
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the redox profile of the 6,6′-biazulenic platform through functionalization along its molecular axis†
Shaun R.
Kelsey
 a,
Georgii
Griaznov
a,
Andrew D.
Spaeth
a,
Daron E.
Janzen
b,
Justin T.
Douglas
c,
Ward H.
Thompson
a and
Mikhail V.
Barybin
*a
a,
Georgii
Griaznov
a,
Andrew D.
Spaeth
a,
Daron E.
Janzen
b,
Justin T.
Douglas
c,
Ward H.
Thompson
a and
Mikhail V.
Barybin
*a
aDepartment of Chemistry, University of Kansas, Lawrence, KS 66045, USA. E-mail: mbarybin@ku.edu
bDepartment of Chemistry and Biochemistry, St. Catherine University, St. Paul, MN 55105, USA
cNMR Laboratory, Molecular Structures Group, University of Kansas, Lawrence, KS 66047, USA
First published on 18th April 2024
Abstract
The E1/2 potential associated with reduction of the linearly-functionalized 6,6′-biazulenic scaffold is accurately correlated to the combined σp Hammett parameters of the substituents over >600 mV range. X-ray crystallographic analysis of the 2,2′-dichloro-substituted derivative revealed unexpectedly short C–Cl bond distances, along with other metric changes, suggesting a non-trivial cycloheptafulvalene-like structural contribution.
Originally isolated from plant essential oils in the nineteenth century, azulenic compounds continue to fascinate scientists to this day. The correct structure of the polar azulenic scaffold, C10H8, (μ ≈ 1.1 Debye1) that features fused five- and seven- membered sp2-carbon rings was first recognized by Pfau and Plattner in 1936.2 The X-ray crystallographic confirmation of azulene's molecular structure was reported 20 years later.3,4 Azulenic building blocks are attractive for developing π-functional molecules and materials.5–7 Among the three possible linear biazulenes, 6,6′-biazulene has recently been emerging as a particularly effective oligoazulenic π-linker in the design of molecular electron reservoirs,8 on-chip microsupercapacitors,9 quasi-molecular rectifiers,10 organic and organometallic self-assembled monolayer (SAM) films,8,11 and organic field-effect transistors.12 Since the synthesis of 6,6′-biazulene in 1980,13 access to its derivatives, especially those functionalized along its molecular axis, has been quite limited.11,14,15
Given that azulene's highest occupied and lowest unoccupied molecular orbitals (HOMO/LUMO) have complementary orbital density distributions, their energies can be varied independently, to a first approximation.16–18 This is accomplished by considering the position of attachment of a substituent to the azulenic core, as well as its electron withdrawing/donating ability.19 In this communication, we envisioned a similar approach for tailoring the energetics of the frontier molecular orbitals of the linear 6,6′-biazulenic framework. Indeed, Fig. 1 illustrates the orbital density complementarity between the HOMO and LUMO of 6,6′-biazulene.
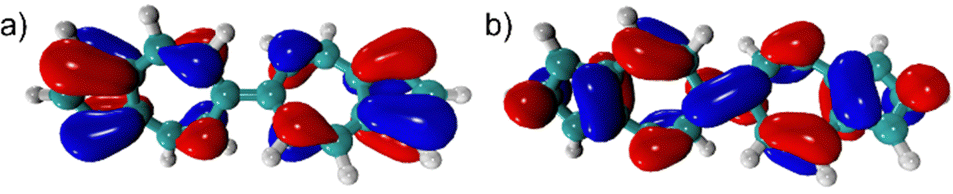 | ||
| Fig. 1 Highest occupied (a) and lowest unoccupied (b) molecular orbitals (HOMO and LUMO) of 6,6′-biazulene (B3LYP functional and cc-pVDZ basis set). | ||
Herein, we demonstrate that the redox potential (E1/2) of the 6,6′-biazulenic scaffold is accurately predictable based on the Hammett σp parameters20 of the substituents X and X′ incorporated along its molecular axis (Fig. 2). To the best of our knowledge, this is the first study unveiling such quantitative redox correlations in the context of azulenic π-systems. In addition, we discuss how dichloro substitution exerts unexpected structural permutations within compound 3a (X = X′ = Cl) in the solid state.
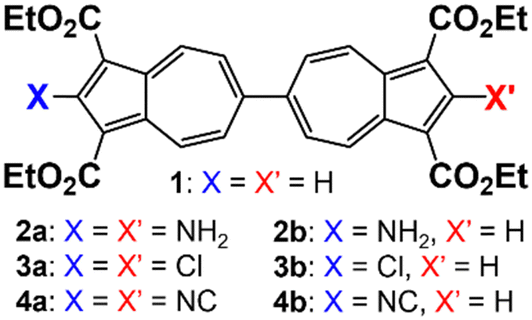 | ||
| Fig. 2 2,2′-Fuctionalized 6,6′-biazulenes considered in this work. | ||
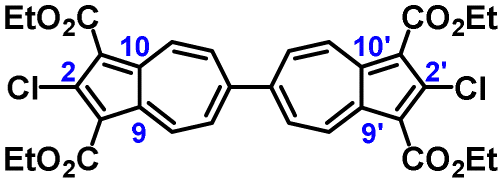
To facilitate synthetic accessibility of 2,2′-functionalized 6,6′-biazulenes, we selected the 1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (1) as the “parent” platform. Rather than employing a toxic organotin reagent to assemble 1via Stille cross-coupling,21 our synthesis of this compound involved simple deamination of the biazulenic derivative 2a (Scheme 1). On the other hand, subjecting 2a to Sandmeyer chlorination afforded dark purple, nearly black, crystalline 2,2′-dichloro-1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (3a) in an 85% yield (Scheme 1). Thus, 3a can be prepared from 2-amino-6-bromo-1,3-diethoxycarbonylazulene22 in two steps with an overall yield of ca. 80% (Scheme S1, ESI†).14 In addition to 3a, we synthesized its unsymmetric congener, 2-chloro-1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (3b), via Sandmeyer chlorination of our recently reported 2-amino-1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (2b) (Scheme 2).8 Both 3a and 3b constitute versatile precursors to a variety of 2-, and 2,2′-functionalized 6,6′-biazulenes.
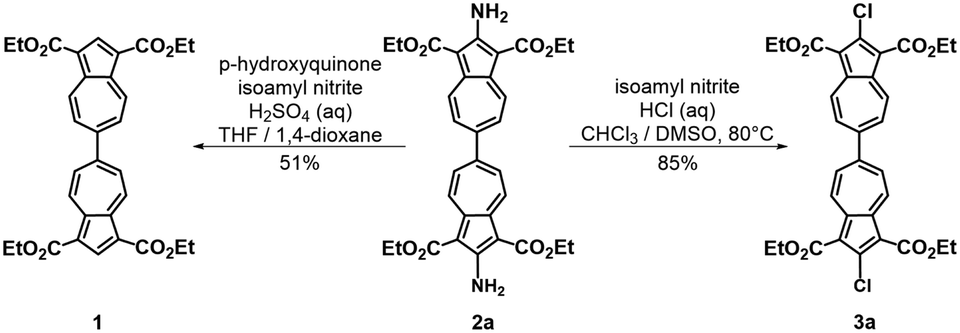 | ||
| Scheme 1 Syntheses of 1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (1) and 2,2′-dichloro-1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (3a). | ||
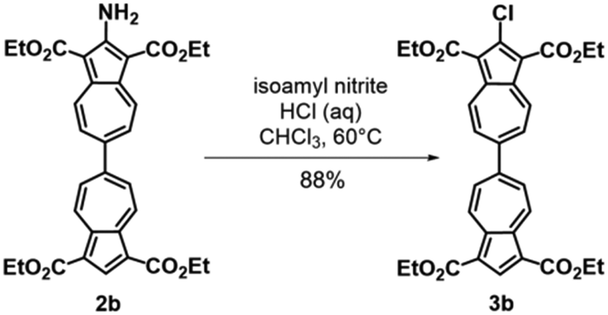 | ||
| Scheme 2 Synthesis of 2-chloro-1,1′,3,3′-tetraethoxycarbonyl-6,6′-biazulene (3b). | ||
Given that unsymmetrically functionalized 3b is a structural hybrid of centrosymmetric 1 and 3a, it is reasonable that its 1H NMR spectrum (Fig. S7, ESI†) appears as a superposition of the 1H patterns observed for the latter compounds (Fig. S1 and S2, ESI†).
We assigned all resonances in the 13C NMR spectrum of 3avia synergistic consideration of its HSQC, HMBC, and 1,1-ADEQUATE23–26 2-D maps (Fig. S3–S6, ESI†). Notably, this is the only unambiguously assigned 13C NMR profile of any 6,6′-biazulene reported in the literature to date.
Our TD-DFT calculations (Table S4, ESI†) corroborate that the lowest energy broad band centered around 500 nm in the electronic absorption spectrum of 3a (Fig. 3a) corresponds to the HOMO → LUMO and HOMO−1 → LUMO transitions. The broad nature of this band can be attributed, in part, to the range of accessible microstates pertaining to the azulene–azulene interplanar angle within 3a in CH2Cl2 solution.11 While the LUMO of 3a involves the entire biazulenic core, 3a's nearly degenerate HOMO and HOMO−1 are each primarily localized on one of the azulenic units (Fig. 3b).
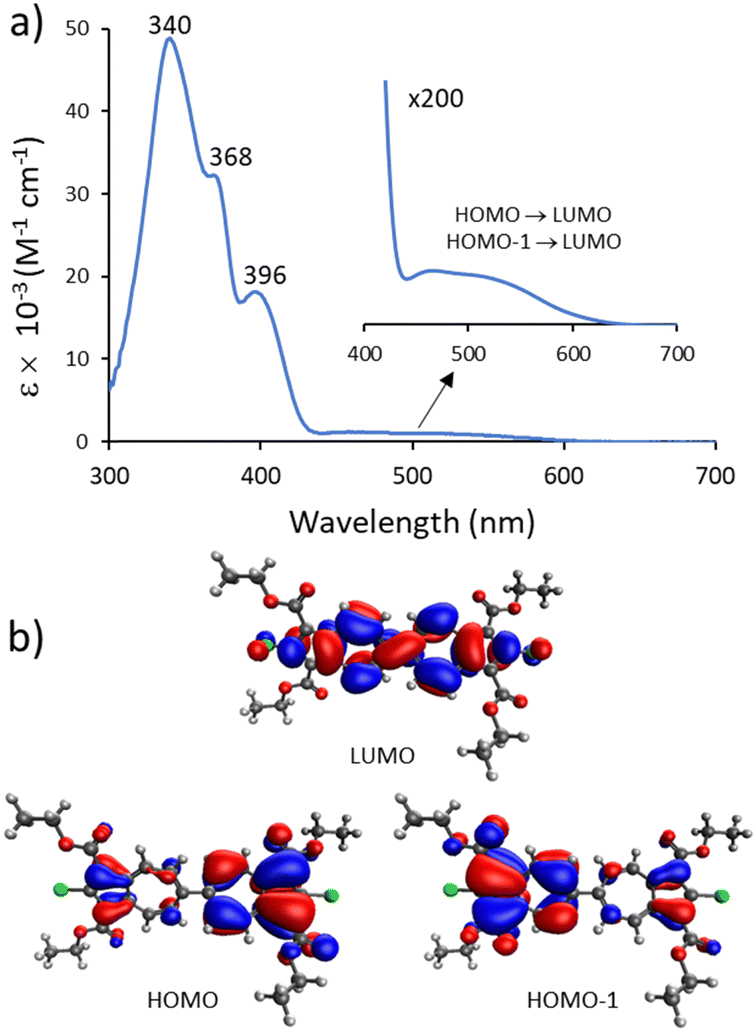 | ||
| Fig. 3 (a) Electronic absorbance spectrum of 3a in CH2Cl2 at 22 °C. (b) TD-DFT calculated molecular orbitals of 3a involved in the S0 → S1 transition. | ||
We have recently shown that 2,2′-functionalized 6,6′-biazulenes undergo reversible one-step, 2e− reduction in CH2Cl2/nBu4N+PF6− solutions under the potential inversion regime27 regardless of whether they feature symmetric or asymmetric substitution along their molecular axis.8 After recognizing a qualitative relationship between the E1/2 values associated with the 2e− reduction of the 6,6′-biazulenic core and the electron donating/withdrawing characteristics of the 2,2′-substituents, we set out to identify a quantitative approach for tuning the 6,6′-biazulenic redox profile (Fig. S13–S16, ESI†). Remarkably, plotting the half-wave redox potential (E1/2) against the combined σp Hammett parameters20 of the substituents X and X′ for the seven 6,6′-biazulenic derivatives listed in Fig. 2 revealed a nearly perfect linear correlation over >600 mV range (Fig. 4 and Table S5, ESI†). While attempts to correlate molecular redox potentials with the Hammett parameters of functional groups have been reported in the past for benzenoid organic/organometallic and ferrocene-based compounds,28–31 this is the first example of invoking such a relationship for a cohort of azulenic derivatives. Notably, a similar trend (Ep,cvs. σp) holds for the family of 2-substituted 1,3-diethoxycarbonyl azulenes shown in Fig. 5 and Table S6 (ESI†). However, the 1e− reduction of the latter compounds is invariably irreversible (Fig. S17–S20, ESI†).11 Coupling any pair of 2-substituted azulenes to form the corresponding 6,6′-biazulene leads to full reversibility of the reduction, with the biazulenic Ep,c being 0.51 ± 0.04 V more positive compared to the average Ep,c of the monoazulenes (Tables S5 and S6, ESI†). This is a consequence of the substantially greater resonance stabilization energy of the 6,6′-biazulenic framework.
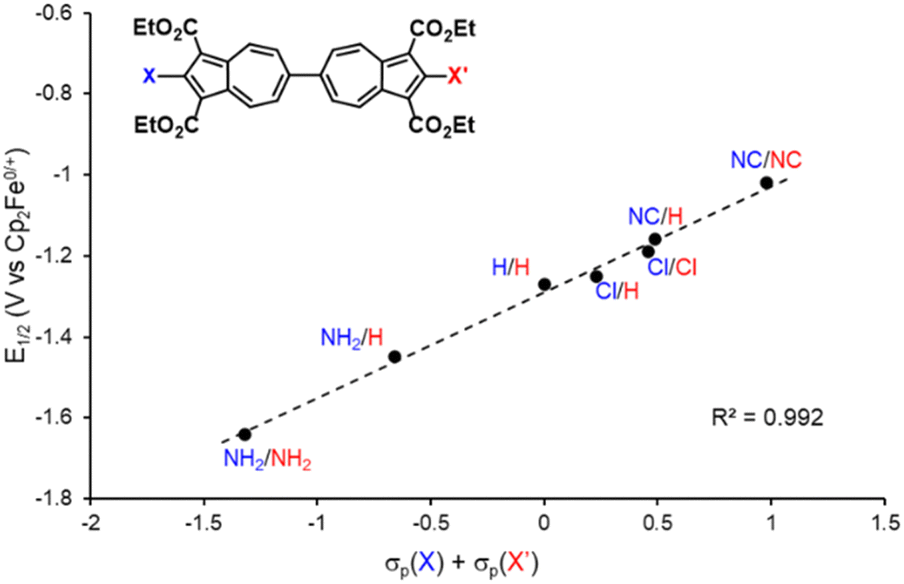 | ||
| Fig. 4 A plot of the 2e− reduction potential (E1/2) for 2,2′-functionalized 6,6′-biazulenes vs. the combined σp Hammett parameters of the substituents X and X′. | ||
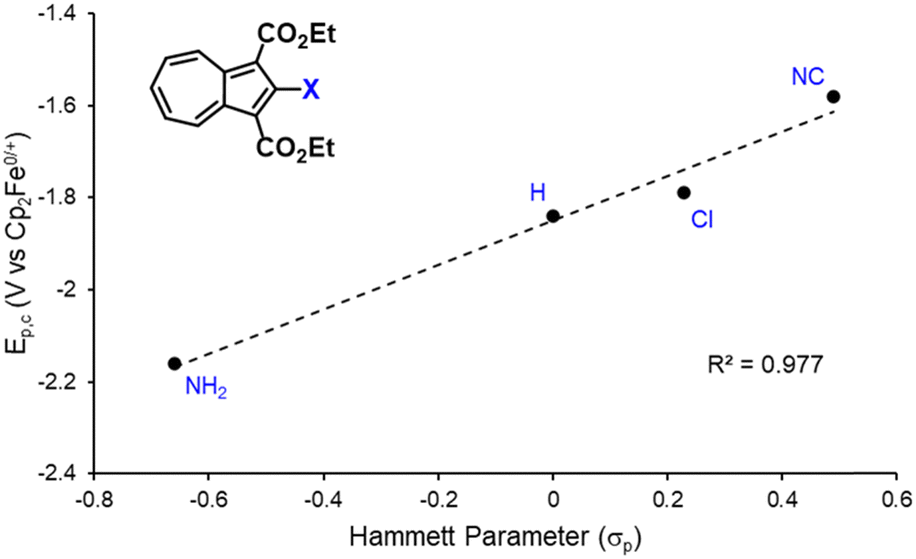 | ||
| Fig. 5 A plot of the reduction potential (Ep,c) for 2-functionalized 1,3-diethoxycarbonylazulenes vs. the σp Hammett parameter of the substituent X. | ||
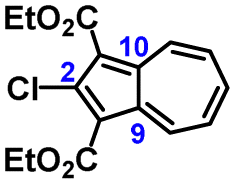
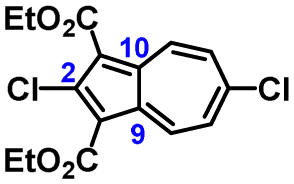
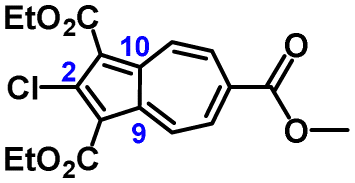
The molecular structure of 3a is illustrated in Fig. 6. The two chemically identical, but crystallographically unique C–Cl bonds, C2–Cl1 and C2′–Cl2, within 3a have the lengths of 1.674(2) Å and 1.682(2) Å, respectively. The longer C–Cl bond belongs to the half of the molecule featuring the shortest Cl⋯O contact (Cl2⋯O6) of 2.816 Å. Both C–Cl bonds in 3a are significantly shorter than any C–Cl bond in all structurally characterized chloroazulenes featuring Cl-substitution at the five-membered ring of the azulenic scaffold (Fig. S22 and Table S14, ESI†). In particular, the C–Cl bonds in 3a are statistically shorter, per the 3-σ criterion,32 than those in the X-ray structures of the other three 2-chloroazulenes known to date (Table 1).33–35
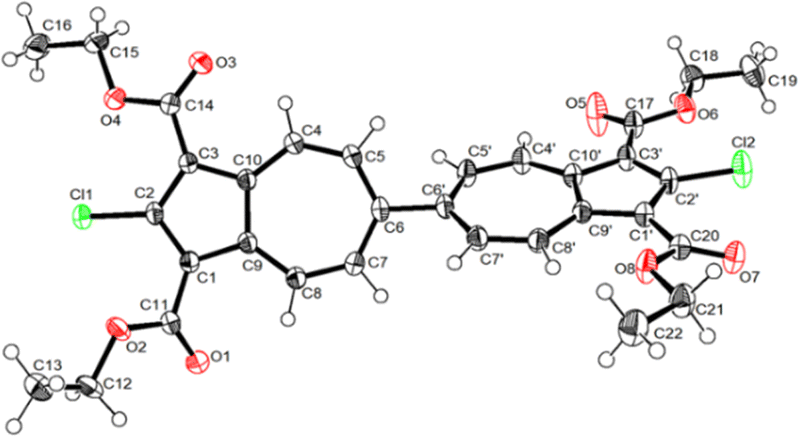 | ||
| Fig. 6 Molecular structure of 3a (50% thermal ellipsoids). Selected interatomic distances (Å) and dihedral angle (deg): C2–Cl1 1.674(2), C2′–Cl2 1.682(2), C6–C6′ 1.478(2), C9–C10 1.437(2), C9′–C10′ 1.440(3), O2⋯Cl1 2.918(2), O4⋯Cl1 2.834(1), O6⋯Cl2 2.816(2), O7⋯Cl2 2.876(2), C5–C6–C6′–C5′ 48.1(2). | ||
| Compound | d(C2–Cl) Å | d(C9–C10) Å | CCDC identifier |
|---|---|---|---|
|
1.674(2) | 1.437(2) | This work |
| 1.682(2) | 1.440(3) | ||
|
1.712(2) | 1.471(2) | HOMRAE |
|
1.714(4) | 1.470(6) | XIGPOU |
|
1.709(2) | 1.470(3) | AYIDIX |
The C–Cl bond contractions in 3a may be rationalized by invoking the minor heptafulvalene-like zwitterionic resonance forms depicted in Fig. 7, which show C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cl double-bond character. This argument mirrors the crystallographic analysis of the chlorodiphenylmethylium cation by Laub et al.36 The short C–Cl bond distance of 1.668(8) Å within this carbocation, which is statistically indistinguishable from those in 3a, was attributed to chlorine back-donation exerting partial double-bond CCl+ character (Fig. S23, ESI†).36 Unusually short C–Cl bonds, including various C–Cl multiple bonding scenarios, continue to attract both experimental and theoretical interest.37–39
Cl double-bond character. This argument mirrors the crystallographic analysis of the chlorodiphenylmethylium cation by Laub et al.36 The short C–Cl bond distance of 1.668(8) Å within this carbocation, which is statistically indistinguishable from those in 3a, was attributed to chlorine back-donation exerting partial double-bond CCl+ character (Fig. S23, ESI†).36 Unusually short C–Cl bonds, including various C–Cl multiple bonding scenarios, continue to attract both experimental and theoretical interest.37–39
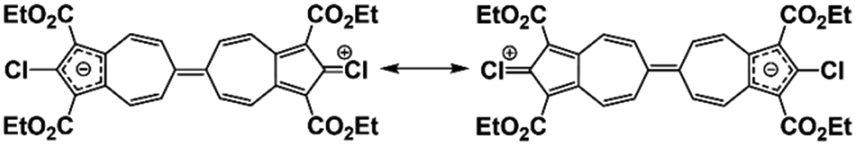 | ||
| Fig. 7 Minor zwitterionic resonance forms of 3a. | ||
The C6–C6′ bond distance of 1.478(2) Å within 3a is markedly shorter than those in all other crystallographically characterized 6,6′-biazulenes (Table 2).8,11,12,40 In addition, as summarized in Table 2, the C–C bonds at the fusion of the five- and seven-membered rings in 3a are substantially contracted as well. Both of these observations are consistent with a nontrivial heptafulvalene-like contribution to the structure of the biazulenic scaffold of 3a.41 Moreover, the azulene–azulene interplanar angles in dichloro 3a and diisocyano 4a, which share the same 1,1′,3′3′-tetraethoxycarbonyl-6,6′-biazulenic core, are 48.1° and 66.9°,11 respectively, although such comparison should be viewed cum grano salis as this torsion parameter is undoubtedly sensitive to crystal packing.
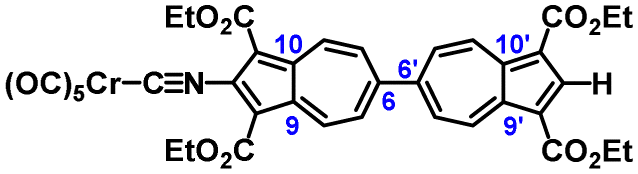
In summary, we demonstrated that the redox profile of the 6,6′-biazulenic scaffold functionalized along its molecular axis is quantitatively tuneable within a wide window of potentials. This was accomplished by considering, for the first time in the context of azulenic systems, the well documented σp Hammett parameters20 reflecting the net electronic influence of the substituents. We anticipate that the facile access to the crystallographically unusual dichloro derivative 3a offers not only new opportunities to engage the 6,6′ architecture as an attractive molecular template in the realm of organic charge transfer and/or conductive materials, but also to explore unorthodox approaches for pursuing C–Cl multiple bonding.
This research was funded by the US National Science Foundation grant CHE-1808120 to M. V. B. S. R. K. acknowledges the support provided by the NSF Graduate Research Fellowship under grant DGE-1940699 and the Madison and Lila Self Graduate Fellowship at the University of Kansas. Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grants (S10OD016360 and S10RR024664), NSF MRI funding (CHE-1625923 and CHE-9977422), and an NIH Center Grant (P20 GM103418). The calculations were performed at the University of Kansas Center for Research Computing (CRC), including the BigJay cluster resource funded through NSF Grant No. MRI-2117449. X-ray crystallographic resources were provided by an NSF-MRI award #1125975. This manuscript is dedicated to Prof. John E. Ellis of the University of Minnesota.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. G. Anderson and B. M. Steckler, J. Am. Chem. Soc., 1959, 81, 4941–4946 CrossRef CAS.
- A. St. Pfau and P. A. Plattner, Helv. Chim. Acta, 1936, 19, 858–879 CrossRef CAS.
- J. M. Robertson and H. M. M. Shearer, Nature, 1956, 177, 885 CrossRef CAS PubMed.
- J. M. Robertson, H. M. M. Shearer, G. A. Sim and D. G. Watson, Nature, 1958, 177–178 CrossRef CAS.
- H. Katagiri, in Advances in Organic Crystal Chemistry, ed. H. Uekusa and M. Sakamoto, Wiley, 2020, pp. 341–358 Search PubMed.
- J. Huang, S. Huang, Y. Zhao, B. Feng, K. Jiang, S. Sun, C. Ke, E. Kymakis and X. Zhuang, Small Methods, 2020, 4, 2000628 CrossRef CAS.
- H. Xin, B. Hou and X. Gao, Acc. Chem. Res., 2021, 54, 1737–1753 CrossRef CAS PubMed.
- P. T. Connelly, J. C. Applegate, D. A. Maldonado, M. K. Okeowo, W. C. Henke, A. G. Oliver, C. L. Berrie and M. V. Barybin, Dalton Trans., 2023, 52, 11419–11426 RSC.
- C. Yang, K. S. Schellhammer, F. Ortmann, S. Sun, R. Dong, M. Karakus, Z. Mics, M. Löffler, F. Zhang, X. Zhuang, E. Cánovas, G. Cuniberti, M. Bonn and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3920–3924 CrossRef CAS PubMed.
- S. Sun, X. Zhuang, L. Wang, B. Zhang, J. Ding, F. Zhang and Y. Chen, J. Mater. Chem. C, 2017, 5, 2223–2229 RSC.
- T. R. Maher, A. D. Spaeth, B. M. Neal, C. L. Berrie, W. H. Thompson, V. W. Day and M. V. Barybin, J. Am. Chem. Soc., 2010, 132, 15924–15926 CrossRef CAS PubMed.
- Y. Shibuya, K. Aonuma, T. Kimura, T. Kaneko, W. Fujiwara, Y. Yamaguchi, D. Kumaki, S. Tokito and H. Katagiri, J. Phys. Chem. C, 2020, 124, 4738–4746 CrossRef CAS.
- M. Hanke and C. Jutz, Synthesis, 1980, 31–32, DOI:10.1055/s-1980-28911.
- An alternative synthesis of 3a from 2-amino-6-bromo-1,3-diethoxycarbonylazulene, albeit in a three-step sequence with an overall ca. 45% yield, has been recently reported: B. Hou, Z. Zhou, C. Yu, X. S. Xue, J. Zhang, X. Yang, J. Li, C. Ge, J. Wang and X. Gao, ACS Macro Lett., 2022, 11, 680–686 CrossRef CAS PubMed.
- Y. Yamaguchi, M. Takubo, K. Ogawa, K. Nakayama, T. Koganezawa and H. Katagiri, J. Am. Chem. Soc., 2016, 138, 11335–11343 CrossRef CAS PubMed.
- R. S. H. Liu and A. E. Asato, J. Photochem. Photobiol., C, 2003, 4, 179–194 CrossRef CAS.
- R. S. H. Liu, J. Chem. Ed., 2002, 79, 183–185 CrossRef CAS.
- R. P. Steer, J. Photochem. Photobiol., C, 2019, 40, 68–80 CrossRef CAS.
- R. E. Robinson, T. C. Holovics, S. F. Deplazes, D. R. Powell, G. H. Lushington, W. H. Thompson and M. V. Barybin, Organometallics, 2005, 24, 2386–2397 CrossRef CAS.
- R. W. Taft, A. Leo and C. Hansch, Chem. Rev., 1991, 91, 165–195 CrossRef.
- S. Ito, T. Okujima and N. Morita, J. Chem. Soc., Perkin Trans. 1, 2002, 1896–1905 RSC.
- T. C. Holovics, R. E. Robinson, E. C. Weintrob, M. Toriyama, G. H. Lushington and M. V. Barybin, J. Am. Chem. Soc., 2006, 128, 2300–2309 CrossRef CAS PubMed.
- G. E. Martin, Annual Reports on NMR Spectroscopy, Elsevier, 2011, ch. 5, vol. 74, pp. 215–291 Search PubMed.
- B. D. Hilton, K. A. Blinov and G. E. Martin, J. Nat. Prod., 2011, 74, 2400–2407 CrossRef PubMed.
- M. M. Senior, R. T. Williamson and G. E. Martin, J. Nat. Prod., 2013, 76, 2088–2093 CrossRef CAS PubMed.
- A. V. Buevich, R. T. Williamson and G. E. Martin, J. Nat. Prod., 2014, 77, 1942–1947 CrossRef CAS PubMed.
- D. H. Evans, Chem. Rev., 2008, 108, 2113–2144 CrossRef CAS PubMed.
- L. Wang, Z. L. Xie, B. T. Phelan, V. M. Lynch, L. X. Chen and K. L. Mulfort, Inorg. Chem., 2023, 62, 14368–14376 CrossRef CAS PubMed.
- J. C. Dickenson, M. E. Haley, J. T. Hyde, Z. M. Reid, T. J. Tarring, D. A. Iovan and D. P. Harrison, Inorg. Chem., 2021, 60, 9956–9969 CrossRef CAS PubMed.
- W. C. Henke, D. Lionetti, W. N. G. Moore, J. A. Hopkins, V. W. Day and J. D. Blakemore, ChemSusChem, 2017, 10, 4589 CrossRef CAS PubMed.
- N. Vo, N. L. Haworth, A. M. Bond and L. L. Martin, ChemElectroChem, 2018, 5, 1–14 CrossRef.
- F. Pukelsheim, Am. Stat., 1994, 48, 88–91 CrossRef.
- S. Förster, W. Seichter, R. Kuhnert and E. Weber, J. Mol. Struc., 2014, 1075, 63–70 CrossRef.
- K. J. Scheetz, A. D. Spaeth, A. S. Vorushilov, D. R. Powell, V. W. Day and M. V. Barybin, Chem. Sci., 2013, 4, 4267–4272 RSC.
- B. B. Ling, CSD Commun., 2016, CCDC 888557 DOI:10.5517/ccdc.csd.ccytm4h.
- T. Laube, E. Bannwart and S. Hollenstein, J. Am. Chem. Soc., 1993, 115, 1731–1733 CrossRef CAS.
- F. Fantuzzi, B. Rudek, W. Wolff and M. A. C. Nascimento, J. Am. Chem. Soc., 2018, 140, 4288–4292 CrossRef CAS PubMed.
- R. Kalescky, E. Kraka and D. Cremer, Int. J. Quantum Chem., 2014, 114, 1060–1072 CrossRef CAS.
- M. Göbel, B. H. Tchitchanov, J. S. Murray, P. Politzer and T. M. Klapotke, Nat. Chem., 2009, 1, 229–235 CrossRef PubMed.
- Not included in Table 2 is the structure of 2,2′-dibromo-1,1′-dibutyl-6,6′-biazulene that is searchable in CCDC (NAPSUW) but suffers from low precision {e.g., d(C6–C6′) = 1.50(2) Å}, unacceptable data/refinement quality (R-factor = 11.7%), and lack of a published synthesis: B. Hou, CSD Commun., 2021, CCDC 2127392.
- R. Thomas and P. Coppens, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1800–1806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic and analytical data, details of the electrochemical, X-ray crystallographic and DFT studies. CCDC 2300779. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00656a |
| This journal is © The Royal Society of Chemistry 2024 |