
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
AI in computational chemistry through the lens of a decade-long journey
Pavlo O.
Dral

State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, and Innovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen University, Xiamen, Fujian 361005, China. E-mail: dral@xmu.edu.cn
First published on 27th February 2024
Abstract
This article gives a perspective on the progress of AI tools in computational chemistry through the lens of the author's decade-long contributions put in the wider context of the trends in this rapidly expanding field. This progress over the last decade is tremendous: while a decade ago we had a glimpse of what was to come through many proof-of-concept studies, now we witness the emergence of many AI-based computational chemistry tools that are mature enough to make faster and more accurate simulations increasingly routine. Such simulations in turn allow us to validate and even revise experimental results, deepen our understanding of the physicochemical processes in nature, and design better materials, devices, and drugs. The rapid introduction of powerful AI tools gives rise to unique challenges and opportunities that are discussed in this article too.
 Pavlo O. Dral | Pavlo O. Dral is a Full Professor in the College of Chemistry and Chemical Engineering at Xiamen University, which he joined in 2019. His research is focused on AI for quantum chemistry and dynamics. Dral is a founder of the MLatom package for AI-enhanced computational chemistry and a co-founder of the Xiamen Atomistic Computing Suite. He did his PhD with Tim Clark in 2010–2013 followed by a post-doc with Walter Thiel in 2013–2019 in Germany. Dral was awarded an Outstanding Youth (Overseas) by the National Natural Science Foundation of China in 2021. More information is available on https://dr-dral.com. |
Introduction
AI is now widely recognized as a powerful technology permeating daily lives from publicly available chatbots to more specialized business, technology, and research tools. Computational chemistry is no exception. AI has appeared as a long-sought technology for computational chemists who are always in need of faster and more accurate tools to increase the reliability and scale of atomistic simulations. It is not just the numbers that we are interested in. AI allows us to look at physicochemical problems from a different perspective to obtain useful insights and also approach the method development from a radically new angle.The author started his journey in the field of AI in computational chemistry in 2013 with a firm belief that AI is the tool to solve many pertinent problems in simulations. Now is the time to reflect on how the field developed over the decade and where it is heading. This article approaches it through the lens of the author's contributions1–46 put in the broader context of the current state of affairs in the field.
During the journey, we introduced and explored many concepts to improve the quantum mechanical (QM) and dynamics methods with machine learning (ML).1–7 We also developed practical tools to perform AI-enhanced simulations ranging from efficient and accurate geometry optimizations and thermochemical calculations to molecular dynamics and spectra generation.4,5,8–19 These tools include new methods, software packages, and their deployment on a cloud computing platform to make them accessible through a web browser. The training of students and researchers is also important as proficiency in AI tools for computational chemistry becomes one of the most sought-after skills. Our contributions can be viewed as the intimately interconnected triad of developing AI methods and concepts breaking through the limitations of traditional quantum chemistry, providing tools, and training and popularization. Each element of the triad influences the other, e.g., feedback from students highlights the current bottlenecks and helps to improve the methods and tools. Below, I discuss these topics starting with a concrete and simple example for motivation.
Getting the numbers and physics right
QM provides a theoretically rigorous way of getting the numbers right in atomistic simulations, all we need to do is ‘just’ to solve the numerical problem based on the Schrödinger equation. Such accurate calculations are possible, with, e.g., full configuration interaction (FCI) with a large, ideally complete, basis set. Take for example the tiny H2 molecule, we can optimize its geometry with the FCI/aug-cc-pV6Z method which would get us a bond length of 0.7415 Å agreeing precisely with the experimental value of 0.7414 Å.47 However, calculations even for such a small system would take us 5 CPU days to do with common software on a desktop computer.47 Then, how do we treat the larger systems of practical interest, which may contain dozens (e.g., small organic molecules) to millions of atoms (e.g., for enzymatic processes)? Traditionally, the only way was to carry out approximations to run calculations faster, leading to a vast sea of QM methods.47 (For larger-scale simulations even further approximations are required leading to the proliferation of molecular mechanics force fields, coarse-grained models, and hybrid QM/MM approaches.) Many such successful QM approximations are density functional theory (DFT) methods such as B3LYP,48,49 which is still one of the most popular methods around and, if DFT is still too expensive, a last resort of a computational chemist to treat the system quantum mechanically is to use fast semi-empirical QM (SQM) methods.50 If one can afford it, the good practice is to use the gold-standard,51–53 much-slower-than-B3LYP, CCSD(T) method54 at least for single-point calculations after geometry optimizations using a cheaper DFT method (for the H2 example CCSD(T) is equivalent to FCI). The cost of making more and more approximations is, in general, the decreased accuracy. In the case of the H2 molecule, with common DFT or SQM we can get its bond length anywhere from ca. 0.6 to 0.8 Å. In other words, we cannot guarantee precise results for large systems with common affordable QM approaches and our main hope is to get the physics at least qualitatively right, i.e., for a chemist, it might be good enough to find the roughly correct geometry of the stable molecular configurations and know its energy relative to other molecular configurations. The only way for a computational chemist to verify the simulations was to compare the observables with an experiment or better theory, if available.Now the above lengthy discussion about rather trivial knowledge was with the purpose to show that ML is a game changer as it provides us with a radically different way to approach the computational chemistry simulations. For the H2 example, we can get the same bond length of 0.7415 Å with ML as with FCI but in a fraction of second.29 Of course, we needed to train on FCI data, but once it is done, the ML model can be perused again and again for different kinds of simulations, e.g., for molecular dynamics (MD) which would be much costlier than geometry optimization with the reference method. This is the power of ML: it allows us to perform very fast simulations without compromising or compromising less on the accuracy. All we need is enough accurate data and good ML algorithms or combinations of ML and QM models. The community is making strides in all these respects.
Below, I will start by introducing the general-purpose AI-based methods that computational chemists can readily apply, then move on to address underlying and new concepts and discuss how to apply AI to solving specific problems.
General-purpose AIQM1 method
One of the best examples from our research is the AIQM1 method which leverages ML to improve the approximated SQM method to get the right numbers and physics in real-world applications at a low computational cost (Fig. 1).14 AIQM1 is more accurate than common DFT approaches such as B3LYP/6-31G* while being orders of magnitude faster because AIQM1's speed is close to that of the SQM methods. AIQM1 can and should be used instead of the slower and less accurate common DFT approaches whenever it is possible (the current recommendation is to use AIQM1 for the neutral closed-shell organic molecules although it also has decent accuracy in many other cases). I cannot emphasize enough that, in the age of AI in computational chemistry, there are fewer and fewer reasons to use slower and, often less accurate, non-AI methods instead of existing AI-based counterparts.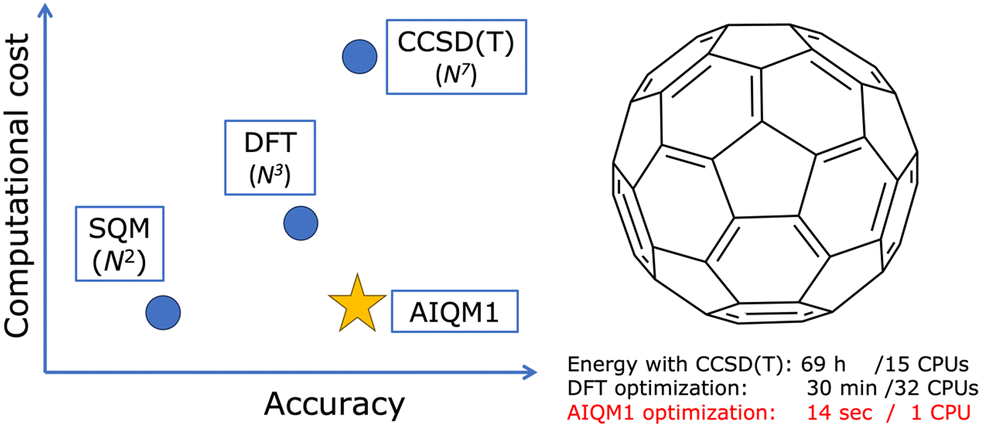 | ||
| Fig. 1 Simplified schematic showing the place of AIQM1 among the traditional quantum mechanical methods: semi-empirical (SQM), density functional theory (DFT), and coupled cluster exemplified by CCSD(T). | ||
To give a perspective: the geometry optimization of the C60 molecule takes just 14 seconds on a single CPU with AIQM1 but needs 30 min on 32 CPU cores with a DFT approach (ωB97XD/6-31G*). At the coupled cluster level, we cannot even get its optimized geometry and can only afford to get energy after spending 70 hours on 15 CPUs and using linear-scaling approximation. The quality of the optimized geometries at AIQM1 is essentially at the coupled cluster level and for many types of bond lengths, we achieve picometer and sub-picometer accuracy (Fig. 2a–c).14 AIQM1 even works in challenging cases when, e.g., B3LYP provides a qualitatively wrong structure like the erroneous cumulenic cyclo-C18 structure of the compound known to be polyynic (Fig. 2d).14 AIQM1 allowed us to improve upon the imprecise X-ray structural determination of the much bigger polyynic compound which would be prohibitive to optimize at the coupled cluster level (Fig. 2e and f).14
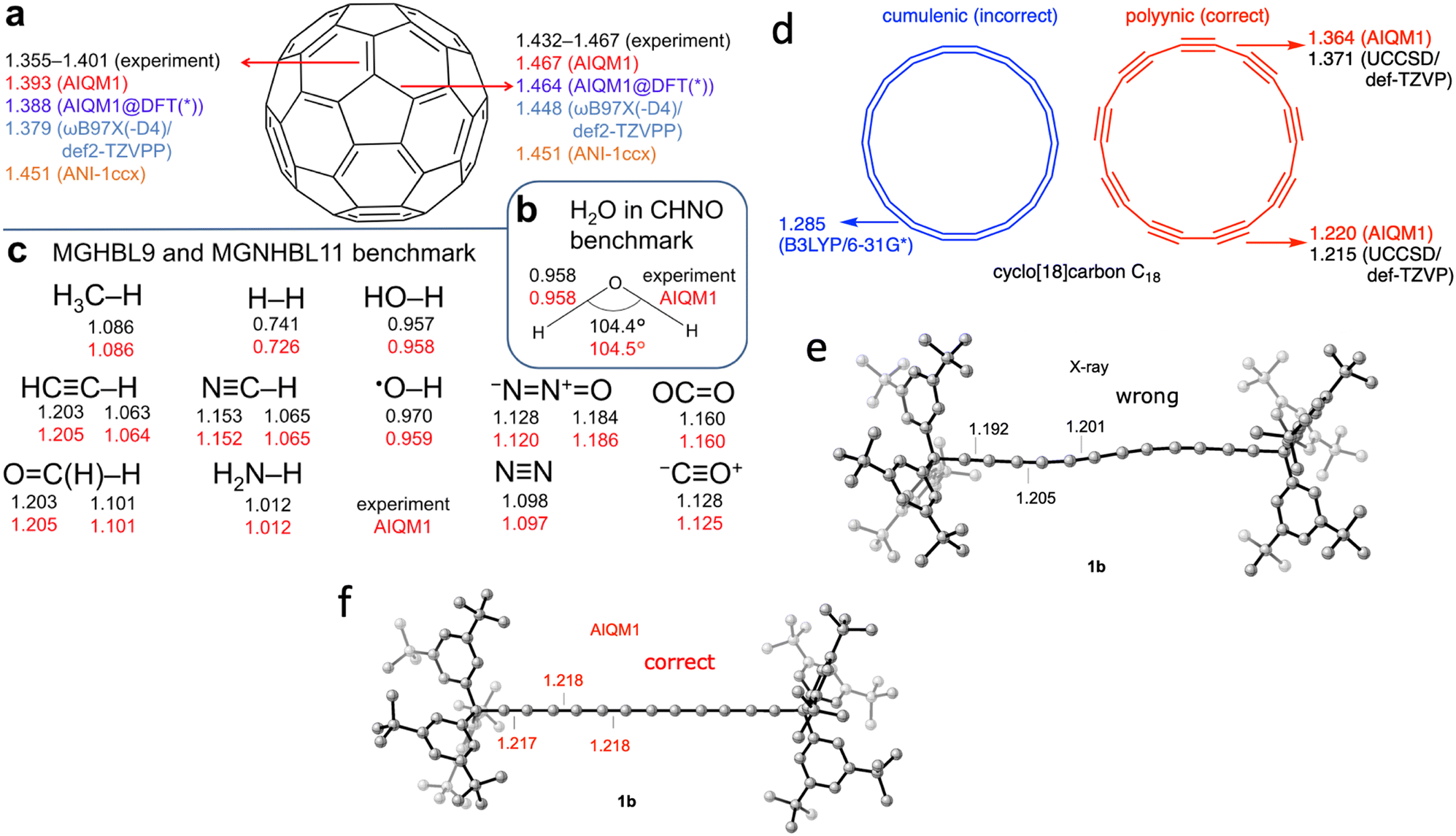 | ||
| Fig. 2 AIQM1 can accurately predict geometries as compared with experiment and the coupled-cluster method (a)–(d). In the case of polyynes, AIQM1 (f) revises experimental bond lengths (e). Adapted with permission from ref. 14. Copyright 2021, the Authors. | ||
Not just geometries are good, energies and forces are also particularly accurate with AIQM1, and for many properties such as reaction energies and enthalpies, isomerization energies, heats of formation, and rotational barriers, it is on par with coupled-cluster-level approaches.12,14 In particular, heats of formation are determined with chemical accuracy12,14 (errors below the coveted 1 kcal mol−1) which is particularly impressive, because, e.g., DFT is known to struggle with these types of calculations and the main approach to make DFT work is to exploit as much as possible the error cancellation by careful construction of the isodesmic reactions and similar schemes.55 AIQM1 gets the heats of formation right much faster without relying on error cancellations.
