
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing Cu–ligand interaction for efficient CO2 reduction towards multi-carbon products†
Jingyi
Chen‡
a,
Lei
Fan‡
a,
Yilin
Zhao
 a,
Haozhou
Yang
a,
Di
Wang
a,
Bihao
Hu
a,
Shibo
Xi
b and
Lei
Wang
*ac
a,
Haozhou
Yang
a,
Di
Wang
a,
Bihao
Hu
a,
Shibo
Xi
b and
Lei
Wang
*ac
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore. E-mail: wanglei8@nus.edu.sg
bInstitute of Sustainability for Chemicals, Energy and Environment, A*STAR, 1 Pesek Road, Jurong Island, 627833, Singapore
cCentre for Hydrogen Innovations, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore
First published on 20th February 2024
Abstract
Electrochemical CO2 reduction (CO2R) to valuable products provides a promising strategy to enable CO2 utilization sustainably. Here, we report the strategy of using Cu-DAT (3,5-diamino-1,2,4-triazole) as a catalyst precursor for efficient CO2 reduction, demonstrating over 80% selectivity towards multicarbon products at 400 mA cm−2, with intrinsic activity over 19 times higher than that of Cu nanoparticles. The catalyst’s active phase is determined to be metallic copper wrapped with the DAT ligand. We attribute this enhanced CO2R performance to the accelerated steps of *CO adsorption and C–C coupling induced by the closely cooperated DAT ligand.
Electrochemical CO2 reduction (CO2R) has gained increasing attention as a promising approach to tackle the challenges of CO2 emissions, owing to its ability of converting CO2 into value-added chemicals when powered by renewable electricity.1,2 Among numerous metal-based electrocatalysts,3 Cu-based materials possess moderate intermediate CO binding strength, favouring the formation of multi-carbon (C2+) products (i.e., ethylene, ethanol).4,5 Several strategies have been employed to further promote the formation of C2+ products, such as organic modification,6–9 alloying10,11 and introducing a dopant,12,13etc. Recently, gas diffusion electrode (GDE)14,15 based CO2R has largely alleviated the severe CO2 diffusion limitations in conventional H-cells, enabling CO2R at an industrially relevant current density.16–19 However, a highly alkaline environment is typically necessary for achieving high selectivity towards C2+ products in GDE reactors.20 Consequently, severe CO2 loss, carbonate salt formation and catalyst structural damage can occur, leading to impeded CO2 diffusion and deteriorated activity.21–23 In contrast, neutral electrolytes (i.e., KHCO3, KCl) can preserve the catalyst integrity and mitigate salt precipitation problems, allowing stable operation of CO2R.24 Herein, we demonstrate the optimization of the microenvironment for CO2R in neutral electrolytes through the introduction of an amino group-rich ligand, resulting in substantially enhanced activity towards C2+ products.
We first conducted CO2R measurements based on the physical mixture of commercial copper nanoparticles (CuNPs) and 3,5-diamino-1,2,4-triazole (DAT), a ligand rich in amino groups, in a typical flow cell in 1 M KHCO3 (Fig. 1a). As shown in Fig. 1b and Fig. S1 (ESI†), the C2+ products selectivity increased significantly (i.e., from <47% to >65%) as the DAT to CuNP ratio increases, indicating the profound promotion effect attributed to the DAT ligand. Notably, the overpotential at 300 mA cm−2 also decreased by as much as ∼100 mV after the introduction of DAT, indicating the enhanced intrinsic activity towards C2+ products (all electrode potentials mentioned here are vs. RHE, unless otherwise defined). Energy-dispersive X-ray spectroscopy (EDS) (Fig. S2, ESI†) suggests the even dispersion of N and Cu elements after CO2R, indicating that the ligand effect is originated from the intimate contact between the DAT ligand and the Cu active-sites. However, it is obvious that the interface between the DAT ligand and Cu particles is not optimized (Fig. S3, ESI†). Hence, to further promote the CO2R performance, particularly the activity and selectivity towards C2+ products, we speculate that it would be beneficial to employ the Cu(II)-DAT-based complex as a catalyst precursor for CO2R, so that maximized contact between Cu active-sites and the DAT ligand can be achieved.
