
Kinetic isotope effect offers selectivity in CO2 reduction†
Suman
Patra‡
,
Sayan
Atta‡
,
Soumili
Ghosh
,
Amit
Majumdar
 * and
Abhishek
Dey
*
* and
Abhishek
Dey
*
School of Chemical Sciences Indian Association for the Cultivation of Science 2A & 2B, Raja SC Mullick Road, Kolkata, WB 700032, India. E-mail: icam@iacs.res.in; abbeyde@gmail.com
First published on 5th April 2024
Abstract
A binuclear Ni complex with N,O donors catalyzes CO2 reduction via its Ni(I) state. The product distribution when H2O is used as a proton source shows similar yields for CO, HCOOH and H2. However, when D2O is used, the product distribution shows a ∼65% selectivity for HCOOH. In situ FTIR indicates that the reaction involves a Ni–COO* and a Ni–CO intermediate. Differences in H/D KIEs on different protonation pathways determine the selectivity of CO2 reduction.
The reduction of CO2 to chemicals having commercial value has taken centre stage in multi-electron multi-proton catalysis. Apart from the obvious appeal of the chemical conversion of a greenhouse gas to value added chemicals, the inherent challenges in the reaction have attracted the attention of several research groups across the world. Substantial progress has been made in the last few years and several molecular and material catalysts have been developed for this purpose and several of these show considerable reactivity.1–14
Reduction of CO2 selectively to any of the several possible products poses a challenge. The 2e−/2H+ reduction of CO2 can produce either HCOOH or CO and competes with the reduction of protons to form H2.15–20 Several groups have contributed to the current understanding of the mechanism of the reactions involved.8,21–26 The reduced metal centre binds either CO2 or H+ to produce M–CO2 or M–H species (Scheme 1). The M–H species can be further protonated to generate H2 or it can transfer the H− to CO2 to generate formate. The M–CO2 species on the other hand generates a M–COOH species upon protonation and then can undergo either a M–C bond cleavage to generate formate or a C–O bond cleavage to generate CO. These species have been observed chemically as well as by using spectro-electrochemistry and characterized.6,20,27
 | ||
| Scheme 1 Selectivity for 2e−/2H+ reduction of CO2via the M–COOH pathway. | ||
Selectivity in 2e−/2H+ reduction has been demonstrated by several groups and it has been proposed that the electronic structure of the M–COOH species plays a vital role in the process.26 When the M–C bond is very covalent, the protonation of the OH-group is favored resulting in C–O bond cleavage and release of CO, while in cases where the M–C bond is not very covalent, the M–C bond cleaves generating HCOOH. The covalency of the M–C bond can be tuned by the choice of the metal as well as the spin states. Using a ligand that has a protonation site has provided relief from the competing HER.20,28,29
In addition to the design of the ligand and the choice of metal controlling the electronic structure of the catalyst, selectivity can also be accessed by kinetic control. For example, at lower concentrations of protons (as well as by using weaker proton sources), the HER is naturally suppressed. In a curious case, the product distribution of 2e−/2H+ CO2 reduction by Pd/C was demonstrated to depend on the isotope of the proton source used. In situ X-ray characterization verifies the formation of Pd–H species. Substituting H2O with D2O reduces the rate of the competing hydrogen evolution reaction (HER) due to lower D+ concentration and enhances the carbon dioxide reduction reaction (CO2RR), leading to a shift from 48% CO in H2O to 76% CO in D2O.30 Clearly, the kinetic isotope effect (KIE) involved in the reduction of CO2 was different from the KIE involved in the reduction of H+ leading to different selectivities in H2O and D2O. Similar changes in product distribution were observed during CO2 reduction by cobalt phthalocyanine immobilized on a coordinating polymer support.31 A significant kinetic isotope effect (KIE) was reported for the decomposition of HCOOH to form CO2 and H2 by an Ir/Ru complex.32 Another study of isotope-controlled selectivity by quantum tunneling results in different chemical reactivity of cyclopropylmethylcarbene.33 While isotope effects have been reported in the kinetics of electrochemical CO2 reduction using molecular catalysts, changes in selectivity have not been reported and whether such changes in selectivity can be obtained in a molecular catalyst under homogeneous conditions are yet to be evaluated.