A particularly important property of AIQM1 is that it provides robust means for uncertainty quantification (UQ) in contrast to non-AI QM approaches. The magnitude of uncertainty is determined by how large is the deviation between eight neural networks in AIQM1.12 The UQ allows us to weed out unreliable predictions and treat them separately. On the other hand, the confident AIQM1 predictions with low uncertainty are a robust tool for detecting errors in experimental heats of formation.12
As we have seen, AIQM1 is not just faster and more accurate than common DFT approaches such as those based on B3LYP, but it can even validate and revise experiments. This is a watershed moment for computational chemistry.
AIQM1 is a QM method, not a pure AI approach and, thus, it can also be applied in other typical QM application areas such as the calculation of excited-state properties. Its performance for the vertical excitations is on par with common TD DFT methods but the speed is higher.14 The AIQM1/MRCI (multi-reference configuration interaction) or CIS (configuration interaction singles) methods may come in handy for excited-state geometry optimizations and dynamics.14,24 For example, we used AIQM1 to investigate large cycloparaphenylene nanolassos and showed that it could explain well not just ground-state properties but also correctly predict the fluorescence quenching in their complexes with fullerenes.24 This means that AIQM1 may be a good choice for, e.g., screening of better materials exhibiting aggregation-induced emission or for investigating photocatalytic processes.
Another example is the simulation of IR spectra, which requires the prediction of the dipole moments that are available at the AIQM1 level.9 Such spectra have very good accuracy in terms of peak positions and can be obtained faster than with DFT approaches.9
What AIQM1 is and why it works
I have started with AIQM1 to give a taste of how AI is changing computational chemistry. But what is AIQM1 and why does it work so well? The method can be used for a good illustration of key concepts such as the Δ- and transfer learning and the importance of good-quality data, QM, and ML models (Fig. 3).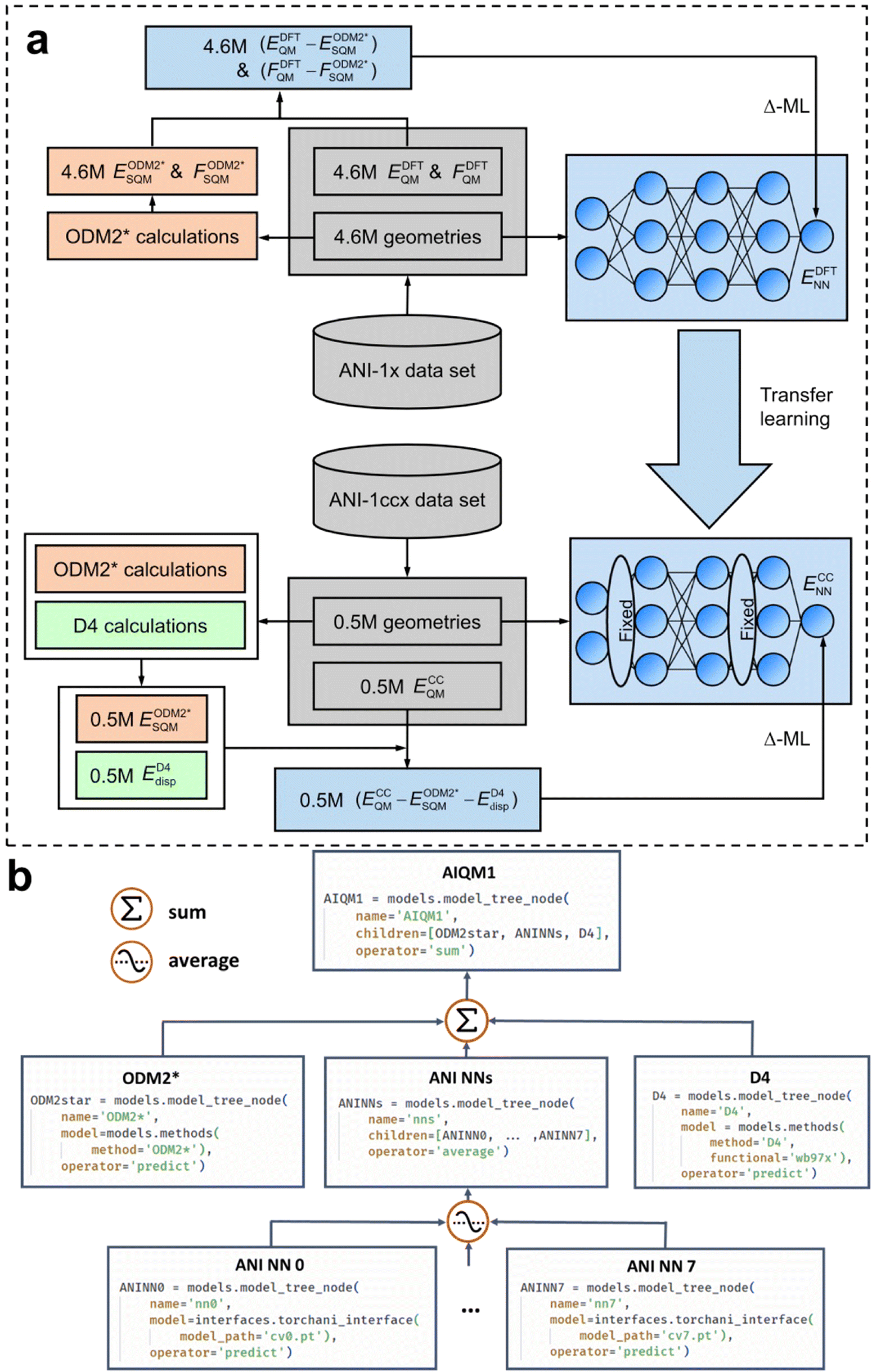 | ||
| Fig. 3 The workflow of the design of the AIQM1 method: (a) the training procedure, (b) the final composition and implementation using ML atom's model tree concepts in Python. (a) Adapted with permission from ref. 14. Copyright 2021, the Authors. (b) Reproduced with permission from ref. 10 under the CC-BY 4.0 license. Copyright 2024, the Authors. | ||
Fig. 3 shows an example of AIQM1 operational process and important stages of the Δ-learning. In the training stage (Fig. 3a), we need to generate the data containing differences between the target and the baseline QM methods. In the case of AIQM1, it is ultimately the differences between the coupled-cluster (CC, ECCQM) and semi-empirical (SQM, 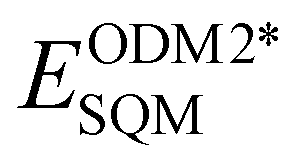 ) energies (note that the D4-dispersion corrections ED4disp were also subtracted from the data but this is a technical detail not important for discussing Δ-learning). Then the chosen ML model is trained on these differences (in the case of AIQM1 it is more complicated as it was done in two stages involving transfer learning, see below). In AIQM1, eight ANI-type56 NNs were trained on different subsets of the data.
) energies (note that the D4-dispersion corrections ED4disp were also subtracted from the data but this is a technical detail not important for discussing Δ-learning). Then the chosen ML model is trained on these differences (in the case of AIQM1 it is more complicated as it was done in two stages involving transfer learning, see below). In AIQM1, eight ANI-type56 NNs were trained on different subsets of the data.
In the prediction stage (Fig. 3b), the baseline QM method should be used to predict the energies. The ML model is then used to predict the differences that are added to the baseline energies to approximate the target QM energies. In AIQM1, the differences are calculated as the average of the eight NN predictions (and D4 corrections are added back too). The resulting energies approximate the coupled-cluster level.
Derivative properties such as forces or Hessians for geometry optimizations, dynamics, or thermochemistry calculations can be readily obtained in AIQM1 by adding the corresponding derivative properties obtained at each component (SQM, NN, and D4). Hence, one can, e.g., obtain geometries at the coupled-cluster level at a cost moderately exceeding the cost of the SQM method which is the slowest among all the components.
How transfer learning works can also be shown in an example of AIQM1. There, the NNs are first trained on the differences between the DFT and SQM properties (energies and forces). These differences are less costly to generate and hence more abundant than available differences between the coupled-cluster and SQM properties. These NNs are then fine-tuned by optimizing a reduced subset of the NN weights on the differences between the coupled-cluster and SQM energies to yield the final NN-correcting models as described above.
Why Δ-learning is the key for AIQM1's versatility and robustness
AIQM1 is a nice example to explain why Δ-learning is so powerful while being a relatively simple concept. In AIQM1, NNs of ANI-type are used to correct the predictions of the SQM approach based on the ODM2 method. ODM2 was shown to be the best for heats of formation and several other properties among other SQM methods, but it was still far from achieving chemical accuracy.59 Needless to say, as an SQM method, ODM2 is generally not as robust as common DFT approaches. A slightly different variant of ANI-type NNs was trained several years before the use of AIQM1 on the same data and using a similar transfer learning procedure which resulted in the pure ML method ANI-1ccx.63 This method is, hence, even faster than AIQM1 while achieving similar accuracy, i.e., approaching the gold-standard coupled cluster level for many applications such as relative energies. Nevertheless, ANI-1ccx is only applicable to predicting the energies and forces of the neutral closed-shell species in an electronic ground state and is known to have qualitative failures for, e.g., optimizing the C60 fullerene where it erroneously predicts all equal bond lengths. ODM2, while being less accurate than ANI-1ccx for many properties, is usually at least qualitatively correct as the underlying QM formalism properly captures the physics of the problem. ANI-1ccx is more accurate than ODM2 for species similar to those in ANI-1ccx training set but may fail even qualitatively for other applications as no physics is explicitly built into it. AIQM1 combines the best of two worlds and is much more accurate than ODM2 and much more stable for capturing correct physics than ANI-1ccx. AIQM1 is noticeably more accurate than ANI-1ccx even for the tasks the latter method is good at. The improved robustness of AIQM1 compared to both ODM2 and ANI-1ccx was particularly obvious when benchmarked on transition states, which were not in the training data of the NNs.35 Thus, using the combination of the baseline QM methods and ML corrections yields a hybrid model that inherits correct physical behavior from the QM baseline and improved accuracy from ML.30Historically, the development of Δ-learning was inspired by an early demonstration of the capability of ML to learn the DFT-level atomization energies for chemically diverse species.30 While the demonstration was pioneering and impressive at the time, it was quickly realized that even the SQM methods were better.64 Instead of being disappointed by the not-so-good performance of ML, in 2013, we saw an opportunity to combine two computationally fast approaches SQM (or other QM) and ML. I must also mention that Δ-learning can find its analogy in similar works predating its publication. A notable example is studies on using NNs to correct heats of formation predicted by DFT to target the experimental values as early as 2003.65,66 Back in 2015, we demonstrated proof-of-concept that the Δ-learning approach is very general and not limited to specific combinations of the baseline and target levels. Since 2013, many improved models and data sets emerged which allowed us to eventually also develop the generally applicable method AIQM1. I will discuss such ML models and data issues later, after looking at how Δ-learning is used, and other concepts and methods combining ML and QM.
Where Δ-learning stands in AI-enhanced computational chemistry
The original motivation behind the Δ-learning concept was the creation of the general-purpose method such as AIQM1. An increasing number of methods related to AIQM1 were developed independently following similar ideas. Some of them such as QDπ67 and DFTB/ChIMES68 also use the SQM method (often based on GFN2-xTB rather than ODM2) and different types of ML corrections, either based on another NN or even splines. Typically, these methods target the DFT-level accuracy while AIQM1 was designed to approximate the coupled-cluster level. Other methods correct DFT to experiment.65,66,69,70Δ-learning usually refers to hybrid models where 3D structural information and elemental composition are supplied as descriptors (input) to the correcting ML model. However, nothing restricts us from using other descriptors, e.g., derived from the baseline QM model. In the end, why not reuse more information from the baseline model when it is available for free? While models such as AIQM1 use only such structural and composition information, other models such as OrbNet Denali71 use features from the QM baseline.30 Both types of strategies were shown to work, at least as a proof-of-concept, for correcting the approximate ab initio QM baseline (e.g., Hartree–Fock) to a more accurate coupled cluster level, i.e., basically learning the correlation energy (an idea which was also quite old72).6,73
Δ-learning is also often used to create hybrid ML/QM models for solving specific problems, e.g., running dynamics.74 This, however, requires prior training of such models and therefore is less straightforward to use than out-of-the-box general-purpose approaches.
Deeper integration of AI with QM: alternatives to Δ-learning
While Δ-learning is a powerful concept, ML only corrects the predictions of an approximate QM baseline method a posteriori. We can use ML in a very different way by improving the QM model and then get better results with such a hybrid model.One of the concepts is improving the QM Hamiltonian with ML. We demonstrated its possibility in 2015 (the same time as Δ-learning) for an SQM approach by correcting the semi-empirical parameters entering the Hamiltonian to make parameters adjustable for each molecule rather than using the fixed set of parameters.7 We showed improved accuracy for such models for atomization energies relative to the DFT target. With time, many independently developed integrated approaches emerged, some of which also improved the SQM Hamiltonian by treating it as a layer of NNs75 or by using splines.76
Ab initio QM and DFT methods can also be similarly improved with ML, on the levels of the Hamiltonian, (Kohn–Sham) molecular orbitals, and functionals.77–79 On the DFT side, several general-purpose ML-improved DFT functionals were proposed, e.g., DM2180 and CF22D.81 Of course, these approaches are slower than ML-improved SQM ones, but they have the potential to be more robust because of the underlying less approximate physical models.
Since accurate ab initio methods such as different variants of coupled cluster and configuration interaction are very slow, there is a high incentive to make them faster by, e.g., accelerating their convergence and selecting the most important configurations.82–84
One of the biggest departures from the common QM methods is using ML to get the best wavefunction for the system.85,86 Impressive results were shown in this respect by obtaining more accurate values than with coupled cluster and recent works87,88 showed that it is possible to also obtain excited-state energies. Although these methods are still very computationally expensive, attempts are made in the direction of general-purpose faster approaches.89 The developments in this field are important to keep an eye on.