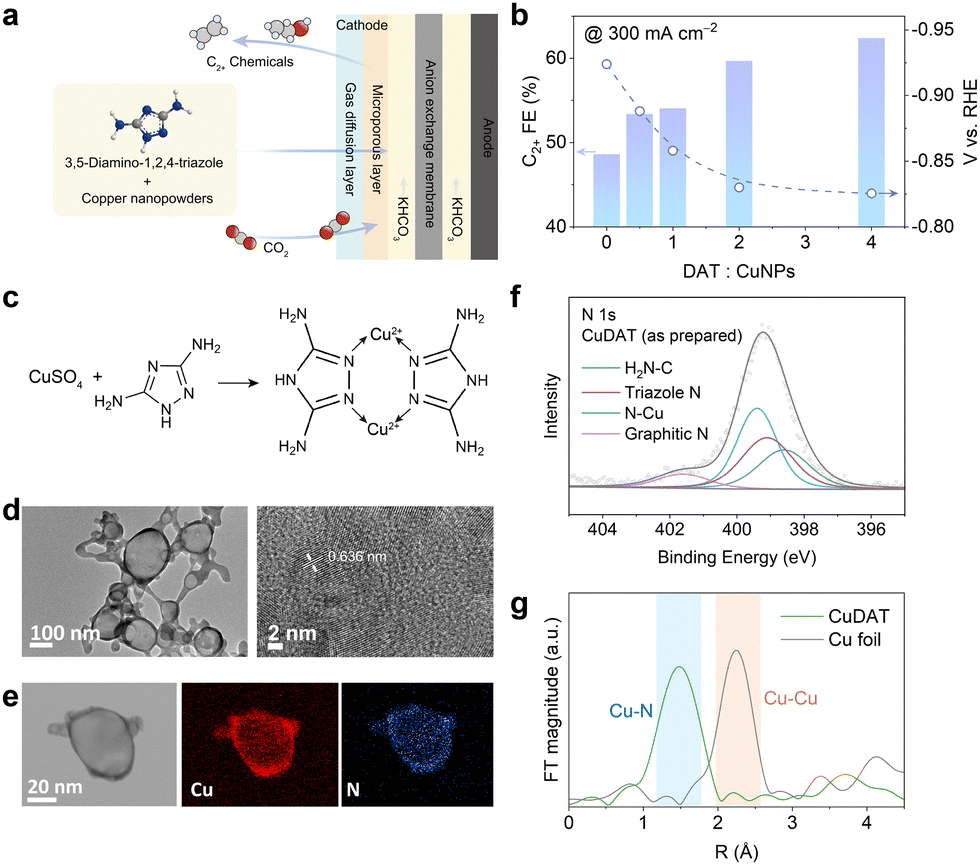 | ||
| Fig. 1 (a) Schematic of CO2R in a flow cell using a physical mixture of CuNPs and DAT ligand. (b) C2+ selectivity and cathodic potential at 300 mA cm−2 with different ratios of DAT to CuNPs. Increasing ligand content leads to enhanced C2+ selectivity and reduced overpotential. (c) Synthetic approach and chemical structure of CuDAT. (d) TEM images, (e) EDS mapping and (f) N 1s XPS of the as-synthesized CuDAT. (g) Cu K edge FT-EXAFS of as-prepared CuDAT and Cu foil. | ||
We then prepared an organometallic complex using Cu2+ (CuSO4) as the metal center and DAT as the chelating ligand based on a modified procedure,25 and the obtained complex is denoted as CuDAT (Fig. 1c).26,27 Transmission electron microscopy (TEM) presents the morphology of the CuDAT particles (Fig. 1d), with a diameter of ∼100 nm and lattice spacing of ∼0.636 nm. The crystallinity is further elucidated by XRD (Fig. S4, ESI†). Besides, the EDS mapping (Fig. 1e) confirms the uniform dispersion of Cu and N. X-ray photoelectron spectroscopy (XPS) suggests that the Cu centers remain as Cu2+, with Cu 2p1/2 and 2p3/2 located at 953.6 eV and 934.6 eV, respectively (Fig. S5, ESI†).28,29 Additionally, N species from the DAT ligand (–NH2 and the triazole heterocycle) and the Cu–N bond can be clearly deconvoluted (Fig. 1f), in agreement with the proposed structure.30,31 Moreover, Fourier transformation of the extended X-ray absorption fine structure (FT-EXAFS) consolidates the Cu–N bond around R = 1.5 Å (Fig. 1g),32 suggesting the coordination between Cu2+ and DAT. The complex assembly is also manifested by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) (Fig. S6, ESI†), exhibiting the characteristic peaks of –NH2 stretching at 3400 cm−1, –NH stretching at 3300 cm−1, –NH2 bending at 1640 cm−1 and C–NH2 stretching at 1050 cm−1, in line with the characteristic signals of the DAT ligand.33
The CO2R performance of CuDAT was assessed in a flow cell (Fig. 2a). Notably, CuDAT exhibited high faradaic efficiency (FE) of >80% towards C2+ products at 400 mA cm−2, which is comparable, if not higher, compared to the state-of-the-art CO2R electrocatalysts under neutral conditions.34 In contrast, CuNPs only delivered <60% FE (C2+) under the same conditions (Fig. S7, ESI†). Additionally, CuDAT exhibited much lower overpotential compared to CuNPs at the same C2+ current density (Fig. 2b). To assess the intrinsic activity of CuDAT, we estimated the electrochemical active surface area (ECSA) of both CuDAT and CuNPs (Fig. S8, ESI†).35 Strikingly, at a modest overpotential of −0.86 V vs. RHE, the ECSA-normalized jC2+ of CuDAT is over 19 times higher than that of CuNPs (Fig. S9, ESI†). Moreover, CuDAT also demonstrated promising CO2R stability at 200 mA cm−2 (Fig. S10, ESI†). Overall, we demonstrate that applying an organometallic complex derived catalyst could be an effective strategy in enhancing the intrinsic activity and selectivity of Cu for C2+ products.
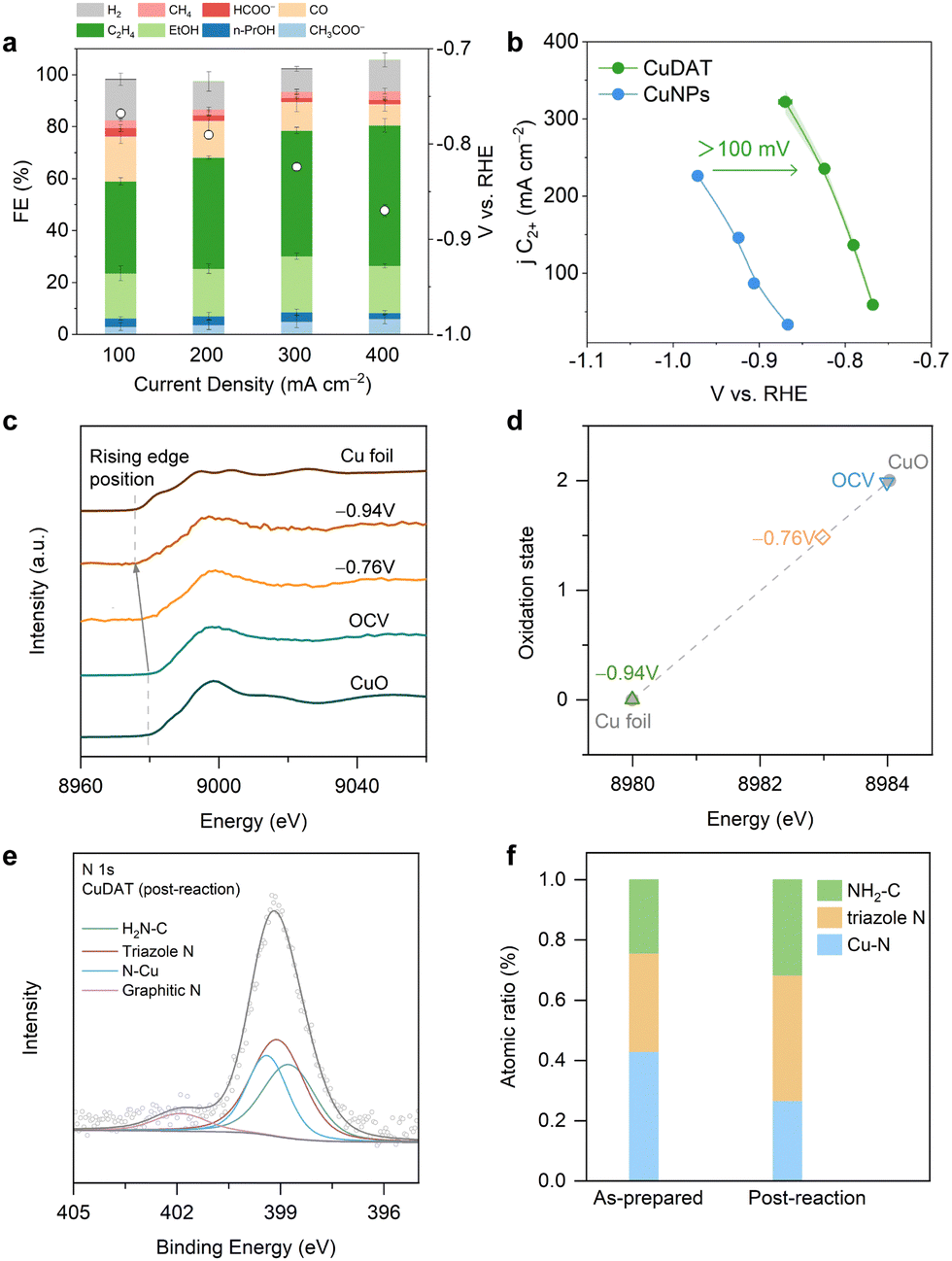 | ||
| Fig. 2 (a) CO2R performance on CuDAT in 1 M KHCO3. (b) jC2+ on the function of the overpotential for CuDAT and CuNPs. (c) Operando Cu K-edge XANES for CuDAT during CO2R in a flow cell. (d) Oxidation state, deduced from the rising edge energy of Cu K edge XANES in CuDAT during CO2R at different overpotentials. (e) N 1s XPS of the post-CO2R CuDAT and (f) atomic percentage of N species for as prepared and post-reaction CuDAT. Triazole N and amino N were largely reserved. | ||
To explore the changes in the chemical state of Cu in CuDAT during CO2R, we performed operando X-ray absorption fine structure (XAFS) experiments using the same flow cell. As shown in Fig. 2c, the rising edge energy of the Cu K-edge X-ray absorption near edge structure (XANES) decreases gradually from open circuit voltage (OCV) to −0.94 V, reflecting the transition of oxidized Cu to its metallic state (Fig. 2d). This is further evidenced by the SEM and TEM images (Fig. S11, ESI†), in which drastic morphological changes occurred after CO2R. Crucially, after CO2R, the Cu (111) lattice is identified and forms aggregated particles ∼5 nm (Fig. S12, ESI†). Additionally, the post-reaction CuDAT sample exhibits a discernible Cu (111) peak on the XRD spectrum, providing further confirmation of the transition from Cu2+ to metallic Cu (Fig. S13, ESI†). The increased peak width is indicative of the small size of the Cu nanoparticles based on the Scherrer equation.36 In comparison with the XRD pattern of as-prepared CuDAT, the original crystallinity is disrupted, underscoring the structural reconstruction that occurs during CO2R (Fig. S14, ESI†). Additionally, cross-section EDS mapping reveals the uniform dispersion of N and Cu elements, indicating that N-containing ligands are retained (Fig. S15, ESI†). Moreover, we found the existence of triazole N at 399.1 eV, amino N at 398.6 eV and N coordinated with Cu at 399.4 eV (Fig. 2e and f). Furthermore, FTIR spectra exhibit characteristic peaks of –NH2 stretching at 3400 cm−1, –NH2 bending at 1640 cm−1, and C–NH2 stretching at 1050 cm−1 (Fig. S16, ESI†).37 Taken together, we conclude that during CO2R, the Cu centers were reduced and formed small Cu clusters/particles, while the DAT ligand remained intact and maintained intimate interaction with the Cu active sites.
