
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative mass spectrometry imaging: therapeutics & biomolecules
Joseph H.
Holbrook
ab,
Gabrielle E.
Kemper
b and
Amanda B.
Hummon
 *abc
*abc
aOhio State Biochemistry Program, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: hummon.1@osu.edu
bDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA
cComprehensive Cancer Center, The Ohio State University, Columbus, Ohio 43210, USA
First published on 22nd January 2024
Abstract
Mass spectrometry imaging (MSI) has become increasingly utilized in the analysis of biological molecules. MSI grants the ability to spatially map thousands of molecules within one experimental run in a label-free manner. While MSI is considered by most to be a qualitative method, recent advancements in instrumentation, sample preparation, and development of standards has made quantitative MSI (qMSI) more common. In this feature article, we present a tailored review of recent advancements in qMSI of therapeutics and biomolecules such as lipids and peptides/proteins. We also provide detailed experimental considerations for conducting qMSI studies on biological samples, aiming to advance the methodology.
 Joseph H. Holbrook | Joseph H. Holbrook received his BS in Chemistry from Eastern Kentucky University in 2020. He is currently a PhD candidate in the Ohio State Biochemistry Program under the supervision of Dr Amanda B. Hummon. His current research interests include drug efficacy studies utilizing mass spectrometry imaging in different model systems. |
 Gabrielle E. Kemper | Gabrielle E. Kemper is currently an undergraduate student at The Ohio State University and will receive her BS in Chemistry in 2024. She currently works in collaboration with Joseph H. Holbrook on drug efficacy studies. Her future plans include applying to medical school following completion of her degree. |
 Amanda B. Hummon | Amanda Hummon earned her AB in chemistry at Cornell University and her PhD in analytical chemistry at the University of Illinois, in the laboratory of Prof. Jonathan Sweedler. She completed her postdoctoral work at the National Cancer Institute. She is currently a Professor in the Department of Chemistry and Biochemistry and the Comprehensive Cancer Center at The Ohio State University. Her laboratory develops mass spectrometric methods to explore cancer tissues, cell cultures, and organoids. |
Introduction
Mass spectrometry imaging (MSI) is a widely used analytical technique that provides information on the spatial distribution of analytes in a sample. Mass spectrometry (MS) analysis generates detailed chemical information for a sample, whereas MSI can provide similar information but in a spatially defined manner as depicted in a “chemical heatmap”. In general, MSI works by accumulating a range of mass-to-charge (m/z) values that correlate to a specific location, or pixel, on a sample. After an imaging analysis is complete, all spectra are combined to create a “chemical heatmap”, where each pixel of the map is associated with a corresponding mass spectrum. This map provides spatial information for analytes of interest, within the given m/z range.1,2MSI is complementary to other methods commonly used for spatial analysis of biological samples, including hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) staining. Both methods are utilized in detecting and locating various biomolecules within tissues and even to the single cell level. Despite the widespread use of both techniques, neither method can provide ample molecular/chemical information on biomolecules of interest.3 MSI can provide simultaneous quantification of many different analyte compounds, unlike H&E and IHC staining.4 MSI also has an added advantage over these traditional techniques in that it is a label-free technique and can study a broad range of analyte compounds simultaneously within one imaging run.5
There are several different techniques used for MSI studies that can be classified into hard and soft ionization techniques. Hard ionization refers to adding an excess amount of internal energy into a molecule and leading to extensive fragmentation of the molecule. This type of ionization is extremely useful for structural characterization of molecules.6 Soft ionization techniques use less energy and result in less fragmentation of molecules; thus, the target molecule remains intact for analysis. Matrix-assisted laser desorption/ionization (MALDI) is one of the most popular approaches for conducting soft ionization. MALDI-MS has been applied to a variety of applications such as mass spectral fingerprinting of bacteria7 and the characterization of polymers,8 proteins,9 and peptides,10 to name a few. The process for MALDI-MS is like MALDI-MSI where a laser strikes a sample containing matrix, which then generates ions for subsequent detection to provide molecular information about the sample. However, a crucial distinction between MALDI-MS and MALDI-MSI lies in the spatial information. While MALDI-MS provides molecular information about the sample it does not provide this information in a spatially defined manner like MALDI-MSI grants, as shown in Fig. 1. MALDI-MSI, combines all the spectra collected over the sample region to create an ion image, which is information that is not achieved by MALDI-MS alone. MALDI has bolstered in popularity for imaging and the technique has been applied on a variety of sample types including tissues,11,12 3D cell cultures, such as spheroids and organoids,13–15 and even single-cell imaging.16,17 Another extensively used soft ionization technique for imaging is desorption electrospray ionization (DESI). DESI has the added benefit of using a liquid-spray extraction which allows for samples to be analyzed in ambient conditions.18 Other, less extensively used MSI techniques include secondary ion mass spectrometry (SIMS) and laser ablation inductively coupled plasma (LA-ICP). SIMS is a hard ionization technique used for imaging that displays high spatial resolution; albeit, at the cost of analyte fragmentation.19 LA-ICP based imaging also displays higher spatial resolution but provides mainly topological elemental information.20 Interestingly, nanoparticle-enhanced LDI-MS, an ionization technique similar to MALDI, has recently been applied for increasing metabolite sensitivity in both breast cancer and neuromyelitis optica spectrum disorder.21,22 This ionization technique is efficient for studies involving lower molecular weight metabolites as MALDI matrices can result in ion suppression and matrix peaks.23 All of these techniques have many variants which allow researchers to tailor their ionization source for specific applications.24
 | ||
| Fig. 1 Classic workflow for (a) MALDI-MS and MALDI-MSI and (b) DESI-MSI on tissue. Depicted is one of the possibilities for the addition of internal standards into the matrix for MALDI application or mixed in with the solvent for DESI application. | ||
Despite their differences, these techniques have the same general experimental flow of using a laser or liquid-spray to raster over a sample causing the formation of ions which then proceed to the mass spectrometer for subsequent detection. While the flow of ions to the detector is generally the same for these techniques, the mechanisms for ion formation are different.25 This feature represents a key advantage in mass spectrometry, where different imaging modalities can be used in a complementary fashion since molecules display different propensities for ionization.26,27
The vast majority of MSI studies involve mapping the distribution of therapeutics and biomolecules, including lipids,28–30 proteins,31–33 peptides,34 carbohydrates,35,36 and other metabolites.37
Biomolecules display heterogeneity; both across different classes of molecules and within subclasses.38,39 It is this inherent heterogeneity that has made biomolecules increasingly popular to study by MSI, as differential expression of potential biomarkers is often correlated to disease states.40,41 Therapeutics are often employed for prevention and treatment of diseases, in hopes of altering the expression of biomolecules back to a normal healthy state.42 In short, endogenous species can be altered in response to an exogenous drug treatment.42,43 This property has made MSI of exogenous therapeutics increasingly popular to study, as investigators can map therapeutics spatially and monitor how endogenous species are altered.27,44 Since biomolecules are involved in many key biological processes and therapeutic intervention can influence their expression, they have become popular targets for biomarker discovery.45 Numerous MSI studies have focused on increasing both the sensitivity and specificity of measurements for biomolecules.46–49 Distinguishing how a disease state or therapeutic intervention impacts biomolecule expression and localization can give us further insight into both the pharmacokinetics50 and pharmacodynamics.51
Quantitative mass spectrometry studies often rely on some form of separations such as gas or liquid chromatography (GC or LC) coupled to a mass spectrometer. There are many protocols and techniques that describe how to perform quantification of therapeutics,52,53 lipids,54,55 proteins,56 and carbohydrates,57 using separations. Quantification using LC-MS is considered the gold standard in quantification of biomolecules, as the technique is robust and routinely utilized. MSI offers a complementary alternative to traditional LC-MS, where spatial information can be gained.58 When working with separations, the user must routinely check column stability and perform quality control checks for both peak integrity and retention time shifting. With improvements in mass spectrometry instrumentation, sensitivity, specificity, and data analysis, MSI has emerged as a potential alternative for quantitative studies, termed quantitative MSI (qMSI).59 qMSI has emerged within the field of mass spectrometry as it allows for relative and absolute values of concentration of specific analytes within a spatial context. MSI has been used extensively in pharmacokinetic and toxicology studies to determine drug and associated metabolite localization within tissues. Conducting qMSI allows for the determination of drug or metabolites concentration in the tissue along with the spatial localization in one workflow without the need of extended validation with other analytical techniques.60 Reduced usage of other analytical techniques allows for higher throughput of novel therapeutics within the therapeutic development pipeline and could contribute to the discovery of biomolecular targets in a timelier manner. Compared to LC-MS, MSI typically has more user-friendly protocols and allows for direct analysis of a sample, without the need for any digestion or fractionation, and provides spatial localization information. It is important to note, spatial omics, where LC-MS and MSI studies are conducted in tandem, has been increasing in popularity.61,62 Spatial omics aids in validating the identification of analytes, provides quantitative information, and grants spatial information for a given sample.
Many qMSI experiments describe relative quantification, which is dependent on the use of internal standards or endogenous molecules to assess the concentration of other molecules within a sample.63,64 Relative quantification studies aid in providing relative concentration changes in addition to the spatial localization of subclasses of biomolecules within samples.65 These types of studies have even been used in pharmacokinetic and pharmacodynamic studies of therapeutics and associated metabolites.66 The capabilities of MSI have improved to the point of obtaining absolute quantification. Absolute quantification involves an intense sample preparation, as it must address differences in both ionization and extraction efficiency of molecules and consider signal suppression caused by “matrix effects”.63,67 Additionally, absolute quantification is comparable to relative quantification in that both must contend with a lack of available standards.
In this feature article, we describe the most recent advancements made in qMSI studies of therapeutics and biomolecules with a focus on MALDI & DESI systems. The following sections will contain considerations for both method development and data analysis. Additionally, we present detailed sections focused on qMSI of therapeutics, lipids, and peptides/proteins. Within, we provide prime examples of qMSI employed by our and other research groups. Finally, the scope and limitations of qMSI will be discussed in further detail. We would also like to highlight other excellent reviews on the topic of qMSI of biomolecules.63,68,69
Experimental considerations
MALDI matrices
Matrix selection is an integral part of the initial setup of a MALDI experiment. There are multiple factors that must be considered when selecting an adequate matrix including, but not limited to: analyte of interest, m/z range, instrument polarity, and crystal size.70,71 Some common MALDI matrices include 2,5-dihydroxybenzoic acid (DHB), α-cyano-4-hydroxycinnamic acid (CHCA), 9-aminoacridine (9-AA), 1,5-diaminonaphthalene (DAN), N-(1-naphthyl)ethylenediamine dihydrochloride (NEDC), norharmane (NRM), and 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB). A list of matrices can be found in Table 1, along with examples of biological molecules that readily ionize with the matrices in MALDI-MSI analysis.| Matrix (Abbreviation) | Common biological targets | Polarity (Ref.) |
|---|---|---|
| 1,5-Diaminonaphthalene (DAN) | Lipids | +72,73, −72–74 |
| 2′,5′-Dihydroxyacetophenone (DHA) | Lipids,75,76 proteins,73 peptides76 | +,73,75,76 −76 |
| 2′,5′-Dihydroxybenzoic acid (DHB) | Lipids,75,77 peptides77 | +75,77 |
| 9-Aminoacridine (9-AA) | Lipids,74 metabolites,78 proteins79 | −74,78,79 |
| α-Cyano-4-hydroxycinnamic acid (CHCA) | Lipids,72N-glycans,80,81 proteins82 | +72,80–82 |
| N-(1-naphthyl)ethylenediamine dihydrochloride (NEDC) | Lipids,46,83 metabolites84 | +,46 −46,83,84 |
| Norharmane (NOR) | Lipids | +85,86 |
| Sinapinic acid (SA) | Proteins, peptides | +87,88 |
| Trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) | Lipids | +70,89 |
MALDI-MSI of lipids benefits from measurements conducted in both positive and negative polarity, by increasing lipid class coverage in biological samples. Positive polarity measurements readily display phosphatidylcholine (PC) and sphingomyelin (SM) lipid species due to their inherent positive charge and have been described using numerous matrices.90 For example, Jalaludin and colleagues, examined exosomal lipid content using a variety of matrices including DHB, CHCA, and SA in positive mode.91 They found PC and SM lipids to be some of the most abundant lipids present in positive mode measurements, but noted that this detection can be influenced by both matrix selection and sample preparation. Negative polarity measurements display mainly phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidylglycerol (PG) using matrices such as DAN, 9-AA, DHA, and others.75,90 Recently, Angerer and colleagues, empirically determined that DAN matrix is best suited for high spatial resolution MALDI-MSI and tandem-MS of lipids in negative polarity.72 DAN matrix predominantly displayed PE lipids, but also exhibited broad lipid coverage in negative polarity over other traditional matrices.