From Δ-learning to pure ML approaches
All the above discussions are mainly concerned with how to obtain more accurate (or faster) QM-based approaches by using ML. The ultimately faster solution is to use pure ML and not to perform any QM calculations during the production simulations. The most common application of pure ML models in computational chemistry is to learn the potential energy (and forces) for atomistic systems, i.e., building the so-called machine learning (interatomic) potentials (MLPs).29,40,62,90–92 Before considering the MLP models in general, let us first discuss how we can learn from the Δ-learning concept. This is admittedly an unusual way of discussing AI in computational chemistry as typically MLPs are considered first as simpler. However, it is beneficial in the context of the practical considerations which are quite general: we want a model that is as accurate as possible but also as robust as possible. While we want to discard the use of the baseline QM model, we want to preserve the benefit of its correct physical behavior. This indeed is possible to a good extent because we can generate much more data at the baseline, faster QM method and use the slow target QM method for generating only a fraction of data. Then the hope is that the correct physics will be learned statistically from more abundant data at the baseline QM level.How we proceed from here depends on the approach we choose. One of them is simply training the surrogate baseline ML model on more plentiful data at the baseline QM level and then training a correcting model on the difference between the target and baseline QM levels – ala Δ-learning but without any QM calculations needed for prediction. Indeed, in the literature, such an approach is also often referred to as Δ-learning,93 but I would prefer not to use this term for such ‘pseudo-hybrid’ models. The reason is that unless the surrogate baseline ML model is essentially indistinguishable from the real QM baseline method, the resulting hybrid model might still suffer from unphysical behavior and reduced accuracy. For example, the use of a surrogate ML baseline model plus ML correction did not yield the model better than the ANI-1ccx method,63 but the use of the real QM baseline (ODM2) yielded the AIQM1 method with substantially improved performance. The better name for such pseudo-hybrid approaches using the surrogate baseline ML models is hierarchical ML – the concept introduced by us in 2020 for generalizing Δ-learning to an arbitrary number of corrections and learning baseline QM too.4 An alternative way of training ML models on the data from several levels is the so-called multilevel combination quantum machine learning (CQML) technique.94 It can be used to create ML models exploiting the implicit correlation between multiple QM levels in the data.
Another approach to learning from multiple levels is aforementioned transfer learning, when the model is first trained on the more abundant data at the baseline QM level and then it is tuned on the smaller data at the target QM level.30 This approach was used in ANI-1ccx, where the model was pre-trained on the DFT baseline level and then transferred to the coupled-cluster level.63 Interestingly, the alternative formulation using two models (a surrogate ML baseline trained on DFT and ML correction to the coupled-cluster level as in hierarchical ML) yielded similar results to the single model obtained via transfer learning.63 Hence, hierarchical ML might be easier to implement as no changes in the training are required but transfer learning may lead to a more efficient solution as it has fewer models.
Transfer learning is also useful for repurposing the ML model from one class of molecules to another, related one as well as across different QM levels. ML approaches can further benefit from unsupervised pre-training. An example of such an ML model is PorphyBERT pre-trained on porphyrin-based dyes and transferred to more accurate DFT properties of metalloporphyrins.95
Despite the increasing number of studies utilizing transfer learning, relatively little research is done in exploring its advantages and limitations in the context of repurposing the models for different levels and molecules. Several studies indicate that while it is possible, the design of the original and target data sets in terms of size and composition should be done carefully for better performance.96,97
Nothing prevents us from combining several approaches into one. It indeed might be highly beneficial and was done, as we have seen, in AIQM1. There, the Δ-learning correction part was also trained using the transfer learning approach by first pre-training the NNs on the differences between the DFT and SQM levels and later transferring to the differences between the coupled cluster and SQM levels.
Zoo of machine learning potentials
I finally move on to describe the machine learning potentials (MLPs) which are also typically the constituent parts of the Δ-learning-based hybrid ML/QM models. We have already mentioned multiple times the ANI-1ccx method. It is one of the successful manifestations of the so-called universal MLPs, i.e., the models that were pre-trained on the big data containing chemically diverse species and applicable out of the box for simulations where its target QM method could be used. ANI-1ccx is constructed from the feed-forward NNs for each chemical element and the predictions for each atom are summed up to produce the total energy; it requires the local descriptor describing the environment around each atom within the cutoff. Such architecture was introduced in the Behler–Parrinello high-dimensional NNs applied for specific applications.61,62These models are just several examples of the many potentials in the rapidly growing zoo of MLPs.40,62,90 Some of them are based on NNs but by far not all. Many of the MLPs are based on kernel methods (kernel ridge regression and Gaussian process regression) and even linear models and polynomials.29,40,62,90–92 Our contribution to the zoo was the KREG-based models introduced in 2017 and based on the KRR and the global descriptor which describes the entire molecule.5,15,28 These models learn total energies without splitting them into atomic contributions. KREG models are most similar to independently introduced GDML-based models98,99 which also use the global descriptor and the KRR algorithm but only learn from forces rather than energies. The KREG models were first designed only for energies and later extended to also learn forces.28 The technical detail that is important to mention is that the inclusion of the force information into the data greatly improves the accuracy of the MLPs.40,100 In kernel methods, it can be done by including the terms corresponding to each force component in the training data explicitly at the level of the regression function.29 In NNs, it is typically done at the level of including the error for the forces in the loss function used during the fitting of NN weights.15
The descriptor plays a big role. In addition to the aforementioned local and global descriptors, which can be combined with different algorithms, the descriptor can be ‘learned’ in, e.g., message-passing NNs.101,102 Furthermore, building equivariant models via using the ‘equivariant’ local descriptors was shown to lead to improved accuracy, although the training time substantially increases compared to models based on feed-forward NNs.103,104
Most of the introduced MLPs were tested on the standard benchmark sets and were used in specific applications. To make them more practical for computational chemists, one can provide pre-trained ‘universal’ models such as ANI-1ccx and now it becomes a hot topic when many groups provide such models trained on varying numbers of chemical elements and data sets of different compositions and data at various QM levels.63,105–108 Universal models can have hiccups as we saw before for ANI-1ccx which has not-so-good performance for transition states. However, they are getting better and better, and, e.g., a recently introduced ANI-based model was shown to describe reactions quite well.109 While the universal MLPs are getting better, MLPs will continue to be developed and trained for specific applications for the foreseeable future.
The good thing is that it seems that the construction of MLPs is becoming quite mature if not routine for many applications. For example, MLPs based on permutationally invariant polynomials have very efficient implementations and were used in many specific applications for studying PESs, chemical dynamics, and vibrational levels of small to medium-sized systems.110
Still, the choice of the appropriate MLP model might be not easy and requires careful consideration of their accuracy, the computational cost for training and prediction, and how these performance metrics change with increasing number of training points as well as the performance for actual simulations. We made a careful analysis of all these factors to give recommendations for the choice of MLPs for learning single-molecule PESs in 2021 (Fig. 4).40 The field of MLPs is improving fast though, so newer models, such as equivariant ones, should be now considered in addition to those we benchmarked. In principle, there is an urgent need for standard, independent, and regular tests for MLPs. The challenge is that even terminology is not standardized. For example, when researchers train an NN-based model on the training data, they might set aside the hold-out test set on which they report the errors. However, training NN is done by performing the backpropagation on the so-called ‘training’ set and monitoring the performance on the validation data sets. The problem now is that the ‘training’ set used for backpropagation is the subset of the ‘total budget’ of the training points which also includes the validation data. If this sounds confusing – it is. That is why we consistently use the term ‘sub-training’ for the set used for backpropagation in NNs (and finding regression coefficients in the kernel methods). This resolves the confusion but unfortunately not followed uniformly in the literature. I hope that the reader of this paper might adopt this terminology of sub-training and validation sets being subsets of the training set.
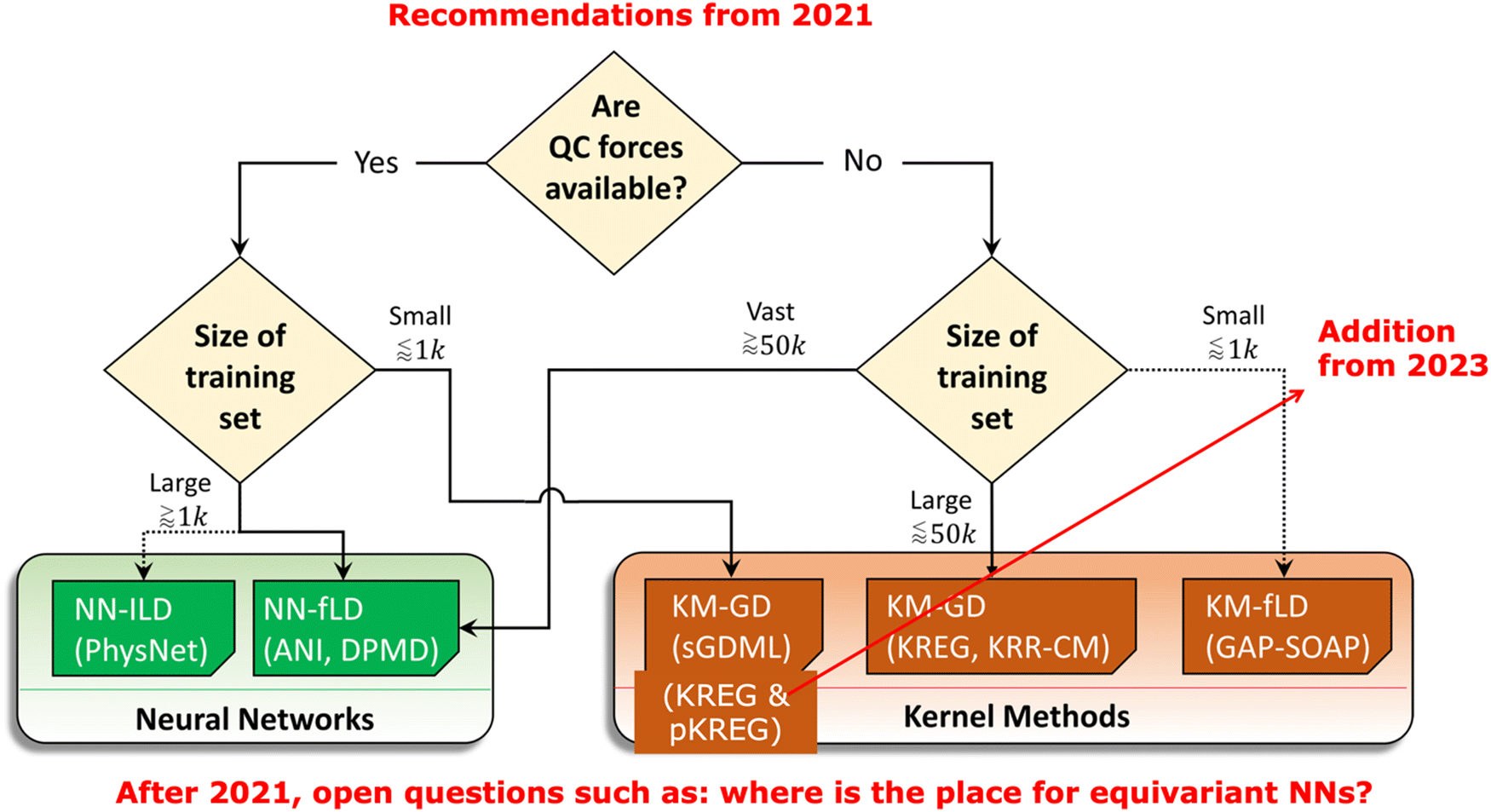 | ||
| Fig. 4 Old recommendation from 2021 for choosing MLPs for learning single-molecule PESs based on considerations concerning accuracy, training, and prediction time. Since 2021, many additions should be considered. Adapted with permission from ref. 40. Copyright 2021, the Authors. | ||
The key to making the use of MLP routine is also providing the means to sample required data at the QM level. Here, various strategies based on ideas behind active learning are explored. Many of them use the deviation between the different MLPs trained for the same task as the indicator of uncertainty while others judge uncertainty differently (i.e., from the variance given by the Gaussian process regression or using the Bayesian NNs).111–114 In any case, active learning selects points in a loop consisting of the simulations, identification of uncertain points, labeling them, training the model, and repeating the procedure until the satisfactory simulations.115 Examples of successful active learning procedures and software include the construction of the aforementioned ANI data sets and models105 as well as approaches based on DPMD116 including the deep potential generator (DP-GEN) procedure117 and algorithms exploiting meta-dynamics.118 Our active learning implementations based on MLatom are also on the way.
One of the pertinent problems of the MLPs is the correct treatment of the large systems which are likely to include important long-range interactions such as dispersion, electrostatics, and induction terms.119 In principle, if MLPs are directly learning the whole system, e.g., with the global descriptors like in sGDML120 or KREG,28 then they can also learn the long-range interactions within the system. Nevertheless, most MLPs applied to very large systems heavily rely on nearsightedness approximations and only treat the local part of the system within some cutoff. This inevitably leads to the loss of accuracy when the properties of the whole system are reconstructed from such MLPs. Lot of progress is made in this respect although many challenges remain too.119 Dispersion interactions can be either added explicitly via, e.g., D4 corrections, similarly to the typical practice in DFT and as is done in ANI-1x-D4 and ANI-2x-D4 methods121 (also implemented in MLatom),10 or they can be attempted to be implicitly learned by MLP from the data (as ANI-1ccx does to some extend).63,119 Both approaches are approximations and can be insufficient.119 Electrostatics is also a challenge to learn, and different approximations were suggested too, i.e., some of them rely on learning charges (e.g., to reproduce the dipole moments from the reference QM calculations)102 and others on self-consistent or message-passing framework iteratively refining the charge distribution until the lower-energy solution is found.119 While many approaches rely on learning point charges,62 an alternative was suggested based on maximally localized Wannier centers.122 One of the interesting directions for exploring large systems is the incorporation of ML into QM/MM and ONIOM schemes.119
Software for computational chemists
It is apparent from the above very incomplete overview that the number and the quality of computational chemistry tools are rapidly increasing, at a much faster pace than ever in its history. Even specialists can no longer keep up with all the developments. In addition, method development can benefit end users only if they can access the software implementing the new methods which ideally should be also easy to use. While in the AI for computational chemistry, we see the laudable trend of making the software open source, the problem is exacerbated by the fact that the computational methods are scattered in different software packages with often incompatible formats of input and output. The AI requires a data-oriented approach to software design with consistent support of the databases and options for training and using ML models. The more traditional QM software is mostly designed to enable calculations of the quantum mechanical properties of a single system at a time. The clash in the software design strategies is also aggravated by the use of different programming languages making it more difficult for developers to merge the two software types. Despite all the difficulties, many solutions and roadmaps to potential solutions begin to emerge.123,124 Particularly the development of packages like ASE allows bridging together different types of models to enable common simulations such as molecular dynamics (MD) and geometry optimizations.125We also provide a software solution for AI-enhanced computational chemistry – an open-source MLatom package that was started in 2013 as a standalone package for creating and using generic ML models for users without programming experience.17,19 In 10 years, it was completely redesigned to support the range of QM (ab initio, DFT, SQM), ML (popular MLPs such as MACE,104 ANI-type NNs, generic KRR models), and the hybrid ML/QM models (AIQM1 and user-customized) via interfaces to many third-party software packages.10,15 While it can be used as originally intended by users without programming experience for their applications, MLatom is at the same time a versatile Python package enabling the developers to seamlessly build their computational workflows.10 Importantly, we lower the barrier and democratize the use of AI in computational chemistry by providing access to MLatom through the web user interface of the XACS (Xiamen Atomistic Computing Suite) cloud computing.126
MLatom can use the QM, ML, and hybrid ML/QM models to perform typical computational chemistry simulations including geometry optimizations, frequencies and thermochemistry calculations, molecular and quantum dynamics, and supports different types of spectroscopies (Fig. 5).10 Below we discuss how AI makes dynamics and spectroscopy faster and even more accurate.
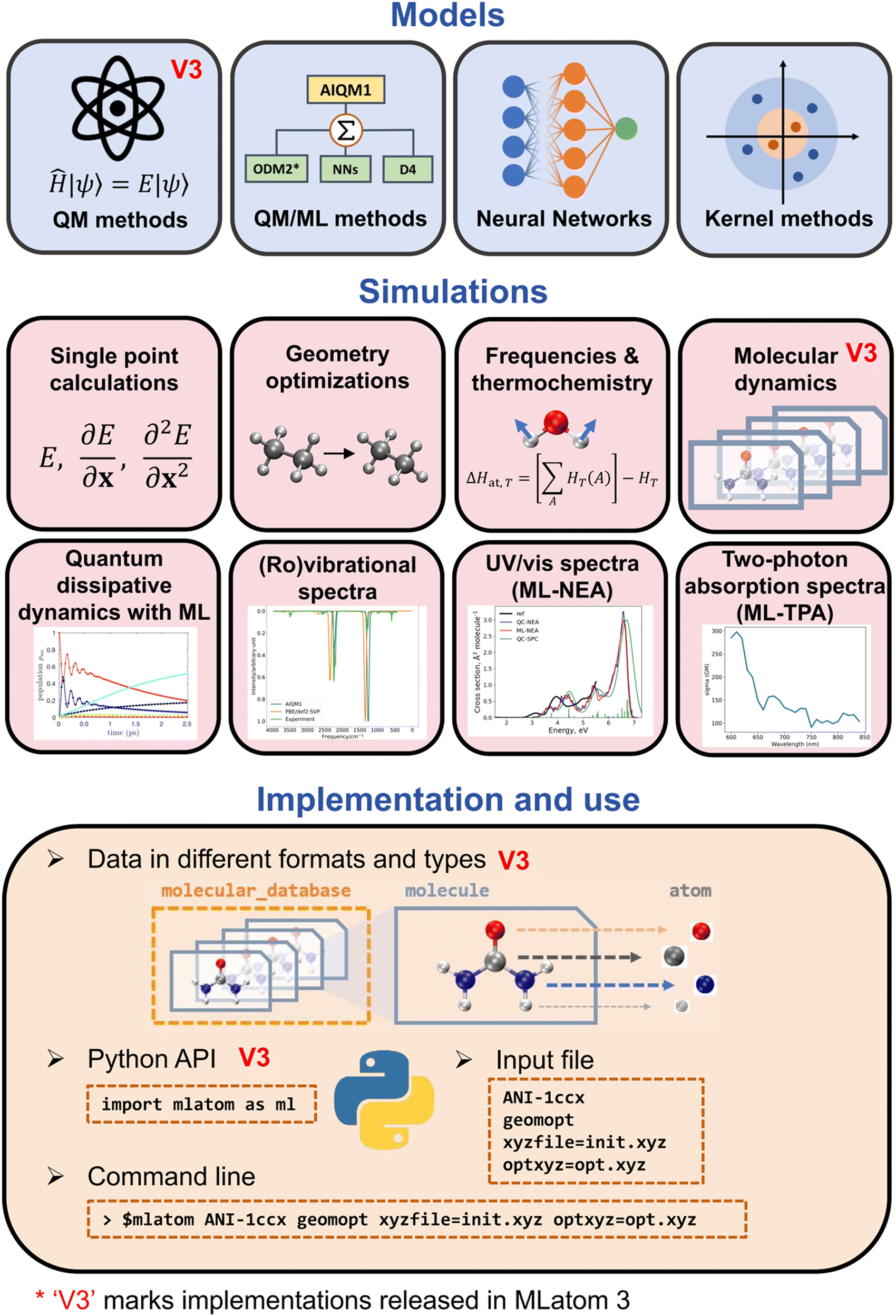 | ||
| Fig. 5 Overview of MLatom functionality. Reproduced with permission from ref. 10. Copyright 2024, the Authors. The plot in panel “Quantum dissipative dynamics with ML” is adapted with permission from ref. 3. Copyright 2022, the Authors. The plot in panel “UV/vis spectra (ML-NEA)” is adapted from ref. 15. Copyright 2021, the Authors. | ||
Dynamics
Dynamics is one of the costliest simulation types in computational chemistry as it requires computationally expensive evaluations of the required properties (e.g., forces in molecular dynamics) at each of the many time steps. Hence, dynamics was one of the first targets for ML, which enables simulations otherwise prohibitively expensive.127 Dynamics is also a nice example of how ML can be used completely differently: either as a surrogate model for the reference (e.g., QM) methods or learning dynamics directly.Accelerating dynamics with surrogate ML models
Using ML as a surrogate potential for fast prediction of forces to propagate MD was one of the first successful applications of ML in computational chemistry.127 The motivation to accelerate MD led to the development of numerous MLPs. This is hence also one of the most mature fields. Hybrid ML/QM models such as AIQM1 can also be used to get more accurate and stable dynamics which might be also sufficiently fast if the baseline QM is fast enough (e.g., if it is SQM method). The relationship between the accuracy of the models on the test set and the stability of the dynamics is not as straightforward though, and one should always perform careful checks and design of the model.9,128–130The use of ML is not limited to classical, ground-state MD. ML was applied to PIMD too.99 Our interests often involved the excited-state nonadiabatic dynamics, which is another area where AI models are subject to intense development.20,27,41,131 Despite all the efforts, ML nonadiabatic dynamics is much less easy to perform than ground-state ML-accelerated dynamics. The reason is that the learning problem is much more difficult, particularly in case of the mixed quantum-classical dynamics based on the trajectory surface-hopping formalism. It needs an accurate description of different adiabatic surfaces and also an accurate evaluation of the hopping probability which ideally requires evaluation of the nonadiabatic coupling vectors (NACVs). The accuracy of the ML models is usually lower for excited-state energies than for ground-state energies and the ML should properly capture intricate details of the excited-state PESs and the gap between them.131 When NACVs are evaluated, they need to be phase-corrected and also the geometries with large-magnitude NACVs properly sampled.18,27,42,131 ML techniques for these tasks are constantly improved. In some cases, it might be a good idea to simplify the learning problem, e.g., the calculation of NACVs can be avoided with the Zhu–Nakamura or Landau–Zener approximations.18,27,42,46,131 As we and others independently showed, it might be easier to set up more robust simulations by simply invoking the reference QM simulations at the regions with a low energy gap.18,46,132
Another type of dynamics is the quantum dissipative dynamics of open quantum systems. It is rather different from MD, but the algorithm is analogous in a way that every time step depends on the previous time steps. Here, NN approaches for learning time series were successfully applied to achieve accurate results at a fraction of the cost.133 Our contribution was to demonstrate that even approaches such as KRR and linear regression which would not be typically considered for treating time series offer a good alternative to NNs.37,39 Interestingly, even linear regression might be more accurate than ‘fancy’ NNs such as bidirectional recurrent NNs (BRNNs) for ML-accelerated quantum dynamics (Fig. 6).37 This is a good reminder that often, simpler models might be completely sufficient and even preferable for a particular application. ML for quantum dynamics is a relatively new field and we also introduced an MLQD package8 interfaced with MLatom and data sets22 which can be used to test and develop new models.
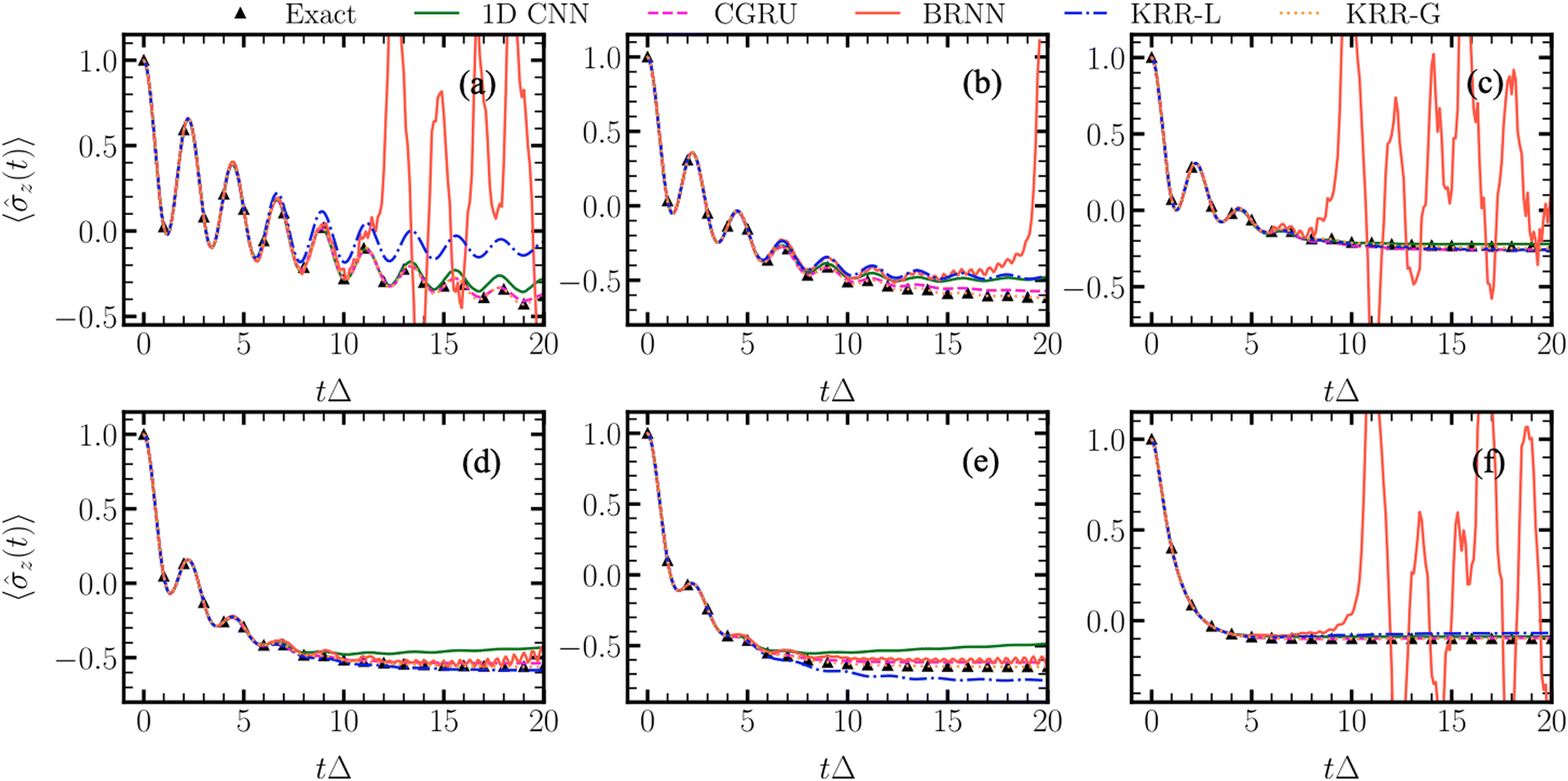 | ||
| Fig. 6 An example is when simpler model (KRR-L corresponding to the ridge linear regression) has better performance than a much more complex one (BRNN, bidirectional recurrent neural network). The plots show how well different algorithms learn the time series from the quantum dissipative dynamics, with the x-axis representing time in units of Δ units and the y-axis – the population difference. CNN denotes convolutional neural network, CGRU – NN based on convolutional gated recurrent unit, BRNN – bidirectional recurrent neural networks, KRR-G – kernel ridge regression with the Gaussian kernel. Reproduced with permission from ref. 37. Copyright 2022, the Authors. | ||
Learning dynamics: the new way of doing computational chemistry
Despite all the accelerations of dynamics with AI, the dynamics was still performed based on the same underlying sequential algorithm, one step at a time. AI provides us an opportunity to rethink how we approach the simulations as now we can, in principle, use ML to learn what we actually need. In the case of dynamics, we need to know the evolution of some property (nuclear coordinates or populations) as a function of time. NNs, as universal approximators,134 can do this.We introduced learning dynamical properties as a function of time and shown the feasibility of this concept for both molecular and quantum dissipative dynamics.1,3 In the case of quantum dynamics, our AI-QD approach successfully learned populations and coherence as a function of time and gave accurate predictions for the test trajectories.3
Before we move on to show how this concept can be applied to MD, we should also mention that AI can predict the entire trajectory section in one shot, instead of predicting each time step separately.2 We introduced and shown the feasibility of this one-shot-learning strategy for quantum dynamics, where we could predict 10-ps long trajectory section in just 70 ms.2
Learning the time evolution of nuclear positions as in MD proved to be much more challenging.1 We showed that it can be done for the simplest systems such as H2 which has periodic oscillations of it bond length and dedicated KRR model can predict its behavior for any arbitrary time.1 For larger systems with many atoms, the learning task is much more difficult, and practically, we could accurately learn molecular dynamics for a finite time segment. These time segments are very long though, much longer than typical MD time steps. We could get reasonable MD quality by predicting up to 10–20 fs with our GICnet model for as large molecules as azobenzene.1 In the case of learning MD trajectories, we could only get a successful result after we constrained the AI to respect the required physics such as the conservation of the total energy and angular momentum.1
Learning trajectories, in fact, is much more than creating a faster MD propagator. In its core, we find an analytical function that represents molecular objects in 4D-spacetime. The nuclear positions are learned for input in 3D space plus a time dimension and predicted likewise. This is in contrast to MLPs that predict an energy for an object learned in 3D space. That is why, we use the term 4D-spacetime models which is a new concept.1 It is different from MD in many respects: 4D-spacetime models can predict nuclear positions at any time (and hence any resolution) in a time segment without the need for iterative propagation making the predictions very fast and readily parallelizable. Sequential predictions for time segments allow the generation of very long MD trajectories at a much-reduced cost, even when compared to MD with MLPs. We can take derivatives of 4D-spacetime models to obtain nuclear velocities and acceleration at different times, from which we can recover forces, making these models more general compared to MLPs.1
In addition, since 4D-spacetime models contain information about the molecular object at different times, they present a unique opportunity to look from a new angle at the chemical reactions. Reactions are contained in the 4D-spacetime models and they can be used to analyze what happens in time when a reactant becomes the product. We showed proof-of-concept that we can predict the reaction pathway and also that we can analyze 4D-spacetime models to understand which structural parameters and velocities lead to chemical reactions.1
Similar to the Δ-learning concept started in 2013, it might take some time until 4D-spacetime models become commonplace and materialize in ready-to-use methods such as AIQM1. In the meantime, they can be used for specific problems as MLPs are used. The concept is quite intuitive and several groups independently studied related approaches, e.g., for interpolating between time steps in MD.135
Geometry optimizations and reaction explorations with AI
Before going to the next topic, I had to at least briefly mention that AI models can be used for accelerating geometry optimizations of minima and transition states.136–140 An increased interest is also now in creating models directly predicting activation energies important for reaction design.33,43,62,78,141–150 Concerning reaction design and exploration, many specialized models are developed also for planning the synthesis and exploration of the reaction mechanisms.33,43,62,78,141–151Spectroscopy
Spectroscopy is one of the major applications of computational chemistry because it gives us a direct way to compare with experimental observables. The direct spectroscopy simulations with QM methods are often very time-consuming and, hence, AI offers itself as a promising solution to reduce the time cost. Interestingly, spectroscopy was a target of ML for quite a long time as an early review from 1990s indicates.152Similar to the dynamics, here AI can be applied using two different strategies. One of them is to create AI counterparts for QM methods and use them in the same way. One example of such strategy which was mentioned before is the use of AIQM1 for running a long Born–Oppenheimer MD trajectory and then post-processing it to obtain the infrared (IR) spectrum.9 Here AI improves the quality of the semi-empirical QM baseline leading to an accurate spectrum at a cost lower than DFT. Pure ML models were also used quite early to generate IR spectra after they were trained using active learning.153 MLPs and 4D-spacetime models were also applied to generate the related power spectra from the MD trajectories.1
Similarly, UV/vis absorption and emission spectra can be simulated by calculating excited-state properties (excitation energies, transition dipole moments or oscillator strengths) for many molecular configurations and different molecules. This is very costly. ML can efficiently learn these properties from a relatively small fraction of the configurations (or molecules) to produce high-precision spectra as was shown in ours and independent studies.16,21,41,154–158 In addition, we developed a procedure based on ML for the automatic selection of the optimal number of sampled configurations (Fig. 7).16 ML-accelerated simulation of the electronic spectrum is also a strong example that the error of ML with respect to the reference QM level is lower than the error of QM with respect to the experiment.16 It has the consequence that the better way to improve the electronic spectrum quality is to choose the better reference QM level rather than trying to improve the ML further.
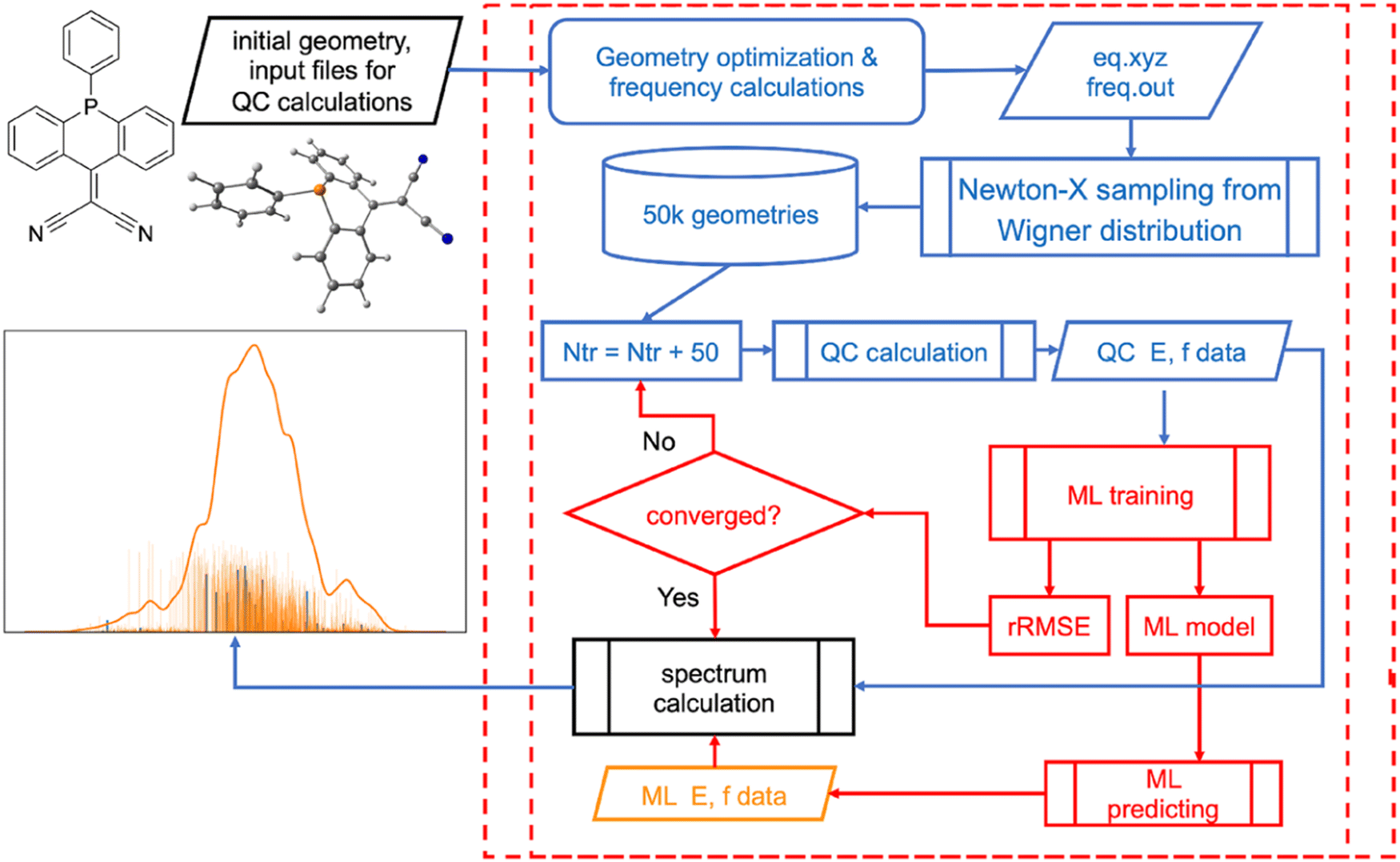 | ||
| Fig. 7 Simulation of precise UV/vis absorption spectra with machine learning (ML) procedure that automatically chooses the optimal number of the training points. Reproduced with permission from ref. 15. Copyright 2021, the Authors. | ||
AI to directly predict spectra
AI can directly predict spectral shapes and intensities.159,160 It can be done for IR and UV/vis absorption and emission spectra mentioned above. This direct strategy might be particularly useful in situations where the previous strategy (using ML as a replacement (or supplement) of QM methods) is too inaccurate and costly for practical applications of, e.g., high-throughput screening of many materials. We had this situation for the two-photon absorption which is a fascinating property of some materials able to absorb two photons and release the photon of higher energy – useful in many applications such as upconverted laser and 3D printing. For this property, we created the ML model directly trained on the experimental data and using inexpensive structural and solvent descriptors.23 This model did not just predict the spectra (two-photon absorption cross sections as the function of the wavelength) with a satisfactory quality similar to DFT but also provided insight which structural parameters are important in the design of the materials with better two-photon absorption (Fig. 8). | ||
| Fig. 8 Learning two-photon absorption required a careful selection of features. Reproduced with permission from ref. 23. Copyright 2023, the Authors. | ||
An inverse problem of elucidating structure from spectra is another important topic where AI is starting to become the center of research.161,162
Materials and drug design
Computational chemistry is also intensively used in materials and drug design. While many of the AI tools described above can aid in such design efforts, specialized AI tools are often necessary to develop for particular tasks and many reviews cover these topics.163 An example of our research is developing specialized models for designing better materials based on mixed metal halides for ammonia separation and storage.38 Finding better materials for this application might help with the pressing issue of energy storage as ammonia can be used as a medium for indirect hydrogen storage. Remarkably, we were able to train ML achieving chemical accuracy for predicting the deammoniation energies which is the key property of such materials.38 The crucial finding was that we can get much better models when we train the models for intermediate QM properties such as ionic polarizabilities and charges which are then fed into the ML model predicting the final target property.38 Similarly, we used ML to learn the two-photon absorption on relatively small experimental data (less than thousand data points) and were able to predict absorption strengths for new compounds which were verified in the lab; this approach has the potential to be used for screening better materials for, e.g., upconverted laser or 3D printing.23Both materials and drug design can follow many strategies. One of them is, as in the above examples, learning the key property of interest and then applying the ML model to screen the database to find the best-performing materials. In the case of drug design, that would mean, for example, using ML for learning, e.g., protein–ligand binding affinities164 or antimicrobial resistance,165 and applying them to screen the databases.166 The other way is to use ML, e.g., generative models and reinforcement learning, to also come up with new potential drug molecules.167 Explainability of ML is also important as it can give insight to experts on what factors are important in the design of new molecules and, e.g., identify the structural classes of antibiotics.168
Nowadays, materials and drug design, and chemistry in general, witness the radical shift to automized lab platforms which increasingly rely on AI for many steps of the process, from hypothesis creation to testing with robots.163,169,170 Which leads to the final topic.
ChatGPT and other elephants in the room
The aforementioned automatization and robotization of the lab processes got a great boost from the large language models such as ChatGPT and its successors, competitors, and related models. They can write snippets of code that can be used by developers of computational chemistry software. These language models can also give suggestions for computational chemists. Currently, there is lots of criticism and concerns about the often-erroneous output from such models but we should keep in mind that these models are just at the beginning of their full potential. ChatGPT-like models might be even too disruptive and even harmful. While I am a big proponent of the use of AI in computational chemistry, I also strongly advocate the ethical, responsible, and cautious use of AI.171 My conviction is that AI should stay as a useful tool helping researchers and not be misused.Conclusions and outlook
It is fitting to close the overview of our 10-year journey in AI in computational chemistry by quoting an attendee of one of my talks who quipped that ‘it looks too good to be true’. Indeed, the reader might get such an impression and I am fully aware of the existing reservations in the more traditionally (quantum mechanically) oriented community. Let me reassure you that AI delivers in computational chemistry what it promises. It is already mature for many applications. Advances in computational chemistry due to AI are breathtaking and they are no longer limited to proof-of-concepts. AI is indeed that good and everyone can check how good AI-enhanced computational chemistry is, even via a web browser.10,19,126 We will see more improvements coming in terms of increased accuracy and efficiency for more and more types of applications as well as for new insights and concepts giving a fresh look at chemistry.Author contributions
This article is a single-author contribution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to thank my collaborators who shaped and contributed to the research overviewed in this article. I acknowledge funding by the National Natural Science Foundation of China (No. 22003051 and funding via the Outstanding Youth Scholars (Overseas, 2021) project), the Fundamental Research Funds for the Central Universities (No. 20720210092), and via the Lab project of the State Key Laboratory of Physical Chemistry of Solid Surfaces. This project is supported by Science and Technology Projects of Innovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM) (No. RD2022070103).References
- F. Ge, L. Zhang, Y.-F. Hou, Y. Chen, A. Ullah and P. O. Dral, Four-Dimensional-Spacetime Atomistic Artificial Intelligence Models, J. Phys. Chem. Lett., 2023, 14, 7732–7743, see also the preprint version at ChemRxiv DOI:10.26434/chemrxiv-2022-qf75v, which contains additional ideas and results.
- A. Ullah and P. O. Dral, One-Shot Trajectory Learning of Open Quantum Systems Dynamics, J. Phys. Chem. Lett., 2022, 13, 6037–6041 CrossRef CAS PubMed.
- A. Ullah and P. O. Dral, Predicting the future of excitation energy transfer in light-harvesting complex with artificial intelligence-based quantum dynamics, Nat. Commun., 2022, 13, 1930 CrossRef CAS PubMed.
- P. O. Dral, A. Owens, A. Dral and G. Csányi, Hierarchical Machine Learning of Potential Energy Surfaces, J. Chem. Phys., 2020, 152, 204110 CrossRef CAS PubMed.
- P. O. Dral, A. Owens, S. N. Yurchenko and W. Thiel, Structure-based sampling and self-correcting machine learning for accurate calculations of potential energy surfaces and vibrational levels, J. Chem. Phys., 2017, 146, 244108 CrossRef PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach, J. Chem. Theory Comput., 2015, 11, 2087–2096 CrossRef CAS PubMed.
- P. O. Dral, O. A. von Lilienfeld and W. Thiel, Machine Learning of Parameters for Accurate Semiempirical Quantum Chemical Calculations, J. Chem. Theory Comput., 2015, 11, 2120–2125 CrossRef CAS PubMed.
- A. Ullah and P. O. Dral, MLQD: A package for machine learning-based quantum dissipative dynamics, Comput. Phys. Commun., 2024, 294, 108940 CrossRef CAS.
- L. Zhang, Y.-F. Hou, F. Ge and P. O. Dral, Energy-conserving molecular dynamics is not energy conserving, Phys. Chem. Chem. Phys., 2023, 25, 23467–23476 RSC.
- P. O. Dral, F. Ge, Y.-F. Hou, P. Zheng, Y. Chen, M. Barbatti, O. Isayev, C. Wang, B.-X. Xue, M. Pinheiro Jr, Y. Su, Y. Dai, Y. Chen, S. Zhang, L. Zhang, A. Ullah, Q. Zhang and Y. Ou, MLatom 3: A Platform for Machine Learning-Enhanced Computational Chemistry Simulations and Workflows, J. Chem. Theory Comput., 2024, 20, 1193–1213 CrossRef CAS PubMed.
- F. Bosia, P. Zheng, A. C. Vaucher, T. Weymuth, P. O. Dral and M. Reiher, Ultra-Fast Semi-Empirical Quantum Chemistry for High-Throughput Computational Campaigns with Sparrow, J. Chem. Phys., 2023, 158, 054118 CrossRef CAS PubMed.
- P. Zheng, W. Yang, W. Wu, O. Isayev and P. O. Dral, Toward Chemical Accuracy in Predicting Enthalpies of Formation with General-Purpose Data-Driven Methods, J. Phys. Chem. Lett., 2022, 13, 3479–3491 CrossRef CAS PubMed.
- M. Barbatti, M. Bondanza, R. Crespo-Otero, B. Demoulin, P. O. Dral, G. Granucci, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, M. Pederzoli, M. Persico, M. Pinheiro Jr, J. Pittner, F. Plasser, E. Sangiogo Gil and L. Stojanovic, Newton-X Platform: New Software Developments for Surface Hopping and Nuclear Ensembles, J. Chem. Theory Comput., 2022, 18, 6851–6865 CrossRef CAS PubMed.
- P. Zheng, R. Zubatyuk, W. Wu, O. Isayev and P. O. Dral, Artificial Intelligence-Enhanced Quantum Chemical Method with Broad Applicability, Nat. Commun., 2021, 12, 7022 CrossRef CAS PubMed.
- P. O. Dral, F. Ge, B.-X. Xue, Y.-F. Hou, M. Pinheiro Jr, J. Huang and M. Barbatti, MLatom 2: An Integrative Platform for Atomistic Machine Learning, Top. Curr. Chem., 2021, 379, 27 CrossRef CAS PubMed.
- B.-X. Xue, M. Barbatti and P. O. Dral, Machine Learning for Absorption Cross Sections, J. Phys. Chem. A, 2020, 124, 7199–7210 CrossRef CAS PubMed.
- P. O. Dral, MLatom: A Program Package for Quantum Chemical Research Assisted by Machine Learning, J. Comput. Chem., 2019, 40, 2339–2347 CrossRef CAS PubMed.
- P. O. Dral, M. Barbatti and W. Thiel, Nonadiabatic Excited-State Dynamics with Machine Learning, J. Phys. Chem. Lett., 2018, 9, 5660–5663 CrossRef CAS PubMed.
- P. O. Dral, F. Ge, Y.-F. Hou, P. Zheng, Y. Chen, B.-X. Xue, M. Pinheiro Jr, Y. Su, Y. Dai, Y. Chen, S. Zhang, L. Zhang, A. Ullah, Q. Zhang and Y. Ou, MLatom A Package for Atomistic Simulations with Machine Learning, Xiamen University, Xiamen, China, https://MLatom.com (accessed March 3, 2024), pp. 2013–2024 Search PubMed.
- L. Zhang, A. Ullah, M. Pinheiro Jr and P. O. Dral, Excited-state dynamics with machine learning, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 329–353 Search PubMed.
- J. Westermayr, P. O. Dral and P. Marquetand, Learning excited-state properties, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 467–488 Search PubMed.
- A. Ullah, L. E. Herrera Rodríguez, P. O. Dral and A. A. Kananenka, QD3SET-1: a database with quantum dissipative dynamics datasets, Front. Phys., 2023, 11, 1223973 CrossRef.
- Y. Su, Y. Dai, Y. Zeng, C. Wei, Y. Chen, F. Ge, P. Zheng, D. Zhou, P. O. Dral and C. Wang, Interpretable Machine Learning of Two-Photon Absorption, Adv. Sci., 2023, 2204902 CrossRef CAS PubMed.
- T. A. Schaub, A. Zieleniewska, R. Kaur, M. Minameyer, W. Yang, C. M. Schüßlbauer, L. Zhang, M. Freiberger, L. N. Zakharov, T. Drewello, P. O. Dral, D. Guldi and R. Jasti, Tunable Macrocyclic Polyparaphenylene Nanolassos via Copper-Free Click Chemistry, Chem. – Eur. J., 2023, 29, e202300668 CrossRef CAS PubMed.
- M. Pinheiro Jr, S. Zhang, P. O. Dral and M. Barbatti, WS22 database: combining Wigner Sampling and geometry interpolation towards configurationally diverse molecular datasets, Sci. Data, 2023, 10, 95 CrossRef PubMed.
- M. Pinheiro Jr and P. O. Dral, Kernel methods, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 205–232 Search PubMed.
- J. Li, M. Vacher, P. O. Dral and S. A. Lopez, in Machine learning methods in photochemistry and photophysics, Theoretical and Computational Photochemistry: Fundamentals, Methods, Applications and Synergy with Experimentation, ed. García-Iriepa, C. and Marazzi, M., Elsevier, 2023, pp 163–189 Search PubMed.
- Y.-F. Hou, F. Ge and P. O. Dral, Explicit Learning of Derivatives with the KREG and pKREG Models on the Example of Accurate Representation of Molecular Potential Energy Surfaces, J. Chem. Theory Comput., 2023, 19, 2369–2379 CrossRef CAS PubMed.
- Y.-F. Hou and P. O. Dral, Kernel method potentials, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 295–312 Search PubMed.
- P. O. Dral, T. Zubatiuk and B.-X. Xue, Learning from multiple quantum chemical methods: Δ-learning, transfer learning, co-kriging, and beyond, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 491–507 Search PubMed.
- P. O. Dral and T. Zubatiuk, Improving semiempirical quantum mechanical methods with machine learning, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 559–575 Search PubMed.
- P. O. Dral, A. Kananenka, F. Ge and B.-X. Xue, Neural networks, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 183–204 Search PubMed.
- Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023 Search PubMed.
- A. E. Clark, P. O. Dral, I. Tamblyn and O. Isayev, Themed collection on Insightful Machine Learning for Physical Chemistry, Phys. Chem. Chem. Phys., 2023, 25, 22563–22564 RSC.
- Y. Chen, Y. Ou, P. Zheng, Y. Huang, F. Ge and P. O. Dral, Benchmark of General-Purpose Machine Learning-Based Quantum Mechanical Method AIQM1 on Reaction Barrier Heights, J. Chem. Phys., 2023, 158, 074103 CrossRef CAS PubMed.
- L. Zhang, S. Zhang, A. Owens, S. N. Yurchenko and P. O. Dral, VIB5 database with accurate ab initio quantum chemical molecular potential energy surfaces, Sci. Data, 2022, 9, 84 CrossRef CAS PubMed.
- L. E. Herrera Rodríguez, A. Ullah, K. J. Rueda Espinosa, P. O. Dral and A. A. Kananenka, A comparative study of different machine learning methods for dissipative quantum dynamics, Mach. Learn. Sci. Technol., 2022, 3, 045016 CrossRef.
- A. de Rezende, M. Malmali, P. O. Dral, H. Lischka, D. Tunega and A. J. A. Aquino, Machine Learning for Designing Mixed Metal Halides for Efficient Ammonia Separation and Storage, J. Phys. Chem. C, 2022, 126, 12184–12196 CrossRef CAS.
- A. Ullah and P. O. Dral, Speeding up quantum dissipative dynamics of open systems with kernel methods, New J. Phys., 2021, 23, 113019 CrossRef CAS.
- M. Pinheiro Jr, F. Ge, N. Ferré, P. O. Dral and M. Barbatti, Choosing the right molecular machine learning potential, Chem. Sci., 2021, 12, 14396–14413 RSC.
- P. O. Dral and M. Barbatti, Molecular Excited States Through a Machine Learning Lens, Nat. Rev. Chem., 2021, 5, 388–405 CrossRef CAS PubMed.
- P. O. Dral, in Quantum Chemistry Assisted by Machine Learning, Advances in Quantum Chemistry: Chemical Physics and Quantum Chemistry, ed. Ruud, K. and Brändas, E. J., Academic Press, 1st edn, 2020, vol. 81, pp. 291–324 Search PubMed.
- P. O. Dral, Quantum Chemistry in the Age of Machine Learning, J. Phys. Chem. Lett., 2020, 11, 2336–2347 CrossRef CAS PubMed.
- W.-K. Chen, X.-Y. Liu, W. Fang, P. O. Dral and G. Cui, Deep Learning for Nonadiabatic Excited-State Dynamics, J. Phys. Chem. Lett., 2018, 9, 6702–6708 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
- S. V. Pios, M. F. Gelin, A. Ullah, P. O. Dral and L. Chen, Artificial-Intelligence-Enhanced On-the-Fly Simulation of Nonlinear Time-Resolved Spectra, J. Phys. Chem. Lett., 2024, 15, 2325–2331 CrossRef CAS PubMed.
- X. Wu and P. Su, Very brief introduction to quantum chemistry, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 3–25 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. O. Dral and J. Řezáč, Semiempirical quantum mechanical methods, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 67–92 Search PubMed.
- J. R. Thomas, B. J. DeLeeuw, G. Vacek, T. D. Crawford, Y. Yamaguchi and H. F. Schaefer, The balance between theoretical method and basis set quality: A systematic study of equilibrium geometries, dipole moments, harmonic vibrational frequencies, and infrared intensities, J. Chem. Phys., 1993, 99, 403–416 CrossRef CAS.
- T. Helgaker, J. Gauss, P. Jørgensen and J. Olsen, The prediction of molecular equilibrium structures by the standard electronic wave functions, J. Chem. Phys., 1997, 106, 6430–6440 CrossRef CAS.
- K. L. Bak, J. Gauss, P. Jørgensen, J. Olsen, T. Helgaker and J. F. Stanton, The accurate determination of molecular equilibrium structures, J. Chem. Phys., 2001, 114, 6548–6556 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, A fifth-order perturbation comparison of electron correlation theories, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- B. Chan, E. Collins and K. Raghavachari, Applications of isodesmic-type reactions for computational thermochemistry, WIREs Comput. Mol. Sci., 2020, 11, e1501 CrossRef.
- X. Gao, F. Ramezanghorbani, O. Isayev, J. S. Smith and A. E. Roitberg, TorchANI: A Free and Open Source PyTorch-Based Deep Learning Implementation of the ANI Neural Network Potentials, J. Chem. Inf. Model., 2020, 60, 3408–3415 CrossRef CAS PubMed.
- J. S. Smith, R. Zubatyuk, B. Nebgen, N. Lubbers, K. Barros, A. E. Roitberg, O. Isayev and S. Tretiak, The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules, Sci. Data, 2020, 7, 134 CrossRef CAS PubMed.
- A. P. Bartók, S. De, C. Poelking, N. Bernstein, J. R. Kermode, G. Csányi and M. Ceriotti, Machine learning unifies the modeling of materials and molecules, Sci. Adv., 2017, 3, e1701816 CrossRef PubMed.
- P. O. Dral, X. Wu and W. Thiel, Semiempirical Quantum-Chemical Methods with Orthogonalization and Dispersion Corrections, J. Chem. Theory Comput., 2019, 15, 1743–1760 CrossRef CAS PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, Extension of the D3 dispersion coefficient model, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy surfaces, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- J. Behler, Four Generations of High-Dimensional Neural Network Potentials, Chem. Rev., 2021, 121, 10037–10072 CrossRef CAS PubMed.
- J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Devereux, K. Barros, S. Tretiak, O. Isayev and A. E. Roitberg, Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning, Nat. Commun., 2019, 10, 2903 CrossRef PubMed.
- J. E. Moussa, Comment on “Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning”, Phys. Rev. Lett., 2012, 109, 059801 CrossRef PubMed.
- J. Wu and X. Xu, The X1 method for accurate and efficient prediction of heats of formation, J. Chem. Phys., 2007, 127, 214105 CrossRef PubMed.
- L. H. Hu, X. J. Wang, L. H. Wong and G. H. Chen, Combined first-principles calculation and neural-network correction approach for heat of formation, J. Chem. Phys., 2003, 119, 11501–11507 CrossRef CAS.
- J. Zeng, Y. Tao, T. J. Giese and D. M. York, QDπ: A Quantum Deep Potential Interaction Model for Drug Discovery, J. Chem. Theory Comput., 2023, 19, 1261–1275 CrossRef CAS PubMed.
- C. H. Pham, R. K. Lindsey, L. E. Fried and N. Goldman, High-Accuracy Semiempirical Quantum Models Based on a Minimal Training Set, J. Phys. Chem. Lett., 2022, 13, 2934–2942 CrossRef CAS PubMed.
- E. M. Collins, K. Raghavachari and A. Fragmentation-Based Graph Embedding, Framework for QM/ML, J. Phys. Chem. A, 2021, 125, 6872–6880 CrossRef CAS PubMed.
- H. Bhattacharjee, N. Anesiadis and D. G. Vlachos, Regularized machine learning on molecular graph model explains systematic error in DFT enthalpies, Sci. Rep., 2021, 11, 14372 CrossRef CAS PubMed.
- A. S. Christensen, S. K. Sirumalla, Z. Qiao, M. B. O'Connor, D. G. A. Smith, F. Ding, P. J. Bygrave, A. Anandkumar, M. Welborn, F. R. Manby and T. F. Miller, 3rd, OrbNet Denali: A machine learning potential for biological and organic chemistry with semi-empirical cost and DFT accuracy, J. Chem. Phys., 2021, 155, 204103 CrossRef CAS PubMed.
- G. M. E. Silva, P. H. Acioli and A. C. Pedroza, Estimating correlation energy of diatomic molecules and atoms with neural networks, J. Comput. Chem., 1997, 18, 1407–1414 CrossRef.
- M. Welborn, L. Cheng and T. F. Miller, III, Transferability in Machine Learning for Electronic Structure via the Molecular Orbital Basis, J. Chem. Theory Comput., 2018, 14, 4772–4779 CrossRef CAS PubMed.
- J. Zhu, V. Q. Vuong, B. G. Sumpter and S. Irle, Artificial neural network correction for density-functional tight-binding molecular dynamics simulations, MRS Commun., 2019, 9, 867–873 CrossRef CAS.
- H. Li, C. Collins, M. Tanha, G. J. Gordon, D. J. Yaron and A. Density, Functional Tight Binding Layer for Deep Learning of Chemical Hamiltonians, J. Chem. Theory Comput., 2018, 14, 5764–5776 CrossRef CAS PubMed.
- F. Hu, F. He and D. J. Yaron, Treating Semiempirical Hamiltonians as Flexible Machine Learning Models Yields Accurate and Interpretable Results, J. Chem. Theory Comput., 2023, 19, 6185–6196 CrossRef CAS PubMed.
- K. T. Schütt, M. Gastegger, A. Tkatchenko, K.-R. Müller and R. J. Maurer, Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions, Nat. Commun., 2019, 10, 5024 CrossRef PubMed.
- J. Westermayr, M. Gastegger, K. T. Schütt and R. J. Maurer, Perspective on integrating machine learning into computational chemistry and materials science, J. Chem. Phys., 2021, 154, 230903 CrossRef CAS PubMed.
- J. Wu, G. Chen, J. Wang and X. Zheng, Redesigning density functional theory with machine learning, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 531–558 Search PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. P. Turban, A. L. Gaunt, J. S. Spencer, A. Matthews, A. Obika, L. Thiry, M. Fortunato, D. Pfau, L. R. Castellanos, S. Petersen, A. W. R. Nelson, P. Kohli, P. Mori-Sanchez, D. Hassabis and A. J. Cohen, Pushing the frontiers of density functionals by solving the fractional electron problem, Science, 2021, 374, 1385–1389 CrossRef CAS PubMed.
- Y. Liu, C. Zhang, Z. Liu, D. G. Truhlar, Y. Wang and X. He, Supervised learning of a chemistry functional with damped dispersion, Nat. Comput. Sci., 2022, 3, 48–58 CrossRef PubMed.
- G. M. Jones, P. D. V. S. Pathirage and K. D. Vogiatzis, Data-driven acceleration of coupled-cluster and perturbation theory methods, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 509–529 Search PubMed.
- J. P. Coe, Machine Learning Configuration Interaction, J. Chem. Theory Comput., 2018, 14, 5739–5749 CrossRef CAS PubMed.
- J. J. Goings, H. Hu, C. Yang and X. Li, Reinforcement Learning Configuration Interaction, J. Chem. Theory Comput., 2021, 17, 5482–5491 CrossRef CAS PubMed.
- S. Battaglia, Machine learning wavefunction, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 577–616 Search PubMed.
- J. Hermann, J. Spencer, K. Choo, A. Mezzacapo, W. M. C. Foulkes, D. Pfau, G. Carleo and F. Noé, Ab initio quantum chemistry with neural-network wavefunctions, Nat. Rev. Chem., 2023, 7, 692–709 CrossRef PubMed.
- M. T. Entwistle, Z. Schatzle, P. A. Erdman, J. Hermann and F. Noe, Electronic excited states in deep variational Monte Carlo, Nat. Commun., 2023, 14, 274 CrossRef CAS PubMed.
- D. Pfau, S. Axelrod, H. Sutterud, I. V. Glehn and J. S. Spencer, Natural Quantum Monte Carlo Computation of Excited States, arXiv, 2023, preprint, arXiv:2308.16848v1 [physics.comp-ph] DOI:10.48550/arXiv.2308.16848.
- M. Scherbela, L. Gerard and P. Grohs, Towards a transferable fermionic neural wavefunction for molecules, Nat. Commun., 2024, 15, 120 CrossRef CAS PubMed.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schutt, A. Tkatchenko and K. R. Muller, Machine Learning Force Fields, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed.
- G. Tallec, G. Laurens, O. Fresse-Colson and J. Lam, Potentials based on linear models, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 253–277 Search PubMed.
- J. Zeng, L. Cao and T. Zhu, Neural network potentials, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 279–294 Search PubMed.
- J. M. Bowman, C. Qu, R. Conte, A. Nandi, P. L. Houston and Q. Yu, Delta-Machine Learned Potential Energy Surfaces and Force Fields, J. Chem. Theory Comput., 2023, 19, 1–17 CrossRef CAS PubMed.
- P. Zaspel, B. Huang, H. Harbrecht and O. A. von Lilienfeld, Boosting Quantum Machine Learning Models with a Multilevel Combination Technique: Pople Diagrams Revisited, J. Chem. Theory Comput., 2019, 15, 1546–1559 CrossRef CAS PubMed.
- A. Su, X. Zhang, C. Zhang, D. Ding, Y. F. Yang, K. Wang and Y. B. She, Deep transfer learning for predicting frontier orbital energies of organic materials using small data and its application to porphyrin photocatalysts, Phys. Chem. Chem. Phys., 2023, 25, 10536–10549 RSC.
- V. Zaverkin, D. Holzmuller, L. Bonfirraro and J. Kästner, Transfer learning for chemically accurate interatomic neural network potentials, Phys. Chem. Chem. Phys., 2023, 25, 5383–5396 RSC.
- N. Hoffmann, J. Schmidt and S. Botti, Transfer learning on large datasets for the accurate prediction of material properties, Digital Discovery, 2023, 2, 1368–1379 RSC.
- S. Chmiela, H. E. Sauceda, I. Poltavsky, K.-R. Müller and A. Tkatchenko, sGDML: Constructing accurate and data efficient molecular force fields using machine learning, Comput. Phys. Commun., 2019, 240, 38–45 CrossRef CAS.
- S. Chmiela, A. Tkatchenko, H. E. Sauceda, I. Poltavsky, K. T. Schütt and K.-R. Müller, Machine learning of accurate energy-conserving molecular force fields, Sci. Adv., 2017, 3, e1603015 CrossRef PubMed.
- A. S. Christensen and O. A. von Lilienfeld, On the role of gradients for machine learning of molecular energies and forces, Mach. Learn.: Sci. Technol., 2020, 1, 045018 Search PubMed.
- K. T. Schütt, H. E. Sauceda, P. J. Kindermans, A. Tkatchenko and K.-R. Müller, SchNet - A deep learning architecture for molecules and materials, J. Chem. Phys., 2018, 148, 241722 CrossRef PubMed.
- O. T. Unke and M. Meuwly, PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges, J. Chem. Theory Comput., 2019, 15, 3678–3693 CrossRef CAS PubMed.
- A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth and B. Kozinsky, Learning local equivariant representations for large-scale atomistic dynamics, Nat. Commun., 2023, 14, 579 CrossRef CAS PubMed.
- I. Batatia, D. P. Kovács, G. N. C. Simm, C. Ortner and G. Csányi, In MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields, Advances in Neural Information Processing Systems, https://openreview.net/forum?id=YPpSngE-ZU, 2022 Search PubMed.
- J. S. Smith, B. Nebgen, N. Lubbers, O. Isayev and A. E. Roitberg, Less is more: Sampling chemical space with active learning, J. Chem. Phys., 2018, 148, 241733 CrossRef PubMed.
- D. Zhang, X. Liu, X. Zhang, C. Zhang, C. Cai, H. Bi, Y. Du, X. Qin, J. Huang, B. Li, Y. Shan, J. Zeng, Y. Zhang, S. Liu, Y. Li, J. Chang, X. Wang, S. Zhou, J. Liu, X. Luo, Z. Wang, W. Jiang, J. Wu, Y. Yang, J. Yang, M. Yang, F.-Q. Gong, L. Zhang, M. Shi, F.-Z. Dai, D. M. York, S. Liu, T. Zhu, Z. Zhong, J. Lv, J. Cheng, W. Jia, M. Chen, G. Ke, W. E, L. Zhang and H. Wang, DPA-2: Towards a universal large atomic model for molecular and material simulation, arXiv, 2023, preprint, arXiv:2312.15492v1 [physics.chem-ph], DOI:10.48550/arXiv.2312.15492.
- S. Takamoto, C. Shinagawa, D. Motoki, K. Nakago, W. Li, I. Kurata, T. Watanabe, Y. Yayama, H. Iriguchi, Y. Asano, T. Onodera, T. Ishii, T. Kudo, H. Ono, R. Sawada, R. Ishitani, M. Ong, T. Yamaguchi, T. Kataoka, A. Hayashi, N. Charoenphakdee and T. Ibuka, Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements, Nat. Commun., 2022, 13, 2991 CrossRef CAS PubMed.
- C. Chen and S. P. Ong, A universal graph deep learning interatomic potential for the periodic table, Nat. Comput. Sci., 2022, 2, 718–728 CrossRef PubMed.
- S. Zhang, M. Makoś, R. Jadrich, E. Kraka, K. Barros, B. Nebgen, S. Tretiak, O. Isayev, N. Lubbers, R. Messerly and J. Smith, Exploring the frontiers of chemistry with a general reactive machine learning potential, ChemRxiv, 2022, preprint DOI:10.26434/chemrxiv-2022-15ct6.
- J. M. Bowman, C. Qu, R. Conte, A. Nandi, P. L. Houston and Q. Yu, The MD17 datasets from the perspective of datasets for gas-phase “small” molecule potentials, J. Chem. Phys., 2022, 156, 240901 CrossRef CAS PubMed.
- G. Imbalzano, Y. Zhuang, V. Kapil, K. Rossi, E. A. Engel, F. Grasselli and M. Ceriotti, Uncertainty estimation for molecular dynamics and sampling, J. Chem. Phys., 2021, 154, 074102 CrossRef CAS PubMed.
- R. Jinnouchi, F. Karsai and G. Kresse, On-the-fly machine learning force field generation: Application to melting points, Phys. Rev. B, 2019, 100, 014105 CrossRef CAS.
- T. Rensmeyer, B. Craig, D. Kramer and O. Niggemann, High Accuracy Uncertainty-Aware Interatomic Force Modeling with Equivariant Bayesian Neural Networks, arXiv, 2023, preprint, arXiv:2304.03694v1 [physics.chem-ph] DOI:10.48550/arXiv.2304.03694.
- L. I. Vazquez-Salazar, E. D. Boittier and M. Meuwly, Uncertainty quantification for predictions of atomistic neural networks, Chem. Sci., 2022, 13, 13068–13084 RSC.
- C. Shang and Z.-P. Liu, Constructing machine learning potentials with active learning, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 313–327 Search PubMed.
- L. Zhang, J. Han, H. Wang, R. Car and W. E, Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS PubMed.
- L. Zhang, D.-Y. Lin, H. Wang, R. Car and W. E, Active learning of uniformly accurate interatomic potentials for materials simulation. Phys. Rev, Materials, 2019, 3, 023804 CAS.
- M. Yang, L. Bonati, D. Polino and M. Parrinello, Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water, Catal. Today, 2022, 387, 143–149 CrossRef CAS.
- D. M. Anstine and O. Isayev, Machine Learning Interatomic Potentials and Long-Range Physics, J. Phys. Chem. A, 2023, 127, 2417–2431 CrossRef CAS PubMed.
- S. Chmiela, V. Vassilev-Galindo, O. T. Unke, A. Kabylda, H. E. Sauceda, A. Tkatchenko and K.-R. Müller, Accurate global machine learning force fields for molecules with hundreds of atoms, Sci. Adv., 2023, 9, eadf0873 CrossRef PubMed.
- D. Folmsbee and G. Hutchison, Assessing conformer energies using electronic structure and machine learning methods, Int. J. Quantum Chem., 2020, 121, e26381 CrossRef.
- L. Zhang, H. Wang, M. C. Muniz, A. Z. Panagiotopoulos, R. Car and W. E, A deep potential model with long-range electrostatic interactions, J. Chem. Phys., 2022, 156, 124107 CrossRef CAS PubMed.
- H. J. Kulik, T. Hammerschmidt, J. Schmidt, S. Botti, M. A. L. Marques, M. Boley, M. Scheffler, M. Todorović, P. Rinke, C. Oses, A. Smolyanyuk, S. Curtarolo, A. Tkatchenko, A. P. Bartók, S. Manzhos, M. Ihara, T. Carrington, J. Behler, O. Isayev, M. Veit, A. Grisafi, J. Nigam, M. Ceriotti, K. T. Schütt, J. Westermayr, M. Gastegger, R. J. Maurer, B. Kalita, K. Burke, R. Nagai, R. Akashi, O. Sugino, J. Hermann, F. Noé, S. Pilati, C. Draxl, M. Kuban, S. Rigamonti, M. Scheidgen, M. Esters, D. Hicks, C. Toher, P. V. Balachandran, I. Tamblyn, S. Whitelam, C. Bellinger and L. M. Ghiringhelli, Roadmap on Machine learning in electronic structure, Electron. Struct., 2022, 4, 023004 CrossRef CAS.
- R. Di Felice, M. L. Mayes, R. M. Richard, D. B. Williams-Young, G. K. Chan, W. A. de Jong, N. Govind, M. Head-Gordon, M. R. Hermes, K. Kowalski, X. Li, H. Lischka, K. T. Mueller, E. Mutlu, A. M. N. Niklasson, M. R. Pederson, B. Peng, R. Shepard, E. F. Valeev, M. van Schilfgaarde, B. Vlaisavljevich, T. L. Windus, S. S. Xantheas, X. Zhang and P. M. Zimmerman, A Perspective on Sustainable Computational Chemistry Software Development and Integration, J. Chem. Theory Comput., 2023, 19, 7056–7076 CrossRef CAS PubMed.
- A. Hjorth Larsen, J. Jorgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dulak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiotz, O. Schutt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The atomic simulation environment-a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- Xiamen Atomistic Computing Suite (XACS), Xiamen University, https://XACScloud.com: 2022-2024 Search PubMed.
- J. Behler, R. Martoňák, D. Donadio and M. Parrinello, Metadynamics simulations of the high-pressure phases of silicon employing a high-dimensional neural network potential, Phys. Rev. Lett., 2008, 100, 185501 CrossRef PubMed.
- A. M. Tokita and J. Behler, How to train a neural network potential, J. Chem. Phys., 2023, 159, 121501 CrossRef CAS PubMed.
- A. M. Miksch, T. Morawietz, J. Kästner, A. Urban and N. Artrith, Strategies for the construction of machine-learning potentials for accurate and efficient atomic-scale simulations, Mach. Learn. Sci. Technol., 2021, 2, 031001 CrossRef.
- J. D. Morrow, J. L. A. Gardner and V. L. Deringer, How to validate machine-learned interatomic potentials, J. Chem. Phys., 2023, 158, 121501 CrossRef CAS PubMed.
- J. Westermayr and P. Marquetand, Machine learning and excited-state molecular dynamics, Mach. Learn.: Sci. Technol., 2020, 1, 043001 Search PubMed.
- D. Hu, Y. Xie, X. Li, L. Li and Z. Lan, Inclusion of Machine Learning Kernel Ridge Regression Potential Energy Surfaces in On-the-Fly Nonadiabatic Molecular Dynamics Simulation, J. Phys. Chem. Lett., 2018, 9, 2725–2732 CrossRef CAS PubMed.
- A. A. Kananenka, K. Yao, S. A. Corcelli and J. L. Skinner, Machine Learning for Vibrational Spectroscopic Maps, J. Chem. Theory Comput., 2019, 15, 6850–6858 CrossRef CAS PubMed.
- I. Goodfellow; Y. Bengio and A. Courville, Deep Learning, MIT Press: 2016 Search PubMed.
- L. Winkler, K.-R. Müller and H. E. Sauceda, High-fidelity molecular dynamics trajectory reconstruction with bi-directional neural networks, Mach. Learn. Sci. Technol., 2022, 3, 025011 CrossRef.
- A. Denzel and J. Kästner, Gaussian process regression for geometry optimization, J. Chem. Phys., 2018, 148, 094114 CrossRef.
- I. Fdez Galván, G. Raggi and R. Lindh, Restricted-Variance Constrained, Reaction Path, and Transition State Molecular Optimizations Using Gradient-Enhanced Kriging, J. Chem. Theory Comput., 2021, 17, 571–582 CrossRef PubMed.
- A. W. Mills, J. J. Goings, D. Beck, C. Yang and X. Li, Exploring Potential Energy Surfaces Using Reinforcement Machine Learning, J. Chem. Inf. Model., 2022, 62, 3169–3179 CrossRef CAS PubMed.
- K. Ahuja, W. H. Green and Y. P. Li, Learning to Optimize Molecular Geometries Using Reinforcement Learning, J. Chem. Theory Comput., 2021, 17, 818–825 CrossRef CAS PubMed.
- R. Lindh and I. Fdez. Galván, Molecular structure optimizations with Gaussian process regression, in Quantum Chemistry in the Age of Machine Learning, ed. P. O. Dral, Elsevier, Amsterdam, Netherlands, 2023, pp. 391–428 Search PubMed.
- D. Kuntz and A. K. Wilson, Machine learning, artificial intelligence, and chemistry: how smart algorithms are reshaping simulation and the laboratory, Pure Appl. Chem., 2022, 94, 1019–1054 CrossRef CAS.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Machine learning for molecular and materials science, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- O. A. von Lilienfeld, K.-R. Müller and A. Tkatchenko, Exploring chemical compound space with quantum-based machine learning, Nat. Rev. Chem., 2020, 4, 347–358 CrossRef PubMed.
- S. Manzhos, Machine learning for the solution of the Schrödinger equation, Mach. Learn. Sci. Technol., 2020, 1, 013002 CrossRef.
- J. Zhang, Y.-K. Lei, Z. Zhang, J. Chang, M. Li, X. Han, L. Yang, Y. I. Yang and Y. Q. Gao, A Perspective on Deep Learning for Molecular Modeling and Simulations, J. Phys. Chem. A, 2020, 124, 6745–6763 CrossRef CAS PubMed.
- T. Mueller, A. Hernandez and C. Wang, Machine learning for interatomic potential models, J. Chem. Phys., 2020, 152, 050902 CrossRef CAS PubMed.
- Z. J. Baum, X. Yu, P. Y. Ayala, Y. Zhao, S. P. Watkins and Q. Zhou, Artificial Intelligence in Chemistry: Current Trends and Future Directions, J. Chem. Inf. Model., 2021, 61, 3197–3212 CrossRef CAS PubMed.
- T. Zubatiuk and O. Isayev, Development of Multimodal Machine Learning Potentials: Toward a Physics-Aware Artificial Intelligence, Acc. Chem. Res., 2021, 54, 1575–1585 CrossRef CAS PubMed.
- K. T. Schütt, S. Chmiela, O. A. von Lilienfeld, A. Tkatchenko, K. Tsuda and K.-R. Müller, Machine Learning Meets Quantum Physics, Springer, Cham, 2020 Search PubMed.
- V. L. Deringer, A. P. Bartok, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csanyi, Gaussian Process Regression for Materials and Molecules, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- M. Meuwly, Machine Learning for Chemical Reactions, Chem. Rev., 2021, 121, 10218–10239 CrossRef CAS PubMed.
- J. Zupan and J. Gasteiger, Neural networks: A new method for solving chemical problems or just a passing phase?, Anal. Chim. Acta, 1991, 248, 1–30 CrossRef CAS.
- M. Gastegger, J. Behler and P. Marquetand, Machine learning molecular dynamics for the simulation of infrared spectra, Chem. Sci., 2017, 8, 6924–6935 RSC.
- J. Westermayr and P. Marquetand, Machine Learning for Electronically Excited States of Molecules, Chem. Rev., 2021, 121, 9873–9926 CrossRef CAS PubMed.
- J. Westermayr and P. Marquetand, Deep learning for UV absorption spectra with SchNarc: First steps toward transferability in chemical compound space, J. Chem. Phys., 2020, 153, 154112 CrossRef CAS PubMed.
- S. Ye, W. Hu, X. Li, J. Zhang, K. Zhong, G. Zhang, Y. Luo, S. Mukamel and J. Jiang, A neural network protocol for electronic excitations of N-methylacetamide, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11612–11617 CrossRef CAS PubMed.
- F. Liu, L. Du, D. Zhang and J. Gao, Direct Learning Hidden Excited State Interaction Patterns from ab initio Dynamics and Its Implication as Alternative Molecular Mechanism Models, Sci. Rep., 2017, 7, 8737 CrossRef PubMed.
- Z. Zou, Y. Zhang, L. Liang, M. Wei, J. Leng, J. Jiang, Y. Luo and W. Hu, A deep learning model for predicting selected organic molecular spectra, Nat. Comput. Sci., 2023, 3, 957–964 CrossRef CAS PubMed.
- K. Singh, J. Munchmeyer, L. Weber, U. Leser and A. Bande, Graph Neural Networks for Learning Molecular Excitation Spectra, J. Chem. Theory Comput., 2022, 18, 4408–4417 CrossRef CAS PubMed.
- P. Kovács, X. Zhu, J. Carrete, G. K. H. Madsen and Z. Wang, Machine-learning Prediction of Infrared Spectra of Interstellar Polycyclic Aromatic Hydrocarbons, Astrophys. J., 2020, 902, 100 CrossRef.
- A. Kotobi, K. Singh, D. Hoche, S. Bari, R. H. Meissner and A. Bande, Integrating Explainability into Graph Neural Network Models for the Prediction of X-ray Absorption Spectra, J. Am. Chem. Soc., 2023, 145, 22584–22598 CrossRef CAS PubMed.
- H. Ren, H. Li, Q. Zhang, L. Liang, W. Guo, F. Huang, Y. Luo and J. Jiang, A machine learning vibrational spectroscopy protocol for spectrum prediction and spectrum-based structure recognition, Fundam. Res., 2021, 1, 488–494 CrossRef CAS.
- E. O. Pyzer-Knapp, J. W. Pitera, P. W. J. Staar, S. Takeda, T. Laino, D. P. Sanders, J. Sexton, J. R. Smith and A. Curioni, Accelerating materials discovery using artificial intelligence, high performance computing and robotics, npj Comput. Mater., 2022, 8 Search PubMed.
- Y. Wang, Q. Jiao, J. Wang, X. Cai, W. Zhao and X. Cui, Prediction of protein-ligand binding affinity with deep learning, Comput. Struct. Biotechnol. J., 2023, 21, 5796–5806 CrossRef CAS PubMed.
- M. C. R. Melo, J. Maasch and C. de la Fuente-Nunez, Accelerating antibiotic discovery through artificial intelligence, Commun. Biol., 2021, 4, 1050 CrossRef CAS PubMed.
- G. Liu, D. B. Catacutan, K. Rathod, K. Swanson, W. Jin, J. C. Mohammed, A. Chiappino-Pepe, S. A. Syed, M. Fragis, K. Rachwalski, J. Magolan, M. G. Surette, B. K. Coombes, T. Jaakkola, R. Barzilay, J. J. Collins and J. M. Stokes, Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii, Nat. Chem. Biol., 2023, 19, 1342–1350 CrossRef CAS PubMed.
- R. I. Horne, M. H. Murtada, D. Huo, Z. F. Brotzakis, R. C. Gregory, A. Possenti, S. Chia and M. Vendruscolo, Exploration and Exploitation Approaches Based on Generative Machine Learning to Identify Potent Small Molecule Inhibitors of alpha-Synuclein Secondary Nucleation, J. Chem. Theory Comput., 2023, 19, 4701–4710 CrossRef CAS PubMed.
- F. Wong, E. J. Zheng, J. A. Valeri, N. M. Donghia, M. N. Anahtar, S. Omori, A. Li, A. Cubillos-Ruiz, A. Krishnan, W. Jin, A. L. Manson, J. Friedrichs, R. Helbig, B. Hajian, D. K. Fiejtek, F. F. Wagner, H. H. Soutter, A. M. Earl, J. M. Stokes, L. D. Renner and J. J. Collins, Discovery of a structural class of antibiotics with explainable deep learning, Nature, 2023, 626, 177–185 CrossRef PubMed.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Autonomous chemical research with large language models, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk, A. Merchant, H. Kim, A. Jain, C. J. Bartel, K. Persson, Y. Zeng and G. Ceder, An autonomous laboratory for the accelerated synthesis of novel materials, Nature, 2023, 624, 86–91 CrossRef CAS PubMed.
- P. O. Dral and A. Ullah, Call for Urgent Regulations on Artificial Intelligence, 2023, preprint DOI:10.20944/preprints202304.0429.v1.
| This journal is © The Royal Society of Chemistry 2024 |