In this manuscript, we report the synthesis of a novel binuclear Ni catalyst, [(Py2ald)2Ni2](BPh4)2 (1(BPh4)2), and explores its application in CO2 reduction by using water as the proton source. The ligand, Hpy2ald (3-{[Bis(2-pyridinylmethyl)amino]methyl}-2-hydroxybenzaldehyde), was synthesized following literature report.34 The catalyst shows no selectivity for either CO or HCOOH and exhibits a competing HER in H2O. However, in D2O the catalyst exhibits 65% selectivity for HCOOH. In situ spectro-electrochemical FTIR provides insight into the mechanism of CO2 reduction.
Cyclic voltammetry (CV) measurements conducted with 0.5 mM 1(BPh4)2 in N2 saturated acetonitrile solution unveiled two closely positioned peaks at −1.82 V and −2.05 V vs. Fc+/Fc0, respectively (Fig. 1A and inset). To identify the associated reduction process, CV was conducted using the free ligand HPy2ald under identical conditions. It was observed that the peak at −2.05 V vs. Fc+/Fc0 is linked to ligand reduction, potentially involving the reduction of the aldehyde group. Supporting evidence for this transformation is found in corroborating Fourier-transform infrared spectroscopy coupled with spectro-electrochemistry (FTIR-SEC) data. For 1(BPh4)2, when the working electrode is held at −2.05 V vs. Fc+/Fc0, the peak corresponding to the –CHO group at 1633 cm−1 diminishes (Fig. 2A, inset), signifying its reduction, while a new peak in the 2500–2700 cm−1 range emerges, corresponding to the formation of the –CH2OH group (Fig. 2A). More importantly, the complete reduction of the –CHO vibration indicates that both –CHO groups are being reduced. The additional peak at −1.82 V vs. Fc+/Fc0 is associated with the Ni(II/I) reduction. The reduction of NiII to NiI was confirmed by FTIR-SEC under one atmosphere CO (Fig. S6, ESI†). NiII does not bind CO, but when FTIR-SEC at −1.85 V vs. Fc+/Fc° was conducted, a clear NiI–CO vibration at 1980 cm−1 was observed. Similar carbonyl species were previously reported in a binuclear cobalt complex bridged by pyrazole.11
 | ||
| Fig. 1 Cyclic voltammetry of 0.5 mM 1(BPh4)2 (green) and free ligand (purple trace) in N2 saturated acetonitrile solution, and its catalytic nature in CO2 saturated acetonitrile in the absence (blue trace) and presence of H2O (red trace). The inset provides a zoomed-in view of the potential range from −0.4 to −2.4 V in an N2 atmosphere (A). Comparison of the CV trace of the Ni-complex in CO2 and N2 saturated acetonitrile in the presence of 5% (V/V) H2O shows electrocatalytic CO2 reduction along with the HER (B). 100 mM tetrabutylammonium perchlorate (TBAP) was used as the supporting electrolyte. | ||
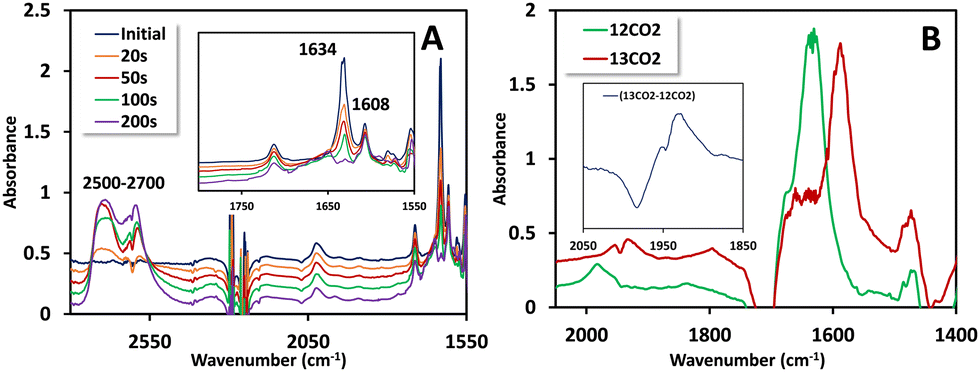 | ||
| Fig. 2 FTIR-SEC spectra of 1(BPh4)2 (4 mM) in N2 saturated acetonitrile while keeping the potential at −2.05 V vs. Fc+/Fc0 showing conversion of –CHO to –CH2OH. The inset shows the zoomed-in view of the 1550–1800 cm−1 region (A); in situ FTIR-SEC spectra (CPE at −2.2 V vs. Fc+/Fc0) of the intermediates of 1(BPh4)2 (4 mM) in acetonitrile using 12CO2 (blue trace) and 13CO2 (red trace) as substrates. The inset shows the zoomed-in difference spectra (13CO2–12CO2) of the 1850–2050 cm−1 region, showing a shift of 44 cm−1 (B). 100 mM tetrabutylammonium perchlorate (TBAP) was used as the supporting electrolyte. | ||
In the presence of saturated CO2 and 5% (V/V) H2O, a strong reduction current is observed at −2.2 V vs. Fc+/Fc0 with a pre-catalytic feature at −1.65 V vs. Fc+/Fc0 which indicates CO2 binding to the reduced NiI (Fig. 1A). The electrocatalytic current at −2.2 V vs. Fc+Fc0 is not observed in the absence of CO2 and represents electrocatalytic CO2 reduction (Fig. 1B). These observations lead to the conclusion that the species responsible for CO2 binding is Ni(I) but the electrocatalysis proceeds after further reduction of NiIII–CO2−/NiIII–COOH (represented as NiIII–COO*). In the presence of 5% (V/V) H2O in an N2 atmosphere, a catalytic current is observed with an onset at −2.3 V vs. Fc+/Fc0, indicating the occurrence of the hydrogen evolution reaction (HER). The absorption spectra of 1(BPh4)2 before and after CPE show an 18% reduction of the bridging phenolate to Ni charge transfer, not present in a monomer (Fig. S11 and S12, ESI†) after 30 minutes.
To delve into the reaction mechanism and identify the intermediates involved, FTIR-SEC was carried out in a CO2-saturated acetonitrile solution in an OTTLE cell. While keeping the working electrode at −2.2 V vs. Fc+/Fc0, the FTIR-SEC results unveiled the presence of two isotope-sensitive vibrations associated with 13CO2 (Fig. 2B). A peak observed at 1633 cm−1, which corresponds to a NiI–COO* intermediate, shifted to 1588 cm−1 when a heavier isotope of CO2 (13CO2) was used. Another peak at 1980 cm−1 was also observed, corresponding to the NiI–CO species previously observed, which is a likely precursor to the carbon monoxide gas detected during the gas analysis. This peak shifted to 1936 cm−1 when 13CO2 was used (Fig. 2B, inset). Based on these observations, a plausible mechanism is proposed (Scheme 2). Further evidence for the mechanism is marshalled from kinetic isotope effects discussed later.
 | ||
| Scheme 2 Reaction mechanism of different pathways of CO2/CO (black arrow) and CO2/HCOOH (blue arrow) along with the HER (green arrow). The relative KIE (KIE*) is shown for all the reaction pathways. | ||
Controlled potential electrolysis (CPE) was conducted using a glassy carbon electrode with a larger surface area (2 cm2) (Fig. 3A). After electrolysis of 0.5 mM of 1(BPh4)2 in CO2 saturated acetonitrile solution in the presence of 5% (V/V) H2O, the gaseous products generated were collected from the headspace and were subjected to analysis using gas chromatography (GC) equipped with a thermal conductivity detector (TCD). The GC-TCD analysis revealed the presence of two gases: hydrogen and carbon monoxide (Fig. 3B and Fig. S8, ESI†). The faradaic yields were determined to be 32% for hydrogen and 23% for carbon monoxide. An additional 36% of the product was identified as formate, when the solution, after electrolysis, was extracted with water and subsequently analysed by ion chromatography (Fig. 3C and Fig. S9, ESI†). Reduction of the metal may lead to formation of Ni nanoparticles on the electrode. The rinse test of the working electrode after CPE, however, does not show any catalytic activity (Fig. S10, ESI†). The results from the CPE indicate that 1(BPh4)2 exhibits no particular selectivity for either of the three 2e−/2H+ reduced products, i.e. H2, CO and HCOOH (Fig. 3D). The mechanistic pathway likely involved in 2e− CO2 reduction (Scheme 1) by 1(BPh4)2 would include key intermediates like Ni–COOH and Ni–CO. The Ni–COOH species would lead to CO after a C–O bond cleavage which upon protonation of the C-atom of the NiI–COOH species will release HCOOH. Formation of H2 entails protonation of the reduced Ni centre (Scheme 1) forming a Ni–H species. The same Ni–H species can produce HCOOH by reducing CO2 (Scheme 1). The different possible pathways involved may or may not involve protonation in the rate determining step (rds). A rds where protons are involved generally show the H/D KIE. In fact, CO2 reduction as well as the competing hydrogen evolution has been reported to exhibit H/D KIEs30,31,35–37 and, in general, pathways involving metal hydrides exert substantial KIEs. The KIEs can both offer insight into the mechanism involved and result in changes in product distribution, i.e. selectivity.