MALDI analysis of peptides and proteins has primarily involved the use of sinapinic acid (SA) for large proteins and CHCA matrix for peptide mapping.92,93 Recently, Gu and colleagues compared SA and CHCA matrices to a more novel 4-hydroxy-3-nitrobenzonitrile matrix in the analysis of organic molecules, peptides, and proteins.94 They found that 4-hydroxy-3-nitrobenzonitrile, in comparison to the more traditional matrices, displayed lower background signal and was comparable to other matrices in the analysis of biological tissues. Similarly, Park et al., used both CHCA and DHB for relative quantification of proteins and peptides by constructing calibration curves for the proportion of analyte-to-matrix ion abundance and the analyte-to-matrix ratio.95 This observation for temperature-selected MALDI is amendable for the use of no internal standard and provides a rapid and inexpensive relative quantification of analytes in MALDI analysis. Most recently, the use of MALDI high-plex immunohistochemistry (MALDI-IHC) was employed for the analysis of intact proteins using CHCA matrix.96 In this study, photo-cleavable mass tags are attached to antibodies for increased specificity of protein targets relative to traditional in situ tryptic digestion. Using these tags and CHCA as a matrix, this team was able to obtain 5 μm spatial resolution imaging of proteins in a semi-quantitative manner.
While not the focus of this feature article, carbohydrates are another significantly important biomolecule for conducting qMSI. Carbohydrate analysis has often involved the use of soft ionization techniques such as MALDI.97 The most conventional matrices used for carbohydrate analysis include DHB and CHCA.97,98 For example, Angel and colleagues published a robust protocol on the imaging of N-glycans and peptides using the same formalin-fixed paraffin embedded (FFPE) tissue section using CHCA as a matrix.99 In this study, they were able to co-localize N-glycans and peptides for region specific information on the same tissue section. Similarly, the Heeren research group was able to perform N-glycan imaging with CHCA matrix in fresh frozen (FF) tissue sections.80 In their study, they optimized a protocol for FF samples that was comparable to the signal obtained to the more traditional FFPE samples imaged for N-glycans.
MALDI matrix application
In general, it is recommended to nebulize or sublimate the matrix onto tissues that are mounted on indium-tin-oxide (ITO) coated slides rather than airbrush or dry drop methodologies.71,100 Nebulization and sublimation are two of the most consistent methods for matrix application as they both apply a homogenous coating of matrix over the slide; nebulization of a matrix is shown in Fig. 1.71,101 Homogenous application of the matrix is essential in mitigating matrix “hotspots” caused by uneven matrix spray and analyte delocalization.59,102 These hotspots can create artifacts in analyte distribution and concentration throughout a sample, which is why it is crucial to use a matrix application that is both homogenous and reproducible.103 Similarly, when conducting MALDI-qMSI, the standard must be homogenously present on/in the sample for reliable construction of calibration curves. Any delocalization of internal standard on a tissue will result in unreliable comparisons between analytes of interest and the standard.Additionally, crystal sizing is another important aspect to consider when choosing a matrix application device. Different matrix application devices can result in diverse matrix crystal sizes which influences the spatial resolution of an image and analyte delocalization.103,104 Single cell spatial resolution is now obtainable with the use of nebulization or sublimation where matrix crystal sizes are as small as 1-to-30 μm in size, depending on the matrix and application device.104 In comparison, most mammalian cells typically have a diameter of 10 to 100 μm.105 Therefore, it is advisable to use a robotic sprayer for nebulization of matrix or a sublimation device for uniform distribution of small matrix crystals across a sample. Using these devices mitigates irregularities between users and allows for uniform application of internal standards for both relative and absolute quantification studies.
Solvent selection: MALDI-MSI
Solvent selection is another key aspect of MALDI-MSI and DESI-MSI. Common organic solvents used in MSI include acetonitrile (ACN) and methanol (MeOH), where the ratios of each solvent are dependent upon the target biomolecule.106,107 For MALDI, the matrix must be readily soluble in the solvent and minimize the delocalization of analytes within the sample upon matrix application.108 Sun et al., thoroughly examined the effect of solvent selection in a MALDI based system and how it can impact the sensitivity of lipid detection.109 In their study, they examined several different combinations of matrix and solvent types and found MALDI-MS spectra of lipids was extremely dependent on the properties of solvent used for dispersing the matrix and dissolving the lipids. Interestingly, they also described that 9-AA matrix displays only a single, [M + H]+, peak for certain lipid classes, while other matrices display multiple adducts, adding to the complexity of the spectra. They determined this was due to the interaction of analytes with the matrix being a solvent dependent process which directly contributes to the enhancement of the ionization/desorption efficiency. This effect on sensitivity likely reflects solvent-mediated alterations to dipole–dipole interactions and hydrogen bonding of analytes with the matrix and provides further evidence for empirically determining the best matrix and solvent for your analyte of interest. Additionally, Leopold and colleagues detailed the importance of understanding solvent volatility in relation to “dried droplet” experiments for creating homogenous matrix/analyte mixtures.70 If different solvents and solvent ratios are used, these factors can influence the co-crystallization for the matrix and analyte, rendering these types of experiments unsuitable for quantitative analysis. Several groups have found much success in MALDI-MSI of biomolecules using different solvent types. For example, Shimma and Sugiura, demonstrated the influence that solvent selection has on analyte sensitivity in MALDI-based analysis.110 Comparing ethanol (EtOH), ACN, and MeOH solvents, they found the sensitivity of the signal for the therapeutic olaparib to be five times greater in an ACN-based solvent than a MeOH-based solvent in mouse liver. Therefore, we suggest it is best to empirically determine the most suitable solvent for the combination of matrix and target analyte to ensure homogenous crystal deposition, high sensitivity, and reproducibility.Solvent selection: DESI-MSI
Solvent selection for DESI-based experiments is also an essential step in the experimental planning process. Similarly to MALDI, typical solvents used in DESI-MSI experiments include ACN and MeOH mixed with varying amounts of H2O both with and without an acid.111 As DESI is a liquid extraction technique, the solvent can directly influence the extraction efficiency of certain classes of biomolecules, like lipids for example.112 Comparison of solvent compositions’ effect on extraction efficiency for proteins/peptides was carried out by Towers and colleagues.113 In this study, they examined organic solvent solution concentrations in the range of 50 to 90% for both ACN and MeOH and how solvent composition affects the ionization efficiency of hemoglobin subunits for DESI-MSI. They found 80% organic solvent to be the optimal concentration and that ACN outperformed MeOH in detection sensitivity for hemoglobin subunits. Additionally, for quantitative studies, internal standards can be incorporated into the extraction solvent for direct online comparison of analytes of interest in real time as shown in Fig. 1.111,114 For example, Lanekoff et al., used four lipid standards added simultaneously to the DESI-solvent, in doing so, direct comparisons between ion abundancies and extent of matrix effects could be determined in mouse brain tissue.115 In comparison to MALDI studies, DESI solvent composition should be empirically determined based on the biological target of interest and, if available, prior research studies. Lastly, for both MALDI and DESI studies, the field of fluidics has emerged for MS analysis of biomolecules at the subcellular level and displays great promise for increasing the sensitivity of molecular targets.116,117 Incorporation of fluidics to MS is very compelling for applications dealing with small sample sizes, increasing analyte sensitivity, and the potential addition to MSI workflows.118Normalization methodologies
Data normalization is a key step in the experimental pipeline for MSI studies. Normalization involves incorporating a scaling factor across the entirety of the acquired MS data set to either reduce or expand the range of the intensity scale to be compared.119 Data normalization is important because it can account for and remove artifacts within the imaging data set. These artifacts include but are not limited to heterogeneous crystal sizing and application, differential abundance of biomolecules within a sample, salt content of the sample, and ion source contamination.59,119,120 The most widely used normalization strategies included total ion count (TIC) or root mean square (RMS) global normalization, which are calculated across the full compiled spectrum. Global normalization strategies have been used for relative quantification studies, but fail to take into account matrix effects, such as differential distributions of salt adducts and heterogeneous sample types having diverse molecules that display altered propensities for ionization.63 Therefore, for targeted quantitative studies, it is advisable to use labeled internal standards as reference m/z peaks for normalization of the data set. This type of normalization is easily accomplished by a variety of techniques such as direct on-tissue spraying of the standard before matrix application, having the standard mixed in with the matrix, or having the internal standard spiked into a reference sample as shown.69 Porta and colleagues detailed the benefits of using labeled internal standards; they compensate for differences in ionization efficiencies, co-crystallization, and analyte extraction efficiencies in a given sample.121 The drawbacks from using internal standards as a means for data normalization is the analytes of interest must be known and the experiment conducted in a targeted manner such that the standards are within the same mass range and of similar chemical makeup of the molecule of interest. This downside is becoming less of a problem with further development of isotopically labeled standards for a variety of biomolecules becoming increasingly available.qMSI of therapeutics
Most therapeutics within the clinical pipeline fail to make it to market.122 Analysis from clinical datasets describe four main reasons for why most drugs are clinical failures including lapses in clinical efficacy, wide deviations in toxicity, modest drug-like properties as detailed by Lipinski's rules,123 and poor execution of experimental planning.122 Therefore, techniques aimed at addressing these issues are sought after, due to the longevity and expenses related to drug discovery and development. MSI is a heavily utilized technique for visualizing therapeutics in a label-free manner within a wide range of biological samples. Not only can MSI provide spatial information for therapeutics and their metabolites, but the technique can also help to identify any differentially expressed biomolecules, which may be caused by drug treatment.Both MALDI-MSI and DESI-MSI have been used effectively for therapeutic penetration analyses in tissues and 3D cell cultures.44,124,125 Most studies focus on direct comparisons between a treated and an untreated sample, providing relative quantification of therapeutics and metabolites. Our research group has focused on therapeutic and metabolite detection using MALDI-MSI since 2011,126 using 3D cell culture models as a means of recapitulating the tumor microenvironment and examining the penetration capabilities of chemotherapeutics.127,128 In 2022, President Biden signed the FDA Modernization Act 2.0 into law, which advocates for the use of alternatives to animal models in drug testing, including the use of 3D cell cultures for drug efficacy studies.129 The combination of 3D cell culture models and MSI can facilitate drug development as the spatial localization of the therapeutic and metabolites can be visualized in addition to potential differential abundances of biological molecules related to the drug treatment.130,131
We have examined the penetration and metabolism of irinotecan (IR), a topoisomerase I inhibitor, in 3D cell culture models, such as a colorectal cancer spheroid model and organoids using MALDI-MSI.13,132 This study mapped the therapeutic, irinotecan, and the active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), in a colorectal cancer spheroid model,13 thus describing relative quantitation of the analytes within the spheroid and providing pharmacokinetic insights for the drug distribution. More recently, we have expanded the development of the spheroid model system and drug delivery systems to further recapitulate in vivo conditions by incorporation of 3D printed fluidic devices,133 introduction of dietary restrictions,134 and drug delivery with liposomes.135 Each of these studies relied on MALDI-MSI for relative quantitation of IR and associated metabolites, as a clinically relevant chemotherapeutic for spheroid model development. In a more recent publication, we describe absolute quantification of IR in a time-dependent penetration study using a spheroid model system and MALDI-qMSI.136 To create the calibration curves, control spheroids were homogeneously sprayed with different concentrations of IR solutions to establish the limit-of-detection (LOD) of IR on the sample as shown in Fig. 2. After spraying with IR solutions, deuterated IR was then applied as an internal standard. Using the generated calibration curve with an R2 = 0.9643, we then applied spatial segmentation to distinguish the concentration of IR throughout the dosage interval in a spheroid region specific manner. We concluded from this study, after a 48-hour incubation period with 20.6 μM, the concentration of irinotecan throughout the spheroid was 16.9 μM. This experiment is the first of its kind to show MALDI-qMSI of a therapeutic in a single spheroid.