In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was carried out to probe the reaction intermediates (Fig. 3a, Fig. S17, ESI†). Absorption bands located at ∼2050 cm−1 were found corresponding to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching mode of *COL (linearly bonded CO).38 After emerging at −0.5 V, the band intensity quickly decreases from −0.6 V to −1.1 V, indicating the rapid consumption of the *CO intermediate due to the C–C coupling towards C2+ products. Moreover, the *CO band position undergoes blue-shift first followed by red-shift along with the increase of overpotential,39 which is caused by both the changes in *CO coverage and the Stark shift effect.40,41 Besides, the *COL absorbance on CuNPs increases gradually from −0.5 V to −0.8 V (Fig. 3b). In contrast, *COL on CuDAT shows a sharp increase and quickly reaches its maximum at −0.65 V, indicating the facilitated *CO adsorption on CuDAT compared to that of CuNPs. Passing the *COL absorbance maximum, CuDAT exhibits a more rapid decrease of the *CO band intensity than CuNPs, suggesting the accelerated rate for producing multi-carbon products.42,43 Moreover, the *CO band on CuDAT appears at lower wavenumber, consolidating its favoured *CO adsorption (Fig. 3c).44
O stretching mode of *COL (linearly bonded CO).38 After emerging at −0.5 V, the band intensity quickly decreases from −0.6 V to −1.1 V, indicating the rapid consumption of the *CO intermediate due to the C–C coupling towards C2+ products. Moreover, the *CO band position undergoes blue-shift first followed by red-shift along with the increase of overpotential,39 which is caused by both the changes in *CO coverage and the Stark shift effect.40,41 Besides, the *COL absorbance on CuNPs increases gradually from −0.5 V to −0.8 V (Fig. 3b). In contrast, *COL on CuDAT shows a sharp increase and quickly reaches its maximum at −0.65 V, indicating the facilitated *CO adsorption on CuDAT compared to that of CuNPs. Passing the *COL absorbance maximum, CuDAT exhibits a more rapid decrease of the *CO band intensity than CuNPs, suggesting the accelerated rate for producing multi-carbon products.42,43 Moreover, the *CO band on CuDAT appears at lower wavenumber, consolidating its favoured *CO adsorption (Fig. 3c).44
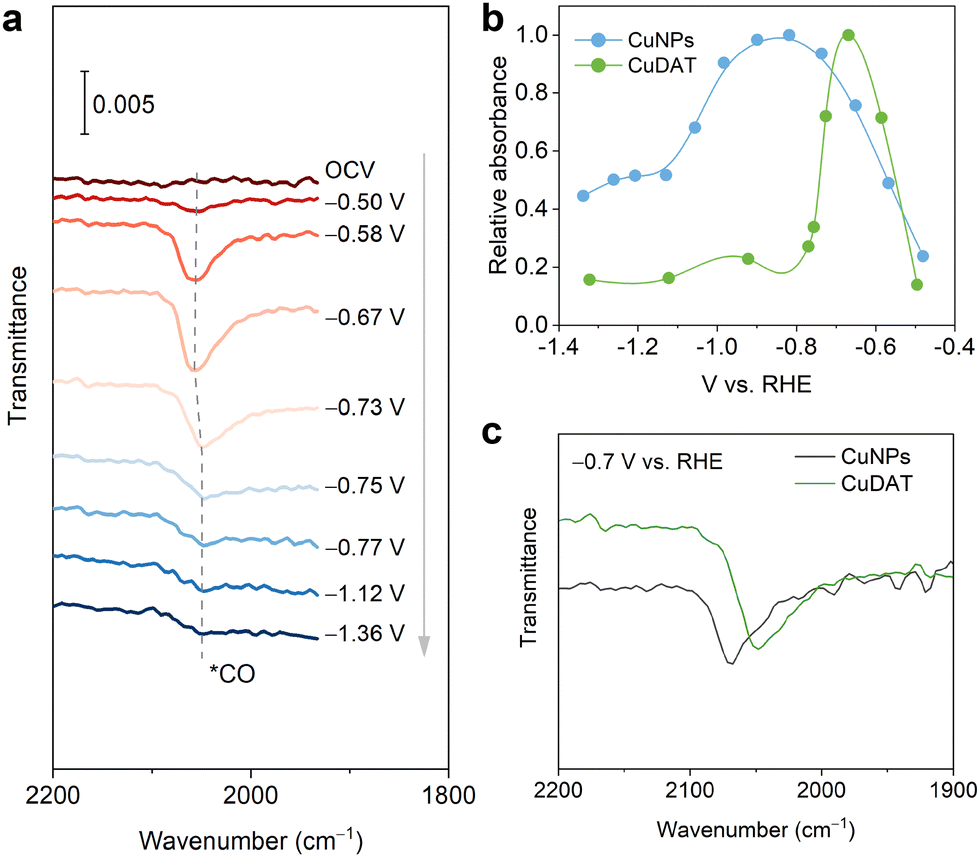 | ||
| Fig. 3 (a) In situ ATR-SEIRAS of CuDAT. (b) Relative absorbance of the *COL band for CuDAT and CuNPs. (c) *COL band in in situ ATR-SEIRAS at −0.7 V vs. RHE. | ||
The preferred *CO adsorption is further confirmed through CO stripping experiments (Fig. S18, ESI†). In comparison to CuNPs, CuDAT displays a CO stripping peak at more positive overpotential, denoting a stronger *CO adsorption on the Cu sites. To underscore the influence of the DAT ligand, we studied in situ ATR-SEIRAS on the physical mixture of CuNPs and DAT ligand as the catalyst composite during CO2R (Fig. S19a, ESI†). Upon the introduction of the DAT ligand, the *COL absorbance band undergoes more rapid emergence and decrease than that of CuNPs as the overpotential increases. This implies the acceleration of both *COL adsorption and C–C coupling (Fig. S19b, ESI†). Additionally, the *COL absorbance band undergoes a redshift with the ligand addition at −0.7 V, further confirming the favoured *CO adsorption after the introduction of the DAT ligand (Fig. S19c, ESI†).
To conclude, we attribute the enhanced C2+ activity of CuDAT to the optimized binding strength of surface *CO, achieved through intimate Cu/ligand contact. This underscores the effectiveness of establishing such close interactions to maximize the promotion effect.
It is worthwhile to develop cost-effective approaches that can directly convert diluted CO2 streams into valuable chemicals and fuels.45,46 However, significant concentration overpotential is anticipated when using a low-concentration CO2 stream, especially at high current densities. Nevertheless, we evaluated CuDAT for CO2R using low-concentration CO2 streams (Fig. 4a, b and Fig. S20, ESI†), as amino-functionalized materials have demonstrated prominent capabilities for local CO2 enrichment.47 At 300 mA cm−2, it is encouraging to observe that CuDAT exhibits both higher CO2R selectivity and significantly reduced overpotential compared to CuNPs across all CO2 partial pressures (pCO2). Specifically, CuDAT maintains high CO2R selectivity (>80%) at 0.2 atm of CO2 (Fig. 4c). Additionally, compared with CuNPs, CuDAT shows reduced overpotential at 300 mA cm−2 by >30 mV at each pCO2 (Fig. S21, ESI†). Moreover, CuDAT maintains high C2+ selectivity (over 70%) pCO2 of 0.3 atm (Fig. 4d), significantly higher than those of CuNPs. These stark contrasts underscore the notable CO2 enrichment capability of CuDAT. It is worth noting that the C2+ formation during CO2R initially increased and then began to decrease at pCO2 around 0.4 atm for both catalysts (Fig. S22, ESI†). We attribute this phenomenon to potential-dependent changes in selectivity,48 as the higher overpotentials (increased concentration overpotential) are favoured for C2+ formation.
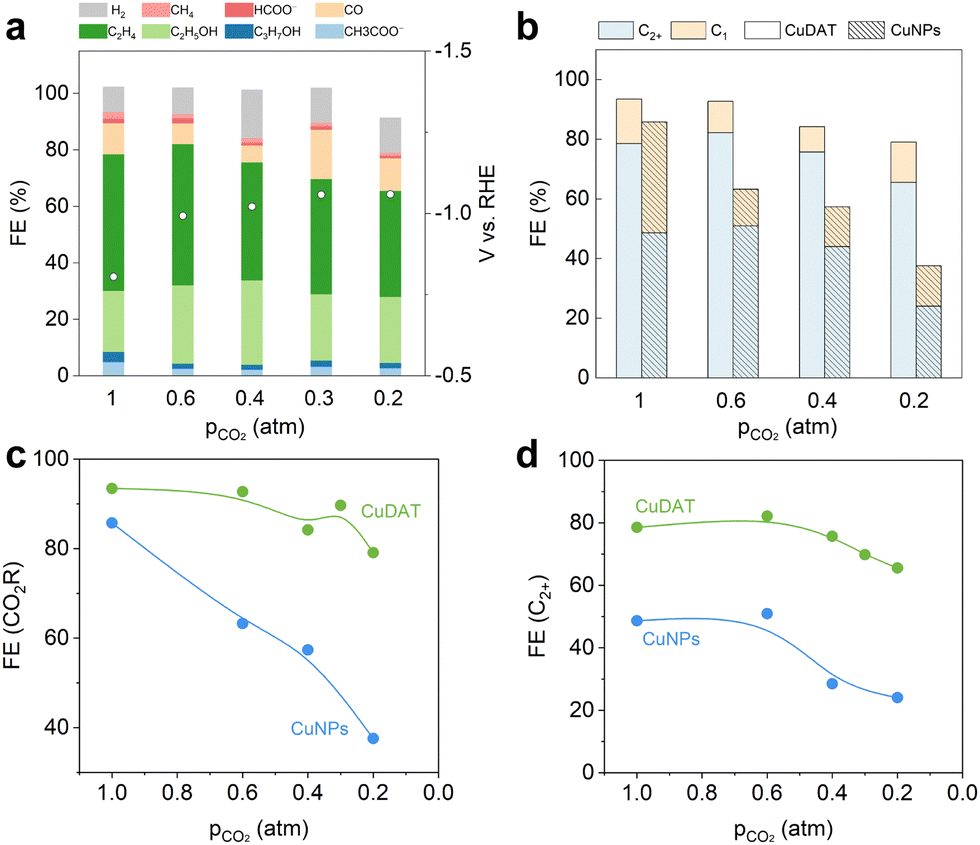 | ||
| Fig. 4 (a) CO2R on CuDAT with different CO2 concentrations at 300 mA cm−2. (b) CO2R performance comparison between CuDAT and CuNPs. (c) CO2R selectivity and (d) FE(C2+) of CuDAT and CuNPs at different CO2 partial pressures. | ||
To summarize, we present a strategy to enhance CO2R towards C2+ products by constructing catalyst precursors through complexation of Cu with organic ligands possessing desirable properties. Owing to the intimate interactions between the Cu active sites and the organic ligands, CuDAT exhibits outstanding CO2R performance towards C2+ products. Based on detailed mechanistic investigations, the enhanced C2+ production on CuDAT is attributed to the facilitated *CO adsorption and accelerated C–C coupling rate, induced by the closely cooperated DAT ligands. Moreover, CuDAT also demonstrates great potential for low-concentration CO2 reduction.
We acknowledge A*STAR (Agency for Science, Technology and Research) under its LCERFI program (award no. U2102d2002). We would also like to acknowledge the support of the National Research Foundation (NRF) Singapore, under the NRF Fellowship (NRF-NRFF13-2021-0007).
Conflicts of interest
There are no conflicts to declare.References
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- C. Yang, Y. Wang, L. Qian, A. M. Al-Enizi, L. Zhang and G. Zheng, ACS Appl. Energy Mater., 2021, 4, 1034–1044 CrossRef CAS.
- J. Chen and L. Wang, Adv. Mater., 2022, 34, e2103900 CrossRef PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ACS Catal., 2019, 9, 7894–7899 CrossRef CAS.
- Y. Zhao, X. Liu, J. Chen, J. Chen, J. Chen, L. Fan, H. Yang, S. Xi, L. Shen and L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218040120 CrossRef CAS PubMed.
- Q. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed.
- J. Chen, X. Liu, S. Xi, T. Zhang, Z. Liu, J. Chen, L. Shen, S. Kawi and L. Wang, ACS Nano, 2022, 16, 13982–13991 CrossRef CAS PubMed.
- J. Chen, L. Chen, J. Chen, D. Wang, Y. Zhao, L. Wen, S. Xi and L. Wang, Appl. Catal., B, 2024, 343, 123551 CrossRef CAS.
- Y. Qiao, G. Kastlunger, R. C. Davis, C. A. G. Rodriguez, A. Vishart, W. Deng, Q. Xu, S. Li, P. Benedek, J. Chen, J. Schröder, J. Perryman, D. U. Lee, T. F. Jaramillo, I. Chorkendorff and B. Seger, ACS Catal., 2023, 13, 9379–9391 CrossRef CAS.
- X. Chen, D. A. Henckel, U. O. Nwabara, Y. Li, A. I. Frenkel, T. T. Fister, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2020, 10, 672–682 CrossRef CAS.
- Y. Lan, G. Niu, F. Wang, D. Cui and Z. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 36128–36136 CrossRef CAS PubMed.
- C. Tang, Z. Chen, Y. Wang, T. Xiao, X. Li, C. Zheng, X. Xu and Z. Sun, ACS Catal., 2022, 12, 11838–11844 CrossRef CAS.
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- L. Chen, J. Chen, L. Fan, J. Chen, T. Zhang, J. Chen, S. Xi, B. Chen and L. Wang, ACS Catal., 2023, 13, 11934–11944 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- M. F. Philips, D. Pavesi, T. Wissink, M. C. Figueiredo, G.-J. M. Gruter, M. T. M. Koper and K. J. P. Schouten, ACS Appl. Energy Mater., 2022, 5, 1720–1730 CrossRef CAS.
- Z.-Z. Niu, F.-Y. Gao, X.-L. Zhang, P.-P. Yang, R. Liu, L.-P. Chi, Z.-Z. Wu, S. Qin, X. Yu and M.-R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- Y. Zhao, X. Zu, R. Chen, X. Li, Y. Jiang, Z. Wang, S. Wang, Y. Wu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2022, 144, 10446–10454 CrossRef CAS PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- R. Lin, J. Guo, X. Li, P. Patel and A. Seifitokaldani, Catalysts, 2020, 10, 473–507 CrossRef CAS.
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 CrossRef CAS.
- X. Zhang, J. Li, Y. Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS PubMed.
- F. R. Brushett, M. S. Thorum, N. S. Lioutas, M. S. Naughton, C. Tornow, H.-R. M. Jhong, A. A. Gewirth and P. J. A. Kenis, J. Am. Chem. Soc., 2010, 132, 12185–12187 CrossRef CAS PubMed.
- E. Aznar, S. Ferrer, J. Borrás, F. Lloret, M. Liu-González, H. Rodríguez-Prieto and S. García-Granda, Eur. J. Inorg. Chem., 2006, 5115–5125 CrossRef CAS.
- S. Fonseca and L. M. C. Pinto, ACS Omega, 2020, 5, 1581–1585 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS PubMed.
- G. Iijima, H. Yamaguchi, T. Inomata, H. Yoto, M. Ito and H. Masuda, ACS Catal., 2020, 10, 15238–15249 CrossRef CAS.
- S. Mukherjee, M. Das, A. Manna, R. Krishna and S. Das, J. Mater. Chem. A, 2019, 7, 1055–1068 RSC.
- N. Zhao, Z. Shi, R. Chenitz, F. Girard and A. Mokrini, Membranes, 2020, 10, 301 CrossRef CAS PubMed.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- J. D. Merline, C. P. Reghunadhan Nair and K. N. Ninan, J. Macromol. Sci. A, 2008, 45, 312–322 CrossRef CAS.
- D. Huang, H. Zhu, Z. Zhao, J. Huang, P. Liao and X. Chen, ACS Catal., 2022, 12, 8444–8450 CrossRef CAS.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Nat. Catal., 2019, 2, 702–708 CrossRef CAS.
- A. Monshi, M. R. Foroughi and M. R. Monshi, World J. Nano Sci. Eng., 2012, 2, 154–160 CrossRef.
- L. Rinawati, R. E. Nugraha, R. M. I. Munifa, U. Chasanah, S. Wahyuningsih and A. H. Ramelan, J. Chem. Pharm. Res., 2015, 7, 85–89 CAS.
- S. Zhu, T. Li, W. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- T. Bürgi, Phys. Chem. Chem. Phys., 2001, 3, 2124–2130 RSC.
- S. Zhu, B. Jiang, W. B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- Y. Hori, O. Koga, H. Yamazaki and T. Matsuo, Electrochim. Acta, 1995, 40, 2617–2622 CrossRef CAS.
- J. Zhang, G. Zeng, S. Zhu, H. Tao, Y. Pan, W. Lai, J. Bao, C. Lian, D. Su, M. Shao and H. Huang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218987120 CrossRef CAS PubMed.
- Y. Xiao, M. Wang, H. Yang, H. Qiu, H. Lu, Y. Da, G. Chen, T. Jiang, W. Fu, B. Hu, J. Chen, L. Chen, Y. Ding, B. Cui, C. Jiang, Z. Sun, Y. Long, H. Yang, Z. Tian, L. Wang and W. Chen, Adv. Energy Mater., 2024, 14, 2302556 CrossRef CAS.
- J. Li, X. Chang, H. Zhang, A. S. Malkani, M. J. Cheng, B. Xu and Q. Lu, Nat. Commun., 2021, 12, 3264 CrossRef CAS PubMed.
- T. Galimova, M. Ram, D. Bogdanov, M. Fasihi, S. Khalili, A. Gulagi, H. Karjunen, T. N. O. Mensah and C. Breyer, J. Cleaner Prod., 2022, 373, 133920 CrossRef CAS.
- C. Breyer, M. Fasihi, C. Bajamundi and F. Creutzig, Joule, 2019, 3, 2053–2057 CrossRef.
- Z. Xiang, S. Leng and D. Cao, J. Phys. Chem. C, 2012, 116, 10573–10579 CrossRef CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05972c |
| ‡ Jingyi Chen and Lei Fan have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |