
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of cyclohepta[b]indoles via gold mediated energy transfer photocatalysis†
Yuan
Zhao
a,
Vladislav A.
Voloshkin
 a,
Ekaterina A.
Martynova
a,
Bholanath
Maity
b,
Luigi
Cavallo
b and
Steven P.
Nolan
*a
a,
Ekaterina A.
Martynova
a,
Bholanath
Maity
b,
Luigi
Cavallo
b and
Steven P.
Nolan
*a
aDepartment of Chemistry and Centre for Sustainable Chemistry Ghent University, Krijgslaan 281, S-3, 9000 Ghent, Belgium. E-mail: steven.nolan@ugent.be
bKAUST Catalysis Center (KCC) King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: luigi.cavallo@kaust.edu.sa
First published on 21st February 2024
Abstract
Photocatalysis involving energy transfer (EnT) has become a valuable technique for building intricate organic frameworks mostly through [2+2]-cycloaddition reactions. Herein, we report a synthetic method leading to functionalized cyclohepta[b]indoles, an important structural motif in natural products and pharmaceuticals, using gold-mediated energy transfer photocatalysis. The scope of this operationally simple and atom-economical strategy is presented. Density functional theory studies were employed in order to gain insights into the mechanism of formation of the cyclohepta[b]indole core.
During the last few decades, the field of photocatalysis has steadily evolved and is currently experiencing a significant surge in activity.1–5 In one of its incarnations, photocatalysis via energy transfer (EnT), which is facilitated by light-absorbing photosensitizers, affords the means to access highly active triplet states. As a result, it now permits reactions previously unattainable under thermal activation. Nowadays it is an important tool used in modern organic synthesis for a large variety of reactions.2b,6–9 Numerous organic sensitizers10–13 and transition metal-based photocatalysts, such as ruthenium,14–16 palladium,17 rhodium,18 and iridium,19–23 have been deployed in EnT photocatalysis. It is known that efficient EnT photosensitizers should possess several key characteristics, namely: (i) high absorption cross-section at the desired wavelength, (ii) efficient intersystem crossing to its triplet state, (iii) significantly long excited triplet state lifetime, and (iv) higher triplet excited-state energy than the targeted substrate.
Recently, we have disclosed two novel photosensitizers [Au(SIPr)(Cbz)] (PhotAuCat I) (SIPr = [N,N-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene]; Cbz = carbazolyl) and [Au(IPr)(Cbz)] (PhotAuCat II) (IPr = [N,N-bis(2,6-diisopropylphenyl)imidazol-2ylidene]).24 With high triplet energy values, namely 67.9 kcal mol−1 for PhotAuCat I and 67.6 kcal mol−1 for PhotAuCat II, these have demonstrated efficiency in diverse [2+2]-photocycloadditions, surpassing state-of-the-art Ir photocatalysts.25–27
Cyclohepta[b]indole serves as a core unit in some natural products as well as in various pharmaceutical compounds.28 For example, arcyriacyanin A isolated from Arcyria nutans inhibits protein kinase C and tyrosine kinase and exhibits cytotoxicity against human cancer cells.29 Additionally, a variety of compounds containing the cyclohepta[b]indole scaffold exhibit important pharmacological effects, such as A-FABP inhibition,30 antitubercular properties31 and SIRT1 inhibition (Fig. 1).32 It is therefore crucial to identify straightforward synthetic routes to this scaffold considering its remarkable biological properties.
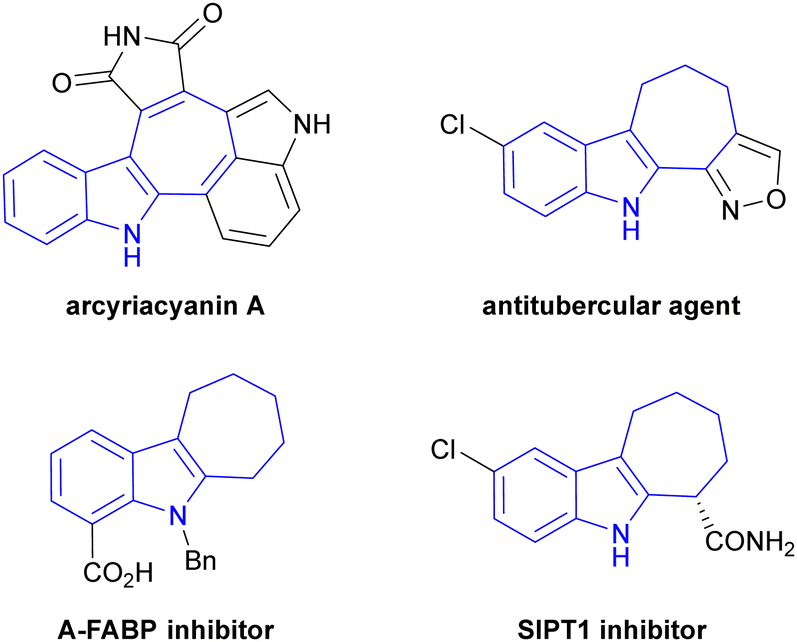 | ||
| Fig. 1 Representative examples of compounds containing a cyclohepta[b]indole scaffold. | ||
A number of methods exist for the synthesis of the cyclohepta[b]indole core,33 including cyclization reactions,34–36 (m+n)-cycloaddition reactions,37–40 and Cope rearrangement.41 However, although these strategies prove viable, most of them involve multi-step routes, high temperature and/or toxic solvents. Noteworthily, there is currently only one example of photochemical synthesis of cyclohepta[b]indole-type scaffolds using direct irradiation in quartz tubes with harsh 254 nm UV light.42
Recently, while exploring the intermolecular [2+2]-cycloaddition of indoles via gold-mediated EnT photocatalysis, we discovered the unexpected formation of cyclohepta[b]indole.27 Considering the great interest in this scaffold, we now further explore this initial observation and report on an efficient protocol for its synthesis via gold-mediated energy transfer photocatalysis from functionalized indoles.
We initiated our investigations with the model substrate, dimethyl-substituted indole 1a. Previously, we found that cyclohepta[b]indole 2a was formed upon irradiation with 365 nm light in the presence of dinuclear gold sensitizer PhotAuCat III (Fig. 2) in a 64% yield in EtOAc after 18 hours (see ESI† for complete optimization studies, Table S1).27PhotAuCat I, being an efficient and easily accessible gold sensitizer, was tested under identical conditions, providing a 74% yield of 2a. An even higher yield (84%) was obtained by using iPrOAc as the solvent. Various solvents, such as EtOAc, Et2O, MTBE, 2-MeTHF, THF, MeOH, and MeCN, were also examined, but afforded the desired product in slightly lower 73–78% yields (see ESI,† Table S1). Noteworthily, when THF was used as a solvent only 8% of the desired product 2a was observed. iPrOAc was found to be the optimum solvent and was used in the screening of other photosensitisers. PhotAuCat II, which has a slightly lower ET value than PhotAuCat I yielded 61% of 2a under the same reaction conditions. Interestingly, when we tested commonly used iridium and organic photosensitizers, namely, [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (PC I) and thioxanthen-9-one (PC II), in both cases no reaction occurred. These results highlight the importance of gold photocatalysts for applications in energy transfer photocatalysis. When utilizing 2 mol% of PhotAuCat I, a noticeable decrease in the product yield was observed, affording a mere 37% of 2a compared to the 84% achieved with 4 mol% catalyst. Control experiments in the absence of photocatalyst and in the absence of irradiation were also conducted and, in both cases, no reaction occurred, indicating the need for both the photocatalyst and light.
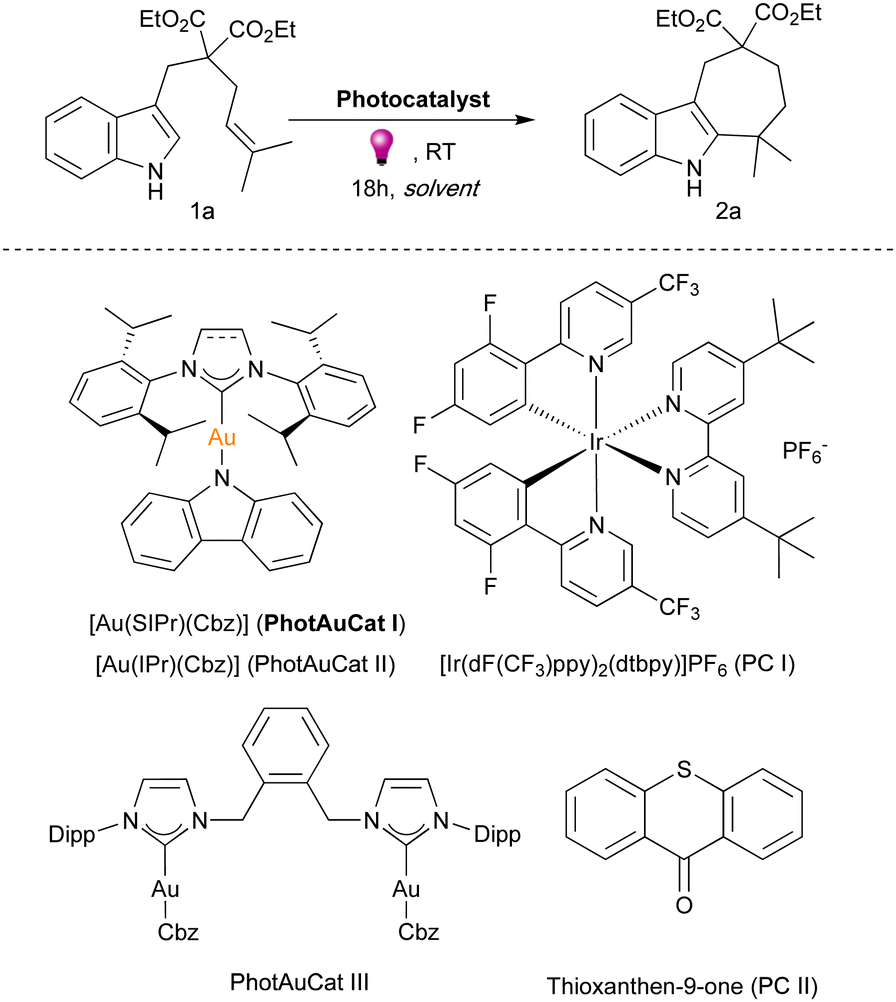 | ||
| Fig. 2 Model reaction studied and photocatalysts tested. | ||
Having optimized the reaction conditions, we next explored the reaction scope with various unprotected indoles (Fig. 3). The presence of two terminal methyl groups on the alkene appears essential for this type of reactivity. We began the exploration of the reaction scope by varying the ester groups on the linker. Noteworthily, ethyl (2a), methyl (2b), bulky tert-butyl (2c), isopropyl (2d) and benzyloxy (2e) ester groups were well tolerated and the desired products were obtained in high yields, 60–95%. Interestingly, changing R from ester to cyano groups led to a lack of reactivity under the optimized reaction conditions. The same result was observed when introducing a p-toluenesulfonamide group (–NTs) in the linker (see ESI†). We then explored various substituents on the indole ring. 3-Methoxy substituted indole proceeded to the cyclized product 2f smoothly in 80% yield as well as indole bearing 4-methoxy group 2g in 81% yield. Different methyl-substituted indoles afforded the desired products 2h–2j in good yields of 62–81%.
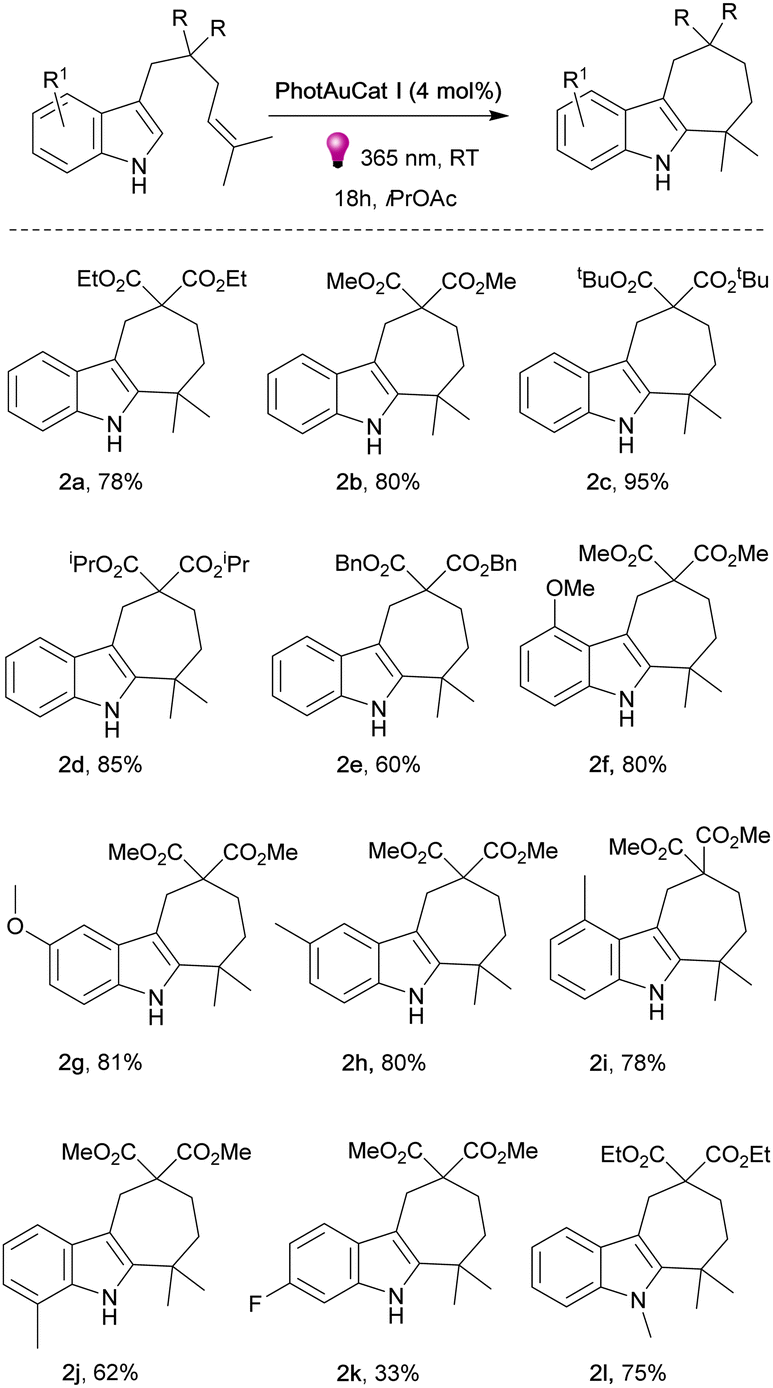 | ||
| Fig. 3 Substrate scope of indole derivatives. Unless otherwise noted, standard reaction conditions: 1a (0.10 mmol) and catalyst (0.004 mmol, 4 mol%) in solvent (2 mL) with 365 nm LEDs at rt for 18 h; yields are of isolated products. | ||
The corresponding product 2k with the 5-fluoro-substituted indole was obtained in 33% yield. 5-Bromo and 6-benzyloxy congeners did not react even by increasing the catalyst loading and prolonging the reaction time (see ESI†). This fact is consistent with previously observed reactivity for bromo-substituted indoles.26 In order to establish whether the absence of an N–H proton can affect the reaction outcome, we synthesized N-methyl indole and performed the reaction with this substrate. The product 2l was isolated with 75% yield showcasing that the presence of an N–H proton is not essential for the transformation to proceed. When an indole with a lone methyl group at the terminal position of the alkene was tested under the same conditions, no reaction occurred (see ESI†). Our earlier observation of the [2+2]-photocycloaddition product formation in cases where the alkene attached to the indole lacks methyl groups serves as compelling evidence for the significant impact of substitution on the terminal position on the product formation.26
Intrigued by the unexpected formation of the cyclohepta[b]indole core instead of the (originally expected) classical [2+2]-cycloaddition product, further mechanistic studies were performed. The absorption spectra of 1a and PhotAuCat I show that only the latter absorbs light at 365 nm (see Fig. S4 in ESI†). To support the involvement of the triplet–triplet energy transfer mechanism, we performed quenching studies. The reaction was completely suppressed in the presence of TEMPO. However, the formation of an adduct of TEMPO was not detected. Based on these observations and previously reported data for gold photocatalysts and for similar indole substrates,26 we hypothesized that the energy transfer proceeds between the excited state photosensitizer and the substrate. However, the reason for the formation of the unexpected ringed product instead of cyclobutane-fused indole remained unclear.
To investigate the reaction mechanism, we employed density functional theory (DFT). An alternative mechanism of cyclization of Ind-1, involving intramolecular hydrogen atom transfer (HAT), was proposed, and analysed to explain the unexpected reactivity (Fig. 4). The analysis begins with the activation of Ind-1 from its singlet spin state to the triplet state TInd-1. This activation is achieved through triplet–triplet energy transfer from the excited triplet state of [Au(SIPr)(Cbz)], facilitated by the higher energy of the triplet state of [Au(SIPr)(Cbz)] (ΔGT = 67.9 kcal mol−1) compared to that of substrate Ind-1 (ΔGT = 62.2 kcal mol−1). The reaction involves a two-step mechanism: (i) C–C bond formation, and (ii) H-atom transfer (HAT). The first step takes place in TInd-1 on the triplet surface viaTTS1–2 with an activation free energy of 10.9 kcal mol−1. This step is energetically feasible due to a moderate barrier and exergonicity (ΔG = −14.5 kcal mol−1). The resulting triplet intermediate TInd-2 undergoes a thermoneutral inter system crossing (ISC) to the open-shell singlet biradical OSInd-2. In the next step, a 1,3-HAT leads to another open-shell intermediate OSInd-3, and eventually to the closed-shell product 2a. This HAT step needs to overcome an energy barrier of 18.6 kcal mol−1via transition state OSTS2–3. Considering this moderate energy barrier, and the high exergonicity (ΔG = −64.0 kcal mol−1), this step is facile and irreversible. Alternatively, the HAT step in TInd-2 along the triplet surface requires a noticeably higher energy barrier of 39.9 kcal mol−1 (Fig. S5 in ESI†), and thus can be discarded. We have also computed the reaction pathway for the formal [2+2]-cycloaddition product from OSInd-2. The process involves a gradual approach of two radical centres to each other, resulting in OSInd-4. This step requires an energy barrier of 23.7 kcal mol−1, which is 5.1 kcal mol−1 higher than the HAT step. Moreover, SInd-4 is 58.1 kcal mol−1 less stable than OSInd-3. Therefore, the higher barrier and the significant destabilization of the product, in line with the experimental observations, reinforce the conclusion that the formal [2+2]-cycloaddition pathway is unproductive.
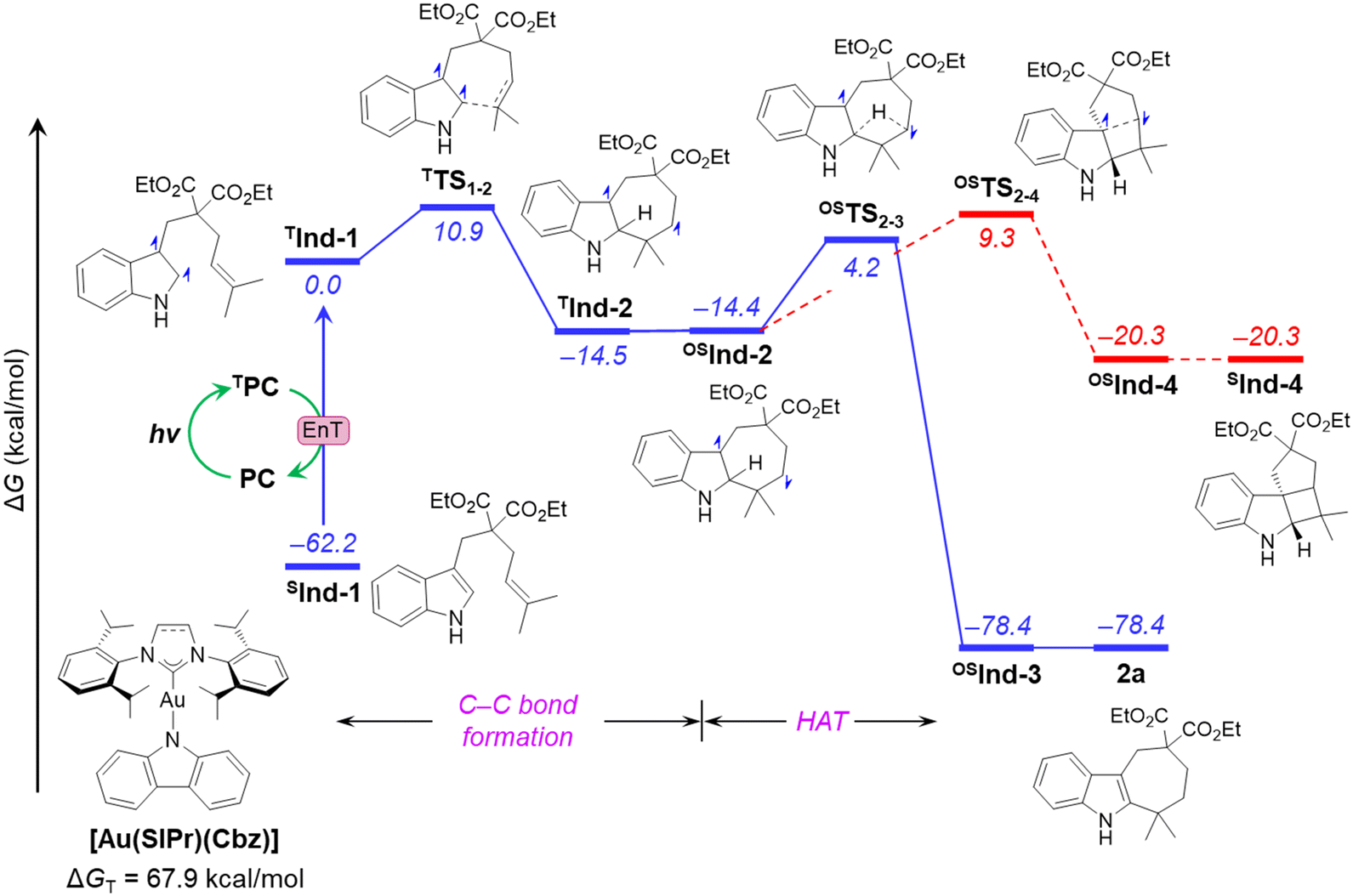 | ||
| Fig. 4 Computed free energy profile of the cyclization reaction. Energy values (in kcal mol−1) are reported at the ωB97X-D(SMD-EtOAc)/def2-TZVPP//ωB97X-D/def2-SVP level of theory. T, triplet; OS, open-shell singlet; S, closed-shell singlet. | ||
We report on the use of [Au(SIPr)(Cbz)] (PhotAuCat I) as a photosensitizer for intramolecular cycloaddition of indoles leading to cyclohepta[b]indoles. A wide scope for this transformation was explored, where the desired products were obtained in good to excellent yields. DFT calculations were conducted and highlight an initial C–C bond formation followed by a hydrogen atom transfer (HAT) step. The method provides a new route to cycloheptane-fused indoles, replacing lengthy and complex protocols. This contribution adds to the reported uses of these gold catalysts as a versatile tool in energy transfer chemistry and catalysis where HAT is now shown to be a compatible process. Further studies aimed at probing the versatility of PhotAuCats are ongoing in our laboratory.
We gratefully acknowledge support from the Special Research Fund (BOF) of Ghent University (project grants to SPN) and the Research Foundation – Flanders (FWO) (G0A6823N). YZ thanks the China Scholarship Council (CSC) (project 202206870016) for a PhD fellowship. LC thanks KAUST for financial support through grants CRG9 no. 4343 and 4701, and the KAUST Core Labs for computational time on the Shaheen HPC platform managed by KSL.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- (a) N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC; (b) S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- B. König, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible light photocatalysis in organic chemistry, WileyVCH, Weinheim, Germany, 1st edn, 2018 Search PubMed.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- W. C. de Souza, B. T. Matsuo, P. M. Matos, J. T. M. Correia, M. S. Santos, B. König and M. W. Paixão, Chem. Eur. J., 2021, 27, 3722–3728 CrossRef CAS PubMed.
- X. Li, R. J. Kutta, C. Jandl, A. Bauer, P. Nuernberger and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 21640–21647 CrossRef CAS PubMed.
- A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed.
- A. B. Rolka and B. Koenig, Org. Lett., 2020, 22, 5035–5040 CrossRef CAS PubMed.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed.
- F. M. Hörmann, C. Kerzig, T. S. Chung, A. Bauer, O. S. Wenger and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 9659–9668 CrossRef PubMed.
- Y.-Q. Zou, S.-W. Duan, X.-G. Meng, X.-Q. Hu, S. Gao, J.-R. Chen and W.-J. Xiao, Tetrahedron, 2012, 68, 6914–6919 CrossRef CAS.
- P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083–6098 RSC.
- N. Hu, H. Jung, Y. Zheng, J. Lee, L. Zhang, Z. Ullah, X. Xie, K. Harms, M.-H. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242–6246 CrossRef CAS PubMed.
- K. A. Rykaczewski and C. S. Schindler, Org. Lett., 2020, 22, 6516–6519 CrossRef CAS PubMed.
- C. Wang and Z. Lu, Org. Lett., 2017, 19, 5888–5891 CrossRef CAS PubMed.
- G. Tan, M. Das, R. Kleinmans, F. Katzenburg, C. Daniliuc and F. Glorius, Nat. Catal., 2022, 5, 1120–1130 CrossRef CAS.
- Y. Liang, R. Kleinmans, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2022, 144, 20207–20213 CrossRef CAS PubMed.
- R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477–482 CrossRef CAS PubMed.
- N. V. Tzouras, E. A. Martynova, X. Ma, T. Scattolin, B. Hupp, H. Busen, M. Saab, Z. Zhang, L. Falivene, G. Pisanò, K. Van Hecke, L. Cavallo, C. S. J. Cazin, A. Steffen and S. P. Nolan, Chem. Eur. J., 2021, 27, 11904–11911 CrossRef CAS PubMed.
- S. G. Guillet, A. A. Logvinov, V. A. Voloshkin, E. A. Martynova and S. P. Nolan, Org. Lett., 2023, 25, 1403–1408 CrossRef CAS PubMed.
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Beliš, K. V. Hecke, C. S. J. Cazin and S. P. Nolan, Chem. Sci., 2022, 13, 6852–6857 RSC.
- X. Ma, V. A. Voloshkin, E. A. Martynova, M. Beliš, M. Peng, M. Villa, N. V. Tzouras, W. Janssens, K. V. Hecke, P. Ceroni and S. P. Nolan, Catal. Sci. Technol., 2023, 13, 4168–4175 RSC.
- E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390–2402 CrossRef CAS PubMed.
- M. Murase, K. Watanabe, T. Yoshida and S. Tobinaga, Chem. Pharm. Bull., 2000, 48, 81–84 CrossRef CAS PubMed.
- T. Barf, F. Lehmann, K. Hammer, S. Haile, E. Axen, C. Medina, J. Uppenberg, S. Svensson, L. Rondahl and T. Lundbäck, Bioorg. Med. Chem. Lett., 2009, 19, 1745–1748 CrossRef CAS PubMed.
- E. Yamuna, R. A. Kumar, M. Zeller and K. J. Rajendra Prasad, Eur. J. Med. Chem., 2012, 47, 228–238 CrossRef CAS PubMed.
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J.-F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, J. Med. Chem., 2005, 48, 8045–8054 CrossRef CAS PubMed.
- J. Gierok, L. Benedix and M. Hiersemann, Eur. J. Org. Chem., 2021, 3748–3758 CrossRef CAS.
- Q. Zeng, K. Dong, J. Huang, L. Qiu and X. Xu, Org. Biomol. Chem., 2019, 17, 2326–2330 RSC.
- L. Zhang, Y. Zhang, W. Li and X. Qi, Angew. Chem., Int. Ed., 2019, 58, 4988–4991 CrossRef CAS PubMed.
- J. Kaufmann, E. Jäckel and E. Haak, Angew. Chem., Int. Ed., 2018, 57, 5908–5911 CrossRef CAS PubMed.
- T. Takeda, S. Harada, A. Okabe and A. Nishida, J. Org. Chem., 2018, 83, 11541–11551 CrossRef CAS PubMed.
- C. Gelis, G. Levitre, J. Merad, P. Retailleau, L. Neuville and G. Masson, Angew. Chem., Int. Ed., 2018, 57, 12121–12125 CrossRef CAS PubMed.
- A. N. Parker, M. C. Martin, R. Shenje and S. France, Org. Lett., 2019, 21, 7268–7273 CrossRef CAS PubMed.
- J. Xu and V. H. Rawal, J. Am. Chem. Soc., 2019, 141, 4820–4823 CrossRef CAS PubMed.
- M. Häfner, Y. M. Sokolenko, P. Gamerdinger, E. Stempel and T. Gaich, Org. Lett., 2019, 21, 7370–7374 CrossRef PubMed.
- D. C. Tymann, L. Benedix, L. Iovkova, R. Pallach, S. Henke, D. Tymann and M. Hiersemann, Chem. Eur. J., 2020, 26, 11974–11978 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00379a |
| This journal is © The Royal Society of Chemistry 2024 |