 | ||
| Fig. 2 qMSI of irinotecan (IR) distribution in HCT 116 spheroids. (A) MSI of IR and the internal standard (IS), d10-irinotecan, detected at 587.286 m/z and 597.349 m/z respectively. Data is normalized against peak area of the IS. (B) Calibration curve of IR quantification. The error bars shown represent the quantification of three biological replicates of spheroids. (C) Penetration curve displaying IR concentration changing as a function of treatment time in whole spheroids. Adapted with permission from ref. 136. Copyright {2023} American Chemical Society. | ||
There has been a plethora of exciting publications recently for using MSI to visualize therapeutic penetration in model systems. For example, Islam and colleagues successfully mapped the therapeutics Imipramine and Chloroquine, along with their metabolites in C57BL/6 male wildtype mice using DESI-MSI, MALDI-MSI, and atmospheric pressure (AP)-MALDI-MSI.137 Their work emphasized the newly developed AP-MALDI-MSI, but also serves as an excellent example of several different ionization techniques being used complementary to one another to elucidate drug and metabolite penetration. Another publication by Traberg and colleagues, describes an innovative mimetic tissue model for skin and incorporates the use of spiked standard homogenates into the mimetic tissue for MALDI-qMSI.138 In their study, they monitored the penetration and concentration of bleomycin, a medication used in the treatment of cancers, using a skin biopsy from a pig. They establish the LOD of bleomycin using the tissue memetic model homogenates for MALDI-MSI and obtained comparable values for the concentration of the drug as determined by LC-MS.
Work by Holm et al., combined the use of DESI- and MALDI-MSI to map the distribution of the immunosuppressant cyclosporine (CsA) in whole body mouse sections and larger organs.139 Their work focused primarily on the use of DESI-MSI, where DESI-MSI was optimized using rat liver homogenate spiked with different concentrations of CsA. Using the homogenates, they determined the LOD of CsA was around 5 μg g−1 and CsA was mainly detected as a Na+ or K+ adduct. Interestingly, they found CsA to be primarily localized to the liver and pancreas, whereas the hydroxy and dihydroxy metabolites were predominantly in the intestines. Additionally, recent work by the Laskin group demonstrated near proportionate concentrations of diclofenac and metabolites between LC-MS and nano-DESI-MSI.140 Quantitative nano-DESI-MSI was achieved by comparison to a deuterated standard incorporated into the solvent spray. Using selected ion monitoring (SIM) during nano-DESI-MSI acquisition allowed for distinguishing the spatial localization of diclofenac metabolites in specific regions within mouse kidney and liver.
We have also examined the efficacy and penetration of other therapeutics using a 3D spheroid model. For example, platinum-based therapeutics, like oxaliplatin, cisplatin, carboplatin were visualized along with metabolites after derivatization with diethyldithiocarbamate using MALDI-MSI.141 In this study, we quantified the concentration of oxaliplatin in different regions of the spheroid model by constructing calibration curves using ultra performance liquid chromatography (UPLC) and coupled this information with the spatial distribution of the drug. We demonstrated the ability to visualize these platinum-based therapeutics, such as oxaliplatin, within 90 minutes of incubation in distinct regions of the spheroid model including the necrotic core. Coupling the information gained from MALDI-MSI with UPLC-MS we were able to monitor drug clearance as oxaliplatin-related species decreased rapidly in 2 hours, where even after 90 minutes, only 7.0% of the initial drug was present in the spheroids providing some pharmacokinetic insight for these drugs in this model. Concurrently, we have focused on MALDI-MSI of protein and peptide therapeutics, ranging from FDA-approved drugs to novel therapeutics. We visualized Cetuximab, a monoclonal antibody that is used for immunotherapy treatment of colorectal cancer, by MALDI-MSI after on-tissue-reduction and -alkylation.142 Cetuximab is a epidermal-growth-factor receptor (EGFR) inhibitor, so by using two different spheroid models that express different levels of EGFR, we examined disparate changes in the pharmacokinetics between the two spheroids using MSI. Similarly, in collaboration with Professor Dehua Pei and his research group, we used MALDI-MSI to examine both the pharmacokinetics and pharmacodynamics of cyclic peptidyl inhibitors within a colorectal spheroid model.143 We found for the cyclic peptidyl inhibitor, PGD97, that once it effectively enters the cytosol, it is rapidly reduced to its active form and subsequently undergoes degradation, with some of the intact peptide still present after 24 hours. This finding was important in determining the efficacy of PGD97 as an inhibitor of the cystic fibrosis transmembrane conductance regulator and associated ligand in the treatment of cystic fibrosis.
qMSI of Lipids
Lipids are essential biomolecules as they are involved in a plethora of biological functions such as providing structural integrity for cells and organelles, transportation of molecules, serving as both an energy source and storage, cell signaling, and a variety of other functions.144,145 This vast functional diversity can be attributed to the broad heterogeneity found within lipid structures.38,146 There are numerous lipid classes such as glycerophospholipids, sphingolipids, sterols, and fatty acyls, to name a few. Within each lipid class are subclasses of lipid molecules. For example, glycerophospholipids can be further disassembled into phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), and sphingomyelin (SM) to name a few of the different types.147 Additionally, each of these lipids display structural heterogeneity, varying in fatty acid chain length and degree of unsaturation which can directly affect lipid function.38 In the context of disease, cancers can regulate the amount of saturated and unsaturated fatty acids within phospholipids located in the plasma membrane of cells to increase resiliency to oxidative stress and chemotherapeutics.148 Additionally, a recent study described the influence of glycerophospholipid expression in the context of chronic obstructive pulmonary disease, where they found differential expression of PC and PE lipids between the disease tissue and control.149 Since lipids play an important part in biological functions and can be indicative of disease onset and progression,150 several imaging modalities have been employed to distinguish the spatial distribution and abundance of lipids, such as MSI.151–153Limited works have been published detailing MALDI-qMSI of lipids.154 Most works have focused on direct comparisons of lipid abundance between a disease state and healthy control tissue.155–158 A recent study by Martín-Siaz and colleagues elegantly demonstrated the blending of MALDI-MSI and HPLC-MS to distinguish how well the two techniques correlate with one another in lipid biomarker discovery in clear cell renal cell carcinoma.159 This study found that both MALDI-MSI and HPLC-MS experiments could spatially classify the lipid signature to distinct regions in the kidney, such as the cortex and medulla. A direct comparison of healthy vs. renal cell carcinoma was made using MALDI-MSI and quantification was conducted using internal standards with HPLC-MS analysis. Their findings highlight the potential of using MALDI-MSI for distinguishing different cell populations in a sample based on the samples lipid profile and how quantification can be conducted complementary to imaging modalities. Jadoul and colleagues demonstrated an innovative method for MALDI-qMSI of lipids within a brain section by using brain homogenates spiked with an internal standard.160 They quantified different PC lipid species spatial distributions in a region specific manner within a mouse brain section as shown in Fig. 3. This study highlights the ability to conduct MALDI-qMSI of lipids and other small endogenous compounds while considering matrix effects caused by imaging a heterogenous tissue. A more recent study by Angelini and coworkers utilized on-tissue derivatization of cholesterol by adapting an enzyme-assisted derivatization for sterol analysis (EADSA) for absolute quantification of cholesterol in a mouse brain tissue.161 In general, the group applied [2H7]cholesterol as an internal standard to mouse brain sections using an automated sprayer, followed by cholesterol oxidase enzyme and Girard-P hydrazine to apply a charge-tagged sterol hydrazone. After incubation, CHCA matrix was applied using an HTX TM-sprayer and endogenous cholesterol was detected using MALDI-MSI and normalized to the applied internal standard. Using this method, they were able to assess, both qualitatively and quantitatively, region specific changes in cholesterol levels between wild-type (WT) and Niemann-Pick C1 disease (Npc1) mice brain sections as a function of age. Similarly, a recent publication by Vandenbosch et al., demonstrates the use of a multi-class internal standard lipid mixture, sprayed homogenously over a mouse brain section for quantification of several different lipid classes in one imaging run using MALDI-MSI.162 They found subtle differences in image quality between normalization to an internal standard and normalizing to the conventionally applied TIC.
 | ||
Fig. 3 Analysis by MALDI-MSI of sections from an intact brain and brain homogenates spiked with a range of PC(16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 d31/18:1) concentrations. (A) A serial section after H&E staining. (B) Definition of ROIs on the section (ROI1: red corpus callosum; ROI2: green cerebral cortex; ROI3: blue lateral septal nucleus; ROI4: grey caudoputamen; ROI5: yellow hypothalamus). (C) Molecular images showing the distribution of different PC ionic species ([M + H]+, [M + Na]+ and [M + K]+) across the intact brain section surface. (D) Molecular image showing the intensity scale corresponding to the [PC(16:0 d31/18:1) + H]+ on the spiked (between 100 and 8000 μg g−1) homogenate sections analysed simultaneously to the intact brain. Reproduced with permission from ref. 160, under Creative Commons License Attribution 4.0 International (CC-BY 4.0), Springer Nature (https://creativecommons.org/licenses/by/4.0/legalcode). :0 d31/18:1) concentrations. (A) A serial section after H&E staining. (B) Definition of ROIs on the section (ROI1: red corpus callosum; ROI2: green cerebral cortex; ROI3: blue lateral septal nucleus; ROI4: grey caudoputamen; ROI5: yellow hypothalamus). (C) Molecular images showing the distribution of different PC ionic species ([M + H]+, [M + Na]+ and [M + K]+) across the intact brain section surface. (D) Molecular image showing the intensity scale corresponding to the [PC(16:0 d31/18:1) + H]+ on the spiked (between 100 and 8000 μg g−1) homogenate sections analysed simultaneously to the intact brain. Reproduced with permission from ref. 160, under Creative Commons License Attribution 4.0 International (CC-BY 4.0), Springer Nature (https://creativecommons.org/licenses/by/4.0/legalcode). | ||
While ongoing work is being conducted on qMSI of lipid species, simultaneously, there has been a push in the imaging community for increased depth in identification and quantification of double bond and stereospecific numbering (sn-) position in lipids.154 Both the degree of saturation and sn-positioning have important biological functions and can even be used for prediction of disease and for diagnostics, making these molecular features essential for further examination.163–165 Born & Prentice described the use of electron induced dissociation (EID) performed with the use of a MALDI source to generate fragments of PC lipids for identification of sn-1 and sn-2 positions, as well as the location of double bonds within the fatty acyl chains.166 Interestingly, the authors demonstrated the ability to characterize and identify PC lipids directly from a rat brain tissue section due to the addition of EID. They later extrapolated this technique further by describing how relative quantification could be performed using this technique on both sodium and potassium adducts in PC lipids.167 This methodology can be used complementary to traditional imaging workflows to provide greater details in lipid characterization in tissue sections. Traditionally, collision induced dissociation (CID) is performed complementary to imaging experiments to provide lipid structural information such as the degree of unsaturation or fatty acyl chain length but lacks the ability to elucidate sn-position and double bond location.168–170 Claes and colleagues combined ozone-induced dissociation (OzID) with CID to demonstrate isomer-resolved MALDI-MSI of lipids in rat brain.168 In this study, ozone was supplied directly into a SYNAPT HDMS G2-Si system where a lipid precursor is trapped and when in the presence of ozone fragmentation occurs at the double bond position. When CID is conducted in tandem with OzID, sn-position can be determined for further characterization of lipid structure. Additionally, sensitivity testing was conducted to establish the LOD of 1.1 fg μm−2 and limit of quantification (LOQ) of 2.2 fg μm−2 of a PC 34:1 standard to establish characteristic OzID fragments that could be used in the remainder of the study. These studies describe innovative methodologies for identification of lipid biomolecules and serve as platforms for increased quantitative studies to be performed using MALDI-qMSI.
Similarly to MALDI-MSI, DESI-MSI of lipids has focused primarily on relative changes in lipid abundance in tissues and more quantitative imaging is forthcoming.171 Research conducted by Mondal et al., detailed the ability to use DESI-MSI to differentiate cancerous versus adjacent normal in samples obtained from 73 breast cancer patients.172 They identified several lipid species that were upregulated in the cancerous tissue, including four diacylglycerol (DG) molecules which were the most unique and intense change in lipid abundance for the entire patient set. They concluded that utilizing DESI-MSI was useful in validation for intraoperative surgical pathology and DG would serve as a unique diagnostic biomarker for predicting breast cancer in patients, as validated by their machine learning experiments. To advance from a more qualitative understanding of lipid distribution to a more quantitative assessment, research groups using DESI-MSI have employed spiking internal standards into their solvent spray. For example, Julia Laskin's research group has published several works detailing the introduction of internal standards to solvent spray to achieve DESI-qMSI with lipid biomolecules. Specifically, the Laskin group developed a high spatial resolution and high throughput protocol for DESI-MSI of lipids that elegantly details all the parameters and considerations needed for obtaining high quality lipid images.173 Within this work, the group detailed how the incorporation of lipid internal standards into the working solvent reflects the actual distribution and concentration of lipid species within uterine tissue, as shown in Fig. 4. This study also focused on matrix effects as a key issue for achieving qMSI and they showed how the incorporated internal standards undergo the same ionization suppression as endogenous lipid species making normalization to the standard more appropriate than traditional normalization to TIC. In a similar study from the Laskin group, the same integration of internal standards to the solvent system was applied to imaging of mouse lung tissues in a comparative fashion with nano-DESI MSI and LC-MS/MS lipidomics.111 The multi-class internal standard lipid mixture spiked in the solvent contained different classes of both lipids and fatty acids for normalization. This study concluded that the direct sampling method of nano-DESI MSI provides close to 50% lipid coverage in comparison to the LC-MS/MS analysis, demonstrating the breadth of lipid coverage obtainable in a high-throughput, spatially defined manner. Recently, Mavroudakis and colleagues modified the extraction solvent in a nano-DESI system with crown ether molecules to better understand alkali metal ion distributions in mouse brain tissue sections with the addition of internal standards for relative quantification.174 Where alkali metal ions play important roles in biological processes, the group examined the interplay between Na+ and K+ concentrations at the elemental level to see how these concentration influence adduct formation in lipid species. In summary, they successfully quantified alkali metals, lipids, and other metabolites and distinguished how they are located spatially in ischemic regions of a mouse stroke model.
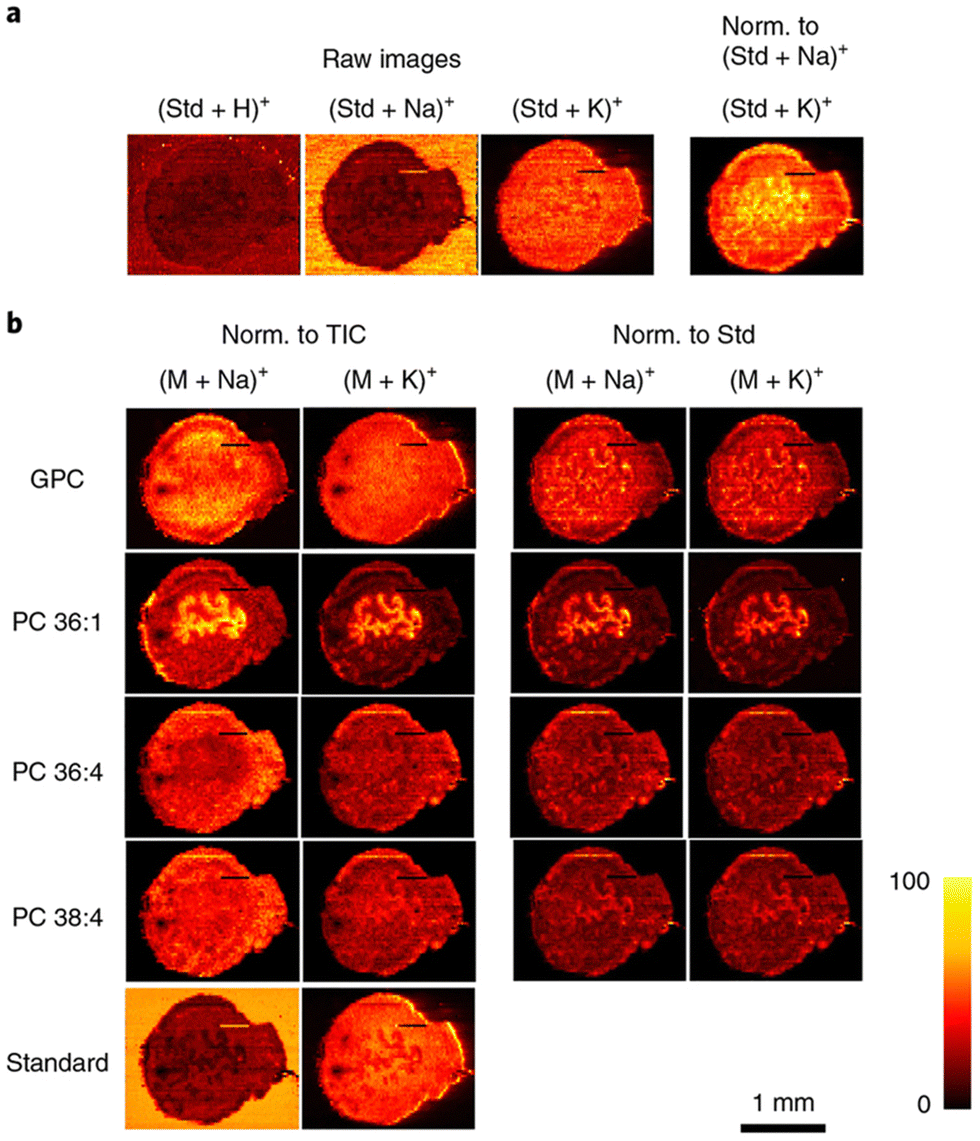 | ||
| Fig. 4 Compensation for matrix effects in nano-DESI MSI by adding internal standards to the solvent in uterine tissues. (a) The use of the internal standard (Std; LPC 19:0) indicates the matrix effects originating from competitive ionization of analytes, as well as variations in the abundance of alkali metal salts. Raw images describe the ion images without any normalization (Norm.). (b) Ion images of [M + Na]+ and [M + K]+ of endogenous GPC, PC 36:1, PC 36:4, PC 38:4, and standard LPC 19:0 (Standard). Ion images are normalized to the TIC in the left two columns and to the standard LPC 19:0 (Std) in the right two columns. By normalizing to the corresponding LPC 19:0 adducts, the same ion images are obtained for [M + Na]+ and [M + K]+ ions of endogenous lipids, which are free of matrix effects and present their actual distribution in uterine tissue. Reproduced with permission from ref. 173, under Creative Commons License Attribution 4.0 International (CC-BY 4.0), Springer Nature (https://creativecommons.org/licenses/by/4.0/legalcode). | ||
qMSI of peptides & proteins
Proteins are considered a fundamental building block of life. Proteins are involved in countless biological roles including regulating gene expression, providing structural integrity in cells, serving as enzymes for catalyzing reactions, immune function, and countless others.175,176 Similarly to lipids, proteins incredibly diverse functional roles can be attributed to the wide structural heterogeneity they display.177 Protein structural diversity can stem from amino acid sequence variability, influence of side chain interactions along the peptide backbone, and post-translational modifications (PTMs).178 Where the structural characteristics translate to biological function, it is clear how any perturbation in protein structure could lead to detrimental outcomes in protein function, leading to disease onset and progression.179 Since dysregulation or modification of proteins can lead to disease; protein structure, involvement in cell signaling, gene regulation, enzyme function, and even their use as potential therapeutics has been studied extensively.180–182 In MS, the gold standard for protein analysis, or proteomics, is LC-MS/MS. While this method is great for both targeted and untargeted studies in identifying and quantifying proteins, it does not provide insight to the localization of proteins in situ.183,184 MSI, specifically qMSI, is continually advancing to provide not only the spatial localization of proteins or related peptides, but also quantitative information as the availability of internal standards, quality controls, and instrumentation improves.Common within the methodology of MALDI-MSI of proteins is an in situ tryptic digestion step prior to matrix application and imaging. On-tissue tryptic digestion is critical because it allows for simplification of the MS analysis. Due to their large size, native proteins display limited compatibility and sensitivity for general MSI runs. However, creating small peptide fragments that correspond to specific proteins is more amendable to MSI analysis and can improve the sensitivity of detection. For example, in a recent study Fiorentino and colleagues describe a comprehensive pipeline for in situ detection of follicle protein content within a mouse ovary by employing trypsin digestion prior to MALDI-MSI analysis.185 Utilizing the same digested tissue, they complemented the MALDI-MSI data with nano-LC-MS/MS and correlate the peptide fragments to specific proteins by the Mascot software and the Swissprot protein database. They successfully employed a spatial proteomics workflow to map proteins of both known and unknown function during the process of folliculogenesis.
One of the benefits of conducting on-tissue tryptic digestion of proteins is that it can lead to more qMSI studies as the peptide abundance can be attributed to the abundance of a specific protein.186 Additionally, in a targeted analysis, isotope-labeled peptide internal standards can be synthesized for improved quantification.187,188 Clemis et al., demonstrated the addition of an isotopically labeled myelin protein tryptic peptide on a rat brain section post on-tissue trypsin digestion which allowed for the normalization of digested myelin protein across the entire brain section.189 The imaging was conducted on a QTrap mass spectrometer with a MALDI source which has the capability for multiple reaction monitoring (MRM) analysis for greater specificity and sensitivity of myelin protein fragments and the internal standard. Using a blank ITO slide, the authors conducted a serial dilution of the standard by aerosol deposition to construct a calibration curve of the ratio of endogenous to labeled peptide prior to qMSI. The authors found that their method supports the accurate quantification of a protein target within a tissue and has the capability to retain the spatial localization of the protein. As previously described, Porta and colleagues stringently monitored their imaging parameters to determine how many pixels are needed for accurate quantification of drugs and peptides using a dried droplet methodology with a MALDI system.121 Within their study, a trypsin digested monoclonal antibody and the associated isotopically labeled peptide standard are spotted together on a MALDI plate to distinguish the appropriate number of pixels for analysis and normalization as shown in Fig. 5. They found at least ten pixels need to be considered to obtain sufficient quantification with standard deviations below 10% for this system. Their analysis was based on similar quantitative proteomic studies where the ratio of an endogenous molecule is often compared to that of an internal standard; in their study the same area or pixels was considered for the peak area of both the digested monoclonal antibody and the internal standard. This study elegantly presents factors that need to be considered for qMSI analysis including but not limited to frequency of the laser, laser power, matrix effects, number of pixels for accurate quantification, and matrix application methods. Recently, Dewez and colleagues used a multilabel peptide approach to correct for any disproportional ionization effects caused by different local environments within a tissue section, which is not entirely taken into account with the use of an external calibration using one standard.190 In their study, they performed a comparison between multilabel and single-label quantification for histone H4 peptides in a pig colon tissue section by MALDI analysis. Utilizing a dynamic concentration for the isotopically labeled internal standards, post on-tissue trypsinization, they calculated the absolute concentration of endogenous H4 peptide in a per-pixel manner. The multilabel analysis proved superior to the single-label as it displayed reduced variance to the predicted concentration of endogenous peptide relative to the single-label. This study highlights the importance of considering tissue heterogeneity for molecules of interest, as concentrations of these molecules can be dynamic and are not often fixed in concentration across an entire tissue section.
 | ||
| Fig. 5 Normalization method for peptide quantification. (a) Size of the region of interest (ROI), (b) MALDI-SRM/MS images for the peptide AEDTAVVYCAR obtained after digestion and derivatization of a monoclonal antibody, its stable isotope-labeled analogue (IS), and after pixel-by-pixel signal ratio (theoretical ratio of 2). Variation of the ratio peptide/IS after (c) pixel-by-pixel normalization and (d) area-by-area normalization as a function of the number of pixels considered per ROI. (e) Inter-spot variability (RSD%) in function of the number of pixels selected per ROI. Reproduced with permission from ref. 121, under Creative Commons License Attribution 4.0 International (CC-BY 4.0), Springer Nature (https://creativecommons.org/licenses/by/4.0/legalcode). | ||
While MALDI-MSI of peptides is considered the gold standard in qMSI analysis of proteins, MALDI-MSI of intact high molecular weight proteins is becoming more feasible. There are currently several techniques being employed for MALDI-MSI of intact proteins, which often involve some sort of functionalization or tagging of the target protein. Recently, MALDI-IHC, or the combination of MALDI-MSI and immunohistochemistry, has been demonstrated for the analysis of a wide variety of protein targets.96,191,192 This approach utilizes the specificity granted by traditional immunohistochemistry analysis by the use of targeted antibodies. These antibodies are functionalized by a photocleavable mass-tag which contain a modified polypeptide motif of a known molecular weight and can be observed by MALDI post ultraviolet radiation to distinguish protein location.192 While this technique allows for the visualization of particular protein targets, the peak heights from the spectral output only detail semi-quantitative data. However, incorporation of internal standards similar in chemical properties to the polypeptide mass-tags could allow for absolute quantification of the protein targets and is currently being explored. Another fascinating technique recently demonstrated by Wang, et al., for MALDI-MSI of protein biomarkers, employs the use of gold nanoparticles functionalized with organic oligomers that serve as mass-tags which are visualized after specific binding of exosome containing proteins to an antibody.193 The authors took advantage of exosome secretion from different cancer cell lines by selectively capturing the exosomes, which all contain different surface protein biomarkers dependent on the cell of origin. Utilizing their antibody-coated gold chip in tandem with mass-tag containing gold nanoparticles they effectively created a quantitative platform to both visualize and classify protein biomarkers on the surface of the exosomes. Both innovative techniques detail the feasibility of whole protein MALDI-MSI analysis and the potential of incorporating more quantitative measurements utilizing these techniques.
Outside of MALDI-MSI, qMSI of peptides and proteins is being conducted by other ionization techniques. Predominantly, liquid extraction surface analysis (LESA) has shown great success for quantification of proteins in tissue.194 Havlikova and colleagues demonstrated how LESA-MS could be used for quantitative imaging of ubiquitin within a rat brain mimetic tissue model by utilizing a dynamic concentration of a spiked isotopically labeled ubiquitin protein.194 Prior to analysis, the authors performed method validation by determining the lower and upper limits of quantification, 68.6 nmol g−1 and 163 nmol g−1 respectively, utilizing the mimetic tissue model. After determining the limits of quantification, a dynamic range of concentrations were applied to the mimetic model and used for construction of a calibration curve for the 9+ charge state of ubiquitin. They then performed the same experimental setup on sagittal sections of rat and mouse brain to determine the concentration of endogenous ubiquitin as 139.641 ± 95.403 nmol g−1 and 90.233 ± 51.512 nmol g−1 respectively. While many additional works from Dr Helen Cooper's research group has focused on LESA-MS usage for imaging of proteins and protein complexes, they have more recently described incorporating nano-DESI as an alternate ambient ionization source.195,196 nano-DESI allows for a nondenaturing solvent to be used for intact protein analysis along with the added advantage of higher resolution imaging relative to LESA-MSI.196 In the first report of native nano-DESI-MSI, Hale & Cooper employed protein standards for LOD determination, which yielded a value of ∼0.4 pmol.195 While relative concentration of protein complexes were reported for the untargeted study, the incorporation of specific internal standards could be used in a more targeted analysis. Thus, not only has this study demonstrated the feasibility of conducting native MSI using nano-DESI, but it also opens the door to future studies involving targeted qMSI.
Conclusions & outlook
Within the past decade, research groups have placed emphasis on advancing the quantitative output of MSI. In this feature article, we have summarized some of the most recent innovative ideas while paying homage to previous foundational works in conducting qMSI for a variety of therapeutics and biomolecules.We emphasize the need to empirically derive the most suitable matrix and solvent for conducting qMSI, which is tailored to the analyte of interest. Additionally, we discuss the homogenous application of matrix for MALDI analysis, which must be applied across the tissue to prevent matrix “hot spot” formation, as well as mitigate analyte delocalization. We focused our discussion of qMSI in the context of the two most utilized ionization sources for imaging, MALDI and DESI. For both ionization sources, we discuss recent advancements in imaging capabilities such as improvements in instrumentation, alternative methods for increasing sensitivity of detection, development of standards, and increased understanding of sample preparation. Such advancements have led to the bolstered usage of MSI for analysis of therapeutics and biomolecules. The enhancement in qMSI capabilities grants future studies to be more accurate and detail the molecular distribution of analytes in a quantitative fashion. The examples we highlighted are foundational for increasing broad-spectrum usage of qMSI. While other important molecules such as carbohydrates were not discussed, they are equally as significant as what was discussed in this feature article. As the usage of MSI expands, we foresee a shift in the MSI community from the more commonly applied direct comparison analysis of analytes to a more quantitative analysis for more accurate conclusions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Institutes of Health (NIH) (R01GM110406, R21AG062144). Additionally, we would like to acknowledge the table of contents figure and Fig. 1 were created with Biorender.com.References
- R. M. Caprioli, T. B. Farmer and J. Gile, Anal. Chem., 1997, 69, 4751–4760 CrossRef CAS PubMed.
- G. Arentz, P. Mittal, C. Zhang, Y.-Y. Ho, M. Briggs, L. Winderbaum, M. K. Hoffmann and P. Hoffmann, in Advances in Cancer Research, ed. R. R. Drake and L. A. McDonnell, Academic Press, 2017, vol. 134, pp. 27–66 Search PubMed.
- G. O’Hurley, E. Sjöstedt, A. Rahman, B. Li, C. Kampf, F. Pontén, W. M. Gallagher and C. Lindskog, Mol. Oncol., 2014, 8, 783–798 CrossRef.
- T. Porta Siegel, G. Hamm, J. Bunch, J. Cappell, J. S. Fletcher and K. Schwamborn, Mol. Imaging Biol., 2018, 20, 888–901 CrossRef CAS.
- M. Holzlechner, E. Eugenin and B. Prideaux, Cancer Rep., 2019, 2, e1229 CrossRef.
- Y. Wang, J. Sun, J. Qiao, J. Ouyang and N. Na, Anal. Chem., 2018, 90, 14095–14099 CrossRef CAS.
- A. Croxatto, G. Prod’hom and G. Greub, FEMS Microbiol. Rev., 2012, 36, 380–407 CrossRef CAS PubMed.
- Z. Wang, Q. Zhang, H. Shen, P. Yang and X. Zhou, Front. Chem., 2021, 9, 698297 CrossRef CAS PubMed.
- P. Chaurand, M. Stoeckli and R. M. Caprioli, Anal. Chem., 1999, 71, 5263–5270 CrossRef CAS.
- T. Liepold, H.-W. Klafki, S. Kumar, J. Walter, O. Wirths, J. Wiltfang and O. Jahn, J. Am. Soc. Mass Spectrom., 2023, 34, 505–512 CrossRef CAS.
- D. M. G. Anderson, Z. Ablonczy, Y. Koutalos, J. Spraggins, R. K. Crouch, R. M. Caprioli and K. L. Schey, J. Am. Soc. Mass Spectrom., 2014, 25, 1394–1403 CrossRef CAS.
- J. L. Moore, N. H. Patterson, J. L. Norris and R. M. Caprioli, Mol. Cell. Proteom., 2023, 100576 CrossRef CAS.
- X. Liu, E. M. Weaver and A. B. Hummon, Anal. Chem., 2013, 85, 6295–6302 CrossRef CAS PubMed.
- Y. Chen, T. Wang, P. Xie, Y. Song, J. Wang and Z. Cai, Anal. Chim. Acta, 2021, 1184, 339011 CrossRef CAS.
- B. Bakker, R. D. W. Vaes, M. R. Aberle, T. Welbers, T. Hankemeier, S. S. Rensen, S. W. M. Olde Damink and R. M. A. Heeren, Nat. Protoc., 2022, 17, 962–979 CrossRef CAS PubMed.
- E. Cuypers, B. S. R. Claes, R. Biemans, N. G. Lieuwes, K. Glunde, L. Dubois and R. M. A. Heeren, Anal. Chem., 2022, 94, 6180–6190 CrossRef CAS.
- K. Ščupáková, F. Dewez, A. K. Walch, R. M. A. Heeren and B. Balluff, Angew. Chem. Int. Ed., 2020, 59, 17447–17450 CrossRef PubMed.
- J. M. Wiseman, D. R. Ifa, Q. Song and R. G. Cooks, Angew. Chem. Int. Ed., 2006, 45, 7188–7192 CrossRef CAS.
- S. Sämfors and J. S. Fletcher, Ann. Rev. Anal. Chem., 2020, 13, 249–271 CrossRef PubMed.
- R. Weiskirchen, S. Weiskirchen, P. Kim and R. Winkler, J. Cheminform., 2019, 11, 16 CrossRef.
- Y. Huang, S. Du, J. Liu, W. Huang, W. Liu, M. Zhang, N. Li, R. Wang, J. Wu, W. Chen, M. Jiang, T. Zhou, J. Cao, J. Yang, L. Huang, A. Gu, J. Niu, Y. Cao, W.-X. Zong, X. Wang, J. Liu, K. Qian and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2122245119 CrossRef CAS.
- W. Chen, H. Yu, Y. Hao, W. Liu, R. Wang, Y. Huang, J. Wu, L. Feng, Y. Guan, L. Huang and K. Qian, ACS Nano, 2023, 17, 19779–19792 CrossRef CAS.
- Y. Tatsuta, Y. Tanaka, A. Ikeda, S. Matsukawa, H. Katano and S. Taira, Mass Spectrom., 2017, 6, S0069 CrossRef PubMed.
- J. G. Swales, G. Hamm, M. R. Clench and R. J. A. Goodwin, Int. J. Mass Spectrom., 2019, 437, 99–112 CrossRef CAS.
- J. C. Vickerman, Analyst, 2011, 136, 2199–2217 RSC.
- N. C. Wamer, C. N. Morse, J. N. Gadient, T. A. Dodson, E. A. Carlson and E. G. Prestwich, J. Am. Soc. Mass Spectrom., 2023, 34, 355–365 CrossRef CAS.
- M. J. He, W. Pu, X. Wang, W. Zhang, D. Tang and Y. Dai, Front. Oncol., 2022, 12, 891018 CrossRef CAS.
- A. P. Bowman, J. F. J. Bogie, J. J. A. Hendriks, M. Haidar, M. Belov, R. M. A. Heeren and S. R. Ellis, Anal. Bioanal. Chem., 2020, 412, 2277–2289 CrossRef CAS.
- A. Roux, P. T. Winnard, M. H. Van Voss, L. Muller, S. N. Jackson, B. Hoffer, A. S. Woods and V. Raman, Mol. Cell. Biochem., 2023, 478, 2567–2580 CrossRef CAS.
- K. D. Duncan, R. Fang, J. Yuan, R. K. Chu, S. K. Dey, K. E. Burnum-Johnson and I. Lanekoff, Anal. Chem., 2018, 90, 7246–7252 CrossRef CAS PubMed.
- M. Giampà, M. K. Andersen, S. Krossa, V. Denti, A. Smith and S. A. Moestue, in Imaging Mass Spectrometry: Methods and Protocols, ed. L. M. Cole and M. R. Clench, Springer US, New York, NY, 2023, pp. 161–172 Search PubMed.
- M.-C. Djidja, J. Chang, A. Hadjiprocopis, F. Schmich, J. Sinclair, M. Mršnik, E. M. Schoof, H. E. Barker, R. Linding, C. Jørgensen and J. T. Erler, J. Proteome Res., 2014, 13, 2297–2313 CrossRef CAS.
- M. Stoeckli, P. Chaurand, D. E. Hallahan and R. M. Caprioli, Nat. Med., 2001, 7, 493–496 CrossRef CAS.
- T. S. Høiem, M. K. Andersen, M. Martin-Lorenzo, R. Longuespée, B. S. R. Claes, A. Nordborg, F. Dewez, B. Balluff, M. Giampà, A. Sharma, L. Hagen, R. M. A. Heeren, T. F. Bathen, G. F. Giskeødegård, S. Krossa and M.-B. Tessem, Proteomics, 2022, 22, 2100223 CrossRef.
- C. T. McDowell, X. Lu, A. S. Mehta, P. M. Angel and R. R. Drake, Mass Spectrom. Rev., 2023, 42, 674–705 CrossRef CAS PubMed.
- C. R. K. Blaschke, C. T. McDowell, A. P. Black, A. S. Mehta, P. M. Angel and R. R. Drake, Clin. Lab. Med., 2021, 41, 247–266 CrossRef.
- M. Shariatgorji, A. Nilsson, E. Fridjonsdottir, T. Vallianatou, P. Källback, L. Katan, J. Sävmarker, I. Mantas, X. Zhang, E. Bezard, P. Svenningsson, L. R. Odell and P. E. Andrén, Nat. Methods, 2019, 16, 1021–1028 CrossRef CAS.
- E. Fahy, D. Cotter, M. Sud and S. Subramaniam, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1811, 2011, 637–647 Search PubMed.
- M. Baumann and S. Meri, Expert Rev. Proteom., 2004, 1, 207–217 CrossRef CAS.
- D. Sun, X. Ren, E. Ari, T. Korcsmaros, P. Csermely and L.-Y. Wu, Brief. Bioinformatics, 2019, 20, 89–101 CrossRef CAS PubMed.
- J. M. Mitchell, R. M. Flight and H. N. B. Moseley, Metabolites, 2021, 11, 740 CrossRef CAS PubMed.
- R. Bhatnagar, N. M. Dixit, E. H. Yang and T. Sallam, Front. Cardiovasc. Med., 2022, 9, 925816 CrossRef CAS.
- N. H. Patterson, B. Alabdulkarim, A. Lazaris, A. Thomas, M. M. Marcinkiewicz, Z. Gao, P. B. Vermeulen, P. Chaurand and P. Metrakos, Sci. Rep., 2016, 6, 36814 CrossRef CAS.
- C. E. Spencer, L. E. Flint, C. J. Duckett, L. M. Cole, N. Cross, D. P. Smith and M. R. Clench, Expert Rev. Proteom., 2020, 17, 827–841 CrossRef CAS.
- A. Taj, A. Rehman and S. Z. Bajwa, in Nanobiosensors, ed. A. Wu. and W. S. Khan, Wiley, Hoboken, NJ, 2020, pp. 73–94 Search PubMed.
- J. H. Holbrook, E. R. Sekera, A. Lopez, B. D. Fries, F. Tobias, K. Akkaya, M. M. Mihaylova and A. B. Hummon, Anal. Chem., 2023, 95, 10603–10609 CrossRef CAS PubMed.
- D. Unsihuay, R. Yin, D. M. Sanchez, M. Yang, Y. Li, X. Sun, S. K. Dey and J. Laskin, Anal. Chim. Acta, 2021, 1186, 339085 CrossRef CAS PubMed.
- F. P. Y. Barré, M. R. L. Paine, B. Flinders, A. J. Trevitt, P. D. Kelly, R. Ait-Belkacem, J. P. Garcia, L. B. Creemers, J. Stauber, R. J. Vreeken, B. Cillero-Pastor, S. R. Ellis and R. M. A. Heeren, Anal. Chem., 2019, 91, 10840–10848 CrossRef.
- M. Feucherolles and G. Frache, Cells, 2022, 11, 3900 CrossRef CAS PubMed.
- J. R. Granborg, A. M. Handler and C. Janfelt, TrAC, Trends Anal. Chem., 2022, 146, 116482 CrossRef CAS.
- W. Tang, J. Chen, J. Zhou, J. Ge, Y. Zhang, P. Li and B. Li, Theranostics, 2019, 9, 932–944 CrossRef CAS.
- F. Saint-Marcoux, F.-L. Sauvage and P. Marquet, Anal. Bioanal. Chem., 2007, 388, 1327–1349 CrossRef CAS PubMed.
- E. N. Fung, P. Bryan and A. Kozhich, Bioanalysis, 2016, 8, 847–856 CrossRef CAS PubMed.
- S. M. Lam, J. Li, H. Sun, W. Mao, Z. Lu, Q. Zhao, C. Han, X. Gong, B. Jiang, G. H. Chua, Z. Zhao, F. Meng and G. Shui, Mol. Biol. Evol., 2022, 39, msac050 CrossRef CAS.
- S. Yang, J. Xue and C. Ye, STAR Protoc., 2022, 3, 101769 CrossRef CAS.
- S. Paramasivan, J. L. Morrison, M. C. Lock, J. R. T. Darby, R. A. Barrero, P. C. Mills and P. Sadowski, J. Proteome Res., 2023, 22, 2018–2029 CrossRef CAS PubMed.
- Q. Zhou, M. R. S. Alvarez, K. Solakyildirim, J. Tena, L. M. N. Serrano, M. Lam, C. Nguyen, F. Tobias, A. B. Hummon, R. C. Nacario and C. B. Lebrilla, Glycobiology, 2023, 33, 2–16 CrossRef CAS PubMed.
- W.-J. Qian, J. M. Jacobs, T. Liu, D. G. Camp and R. D. Smith, Mol. Cell. Proteom., 2006, 5, 1727–1744 CrossRef CAS.
- F. Tobias and A. B. Hummon, J. Proteome Res., 2020, 19, 3620–3630 CrossRef CAS.
- L. Lamont, D. Hadavi, B. Viehmann, B. Flinders, R. M. A. Heeren and R. J. Vreeken, and T. Porta Siegel, Anal. Bioanal. Chem., 2021, 413, 2779–2791 CrossRef CAS PubMed.
- S. T. P. Mezger, A. M. A. Mingels, O. Bekers, R. M. A. Heeren and B. Cillero-Pastor, Anal. Chem., 2021, 93, 2527–2533 CrossRef CAS.
- H. Zhang, D. G. Delafield and L. Li, Nat. Methods, 2023, 20, 327–330 CrossRef CAS.
- D. Unsihuay, D. Mesa Sanchez and J. Laskin, Annu. Rev. Phys. Chem., 2021, 72, 307–329 CrossRef CAS PubMed.
- R. Legouffe, O. Jeanneton, M. Gaudin, A. Tomezyk, A. Gerstenberg, M. Dumas, C. Heusèle, D. Bonnel, J. Stauber and S. Schnebert, Anal. Bioanal. Chem., 2022, 414, 5781–5791 CrossRef CAS PubMed.
- L. Morosi, C. Matteo, M. Meroni, T. Ceruti, I. Fuso Nerini, E. Bello, R. Frapolli, M. D’Incalci, M. Zucchetti and E. Davoli, Talanta, 2022, 237, 122918 CrossRef CAS.
- L. Poncelet, R. Ait-Belkacem, R. Marillier, B. Gomes and J. Stauber, J. Pharm. Biomed. Anal., 2019, 170, 220–227 CrossRef CAS.
- Ying Liu Yu Chen, Yan He Ximei Li, Ying Peng Weiwei Li and Jiang Zheng, Drug Metab. Dispos., 2023, 52, 001069 Search PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Anal. Chem., 2018, 90, 240–265 CrossRef CAS PubMed.
- X. Zhu, T. Xu, C. Peng and S. Wu, Front. Chem., 2022, 9, 782432 CrossRef PubMed.
- J. Leopold, Y. Popkova, K. M. Engel and J. Schiller, Biomolecules, 2018, 8, 173 CrossRef PubMed.
- E. Gemperline, S. Rawson and L. Li, Anal. Chem., 2014, 86, 10030–10035 CrossRef CAS.
- T. B. Angerer, J. Bour, J.-L. Biagi, E. Moskovets and G. Frache, J. Am. Soc. Mass Spectrom., 2022, 33, 760–771 CrossRef CAS PubMed.
- I. Kaya, D. Brinet, W. Michno, M. Başkurt, H. Zetterberg, K. Blenow and J. Hanrieder, ACS Chem. Neurosci., 2017, 8, 2778–2790 CrossRef CAS.
- F. Tobias, K. C. Pathmasiri and S. M. Cologna, Anal. Bioanal. Chem., 2019, 411, 5659–5668 CrossRef CAS.
- J. C. McMillen, J. A. Fincher, D. R. Klein, J. M. Spraggins and R. M. Caprioli, J. Mass Spectrom., 2020, 55, e4663 CrossRef CAS PubMed.
- I. Kaya, H. Zetterberg, K. Blennow and J. Hanrieder, ACS Chem. Neurosci., 2018, 9, 1802–1817 CrossRef CAS PubMed.
- K. Bednarczyk, M. Gawin, M. Chekan, A. Kurczyk, G. Mrukwa, M. Pietrowska, J. Polanska and P. Widlak, J. Mol. Histol., 2019, 50, 1–10 CrossRef PubMed.
- L. Kreutzer, P. Weber, T. Heider, M. Heikenwälder, T. Riedl, P. Baumeister, F. Klauschen, C. Belka, A. Walch, H. Zitzelsberger, J. Hess and K. Unger, Lab. Investig., 2022, 102, 1400–1405 CrossRef CAS.
- R. L. Vermillion-Salsbury and D. M. Hercules, Rapid Commun. Mass Spectrom., 2002, 16, 1575–1581 CrossRef CAS.
- A. Grgic, K. K. Krestensen and R. M. A. Heeren, Sci. Rep., 2023, 13, 2776 CrossRef CAS PubMed.
- R. R. Drake, C. A. West, A. S. Mehta and P. M. Angel, in Glycobiophysics, ed. Y. Yamaguchi and K. Kato, Springer Singapore, Singapore, 2018, pp. 59–76 Search PubMed.
- F. Zubair, P. E. Laibinis, W. G. Swisher, J. Yang, J. M. Spraggins, J. L. Norris and R. M. Caprioli, J. Mass Spectrom., 2016, 51, 1168–1179 CrossRef CAS PubMed.
- M. K. Andersen, T. S. Høiem, B. S. R. Claes, B. Balluff, M. Martin-Lorenzo, E. Richardsen, S. Krossa, H. Bertilsson, R. M. A. Heeren, M. B. Rye, G. F. Giskeødegård, T. F. Bathen and M.-B. Tessem, Cancer Metab., 2021, 9, 9 CrossRef PubMed.
- W. Li, L. Ren, X. Zheng, J. Liu, J. Wang, T. Ji and G. Du, Acta Pharm. Sin. B., 2020, 10, 301–312 CrossRef CAS PubMed.
- M. R. L. Paine, J. Liu, D. Huang, S. R. Ellis, D. Trede, J. H. Kobarg, R. M. A. Heeren, F. M. Fernández and T. J. MacDonald, Sci. Rep., 2019, 9, 2205 CrossRef.
- H. Yang, S. N. Jackson, A. S. Woods, D. R. Goodlett, R. K. Ernst and A. J. Scott, J. Am. Soc. Mass Spectrom., 2020, 31, 2495–2502 CrossRef CAS PubMed.
- R. Guran, L. Vanickova, V. Horak, S. Krizkova, P. Michalek, Z. Heger, O. Zitka and V. Adam, PLoS One, 2017, 12, e0189305 CrossRef.
- M. Andersson, M. R. Groseclose, A. Y. Deutch and R. M. Caprioli, Nat. Methods, 2008, 5, 101–108 CrossRef CAS PubMed.
- X.-L. Pei, X.-N. Liu, J.-L. Du, C. Gong and X. Xu, Int. J. Mass Spectrom., 2020, 455, 116373 CrossRef CAS.
- K. A. Zemski Berry, J. A. Hankin, R. M. Barkley, J. M. Spraggins, R. M. Caprioli and R. C. Murphy, Chem. Rev., 2011, 111, 6491–6512 CrossRef CAS PubMed.
- I. Jalaludin, H.-Q. Nguyen, K.-S. Jang, J. Lee, D. M. Lubman and J. Kim, Rapid Commun. Mass Spectrom., 2023, 37, e9427 CrossRef CAS PubMed.
- H. Choi, D. Lee, Y. Kim, H.Q. Nguyen, S. Han and J. Kim, J. Am. Soc. Mass Spectrom., 2019, 30, 1174–1178 CrossRef CAS PubMed.
- Y. Ucal and A. Ozpinar, J. Mass Spectrom., 2018, 53, 635–648 CrossRef CAS.
- H. Gu, K. Ma, W. Zhao, L. Qiu and W. Xu, Analyst, 2021, 146, 4080–4086 RSC.
- K. M. Park, Y. J. Bae, S. H. Ahn and M. S. Kim, Anal. Chem., 2012, 84, 10332–10337 CrossRef CAS PubMed.
- M. J. Lim, G. Yagnik, C. Henkel, S. F. Frost, T. Bien and K. J. Rothschild, Front. Chem., 2023, 11, 1182404 CrossRef CAS.
- J. Wang, J. Zhao, S. Nie, M. Xie and S. Li, Food Chem., 2023, 399, 133968 CrossRef CAS.
- D. J. Harvey, Mass Spectrom. Rev., 2009, 28, 273–361 CrossRef CAS PubMed.
- P. M. Angel, A. Mehta, K. Norris-Caneda and R. R. Drake, in Tissue Proteomics: Methods and Protocols, ed. M. M. Sarwal and T. K. Sigdel, SpringerNew York, New York, NY, 2018, pp. 225–241 Search PubMed.
- T. Nishikaze, H. Okumura, H. Jinmei and J. Amano, Mass Spectrom., 2012, 1, A0006 Search PubMed.
- N. Shanmugaraj, T. Rutten, A. Svatoš, T. Schnurbusch and H.-P. Mock, J. Am. Soc. Mass Spectrom., 2023, 34, 513–517 CrossRef CAS.
- R. C. Murphy, J. A. Hankin, R. M. Barkley and K. A. Zemski Berry, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2011, 1811, 970–975 CrossRef CAS PubMed.
- C. Tressler, S. Tilley, E. Yang, C. Donohue, E. Barton, A. Creissen and K. Glunde, J. Am. Soc. Mass Spectrom., 2021, 32, 2728–2737 CrossRef CAS.
- K. Ščupáková, B. Balluff, C. Tressler, T. Adelaja, R. M. A. Heeren, K. Glunde and G. Ertaylan, Clin. Chem. Lab. Med., 2020, 58, 914–929 CrossRef PubMed.
- M. B. Ginzberg, R. Kafri and M. Kirschner, Science, 2015, 348, 1245075 CrossRef.
- H. Brandt, T. Ehmann and M. Otto, Rapid Commun. Mass Spectrom., 2010, 24, 2439–2444 CrossRef CAS PubMed.
- H. Hazama, S. Furukawa and K. Awazu, Chem. Phys., 2013, 419, 196–199 CrossRef CAS.
- D. M. G. Anderson, K. A. Floyd, S. Barnes, J. M. Clark, J. I. Clark, H. Mchaourab and K. L. Schey, Anal. Bioanal. Chem., 2015, 407, 2311–2320 CrossRef CAS PubMed.
- G. Sun, K. Yang, Z. Zhao, S. Guan, X. Han and R. W. Gross, Anal. Chem., 2008, 80, 7576–7585 CrossRef CAS.
- S. Shimma and Y. Sugiura, Mass Spectrom., 2014, 3, S0029 CrossRef.
- S. N. Nguyen, J. E. Kyle, S. E. Dautel, R. Sontag, T. Luders, R. Corley, C. Ansong, J. Carson and J. Laskin, Anal. Chem., 2019, 91, 11629–11635 CrossRef CAS PubMed.
- J. Laskin and I. Lanekoff, Anal. Chem., 2016, 88, 52–73 CrossRef CAS PubMed.
- M. W. Towers, T. Karancsi, E. A. Jones, S. D. Pringle and E. Claude, J. Am. Soc. Mass Spectrom., 2018, 29, 2456–2466 CrossRef CAS PubMed.
- L. Mavroudakis and I. Lanekoff, in Imaging Mass Spectrometry: Methods and Protocols, ed. L. M. Cole and M. R. Clench, Springer US, New York, NY, 2023, pp. 107–121 Search PubMed.
- I. Lanekoff, S. L. Stevens, M. P. Stenzel-Poore and J. Laskin, Analyst, 2014, 139, 3528–3532 RSC.
- N. Chantipmanee and Y. Xu, View, 2023, 4, 20220036 CrossRef CAS.
- Z. Yang, B. Li, D. D. Stuart and Q. Cheng, View, 2023, 4, 20220041 CrossRef CAS.
- J. Chen, M. Tang and D. Xu, J. Chromatogr. Open, 2021, 1, 100021 CrossRef.
- S.-O. Deininger, D. S. Cornett, R. Paape, M. Becker, C. Pineau, S. Rauser, A. Walch and E. Wolski, Anal. Bioanal. Chem., 2011, 401, 167–181 CrossRef CAS.
- A. Tu and D. C. Muddiman, Anal. Bioanal. Chem., 2019, 411, 5729–5743 CrossRef CAS PubMed.
- T. Porta, A. Lesur, E. Varesio and G. Hopfgartner, Anal. Bioanal. Chem., 2015, 407, 2177–2187 CrossRef CAS PubMed.
- D. Sun, W. Gao, H. Hu and S. Zhou, Acta Pharm. Sin. B., 2022, 12, 3049–3062 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS.
- S. Guo, K. Li, Y. Chen and B. Li, Acta Pharm. Sin. B., 2022, 12, 2120–2126 CrossRef CAS.
- A. Dannhorn, M. L. Doria, J. McKenzie, P. Inglese, J. G. Swales, G. Hamm, N. Strittmatter, G. Maglennon, S. Ghaem-Maghami, R. J. A. Goodwin and Z. Takats, Metabolites, 2023, 13, 377 CrossRef CAS PubMed.
- H. Li and A. B. Hummon, Anal. Chem., 2011, 83, 8794–8801 CrossRef CAS.
- F. Tobias and A. B. Hummon, J. Mass Spectrom., 2022, 57, e4880 CrossRef CAS PubMed.
- Y. Wang and A. B. Hummon, J. Biol. Chem., 2021, 297, 101139 CrossRef.
- J. J. Han, Artif. Organs, 2023, 47, 449–450 CrossRef.
- R. L. Shortt, Y. Wang, A. B. Hummon and L. M. Jones, J. Am. Soc. Mass Spectrom., 2023, 34, 417–425 CrossRef CAS PubMed.
- M. Machálková, B. Pavlatovská, J. Michálek, A. Pruška, K. Štěpka, T. Nečasová, K. A. Radaszkiewicz, M. Kozubek, J. Šmarda, J. Preisler and J. Navrátilová, Anal. Chem., 2019, 91, 13475–13484 CrossRef.
- X. Liu, C. Flinders, S. M. Mumenthaler and A. B. Hummon, J. Am. Soc. Mass Spectrom., 2018, 29, 516–526 CrossRef CAS.
- G. J. LaBonia, S. Y. Lockwood, A. A. Heller, D. M. Spence and A. B. Hummon, Proteomics, 2016, 16, 1814–1821 CrossRef CAS.
- M. M. Schroll, K. R. Ludwig, G. J. LaBonia, E. L. Herring and A. B. Hummon, J. Am. Soc. Mass Spectrom., 2018, 29, 2012–2022 CrossRef CAS.
- J. K. Lukowski, E. M. Weaver and A. B. Hummon, Anal. Chem., 2017, 89, 8453–8458 CrossRef CAS PubMed.
- Y. Wang and A. B. Hummon, Anal. Chem., 2023, 95, 9227–9236 CrossRef CAS PubMed.
- A. Islam, T. Sakamoto, Q. Zhai, Md. M. Rahman, Md. A. Mamun, Y. Takahashi, T. Kahyo and M. Setou, Pharm., 2022, 15, 1314 CAS.
- A. Traberg, F. E. Pinto, A. C. N. Hansen, M. Haedersdal, C. M. Lerche and C. Janfelt, Pharm., 2022, 15, 1583 CAS.
- N. B. Holm, M. Deryabina, C. B. Knudsen and C. Janfelt, Anal. Bioanal. Chem., 2022, 414, 7167–7177 CrossRef CAS PubMed.
- D. Mesa Sanchez, H. M. Brown, R. Yin, B. Chen, M. Vavrek, M. T. Cancilla, W. Zhong, B. Shyong, N. R. Zhang, F. Li and J. Laskin, Anal. Chim. Acta, 2022, 1233, 340490 CrossRef CAS.
- X. Liu and A. B. Hummon, Sci. Rep., 2016, 6, 38507 CrossRef CAS.
- X. Liu, J. K. Lukowski, C. Flinders, S. Kim, R. A. Georgiadis, S. M. Mumenthaler and A. B. Hummon, Anal. Chem., 2018, 90, 14156–14164 CrossRef CAS PubMed.
- P. G. Dougherty, J. H. Wellmerling, A. Koley, J. K. Lukowski, A. B. Hummon, E. Cormet-Boyaka and D. Pei, J. Med. Chem., 2020, 63, 15773–15784 CrossRef CAS.
- H. Sunshine and M. L. Iruela-Arispe, Curr. Opin. Lipidol., 2017, 28, 408–413 CrossRef CAS.
- R. Guo, Y. Chen, H. Borgard, M. Jijiwa, M. Nasu, M. He and Y. Deng, Molecules, 2020, 25, 4864 CrossRef CAS PubMed.
- A. Bhaduri, E. K. Neumann, A. R. Kriegstein and J. V. Sweedler, JACS Au, 2021, 1, 2261–2270 CrossRef CAS PubMed.
- D. Hishikawa, T. Hashidate, T. Shimizu and H. Shindou, J. Lipid Res., 2014, 55, 799–807 CrossRef CAS.
- V. Natesan and S.-J. Kim, Biomol. Ther., 2021, 29, 596–604 CrossRef CAS.
- Q. Huang, X. Wu, Y. Gu, T. Wang, Y. Zhan, J. Chen, Z. Zeng, Y. Lv, J. Zhao and J. Xie, Front. Mol. Biosci., 2022, 9, 839259 CrossRef CAS PubMed.
- B. Paul, M. Lewinska and J. B. Andersen, JHEP Rep., 2022, 4, 100479 CrossRef PubMed.
- K. C. Pathmasiri, T. T. A. Nguyen, N. Khamidova and S. M. Cologna, in Current Topics in Membranes, ed. M. A. Model and I. Levitan, Academic Press, 2021, vol. 88, pp. 315–357 Search PubMed.
- K. Zhanghao, W. Liu, M. Li, Z. Wu, X. Wang, X. Chen, C. Shan, H. Wang, X. Chen, Q. Dai, P. Xi and D. Jin, Nat. Commun., 2020, 11, 5890 CrossRef CAS PubMed.
- Y. Hu, R.-Q. Zhang, S.-L. Liu and Z.-G. Wang, Biosens. Bioelectron., 2023, 240, 115649 CrossRef CAS PubMed.
- K. M. Engel, P. Prabutzki, J. Leopold, A. Nimptsch, K. Lemmnitzer, D. R. N. Vos, C. Hopf and J. Schiller, Prog. Lipid Res., 2022, 86, 101145 CrossRef CAS.
- F. Tobias, M. T. Olson and S. M. Cologna, J. Lipid Res., 2018, 59, 2446–2455 CrossRef CAS.
- H. J. Kwon, J. Y. Oh, K. S. Lee, H. K. Lim, J. Lee, H.-R. Yoon and J. Jung, Int. J. Anal. Chem., 2022, 2022, 6007158 Search PubMed.
- J. A. Fincher, D. R. Jones, A. R. Korte, J. E. Dyer, P. Parlanti, A. Popratiloff, C. A. Brantner, N. J. Morris, R. K. Pirlo, V. K. Shanmugam and A. Vertes, Sci. Rep., 2019, 9, 17508 CrossRef PubMed.
- R. Tans, S. Dey, N. S. Dey, J.-H. Cao, P. S. Paul, G. Calder, P. O’Toole, P. M. Kaye and R. M. A. Heeren, Front. Immunol., 2022, 13, 862104 CrossRef CAS PubMed.
- L. Martín-Saiz, B. Abad-García, J. D. Solano-Iturri, L. Mosteiro, J. Martín-Allende, Y. Rueda, A. Pérez-Fernández, M. Unda, P. Coterón-Ochoa, A. Goya, A. Saiz, J. Martínez, B. Ochoa, O. Fresnedo, G. Larrinaga and J. A. Fernández, Anal. Chem., 2023, 95, 2285–2293 CrossRef.
- L. Jadoul, R. Longuespée, A. Noël and E. De Pauw, Anal. Bioanal. Chem., 2015, 407, 2095–2106 CrossRef CAS.
- R. Angelini, E. Yutuc, M. F. Wyatt, J. Newton, F. A. Yusuf, L. Griffiths, B. J. Cooze, D. El Assad, G. Frache, W. Rao, L. B. Allen, Z. Korade, T. T. A. Nguyen, R. A. C. Rathnayake, S. M. Cologna, O. W. Howell, M. R. Clench, Y. Wang and W. J. Griffiths, Anal. Chem., 2021, 93, 4932–4943 CrossRef CAS PubMed.
- M. Vandenbosch, S. M. Mutuku, M. J. Q. Mantas, N. H. Patterson, T. Hallmark, M. Claesen, R. M. A. Heeren, N. G. Hatcher, N. Verbeeck, K. Ekroos and S. R. Ellis, Anal. Chem., 2023, 95, 18719–18730 CrossRef CAS PubMed.
- C. Beermann, M. Möbius, N. Winterling, J. J. Schmitt and G. Boehm, Lipids, 2005, 40, 211–218 CrossRef CAS PubMed.
- E. Sokoła-Wysoczańska, T. Wysoczański, J. Wagner, K. Czyż, R. Bodkowski, S. Lochyński and B. Patkowska-Sokoła, Nutrients, 2018, 10, 1561 CrossRef.
- N. Cetraro, R. B. Cody and J. Y. Yew, Analyst, 2019, 144, 5848–5855 RSC.
- M.-E. N. Born and B. M. Prentice, Int. J. Mass Spectrom., 2020, 452, 116338 CrossRef CAS.
- T. Yan, M.-E. N. Born and B. M. Prentice, Int. J. Mass Spectrom., 2023, 485, 116998 CrossRef CAS PubMed.
- B. S. R. Claes, A. P. Bowman, B. L. J. Poad, R. S. E. Young, R. M. A. Heeren, S. J. Blanksby and S. R. Ellis, Anal. Chem., 2021, 93, 9826–9834 CrossRef CAS.
- S. Takashima, K. Toyoshi, T. Yamamoto and N. Shimozawa, Sci. Rep., 2020, 10, 12988 CrossRef CAS.
- R. T. Steven and J. Bunch, Anal. Bioanal. Chem., 2013, 405, 4719–4728 CrossRef CAS PubMed.
- L. S. Eberlin, C. R. Ferreira, A. L. Dill, D. R. Ifa and R. G. Cooks, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1811, 2011, 946–960 Search PubMed.
- S. Mondal, Y. Sthanikam, A. Kumar, A. Nandy, S. Chattopadhyay, D. Koner, N. Rukmangadha, H. Narendra and S. Banerjee, Anal. Chem., 2023, 95, 8054–8062 CrossRef CAS.
- R. Yin, K. E. Burnum-Johnson, X. Sun, S. K. Dey and J. Laskin, Nat. Protoc., 2019, 14, 3445–3470 CrossRef CAS PubMed.
- L. Mavroudakis, K. D. Duncan and I. Lanekoff, Anal. Chem., 2022, 94, 2391–2398 CrossRef CAS PubMed.
- T. Mitsis, A. Efthimiadou, F. Bacopoulou, D. Vlachakis, G. Chrousos and E. Eliopoulos, World Acad. Sci. J., 2020, 2, 3–8 Search PubMed.
- R. Morris, K. A. Black and E. J. Stollar, Essays Biochem., 2022, 66, 255–285 CrossRef CAS.
- C. Sotomayor-Vivas, E. Hernández-Lemus and R. Dorantes-Gilardi, PLoS One, 2022, 17, e0261829 CrossRef CAS PubMed.
- V. M. Prabantu, V. Gadiyaram, S. Vishveshwara and N. Srinivasan, Curr. Res. Struct. Biol., 2022, 4, 134–145 CrossRef CAS.
- U. Baul, D. Chakraborty, M. L. Mugnai, J. E. Straub and D. Thirumalai, J. Phys. Chem. B, 2019, 123, 3462–3474 CrossRef CAS PubMed.
- S. A. Rather, F. A. Masoodi, J. A. Rather, T. A. Ganaie, R. Akhter and S. M. Wani, in Food biopolymers: structural, functional and nutraceutical properties, ed. A. Gani and B. A. Ashwar, Springer International Publishing, Cham, 2021, pp. 299–318 Search PubMed.
- R. Dolcemascolo, L. Goiriz, R. Montagud-Martínez and G. Rodrigo, PLoS Comput. Biol., 2022, 18, e1010087 CrossRef CAS.
- H. Lagass, A. Alexaki, V. Simhadri, N. Katagiri, W. Jankowski, Z. Sauna and C. Kimchi-Sarfaty, F1000Res., 2017, 6, 113 Search PubMed.
- M. Winter, A. Tholey, A. Kristen and C. Röcken, Proteomics, 2017, 17, 1700236 CrossRef PubMed.
- G. Guo, M. Papanicolaou, N. J. Demarais, Z. Wang, K. L. Schey, P. Timpson, T. R. Cox and A. C. Grey, Nat. Commun., 2021, 12, 3241 CrossRef CAS PubMed.
- G. Fiorentino, A. Smith, G. Nicora, R. Bellazzi, F. Magni, S. Garagna and M. Zuccotti, Mol. Hum. Reprod., 2023, 29, gaad006 CrossRef CAS.
- C. Chen, J. Hou, J. J. Tanner and J. Cheng, Int. J. Mol. Sci., 2020, 21, 2873 CrossRef CAS PubMed.
- T. Boström, Nat. Methods, 2016, 13, iv–vi CrossRef.
- F. Calderón-Celis, J. R. Encinar and A. Sanz-Medel, Mass Spectrom. Rev., 2018, 37, 715–737 CrossRef PubMed.
- E. J. Clemis, D. S. Smith, A. G. Camenzind, R. M. Danell, C. E. Parker and C. H. Borchers, Anal. Chem., 2012, 84, 3514–3522 CrossRef CAS PubMed.
- F. Dewez, E. De Pauw, R. M. A. Heeren and B. Balluff, Anal. Chem., 2021, 93, 1393–1400 CrossRef CAS PubMed.
- B. S. R. Claes, K. K. Krestensen, G. Yagnik, A. Grgic, C. Kuik, M. J. Lim, K. J. Rothschild, M. Vandenbosch and R. M. A. Heeren, Anal. Chem., 2023, 95, 2329–2338 CrossRef CAS PubMed.
- G. Yagnik, Z. Liu, K. J. Rothschild and M. J. Lim, J. Am. Soc. Mass Spectrom., 2021, 32, 977–988 CrossRef CAS PubMed.
- Y. Wang, K. Zhang, X. Huang, L. Qiao and B. Liu, Anal. Chem., 2021, 93, 709–714 CrossRef CAS PubMed.
- J. Havlikova, E. C. Randall, R. L. Griffiths, J. G. Swales, R. J. A. Goodwin, J. Bunch, I. B. Styles and H. J. Cooper, Anal. Chem., 2019, 91, 14198–14202 CrossRef CAS PubMed.
- O. J. Hale and H. J. Cooper, Anal. Chem., 2021, 93, 4619–4627 CrossRef CAS.
- E. Illes-Toth, O. J. Hale, J. W. Hughes, N. Strittmatter, J. Rose, B. Clayton, R. Sargeant, S. Jones, A. Dannhorn, R. J. A. Goodwin and H. J. Cooper, Angew. Chem., Int. Ed., 2022, 61, e202202075 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |