) Y. Ahn, D. W. Jang, Y. Cha, M. Kim, K. Ahn and Y. C. Kim, Bull. Korean Chem. Soc., 2013, 34, 107–111 CrossRef CAS.
- (a) D. M. Fischer, H. Lindner, W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 774–780 CrossRef CAS PubMed; (b) S. Shibutani, T. Kodo, M. Takeda, K. Nagao, N. Tokunaga, Y. Sasaki and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 1211–1216 CrossRef CAS PubMed; (c) Y. Zhao, H. Gong, K. Jiang, S. Yan, J. Lin and M. Chen, Macromolecules, 2018, 51, 938–946 CrossRef CAS; (d) H. Zhou, L. Zhang, P. Wen, Y. Zhou, Y. Zhao, Q. Zhao, Y. Gu, R. Bai and M. Chen, Angew. Chem., Int. Ed., 2023, 62, e202304461 CrossRef CAS; (e) Q. Quan, Y. Zhao, K. Chen, H. Zhou, C. Zhou and M. Chen, ACS Catal., 2022, 12, 7269–7277 CrossRef CAS.
- Our previous work of organophotoredox reactions see: (a) K. Tanaka, M. Kishimoto, Y. Tanaka, Y. Kamiyama, Y. Asada, M. Sukekawa, N. Ohtsuka, T. Suzuki, N. Momiyama, K. Honda and Y. Hoshino, J. Org. Chem., 2022, 87, 3319–3328 CrossRef CAS; (b) K. Tanaka, Y. Asada and Y. Hoshino, Chem. Commun., 2022, 58, 2476–2479 RSC; (c) K. Tanaka, Y. Iwama, M. Kishimoto, N. Ohtsuka, Y. Hoshino and K. Honda, Org. Lett., 2020, 22, 5207–5211 CrossRef CAS; (d) K. Tanaka, Y. Hoshino and K. Honda, Shikizai Kyokaishi, 2020, 93, 49–53 CAS; (e) K. Tanaka, D. Omata, Y. Asada, Y. Hoshino and K. Honda, J. Org. Chem., 2019, 84, 10669–10678 CrossRef CAS PubMed; (f) K. Tanaka, Y. Asada, Y. Hoshino and K. Honda, Org. Biomol. Chem., 2020, 18, 8074–8078 RSC; (g) K. Tanaka, Y. Tanaka, M. Kishimoto, Y. Hoshino and K. Honda, J. Org. Chem., 2019, 15, 2105–2112 CAS; (h) K. Tanaka, M. Kishimoto, M. Sukekawa, Y. Hoshino and K. Honda, Tetrahedron Lett., 2018, 59, 3361–3364 CrossRef CAS.
- (a) A. Joshi-Pangu, F. Levesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed; (b) J. Li, C.-Y. Huang, J.-T. Han and C.-J. Li, ACS Catal., 2021, 11, 14148–14158 CrossRef CAS; (c) E. Alfonzo, F. S. Alfonso and A. B. Beeler, Org. Lett., 2017, 19, 2989–2992 CrossRef CAS PubMed; (d) S. Arikawa, A. Shimizu, D. Shiomi, K. Sato, T. Takui, H. Sotome, H. Miyasaka, M. Murai, S. Yamaguchi and R. Shintani, Angew. Chem., Int. Ed., 2023, 62, e202302714 CrossRef CAS; (e) L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS.
- See ESI†.
- (a) S. Chen, Z. Li, K. Hu, W. Feng, G. Mao, F. Xiao and G. Deng, Org. Biomol. Chem., 2023, 21, 1920–1926 RSC; (b) G. Lamanna, C. Faggi, F. Gasparrini, A. Ciogli, C. Villani, P. J. Stephens, F. J. Devlin and S. Menichetti, Chem. Eur. J., 2008, 14, 5747–5750 CrossRef CAS PubMed; (c) B. D. Gliemann, A. G. Petrovic, E. M. Zolnhofer, P. O. Dral, F. Hampel, G. Breitenbruch, P. Schulze, V. Raghavan, K. Meyer, P. L. Polavarapu, N. Berova and M. Kivala, Chem. Asian J., 2017, 12, 31–35 CrossRef CAS; (d) S. Miguez-Lago, B. D. Gliemann, M. Kivala and M. M. Cid, Chem. Eur. J., 2021, 27, 13352–13357 CrossRef CAS PubMed; (e) R. Amorati, L. Valgimigli, A. Baschieri, Y. Guo, F. Mollica, S. Menichetti, M. Lupi and C. Viglianisi, ChemPhysChem, 2021, 22, 1446–1454 CrossRef CAS PubMed; (f) S. Menichetti, S. Cecchi, P. Procacci, M. Innocenti, L. Becucci, L. Franco and C. Viglianisi, Chem. Commun., 2015, 51, 11452–11454 RSC; (g) M. Lupi, O. Salmi, C. Viglianisi and S. Menichetti, Adv. Synth. Catal., 2023, 365, 1705–1712 CrossRef CAS; (h) M. Lupi, M. Onori, S. Menichetti, S. Abbate, G. Longhi and C. Viglianisi, Molecules, 2022, 27, 1160 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS PubMed.
- S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O’Duill, K. Wheelhouse, G. Rassias, M. Medebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS.
- (a) H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed; (b) T. Bortolato, G. Simionato, M. Vayer, C. Rosso, L. Paoloni, E. M. Benetti, A. Sartorel, D. Leboeuf and L. DellAmico, J. Am. Chem. Soc., 2023, 145, 1835–1846 CrossRef CAS PubMed.
- (a) L. Pan, A. S. Kelley, M. V. Cooke, M. M. Deckert and S. Laulhe, ACS Sustainable Chem. Eng., 2022, 10, 691–695 CrossRef CAS PubMed; (b) D. Liu, M.-J. Jiao, Z.-T. Feng, X.-Z. Wang, G.-Q. Xu and P.-F. Xu, Org. Lett., 2018, 20, 5700–5704 CrossRef CAS PubMed; (c) J. Mateos, F. Rigodanza, A. Vega-PeÇaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyl, M. Bonchio and L. DellAmico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS.
- S. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00904e |
| This journal is © The Royal Society of Chemistry 2024 |