 | ||
| Fig. 3 CPE performed at −2.2 V vs. Fc+/Fc0 varying the heavier isotope of the proton source for 1/2 hour using a glassy carbon plate as the working electrode, Pt wire as the counter electrode and Ag/AgCl as the reference electrode. Charge consumption (A) followed by product detection using gas chromatography-TCD (B) and ion chromatography (C) and their faradaic yields (D) are compared for both H2O and D2O (12CO2/H2O – red trace, 12CO2/D2O – blue trace). | ||
In pursuit of a deeper understanding of the mechanism underlying carbon dioxide reduction, which yields both carbon monoxide and formic acid as reduced products, a kinetic isotope effect (KIE) investigation was undertaken. On using heavy water (D2O) instead of H2O, the overall catalytic current was decreased (Fig. 3A). The products detected using GC and IC (ion chromatography) were H2, CO and HCOOH. However, the selectivity for HCOOH increases from 36% to a staggering 63% when D2O is used instead of H2O. The FY of CO and H2 shrinks from 32% and 23% to 12% and 16%, respectively. The charges consumed by H2, CO and HCOOH in H2O were 3.26C, 2.35C and 3.67C, whereas in D2O they were 1.33C, 1.00C and 5.23C, respectively (Table 1). Thus, in D2O the production of H2 and CO is suppressed relative to H2O, whereas the production of HCOOH is enhanced. These results present a unique case where the selectivity between the three products of CDR, namely H2, CO and HCOOH, for a molecular catalyst is shifted drastically by the use of isotopes.
| Charge consumed | H2O | D2O | Relative KIE (H/D) | ||
|---|---|---|---|---|---|
| 10.2 | 8.3 | ||||
| Products | Faradaic yield (%) | Charge (C) | Faradaic yield (%) | Charge (C) | |
| H2 | 32 | 3.26 | 16 | 1.33 | 3.5 |
| CO | 23 | 2.35 | 12 | 1.00 | 3.3 |
| HCOOH | 36 | 3.67 | 63 | 5.23 | 1 |
The results indicate that H2 evolution and CO production are inhibited in the presence of D2O relative to H2O, while HCOOH production is accelerated. Since these reactions are competitive, the absolute currents being consumed cannot be used to derive a H/D KIE for the individual reactions. However, a relative KIE (KIE* in Scheme 2) (Table 1) can be obtained relative to the HCOOH formation reaction. The reduction of CO2 to CO exhibits a relative KIE of 3.3, while the H2 evolution shows a relative KIE of 3.5 relative to any implicit KIE HCOOH may have. Formation of CO requires two protons and it is likely that the second protonation step (protonation of the M–COOH species) is the rds, as suggested by its accumulation during catalysis, resulting in a H/D KIE. The relative KIE observed here is similar to the KIE observed in CO2 reduction to CO by Co–thiolate complexes where the protonation of the –OH end of a M–COOH species is determined to be the rds.17 Similarly, the formation of H2 involves two protonation steps and the second protonation (i.e. protonation of M–H) is likely the rds that earns the H/D KIE. A similar KIE was recorded for H2 evolution by Ni complexes where the protonation of the Ni–H species was the rds.38
The formic acid produced can be formed either via Ni–H attack on CO2 or via C-protonation of the Ni–COOH intermediate. The H/D KIE reported for the hydride pathway is substantial (5–10)37 because of the shift of the hydride from the metal to the CO2 in the TS. The fact that the HCOOH increase in D2O contradicts this possibility in the case of 1(BPh4)2. Rather, the hydrolysis of Ni–COOH via C-protonation seems to be the most logical pathway.
In summary, differential H/D KIEs associated with the two competing 2e−/2H+ CO2 reduction pathways and that of the HER allow obtaining ∼65% selectivity in HCOOH formation. This is the first example where selectivity in CO2 reduction taking advantage of KIEs could be demonstrated in a molecular system.
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
A. D. acknowledges SERB research grant no. CRG/2021/00154 and DST/TMD(EWO)/IC5-2018/03(G). A. M. acknowledges CRG/2022/001931 for funding. S. P., S. A., and S. G. acknowledge the CSIR for senior research fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- H.-R. “Molly” Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- Q. Chang, Y. Liu, J.-H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS PubMed.
- H. Kang, A. Staples-West, A. Washington, C. Turchiano, A. Cooksy, J. Huang and J. Gu, ChemCatChem, 2023, 15, e202300576 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- R. Cao, ChemSusChem, 2022, 15, e202201788 CrossRef CAS PubMed.
- S. Amanullah, P. Saha, A. Nayek, M. E. Ahmed and A. Dey, Chem. Soc. Rev., 2021, 50, 3755–3823 RSC.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Prim., 2023, 3, 61 CrossRef CAS.
- M. Bonchio, J. Bonin, O. Ishitani, T.-B. Lu, T. Morikawa, A. J. Morris, E. Reisner, D. Sarkar, F. M. Toma and M. Robert, Nat. Catal., 2023, 6, 657–665 CrossRef.
- A. Bohn, J. J. Moreno, P. Thuéry, M. Robert and O. Rivada-Wheelaghan, Chem. – Eur. J., 2023, 29, e202202361 CrossRef CAS PubMed.
- S. Amanullah, P. Gotico, M. Sircoglou, W. Leibl, M. J. Llansola-Portoles, T. Tibiletti, A. Quaranta, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2023, e202314439 Search PubMed.
- P. Gotico, Z. Halime, W. Leibl and A. Aukauloo, ChemPlusChem, 2023, 88, e202300222 CrossRef CAS PubMed.
- C. Zhang, P. Gotico, R. Guillot, D. Dragoe, W. Leibl, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2023, 62, e202214665 CrossRef CAS PubMed.
- L. Chen, Z. Guo, X.-G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- S. Dey, M. E. Ahmed and A. Dey, Inorg. Chem., 2018, 57, 5939–5947 CrossRef CAS PubMed.
- M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 1386–1393 CrossRef CAS PubMed.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2020, 5, 72–78 CrossRef CAS.
- S. Amanullah, P. Saha and A. Dey, J. Am. Chem. Soc., 2021, 143, 13579–13592 CrossRef CAS PubMed.
- J. A. Barrett, C. J. Miller and C. P. Kubiak, Trends Chem., 2021, 3, 176–187 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- E. Fujita, Coord. Chem. Rev., 1999, 185–186, 373–384 CrossRef CAS.
- E. Fujita, C. Creutz, N. Sutin and B. S. Brunschwig, Inorg. Chem., 1993, 32, 2657–2662 CrossRef CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- B. Mondal, A. Rana, P. Sen and A. Dey, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef CAS PubMed.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. I. I. I. Miller and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS PubMed.
- M. E. Ahmed, A. Rana, R. Saha, S. Dey and A. Dey, Inorg. Chem., 2020, 59, 5292–5302 CrossRef CAS PubMed.
- J. H. Lee, B. M. Tackett, Z. Xie, S. Hwang and J. G. Chen, Chem. Commun., 2020, 56, 106–108 RSC.
- Y. Liu and C. C. L. McCrory, Nat. Commun., 2019, 10, 1683 CrossRef PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed.
- A. Nandi, D. Gerbig, P. R. Schreiner, W. T. Borden and S. Kozuch, J. Am. Chem. Soc., 2017, 139, 9097–9099 CrossRef CAS PubMed.
- I. A. Koval, D. Pursche, A. F. Stassen, P. Gamez, B. Krebs and J. Reedijk, Eur. J. Inorg. Chem., 2003, 1669–1674 CrossRef CAS.
- N. Devi, C. K. Williams, A. Chaturvedi and J. “Jimmy” Jiang, ACS Appl. Energy Mater., 2021, 4, 3604–3611 CrossRef CAS.
- S. Dey, T. K. Todorova, M. Fontecave and V. Mougel, Angew. Chem., Int. Ed., 2020, 59, 15726–15733 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka, K. Ohkubo, Y. Shiota, K. Yoshizawa, S. Fukuzumi and T. Kojima, Inorg. Chem., 2018, 57, 7180–7190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details, control electrochemical experiments and structural data. CCDC 2208720 and 2310122. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc06336d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |