
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unravelling the structure of CO2 in silica adsorbents: an NMR and computational perspective
Mariana
Sardo
 a,
Tiago
Morais
ac,
Márcio
Soares
a,
Ricardo
Vieira
a,
Marina
Ilkaeva
ab,
Mirtha A. O.
Lourenço
a,
Ildefonso
Marín-Montesinos
a and
Luís
Mafra
*a
a,
Tiago
Morais
ac,
Márcio
Soares
a,
Ricardo
Vieira
a,
Marina
Ilkaeva
ab,
Mirtha A. O.
Lourenço
a,
Ildefonso
Marín-Montesinos
a and
Luís
Mafra
*a
aCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: lmafra@ua.pt
bDepartment of Chemical and Environmental Engineering, University of Oviedo, Av. Julián Clavería 8, 33006 Oviedo, Spain
cDepartment of Chemistry, University of Iceland, Science Institute, Dunhaga 3, 107 Reykjavik, Iceland
First published on 15th March 2024
Abstract
This comprehensive review describes recent advancements in the use of solid-state NMR-assisted methods and computational modeling strategies to unravel gas adsorption mechanisms and CO2 speciation in porous CO2-adsorbent silica materials at the atomic scale. This work provides new perspectives for the innovative modifications of these materials rendering them more amenable to the use of advanced NMR methods.
 Mariana Sardo | Mariana Sardo received a BSc in Physics and Chemistry (2005) and a PhD in Chemistry (2009), both from the University of Aveiro, followed by postdoctoral training at ETH – Zurich (2010). Soon after, she joined the NMR group at CICECO – Aveiro Institute of Materials, Chemistry Department where she is currently an assistant researcher. Her work is focused on the use of solid-state NMR methods for the characterization of pharmaceutical systems and porous materials (in particular, gas–solid interactions in modified porous solid sorbents with potential for gas sorption and micro and mesoporous systems relevant to heterogeneous catalysis). |
 Tiago Morais | Tiago Morais holds both a BSc (2019) and a MSc (2021) degree in Biotechnology from the University of Aveiro. In 2022 he started his PhD in Chemistry on Team SPECKO (CICECO – Aveiro Institute of Materials) under the supervision of Dr Luís Mafra, Dr Mariana Sardo and Dr Mirtha Lourenço. This PhD is being conducted in a co-tutelar agreement with the University of Iceland, under the supervision of Prof. Snorri Th. Sigurdsson. His research is focused on the synthesis, modification and characterization of silica-based materials for CO2 capture, and in the development of labelling strategies (isotope labelling and radicals incorporation) for NMR and DNP studies. |
 Márcio Soares | Márcio Soares got his BSc in Biotechnology (2019) and a MSc degree in Clinical Biochemistry (2021), both from the University of Aveiro. In 2022 he started his PhD studies on Team SPECKO (CICECO – Aveiro Institute of Materials) under the supervision of Dr Luis Mafra, Dr Mariana Sardo and Dr Ildefonso Marin. The focal point of his research lies in conducting computer modelling investigations closely integrated with the synthesis and characterization processes of porous materials. The overarching objective is to elucidate the mechanisms of gas sorption in solid adsorbents utilizing surface-enhanced ex(in)-situ NMR techniques. |
 Ricardo Vieira | Ricardo Vieira received his BSc (2009), MSc (2011) and PhD (2018) in Physics from the University of Coimbra. In 2018, he served as a postdoctoral researcher at the University of Aveiro in the Institute of Nanostructures, Nanomodeling, and Nanofabrication (I3N). Since 2019, he holds a junior research position at the University of Aveiro – CICECO-Aveiro Institute of Materials, focusing on magnetic resonance techniques like solid-state NMR and muon spectroscopy. His research emphasizes the characterization of porous and film materials, particularly gas–solid interactions in modified porous solid sorbents for gas sorption, and micro and mesoporous systems relevant to heterogeneous catalysis. |
 Marina Ilkaeva | Marina Ilkaeva, an Environmental Engineer (2012, South Ural State University), obtained her PhD degree in Material Science (2018) from the University of Oviedo. From 2018 to 2023, she was a researcher at the associated laboratory CICECO of the University of Aveiro. Currently, she is a “Beatriz Galindo” junior research fellow at the University of Oviedo. Her research interests are centered on the application of materials for environmental purposes such as advanced oxidation processes for water remediation, photoredox biomass valorization for the production of valuable chemicals and CO2 capture and conversion. |
 Mirtha A. O. Lourenço | Mirtha Lourenço earned her BSc (2009) and MSc (2010) in Chemistry from the University of Coimbra and a PhD (2016) in Materials Science and Engineering from the University of Aveiro. Following this, in 2018, she served as a postdoctoral researcher at the Italian Institute of Technology – Center for Sustainable Future Technologies. Since 2021, she has held a research position at the University of Aveiro, currently serving as a European Research Area Postdoctoral Fellow (ERA-PF) supported by the European Commission. Her research focuses on developing sustainable porous adsorbents for effective CO2 capture, separation, and conversion and elucidating the structural mechanisms involved in these processes. |
 Ildefonso Marín-Montesinos | Ildefonso Marín-Montesinos received his BSc from the University of Sevilla (2001) and his PhD from the University of Southampton (2007). He then moved to the University of Birmingham for a postdoctoral position. In 2010, he received a “Juan de la Cierva” grant to work with Prof. M. Pons at the Universitat de Barcelona. He then moved to IRIG at CEA Grenoble to work with Dr S. Hediger and Dr G. De Paëpe. Currently, he is a researcher at CICECO-University of Aveiro. His research focuses on the characterization of biomolecular systems and porous solid materials for environmental applications using magnetic resonance techniques. |
 Luís Mafra | Luís Mafra, Principal Researcher at CICECO-Aveiro Institute of Materials, is an expert in solid-state Nuclear Magnetic Resonance (NMR) spectroscopy. In 2006, he obtained his PhD, from the University of Aveiro & University of Caen, specializing in the development of high-resolution NMR methods to observe 1H and quadrupolar nuclei in hybrid materials. His present research interests revolve around the synergetic application of solid-state NMR techniques in tandem with other spectroscopies, diffraction and computational methods, with a primary focus on unraveling the structure of acid sites in heterogeneous porous catalysts, and probing CO2-sorption mechanisms at the surface of porous adsorbents. |
1. Introduction
1.1. Environmental challenges and the development of new sorbent materials for carbon capture
Global warming will continue to increase in the next decades (2021–2040) mainly due to increased cumulative CO2 emissions in nearly all considered scenarios.1,2 80% of the global energy supply still relies on the consumption of fossil fuels, responsible for the increase by almost 2.1 Gt of energy-related CO2 emissions in 2021, making it the largest ever year-on-year increase in energy-related CO2 emissions in absolute terms.3 From 2010–2019, the average annual global greenhouse gas emissions were at their highest level in human history, but the rate of growth has slowed down. Without an immediate reduction in emission levels, across all sectors, limiting global warming to 1.5 °C will not be achieved in the early 2030s.4 To address this, it is crucial to achieve net-zero emissions. The Intergovernmental Panel for Climate Change (IPCC) suggests that agriculture, forestry, and other land use may provide large-scale emission reduction by removing and storing CO2. However, land cannot compensate for delayed emissions reductions in other sectors.1,2 CO2 capture is thus an imperative, which has led to the exploration of various carbon capture technologies. The concentration of CO2 in the gas stream, the pressure of the gas stream and the fuel type (solid or gaseous) are important factors in selecting the capture system.There are three main approaches to CO2 capture, for industrial and power plant applications. Post-combustion5 systems separate CO2 from the flue gases produced by combustion of a primary fuel (coal, natural gas, oil or biomass) in air. Oxy-fuel combustion6 uses oxygen instead of air for combustion, producing a flue gas that is mainly composed of H2O and CO2 and therefore readily captured. Pre-combustion7 systems process the primary fuel in a reactor to produce separate streams of CO2 for storage and H2 which is used as a fuel. Other industrial processes, including the production of low-carbon or carbon-free fuels, employ one or more of these basic capture methods.2 Post-combustion capture is the most popular and mature technology used in the industry, as it requires no modification of existing plants. It includes absorption, adsorption, cryogenic, membrane and hydrate-based separations.8
Adsorption is especially significant as it is capable of separating CO2 from various mixtures such as flue gas (CO2/N2, CO2/H2) and biogas (CO2/CH4). Previous studies have shown that amine-based systems for CO2 absorption are the most suitable for combustion-based power plants for several reasons. These systems are highly efficient for diluted CO2 streams (e.g., flue gases from coal combustion with ca. 10% to 12% CO2 by volume) are currently available and used at the commercial level and bear similarities to other “end-of-pipe” environmental control systems employed in power plants, operating under normal temperature and pressure conditions.9
Aqueous solutions of monoethanolamine have been the benchmark technology for scrubbing CO2 from process gases,10,11 due to their intrinsic high affinity towards CO2 and remain the standard to which other technologies are compared. However, they present high energy consumption requirements and operational issues such as degradation and toxicity. To overcome such limitations, several classes of solid amine adsorbents have emerged, as they combine the advantages of amine solutions and solid porous materials. Due to the high reactivity between loaded amines and CO2, solid amine adsorbents can selectively adsorb CO2 efficiently and quickly, while the CO2 regeneration energy decreases by more than 50% compared to amine solutions.12 These solid adsorbents include activated carbons, ion-exchange resins, zeolites, porous silicates, metal oxides, organic–inorganic hybrid sorbents (e.g., supported amine-modified silicas, metal–organic frameworks (MOFs)), covalent–organic frameworks (COFs), and composite materials.13–27
1.2. Silica materials for CO2 capture applications
Silica sorbents are highly promising in the pursuit of mitigating climate change, particularly in carbon capture for industrial applications, as reported in the extensive reviews dedicated to the topic.8,28Mesoporous silica is a silicon and oxygen-based porous material with pores ranging from 2–50 nm in diameter, making it ideal for the capture and storage of CO2. Its high surface area and large pore volume enable efficient CO2 adsorption, while its chemical stability and resistance to degradation (due to the absence of organic moieties in the structure) make it suitable for use under harsh industrial conditions (e.g. high pressure and temperature). The tuneable pore size and ordered mesoporous structure greatly enhance adsorption performance, while abundant surface silanol groups facilitate the framework modification with amine moieties.29 Amine modified silica-based materials show promising capture capacity at moderate temperatures (40–80 °C), and a relatively low desorption temperature, which decreases the energy cost of regeneration.30 Hence, silica-based materials are widely used and have been the subject of extensive research. To capture CO2 using mesoporous silica, the gas is typically passed through a fixed-bed reactor with the material, where it is adsorbed onto the surface of the pores and then heated to release the CO2 for storage or further use. However, the cost of the material and adsorption capacity is currently a bottleneck in using mesoporous silica for CO2 capture. Additionally, different preparation methods can influence amine loading, thermal stability, and adsorption capacity, which should be selected based on the application. Many researchers are seeking ways to lower costs and improve efficiency, such as using cheaper precursors or developing new, more efficient adsorption materials.
1.3. Which analytical tools are more promising to characterise CO2 adsorbent materials?
Characterising complex adsorbent materials for carbon capture is challenging and often limits the design of improved materials. Currently, various techniques such as single-crystal and powder X-ray diffraction, infrared and NMR spectroscopies are used to understand CO2 capture, each with its own advantages and limitations.31 Solid-state (ss) NMR spectroscopy is an attractive and site-selective technique for investigating CO2 binding modes in adsorbents as it provides atomic-scale insights into the type of chemical species formed upon CO2 adsorption, the local structure, dynamics and diffusion of CO2 species formed at the surface of the sorbent material, without the need for long-range ordering (necessary for X-ray diffraction methods), shedding light on the type of gas-sorption surface mechanisms.This review aims to summarize the most relevant contributions published in the field of silica-based CO2 sorbent materials wherein ssNMR has been explored to investigate at the atomic level the structure and molecular dynamics of CO2 species formed at porous surfaces. Following this general introduction, in which we have provided the motivation underlying the current focus on studying CO2 sorbents based on silica materials, from an ssNMR perspective, the remainder of this feature article is organized into four core sections (2–5) wherein we try to maintain the focus on our research while always trying to make bridges with related works from other authors whenever pertinent. Section 2 describes the available methods applied in the chemical modification of silica materials, including a comparative overview of various modified silica materials with respect to the type of amine, amine density, textural properties, and CO2 capture capabilities. A perspective view of the strategies for the surface modification of silica materials for enhancing NMR signals is also included. Section 3 focuses on the study of CO2 adsorption mechanisms through the combination of ssNMR methods and computer modelling. CO2 adsorption studies performed by ssNMR-assisted techniques are reported in Section 4. Section 5 is dedicated to modelling and simulation approaches applied to micro and mesostructured materials and although we have only provided residual contributions here, we nevertheless attempted to gather topics that are still underexplored or not yet widely used in these materials, including in silico functionalization strategies and computational methods to model pore surface interactions with adsorbates. Section 6 provides a brief overview of the potential applications of CO2 silica chemisorbents and what role may ssNMR have in the design of better and more efficient sorbents.
2. Silica surface modification strategies for CO2 capture applications
2.1. Ordered mesoporous silica-based materials
In 1990, the first periodic mesoporous silica (PMS) materials were discovered, the M41S family.32,33 Despite their interest, these materials had thin walls (0.3–1 nm) and small pores (2–10 nm),34 resulting in poor hydrothermal and mechanical stability. In 1998, the Santa Barbara Amorphous (SBA) family materials35,36 addressed these limitations by exhibiting thicker pore walls (3–6 nm), larger pore sizes (5–30 nm) and a highly ordered mesoscale structure. This led to enhanced diffusion rates of molecules within the pores,35,36 making these materials appealing for incorporating organic functionalities and creating hybrid materials with distinct properties.In 1999, a novel class of hybrid materials, known as periodic mesoporous organosilicas (PMO), was successfully synthesized.37–39 This family of silica-based materials allows the incorporation of high concentrations of organic linkers directly into their pore walls. Notably, a plethora of amine choices can be seamlessly integrated as an integral part of the inorganic-oxide framework. This integration can enhance the formation of CO2 adducts without compromising the textural properties of the adsorbent (e.g., pore volume and surface area). This versatility of introducing functional groups (R) during the condensation reaction of organosilane precursors ((R′O)3Si-R-Si(OR′)3) effectively addresses challenges related to pore-blockage or excessive amine dilution commonly observed in the PMS sorbents. In PMO sorbents, by changing the size of the R group of the organosilane precursor, it is possible to control the functional group type and distance, the silanol density, the hydrophilicity/hydrophobicity, the rigidity/flexibility, and the mechanical/thermal/hydrothermal stability.37–39 Independently of the silica family, introducing organic moieties covalently bonded along the channel pore wall can be achieved through two main distinct methods: (i) co-condensation and (ii) post-modification reactions, which include grafting or anchoring strategies typically at the free silanol groups or at the organic bridge, in the case of PMOs (Fig. 1).40 The details about each method and limitations/advantages thereof are abundantly described elsewhere.41,42
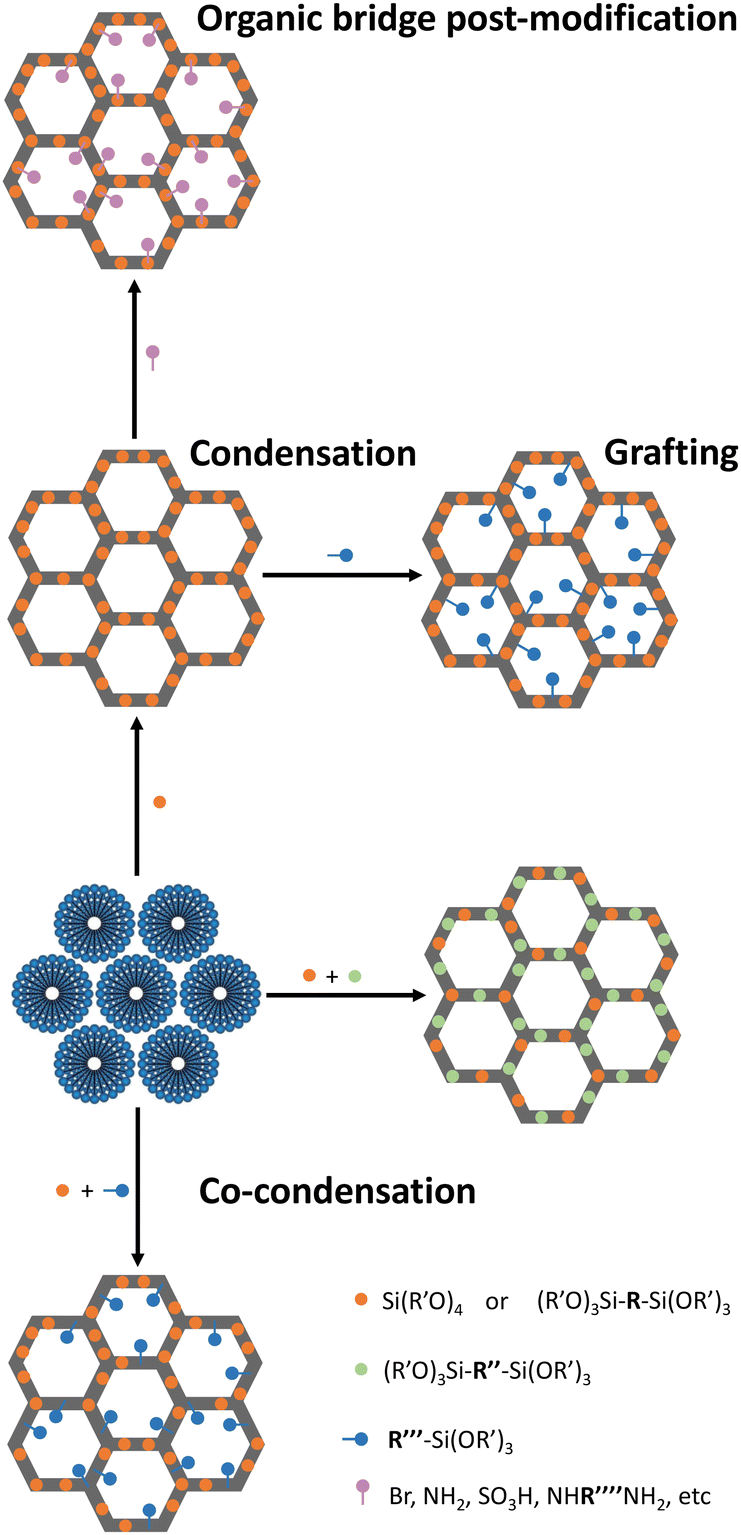 | ||
| Fig. 1 Schematic representation for the functionalization of PMS or PMO. | ||
These methods enable PMS or PMO materials’ tunability to achieve different polarities and/or introduce new reaction centres. These approaches have been employed to study the impact of factors such as amine loading, amine type, and inter-amine functional group distance in developing amine-modified porous silica (AMPS) sorbents specifically designed for CO2 adsorption–separation.37–39
2.2. Surface amine-modification towards enhanced CO2 capture
Despite post-modification through grafting often results in a decrease in the specific surface area depending on the content and size of the group introduced (cf., Table 1),43 this modification methodology is the most applied, because as opposed to the co-condensation method, the fact that the functionalization (grafting) occurs after PMS synthesis, it has minimal impact on mesostructure periodicity, often causing broad pore-size distributions, constraining the shape, size, and selectivity of these materials, as observed in CO2 sorbents prepared by co-condensation of bis((triethoxysilyl)propyl)ethylenediamine (BTEPED) and TEOS (Fig. 2 and Table 1).44| Sample type | Amine group/amine type | Amine loading (mmol g−1) | S BET (m2 g−1) | V p (cm3 g−1) | d p (nm) | CO2 ads. capacity (mmol g−1) | Ads. temp. (°C) | Ratiod CO2 ads./N loading | Ratio CO2 ads/SBET (mmol m−2 × 103) |
|---|---|---|---|---|---|---|---|---|---|
| a Specific surface area. b Pore volume. c Pore diameter. d Amine efficiency is calculated as the ratio between the CO2 adsorption capacity and amine loading. Under dry conditions, the maximum amine efficiency is 0.5. Under moisture conditions, the maximum amine efficiency is 1.0. e Adsorption measurements performed using 5% CO2 gas flow in a TGapparatus; n.p. denotes for non-provided data; abbreviation nomenclature: TEOS (tetraethoxysilane); BTEPED (bis((triethoxysilyl)propyl)ethylenediamine); PE-MCM-41 (pore expanded MCM-41); AP and APTMS (3-(trimethoxysilyl)propylamine); ED, N-3 and TSPED (N-[3-(trimethoxysilyl)propyl]ethylenediamine); DT (N1-(3-trimethoxysilylpropyl)diethylenetriamine); 3-DEAPTES ([3-(diethylamino)propyl]trimethoxysilane); TMMAP ([trimethoxy[3-(methylamino)propyl]silane]); APTES (3-(triethoxysilyl)propylamine); TEIPS (triethoxy-3-(2-imidazolin-1-yl)-propylsilane); DAB (diaminobutane); DAH (diaminohexane); DADD (diaminododecane); DETA (diethylenetriamine); TEPA (tetra-ethylenepentamine); Prim. (primary amine); Sec. (secondary amine); Tert. (tertiary amine). | |||||||||
| Co-condensation | |||||||||
| GD744 | BTEPED + TEOS/Sec. | n.p. | 735 | 0.75 | 5.0 | 1.5 | 0 | — | 2.04 |
| GD544 | 727 | 0.58 | 2.0/5.0 | 1.9 | 2.61 | ||||
| GD344 | 13 | 0.04 | Disordered | 2.1 | 161.54 | ||||
| GD044 | BTEPED/Sec. | 38 | 0.10 | 2.3 | 60.52 | ||||
| Grafting | |||||||||
| MCM-4145 | — | — | 1088 | 0.83 | 2.6 | 0.37 | 45 | — | 0.34 |
| PE-MCM-445 | 894 | 1.28 | 5.1 | 0.34 | 0.38 | ||||
| PE-MCM-AP45 | APTMS/Prim. | 2.43 | 544 | 0.74 | 4.3 | 0.87 | 0.36 | 1.60 | |
| PE-MCM-ED45 | N-3/Prim. + Sec. | 4.00 | 421 | 0.60 | 4.2 | 1.51 | 0.38 | 3.59 | |
| PE-MCM-DT45 | DT/Tert. Prim. + Sec. | 4.86 | 373 | 0.54 | 4.0 | 1.75 | 0.36 | 4.69 | |
| SBA-1546 | — | — | 717 | 0.92 | 7.0 | ∼0.65 | 25 | — | 0.91 |
| APTES@SBA-1546 | APTES/Prim. | 2.82 | 344 | 0.56 | 6.4 | ∼1.05 | 0.37 | 3.05 | |
| 3-DEAPTES@SBA-1546 | DEAPTES Tert. | 1.59 | 318 | 0.46 | 5.8 | ∼0.25 | 0.16 | 0.79 | |
| TMMAP@SBA-1546 | TMMAP/Sec. | 3.09 | 260 | 0.39 | 5.9 | ∼0.85 | 0.28 | 3.27 | |
| N-3@SBA-1546 | N-3/Prim. + Sec. | 3.93 | 241 | 0.39 | 5.5 | ∼1.45 | 0.37 | 6.02 | |
| APTES@PhPMO47 | APTES/Prim. | — | — | — | — | 0.72 | — | — | |
| TSPED@Ph-PMO47 | N-3/Prim. + Sec. | 4.90 | 180 | 0.37 | 7.2 | 2.25 | 0.46 | 12.50 | |
| TEIPS@Ph-PMO47 | TEIPS/Tert. | — | — | — | — | 0.40 | — | — | |
| APTMS@PhPMO48 | APTMS/Prim. | 1.39 | 634 | 0.43 | 2.2 | 0.72 | 0.52 | 1.14 | |
| Organic bridge post-synthetic modification | |||||||||
| DAB-Et-PMO49 | DAB/Prim. + Sec. | 1.81 | 368 | 0.42 | 5.4 | 0.16e | 0 | 0.09 | 60.53 |
| DAH-Et-PMO49 | DAH/Prim. + Sec. | 0.60 | 449 | 0.56 | 5.2 | 0.13e | 0.22 | 0.43 | |
| DADD-Et-PMO49 | DADD/Prim. + Sec. | 0.33 | 450 | 0.46 | 5.7 | 0.14e | 0.42 | 0.29 | |
| DETA-Et-PMO49 | DETA/Prim. + Sec. | 2.48 | 379 | 0.47 | 4.3 | 0.25e | 0.10 | 0.31 | |
| TEPA-Et-PMO49 | TEPA/Prim. + Sec. | 3.04 | 530 | 0.56 | 4.7 | 0.29e | 0.10 | 0.66 | |
| PhPMO48 | — | — | 1004 | 0.69 | 2.5 | 0.37 | 25 | — | 0.55 |
| NH2PhPMO48 | Aniline/Prim. | 1.85 | 924 | 0.70 | 2.4 | 0.40 | 0.22 | 0.37 | |
| Both grafting and organic bridge post-synthetic modification | |||||||||
| APTMS@NH2PhPMO48 | Aniline + APTMS/Prim. | 2.60 | 305 | 0.27 | 2.2 | 0.50 | 25 | 0.19 | 1.64 |
 | ||
| Fig. 2 Schematic illustration of PMS or PMO modifications through (a, b) grafting and (b, c) organic-bridge functionalization, employing various amine sources. | ||
Our group has used this approach to modify SBA-15 with similar loading of amine moieties with distinct bulkiness such as primary [3-aminopropyltriethoxysilane (APTES)], secondary [trimethoxy[3-(methylamino)propyl]silane (TMMAP)], and tertiary ([3-(diethylamino)propyl]trimethoxysilane) (DEAPTES) amines and also with a diamine containing primary and secondary amine groups [N-[3-(trimethoxysilyl)propyl]-ethylenediamine (N-3)] (Fig. 2(a)). The impact of having silica surfaces anchored with such distinct amine moieties critically influences the CO2 adsorption capacity, gas selectivity and CO2 speciation as shown ahead.
Our research aims to study the influence of amine type (e.g., primary, secondary) on CO2 adsorption50 and CO2/CH4 separation,46 the impact of water vapor upon CO2 adsorption on APTES- and DEAPTES-grafted SBA-1551 as well as the CO2-adducts formed in the amine grafted silica sorbents (see Section 3 for further details). The CO2 adsorption capacity increased from tertiary (DEAPTES@SBA-15), secondary (TMMAP@SBA-15), to primary amines (APTES@SBA-15), having the latter the highest ratio between CO2 adsorption and N loading (known as amine efficiency, ratio = 0.37, Table 1).46 The same amine efficiency for CO2 is obtained when the number of amines is almost 40% higher when both primary and secondary amines are used (N-3@SBA-15, Table 1). The selectivity towards CO2 in the CO2/CH4 mixture was higher in the presence of secondary amines (TMMAP@SBA-15 and N-3@SBA-15 sorbents, Fig. 2(a)). Similar conclusions can be perceived in other examples of AMPS45 listed in Table 1.
Alkyl amine-derived PMO (APTMS@PhPMO) obtained by the grafting method (Fig. 2(b)) seems to present a higher CO2 adsorption capacity when compared with similarly derived PMS (Table 1). APTMS@PhPMO shows a CO2 adsorption/N loading ratio twice the one obtained for the homologous SBA-15 material (APTES@SBA-15, Table 1), both tested at 25 °C.48 This enhancement of the adsorption capacity may be credited to the hydrophilic–hydrophobic character of the PMO sorbents, which can lead to a better homogeneity of the APTMS/APTES groups and a diffusion of the CO2 molecules along the pore channels. Nevertheless, it is essential to consider additional factors, including specific surface area, pore size, and pore volume, to ensure a more accurate and realistic comparison. Although this approach has provided materials with interesting properties, it comes with drawbacks such as low organic group loadings, which are often heterogeneously distributed within the pores, frequently inducing substantial pore volume reduction and channel blockage.
Our group also modified the Ph-PMO sorbent by an organic-bridge post-modification reaction through amination of the aromatic groups (NH2PhPMO) and by applying these two synthetic approaches (APTES@NH2PhPMO), Fig. 2(b), to study the influence of the type of amines on the CO2 adsorption–separation from CO2/CH4 gas mixtures.48 In tandem with computational modelling (cf. Section 5), we found that alkyl amines interact more strongly with the CO2 molecules than aromatic amines, such as the electron-donating pair of the N group is less available to react in the aromatic amines than in the case of alkyl amines. We also found that the type of amines is more relevant than their amount in the channels, as all these modifications lead to a decrease in amine efficiency compared with the APTES@PhPMO sorbent (Table 1).48
The organic-bridge post-modification (Fig. 1 and 2(c)) of PMO for CO2 adsorption with different proportions of primary and secondary amines and different chain lengths was also attempted by De Cank et al.49 The authors used a two-step approach to modify the ethylene bridge of Et-PMO with the alkylamines diaminobutane (DAB), diaminohexane (DAH), diaminododecane (DADD), diethylenetriamine (DETA) and tetra-ethylenepentamine (TEPA), Fig. 2(c). The authors found that this approach has advantages compared with the grafting procedure, such as higher thermal stability of the anchored amine groups. Additionally, they observed that the quantities of CO2 adsorbed are higher on PMO sorbents with low nitrogen content, showing the following CO2 adsorption capacity sequence PMO-DADD > PMO-DAH > PMO-DAB, which is the inverse of the modification degree (Table 1). A similar phenomenon is observed for the sorbents modified with tri- and penta-amines: PMO-DETA > PMO-TEPA.49
Developing the next-generation CO2 adsorbents relies on understanding how synthesis conditions impact the structural and textural properties of the sorbents, thereby influencing active adsorption sites at the atomic level. Achieving this requires the preparation of materials with control over the quantity and homogeneity of surface amine coverage. This aspect often implies performing grafting of organic moieties at a controllable intermolecular distance while maintaining the mesostructure. This approach addresses fundamental questions such as unravelling the role of silanol groups in CO2 speciation and uptake, identifying the optimal type of amine functional groups and the necessary degree of amine coverage for maximizing CO2 adsorption/selectivity. Moreover, it also helps to obtain atomic-level evidence about the benefits of cooperation between neighboring functional groups in CO2 speciation/uptake through a combination of ssNMR and computational methods. These methods allow structural elucidation of the CO2 species formed at confined surfaces at atomic resolution to be obtained. However, the bottleneck for future ground-breaking atomic level structural details on the CO2 adsorption mechanisms lies in the development of novel surface modification strategies targeted to boost the capabilities offered by NMR spectroscopy. The next section will provide insights on this subject.
2.3. Future perspectives in surface modification strategies towards the enhancement of NMR signal
Although ssNMR is a powerful technique for studying the structure and dynamics of silica CO2-sorbents, it is often hampered by its inherently low sensitivity. Several strategies are being explored to overcome such limitations: (i) improvements in hardware (e.g., high-field magnets) and pulse sequences, (ii) isotope labelling (such as 13C and 15N), and (iii) use of polarization techniques such as dynamic nuclear polarization (DNP), which consists in transferring polarization of electrons (located in polarizing agents) to the target nuclei (Fig. 3). Regarding (ii) and (iii), both isotopically labelled molecules and the polarizing agents may be incorporated during the synthesis and/or post-modification of silica materials, an activity that is currently being explored by many researchers, including our group.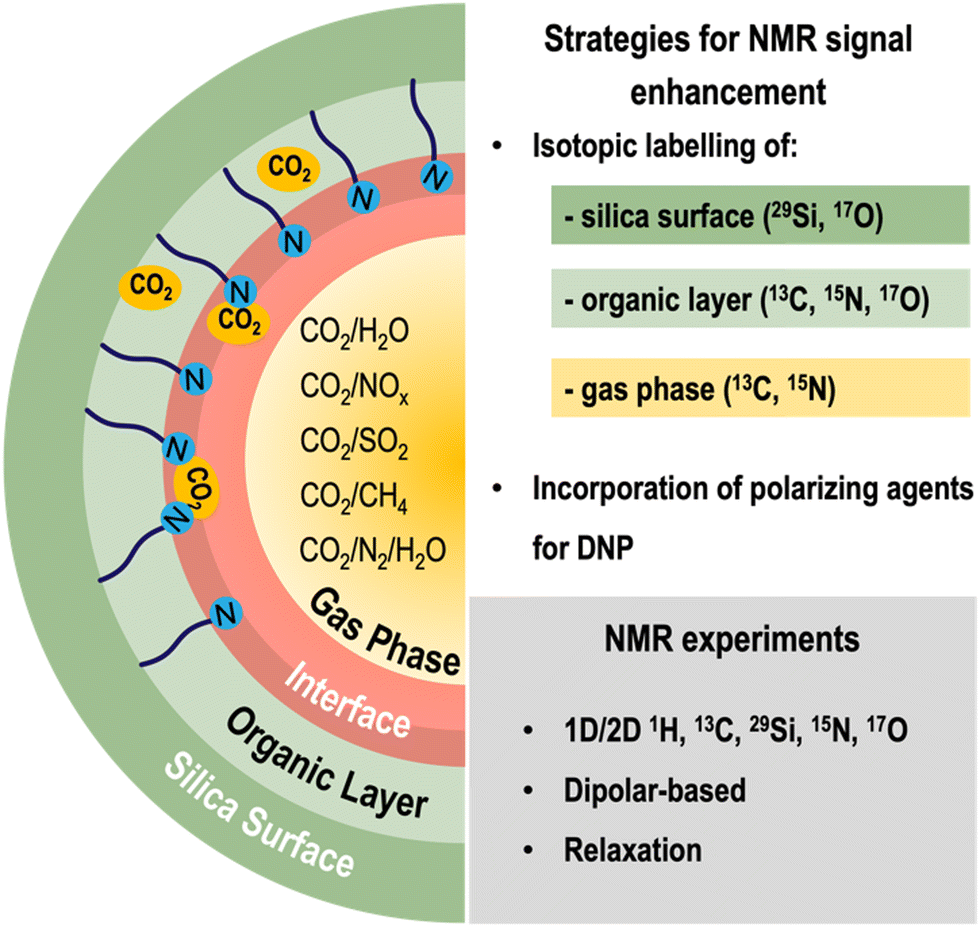 | ||
| Fig. 3 Overview of the methodology used for the ssNMR characterization of gas–solid interactions in silica CO2 adsorbents, involving strategies to enhance the NMR signal (namely isotopic labelling of the silica surface, the organic layer and/or the gas phase, as well as the incorporation of polarizing agents for MAS-DNP studies) as well as the main NMR experiments that may be explored for this application. | ||
Isotope labelling involves exchanging some or all the naturally occurring isotopes of a compound with NMR-active isotopes (e.g., 2H, 13C, 15N, etc.). When studying CO2 adsorption, some dilute chemisorbed CO2 species are hard to detect due to lack of sensitivity; hence, to overcome this limitation, our group resorted to the use of 13C-labelled CO2 (13CO2) loaded materials to study CO2 speciation in AMPS.
Using this approach, it was possible to obtain new information on the nature of CO2 chemisorbed species in AMPS sorbents,50 reinforcing the potential of other isotope labelling schemes such as 15N and 19F into the organic moieties of porous adsorbents. For instance, introducing fluorine atoms may not only confer improved chemical stability to the sorbent52–54 but also significantly enhance CO2 adsorption capacity and selectivity in CO2/N2 gas mixtures, while maintaining the NMR sensitivity conferred by the high natural abundance of 19F isotopes.55 The replacement of hydrogen with fluorine atoms also enhances the hydrophobicity of the materials rendering this choice more attractive for applications under moist conditions where water competes with other gases for the same adsorption sites.
Combining 13C and 17O isotope enrichment of the CO2 molecule may potentially enable the unambiguous assignment of CO2 species that are very challenging to assign using 13C alone as their chemical shifts fall in a very narrow spectral region. However, the potential of 17O NMR in this field might be hindered by the prohibitively high cost of 17O-labeled CO2 (ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 € per L when available). More affordable paths for isotopic labelling of gases are key to get the most of ssNMR applied to gas-loaded sorbents.
000 € per L when available). More affordable paths for isotopic labelling of gases are key to get the most of ssNMR applied to gas-loaded sorbents.
The other strategy used to enhance NMR sensitivity results from the application of DNP methods, profiting from the higher polarization of unpaired electron spins compared to the nuclear spins under the same experimental conditions. However, the process of introducing the polarizing agent, while maintaining the temperature and pressure conditions necessary for the gas adsorption is an enormous challenge and requires dedicated strategies. Traditionally, the polarizing agents, usually organic radicals, are introduced in the system56 either through impregnation methods56,57 or by grafting and co-condensation.58–60 These methodologies must be adapted for the case of gas adsorption studies. For instance, new radical formulations customized for porous materials and/or impregnation protocols under controlled conditions of temperature, pressure and gas composition, need to be developed to target this relevant niche application.
3. Studying CO2 adsorption mechanisms combining NMR and modelling
3.1. CO2 speciation studies
NMR spectroscopy has been widely employed to probe the local structure and dynamics of both the host framework and adsorbed guest molecules within porous materials.28,61–68 In the last few years, ssNMR has become the technique of choice to investigate the nature of CO2 species formed in AMPS materials, mostly under dry conditions.50,51,61,63,69–74 Traditionally, CO2 speciation studies arising from chemisorption exploit chemical shift (CS) analysis that can be performed with accuracy in combination with computer modelling. However, recently, NMR relaxometry in combination with chemical shift anisotropy (CSA) analysis has been explored to elucidate the speciation of the physisorbed fraction based on distinct dynamic behaviour.73,74 In the following section, the most recent and impactful works will be briefly discussed.In a pioneering NMR study that included computer modelling, 13C NMR resonances were assigned for the first time to specific structures of chemisorbed CO2 species on SBA-15 functionalized with propylamine groups, exposed to variable CO2 pressures (from 1.3 mbar to 1 bar) under dry conditions.69 Three distinct chemisorbed CO2 species were identified with different populations determined by the CO2 partial pressure employed (details in the description of the apparatus utilized for variable-pressure ssNMR below). The species include two variants of carbamic acid species (A, B) and one alkylammonium carbamate ion pair (C) (Fig. 4). Note that among the three 13C ssNMR methods used, only the MAS and MultiCP spectra offer quantitative results, with the latter delivering about an 18-fold reduction in experimental time.
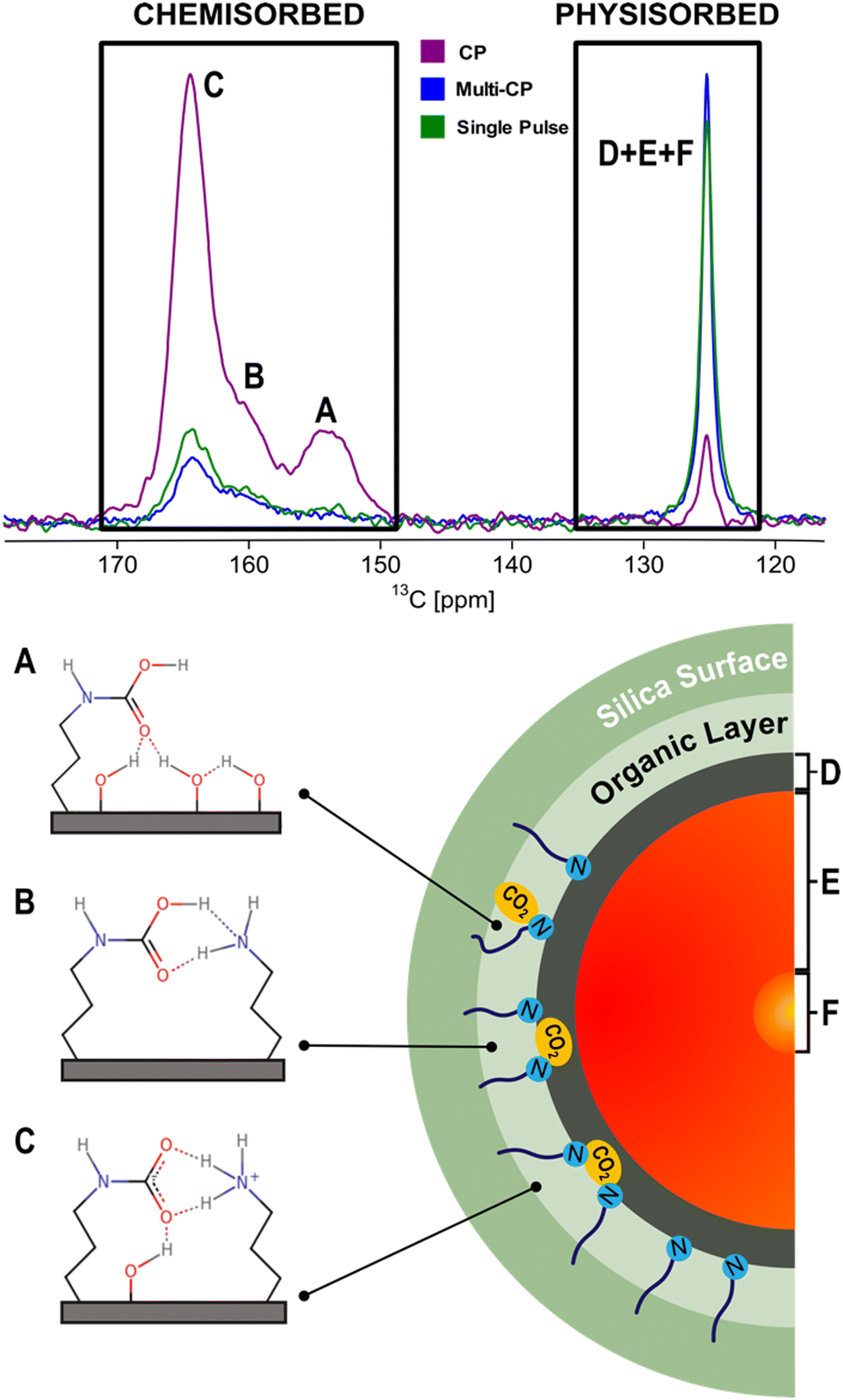 | ||
| Fig. 4 (top) Comparison between the 13C CPMAS (purple), MultiCP (blue) and single pulse (green) NMR spectra of dry APTES@SBA-15 after 13CO2 adsorption (bottom, left). Scheme of the three hypothesised CO2 chemisorbed species (A)–(C) formed in APTES@SBA-15 loaded with 13CO2 (P = 770 torr) as described in previous studies.50,71 (bottom, right) Scheme of a simplified CO2-filled pore of APTES@SBA-15 after adsorption, containing the physisorbed species (D)–(F) shown in our recent work.73Fig. 7 will further detail how the physisorbed CO2 peak is decomposed into three distinct components. Adapted from ref. 73 with permission from ACS, copyright 2021. | ||
The successful completion of this study relied heavily on the precise control over key variables, namely temperature, partial pressure, and humidity.
To regulate these conditions, an experimental setup was constructed (Fig. 6), consisting of a custom-built high-vacuum apparatus linked to a turbomolecular pumping station capable of achieving a vacuum level greater than 10−4 Pa. A borosilicate glass container was connected to the vacuum apparatus and utilized as a chamber to house an NMR rotor. This arrangement facilitated the degassing and heating of zirconia NMR rotors under high vacuum conditions, with temperatures reaching up to 300 °C. The heating process was carried out using a specifically designed oven, which was connected to a power controller, and the temperature was monitored using a thermocouple. The desired gas was introduced into the system through a canister connected to both the vacuum apparatus and the glass container. The pressure within the glass container was measured using a capacitance transducer. Subsequently, our innovative sorption device served as inspiration for further research on CO2 adsorption on amine-functionalized MOFs utilizing ssNMR spectroscopy.75
In 2018, T. Čendak et al. helped to elucidate how the proximity of amine groups affects the formed CO2 species in SBA-15 silica, using 13C CSA NMR experiments.50 Two strategies were employed to modify the average distance between the anchored amine groups. First, the amine surface density was varied by changing the quantity of loaded amine groups, and second, amine moieties were protected with bulky carbamate groups ((3-triethoxysilylpropyl)-tert-butylcarbamate: TESPtBC) to prevent the clustering of the propylamines during grafting. The TESPtBC group is easily removed by heat prior to CO2 adsorption, thus regenerating the initial primary amines with augmented inter-amine spacing. This second approach favoured the formation of isolated propylcarbamic acid species, while the first promoted ion pair species due to the formation of amine aggregates. To determine the protonation state of each chemisorbed CO2 species, we monitored the changes in the principal values of their 13C CSA tensors by means of spinning sideband analysis through 13C cross-polarization (CP) NMR, at low MAS rates (2 kHz). A comparative evaluation of our results versus a repository of 13C CSA tensor parameters comprising 70 protonated and deprotonated carboxylates found in amino acids, facilitated the successful assignment of resonances corresponding to either aggregated or isolated CO2 species.50 In addition, Shimon et al.76 examined the system of a propylamine-functionalized SBA-15 when exposed to CO2. The material was loaded with 1 bar of CO2 and 15N CPMAS NMR experiments were carried out, with both carbamic acid and carbamate being observed in the results. Although CP is not quantitative, it was concluded that both carbamic acid and carbamate were formed after exposure to CO2, with the former desorbing faster than the latter. The authors also noted that, 100 hours after loading the sample with CO2, there was less carbamic acid than initially, while the quantity of carbamate remained constant.
While chemisorbed species often exhibit distinctive 13C CS, the physisorbed CO2 shows a single resonance at 125 ppm (Fig. 4). Our group showed that it is possible to discriminate among three physisorbed CO2 species using 13C NMR relaxation and 13C CSA measurements.73 The 13C MultiCP spectrum of CO2@APTES-SBA15 displays the presence of spinning sidebands, which can be associated with a fraction of CO2 molecules (species D) with fewer degrees of freedom when confined. Deconvolution analysis demonstrates that the signal with spinning sidebands corresponds to a small fraction of the full physisorbed CO2 (15%). Moreover, relaxation studies, namely T1 measurements, showed three distinct superimposed components associated with the single resonance of physisorbed CO2 at 125 ppm. These three CO2 species have T1 values of 2.4 s, 0.2 s and <500 μs, which fall in the range of CO2 species in a solid-like (species D), liquid-like (species E) and gas (species F) state, respectively (Fig. 4). The species D is associated with CO2 molecules directly interacting with the surface of the adsorbent, as confirmed by a 1H–13C HETCOR experiment,73 which clearly showed correlations between the 13C CO2 resonances and the 1H of the propylamine, and silanol groups from the silica surface. The distinct dynamics observed in species E and F could potentially be attributed to the density gradient spanning from the wall to the pore centre. This phenomenon may arise from the distance-dependent interaction between the CO2 molecules and the pore surface. Moreover, the combination of slow MAS/Multi-CP 13C NMR at MAS frequencies of 10 and 2.5 kHz and a T1 saturation-recovery experiment under quantitative conditions provides a toolbox for unambiguous identification and quantification of different CO2 chemi- and physisorbed species confined in AMPS. For the case of CO2@APTES-SBA15, the results showed that 45% of the total adsorbed CO2 was chemisorbed, with species A, B, and C representing 2%, 16%, and 27%, respectively. In the case of the physisorbed fraction, species D, E and F accounted for 8%, 39% and 8%, respectively. The methodology developed in this work is the basis for performing the NMR-assisted adsorption studies described in Section 4, also applicable to any other porous system.
Recently, chemisorbed CO2 species in MOFs and AMPS were studied by 17O ssNMR.77 The authors undertook a computational investigation to determine the utility of 17O ssNMR to probe CO2 capture chemistry. The simulations were supported by 17O NMR experiments on a series of MOFs loaded with C17O2, which indicated the formation of ammonium carbamate chains and a mixed carbamic acid–ammonium carbamate adsorption mode. The study was further extended to AMPS adsorbents: propylamine-SBA15 (Pr-Si) and triamine-SBA15 (Tri-Si) were loaded with 1083 mbar and 993 mbar of C17O2, respectively. The 17O MAS NMR spectra showed broad signals at 177.5 ppm and 28.7 ppm for Tri-Si, and 175.5 ppm and 33.3 ppm for Pr-Si. The signal at ∼175 ppm was assigned to ammonium carbamate oxygens, by comparison with Density Functional Theory (DFT) calculations for AMPS. However, the signals at ca. 30 ppm could not be assigned to any of the previously proposed carbamic acid, ammonium bicarbonate, or silylpropylcarbamate species. Therefore, the authors suggest either a new adsorption mode in AMPS, or isotopic enrichment of oxygen atoms in the silica backbone as possible explanations. This study highlights the utility of 17O NMR measurements for probing CO2 capture modes, opening the door to complement information retrieved from 13C NMR to study CO2 speciation in porous materials. However, as mentioned previously, the 17O enrichment of CO2 is prohibitively expensive.
3.2. Moisture-induced CO2 speciation
Water is a critical component of flue gases with varying proportions depending on the combustion source. The presence of water in flue gases has a major impact on the CO2 speciation formed. Hence, a careful investigation of the role of water is essential to optimise AMPS sorbents for CO2 capture in moist gas mixtures. Moreover, it has been shown that the presence of moisture prevents chemisorbed carbamic acid degradation into urea which leads to sorbent inactivation.78 Despite the importance of the role of water in the CO2 adsorption in silica porous materials, the subject has been weakly explored by NMR.51,70,79,80 This may be related to the difficulty of introducing an accurate proportion of water in the gas mixture.Amongst the few studies addressing the issue, our group investigated the CO2 speciation in SBA-15 derivatized with primary and tertiary amines in the presence of moist gas mixtures.51 The proportion of water was precisely controlled using a dedicated vacuum line. The study revealed that in the case of SBA-15 grafted with primary amines, although the presence of water does not affect CO2 capture, it promotes the conversion of carbamic acid into carbamate ion pairs by shuttling the proton from carbamic acid to a neighbour amine. The dominant CO2 species thus becomes the carbamate ion pair. In the case of tertiary amines, the formation of bicarbonate dominates the chemisorbed species with a small contribution from a carbamic acid species (Fig. 5). The experimental data were complemented by DFT calculations for the prediction of 13C CS for each species.
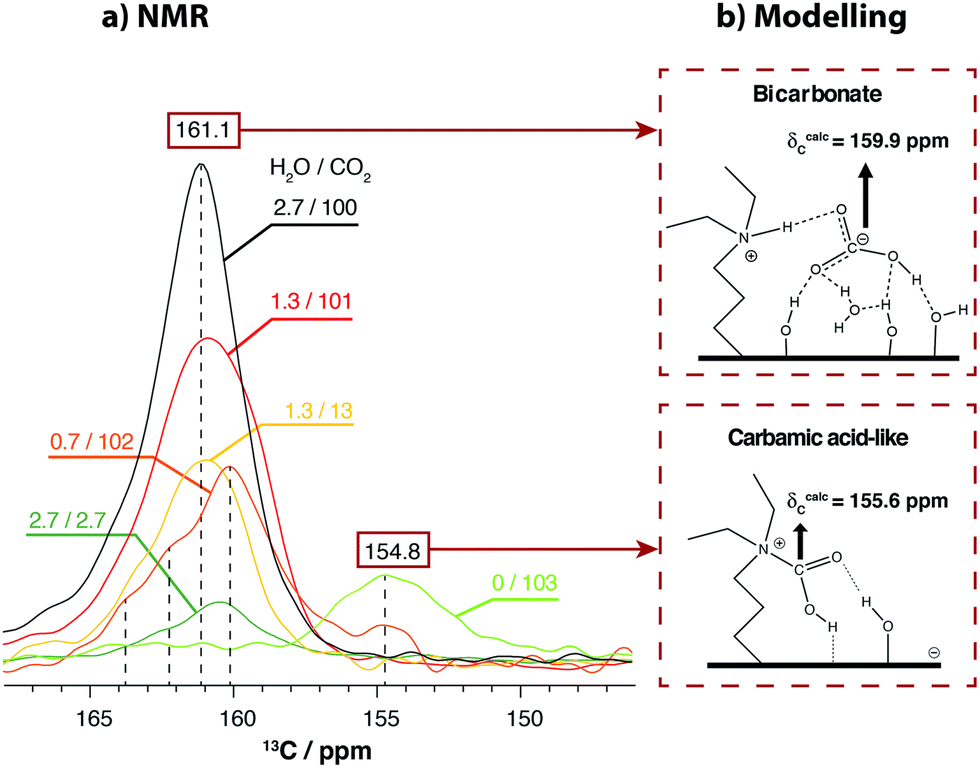 | ||
| Fig. 5 (a) 13C CPMAS NMR spectra of the chemisorbed species formed in DEAPTES@SBA-15 after exposure to different partial pressures of H2O followed by adsorption of 13CO2. Specific partial pressures are given in the figure, in kPa. The spectra were normalized with respect to the intensity of the propylamine carbons to facilitate comparison. (b) 2D representation of the optimised structures for bicarbonate and carbamic acid-like species. Reproduced from ref. 51 with permission from RSC, copyright 2021. | ||
The CO2 speciation in SBA-15 grafted with tertiary amines has also been investigated by Chen et al.80 In this work, the water was introduced into the sample through the preparation of slurries resulting in samples with an uncontrolled proportion of water. The samples were exposed to pure 13CO2 and the resulting system was explored by 13C–1H HETCOR spectroscopy. They conclude that two types of bicarbonates were formed depending on two water environments. One resulting from the interaction with water in the proximity of silanol groups and another from the interaction with bulk water in the pore centre.
The CO2 speciation in deaminated silica has been recently addressed by Szego and co-workers.79 Chromatographic particles of porous silica Davisil LC60 were functionalized with a primary amine (APTES) and a diamine (3-(2-aminoethylamino)propyltriethoxysilane (AEAPTS)). Upon adsorption under both dry and wet conditions, the chemisorption of CO2 was analysed by a diversity of ssNMR methods. The single-pulse 13C and 13C CPMAS NMR spectra showed that the chemisorbed species in the dry samples were carbamic acids and carbamate ammonium ions, while in the wet samples, both species were detected together with HCO3−. The authors demonstrate that the CP efficiency to the HCO3− was reduced as compared to the directly detected 13C NMR. However, 13C CPMAS NMR could still be used for qualitative assessment of the presence of HCO3− without having to cool the samples down. This technique is a promising tool for evaluating the chemisorption of CO2 on mesoporous silica and could be useful for similar samples without isotopic 13C enrichment. Additionally, it was proposed that the presence of H2O in the aminated samples resulted in a sharpening of all signals (1H and 13C) under rapid MAS, suggesting an increased mobility of the species. This sharpening enabled the detection of two distinct types of carbamates formed upon CO2 chemisorption on the diaminated sample, as well as the resolution of most of the signals from the alkyl chains in the 1H domain. The high resolution of the 1H and 13C NMR spectra may provide a better understanding of how AMPS capture CO2 and promote their rational application in fields such as catalysis.
3.3. CO2 adsorption in gas mixtures
Understanding CO2 capture mechanisms in solid sorbents from realistic gas mixtures is an essential step towards the final application of these materials in real scenarios. Also, these studies are important when trying to find a particular application for a given solid porous material. For instance, the adsorption behaviour of a sorbent material will be strongly influenced by the origin of the gas mixture (flue gas, biogas, or direct air capture). In the last years, many studies have emerged focusing on the selective CO2 adsorption from real mixtures with different origins using distinct solid sorbents.46,64,65,81–91 However, few studies have been published investigating the adsorption mechanisms in AMPS by ssNMR. The studies published in this respect will be briefly discussed below, emphasizing the utility of NMR techniques to address the subject.A pioneering work published in 2018 by our group46 demonstrated the potential of SBA-15 materials incorporating amine groups for CH4/CO2 separation, such as found in natural, bio and landfill gases. Aminosilanes of distinct steric hindrances were grafted onto SBA-15 silica and adsorption measurements were conducted up to 1000 kPa (10 bar) at 25 °C using CO2/CH4 mixtures, providing useful insights into potential applications. Remarkably, we observed extremely high CO2/CH4 selectivity with values of 1114 and 15000 for secondary (TMMAP) and mixed primary/secondary (N-3) amine-functionalized SBA-15, respectively. This selectivity was attributed to the reaction of CO2 with surface amines, as confirmed by ssNMR of samples exposed to 13CO2. Furthermore, the primary and secondary amines remain unsaturated with CO2 at pressures below 40 kPa, suggesting that they can be partially regenerated by decreasing pressure. It was also reported that samples containing primary and secondary amines have a higher selectivity than SBA-15 due to their interactions with the polar carbamic acid species on the surface of the pores. Solid-state NMR data confirm this, as it reveals an interaction between the amines and CO2 not seen in SBA-15. At lower pressures, the relatively large mesopores and increased chemisorption of CO2 explain the higher selectivity, while polar interactions between the amines and CO2 further promote CO2 adsorption and decrease CH4 adsorption at higher pressures. Moreover, a preliminary evaluation of the materials' capacity for cyclic separation by pressure modulation revealed that TMMAP@SBA-15 and N-3@SBA-15 samples exhibit higher selectivity for CO2/CH4 separation, but lower working capacities than the primary amine APTES@SBA-15. The latter presents a better compromise between selectivity and working capacity, especially if a vacuum swing is used with a moderate vacuum (10 kPa regeneration pressure). Pacheco et al. in a follow-up work81 studied the influence of water in the separation of CO2/CH4 mixtures using AMPS materials. The adsorption behaviour of primary and tertiary AMPS (APTES@SBA-15 and DEAPTES@SBA-15) for CO2 and CH4 was studied under both dry and wet conditions. It was found that APTES@SBA-15 had a higher CO2 uptake than DEAPTES@SBA-15 due to the type and strength of interactions involving primary amines and CO2 at the pore surface. Meanwhile, DEAPTES@SBA-15 demonstrated an increase in CO2 uptake in the presence of pre-adsorbed water, attributed to the formation of bicarbonate species. This resulted in a much higher selectivity for CO2 in the CO2/CH4 separation for DEAPTES@SBA-15 than for APTES@SBA-15. Further investigations using 13C NMR spectroscopy and DFT calculations confirmed the formation of bicarbonate species when using tertiary amines under moist conditions, and a general stabilisation of the CO2 species formed when using primary amines. It was concluded that the low quantity of water present in a typical biogas stream would not significantly affect the CO2/CH4 separation in the first adsorption cycle but could lead to the observed effects during subsequent cycles. This work stresses the importance of the tandem use of DFT/ssNMR in addressing CO2 speciation in complex mixtures.
Flue gases discharged from coal-fired power plants contain not only carbon dioxide (CO2) but also acid–gas impurities, such as sulphur oxides (SOx) and nitrogen oxides (NOx), with typical concentrations ranging from 0.2 to 0.25 vol% and 0.15 to 0.2 vol%, respectively.92 The presence of these impurities poses various practical challenges in CO2 capture processes, leading to reduced adsorbent lifespan and capacity, adsorbent poisoning, decreased product purity, and increased operational expenses.93,94 While the existing literature predominantly focuses on the development and characterization of adsorbents with favourable CO2 capacities, limited attention has been devoted to the investigation of CO2 mixtures containing these acid-gas impurities. The lack of studies examining complex mixtures is predominantly attributed to the challenges arising from the corrosive and toxic nature of these gases. Razaei et al. examined the irreversible binding of SO2, NO, and NO2 to four supported amine adsorbents, assessing their adsorption capacities and their impact on CO2 adsorption capacities.95 The adsorbents consist of poly(ethyleneimine) and three silane coupling agents with primary, secondary, and tertiary amines, and their performance was evaluated using different characterization techniques, including 13C ssNMR. Under the experimental conditions used, it was observed that primary amines with high amine loadings exhibited a greater affinity for NO compared to their secondary and tertiary amine counterparts. However, the overall NO adsorption on the aminosilica adsorbents was found to be low, resulting in negligible changes in the CO2 capacities of the exposed adsorbent materials. Conversely, all materials displayed a notable adsorption capacity for nitrogen dioxide (NO2). Consequently, all adsorbents treated with NO2 demonstrated a substantial reduction in CO2 capacity, which can be attributed to the deactivation of amine groups caused by the irreversible binding of NO2. In materials with similar amine loadings, secondary amines exhibited a higher affinity for SO2; however, their loss in CO2 capacity after exposure to SO2 was lower than that of primary amines. This suggests that secondary amines are more stable in the presence of SO2, indicating greater desorption of SO2 from secondary amines during the desorption stage. These results highlight the need to reduce SO2 and NO2 concentrations in flue gas prior to the CO2 capture process. Conversely, the presence of NO does not significantly impact the capture efficiency of these materials. This suggests that such materials hold promise for post-combustion CO2 capture from flue gas streams derived from natural gas combustion, which typically exhibit reduced SO2 concentrations but significant NOx concentrations. In a follow-up article, the same authors evaluate the stability of amine adsorbents to SO2 and NOx in post-combustion CO2 capture by performing dynamic, multicomponent adsorption experiments in a fixed bed.96 This study investigates the effects of SO2, NO, and NO2 impurities on the dynamic adsorption capacity of amine-impregnated and amine-grafted silica adsorbents in CO2 adsorption breakthrough experiments where a defined mixture of gases flows through the packed-bed sorbent column. The experiments involve dual-component co-adsorption of SO2/CO2, NO/CO2, and NO2/CO2, as well as three-component SO2/NO/CO2 adsorption. The results indicate that SO2 significantly impacts the dynamic CO2 capacity of aminosilica adsorbents, but the adsorbents remain stable during co-adsorption if the bed is not fully saturated with SO2. NO shows little competitive effect on CO2 adsorption, attributed to the lower affinity of amine-based adsorbents towards NO compared to SO2. The presence of NO2 at low levels in the simulated flue gas has minimal impact on CO2 adsorption. Among the investigated adsorbents, those containing secondary amine groups demonstrate greater stability against SOx and NOx impurities in CO2 capture processes compared to adsorbents with primary amine groups. However, additional studies are required to address crucial aspects such as the potential adsorption mechanisms of these acid gases and the impact on the speciation of chemisorbed and physisorbed CO2 fractions. ssNMR spectroscopy will play a pivotal role in further investigations, as it possesses the capability to differentiate between distinct chemical and dynamic environments, regardless of the crystalline nature of the solid material.
3.4. Correlating CO2 structure and dynamics through NMR relaxation studies
Solid-state NMR has been used extensively to probe dynamics in biomolecular systems providing information about local interactions and the motional modes of molecular groups.97–100 In porous materials, studies of molecular motion have been performed in adsorbed CO2 in MOFs.101 However, NMR studies addressing the molecular dynamics of the different CO2 species in porous silica are scarce.73,74,102–104 The information obtained from these studies, far from being a scientific curiosity, is especially useful to assess important aspects such as stability or activation energies of the distinct CO2 species formed in confinement.Recently, our group has conducted for the first time a comprehensive analysis of the molecular dynamics of chemi- (A, B and C) and physisorbed (D, E, and F) CO2 species in amine-modified SBA-15 using ssNMR to measure their rotating frame spin–lattice relaxation times (T1ρ).74 The chemisorbed species A, B, and C were found to have the shortest T1ρ values (4.7, 1.1 and 1.4 ms, Table 2) as expected due to their highest rigidity. The T1ρ value of 8.1 ms for species D, which is engaged in weak interactions with the silica surface, was found to be intermediate. For the two pure physisorbed CO2 species E and F, the T1ρ values were much higher (10.0 and 44.0 ms, Table 2), reflecting their highly dynamic nature. Additionally, to obtain further insight into the dynamics of CO2 confined in amine-modified SBA-15, rotational correlation times (τc) were estimated following the dependence of T1ρ with the locking field (Table 2). The experimental data were fitted using either the Bloembergen–Purcell–Pound theory,105 for physisorbed CO2 species E, or the Bloch, Wangsness106 and Redfield theory107 for chemisorbed species A–D. The obtained τc values for species D (32 μs) and E (20 μs) are typical for molecular dynamics of a viscous liquid, while the chemisorbed species A–C exhibit longer τc values (162, 62, and 123 μs, respectively), reflecting the higher CO2 molecular rigidity. This analysis permitted the estimation of heteronuclear dipolar coupling constants and reduced CSA values, providing further insight into the nature of the adsorbed CO2. Previous studies combining ssNMR and DFT calculations50,69 coincide with these results, reinforcing the identification of the chemisorbed species A and B as two carbamic acid species, and C as an alkylammonium carbamate ion pair. These results further support the presence of at least two physisorbed species (D and E). However, the fast dynamics of species F made it impossible to carry out this analysis due to instrumental limitations.
| 13CO2 species | Fraction% | T 1/s | T 1ρ/ms | τ c/μs | b IS/Hz | δ/ppm |
|---|---|---|---|---|---|---|
| A | 2(1) | 57(4) | 4.7(1) | 16(3) × 101 | 69(3) × 102 | 7(1) × 101 |
| B | 16(5) | 7(1) | 1.1(1) | 62(2) | 153(3) × 102 | 7(1) × 101 |
| C | 27(8) | 12.8(4 × 10−15) | 1.4(1) | 123(1) | 22(1) × 103 | 49(9) |
| D | 8(1) | 2.4(3) | 8.1(1) | 32(4) | 40(2) × 102 | 6(1) × 101 |
| E | 39(1) | 0.09(2) | 9.4(9) | 20(4) | — | 0.20(3) |
| F | 8(2) | <5 × 10−4 | 44(5) | — | — | — |
4. NMR-assisted adsorption studies
4.1. Conventional approaches and their limitations
The performance of adsorbents for gas uptake is typically evaluated using manometric (also called volumetric) and gravimetric adsorption techniques. These methods provide a graph showing the partial gas pressure (or time) versus adsorbed gas amount at a constant temperature – an isotherm. The isotherm contains important information regarding sorbent's textural properties (specific surface area, pore volume, pore size distribution, etc.) and parameters related to its performance in gas capture (adsorption capacity, selectivity to a certain gas and isosteric enthalpy of adsorption, as the most important ones).108 However, despite their wide use, these conventional methods alone do not shed light on crucial questions such as, what is the contribution of the chemi- or physisorption to the adsorption process under these particular conditions? Neither do they give insight into the nature of adsorption sites. Therefore, to fully understand the performance of solids in gas adsorption applications, the use of additional techniques is mandatory. In this section, alternative techniques for elucidating adsorption properties of materials are discussed giving special attention to the ssNMR-assisted techniques.4.2. Low-field-NMR-assisted gas “relaxorption”
Owing to the sensitivity to probe adsorbate–adsorbate and adsorbate–adsorbent interactions, NMR relaxation methods have been used to quantify adsorbed gases on solids enabling to register adsorption isotherms.109–118 In general, this approach covered a wide range of materials including zeolites, mesoporous silica, activated carbons and MOFs which were probed for the adsorption of CO2, CH4, C2H6, C3H8 and also vapours of MeOH, PrOH and H2O for gas capture/separation and other emerging applications. In most cases, these experiments were limited to the use of low-field magnets and carried out ex situ.109–112,114 In contrast, in situ tests were less common mainly due to the difficulties in designing and building the experimental setup. One alternative was found by integrating NMR with either a sorption analyser or a homemade gas dosing system.115–119 For instance, the adsorption of MeOH vapours was studied in the sorption apparatus coupled with the low-field NMR spectrometer providing simultaneous in situ and in operando measurements. Information on pore filling, structural heterogeneity and host phase transition was obtained.115 Furthermore, due to the unique properties of 129Xe (high sensitivity to the surrounding chemical environment resulting in a significant difference in CSs on 129Xe NMR spectra) it has been widely used as a probe molecule to study adsorbents.120 Its diffusion and adsorption on materials were followed to calculate specific surface area, pore volume, isosteric enthalpy of adsorption of gases on solid materials. To sum up, all these methodologies are limited to follow the physisorbed fraction of the adsorbate, mostly restricted to following 1H signals in the adsorbed molecule due to sensitivity reasons.4.3. CO2 adsorption isotherms by high-field ssNMR-assisted techniques
As mentioned above, the use of NMR methods to collect adsorption data has been mainly limited to the use of low-field NMR. However, our group has tried to explore ssNMR methodologies to overcome these shortcomings as many advantages would arise from applying the standard high-resolution ssNMR features (high-field magnet and MAS), which are crucial to record full adsorption isotherms of confined non-protonated gas molecules. One of the first attempts to apply variable pressure ssNMR under MAS for the CO2 adsorption on mesoporous silica and detect different adsorbed gas adducts was made by our group.69 Moreover, in a follow-up work, it was demonstrated that the quantification of chemisorbed CO2 species is possible by using MultiCP NMR spectroscopy while the physisorbed species may be quantified by means of T1 measurements.73 Very recently, we have also shown that the ssNMR-assisted method is able to record full gas adsorption isotherms as it was reported for 13CO2 adsorption on APTES@SBA-15.121 The scheme with the different stages of our methodology is represented in Fig. 6. Recorded adsorption data were validated by comparison with the data obtained using conventional static manometric techniques (Fig. 7(a)). | ||
| Fig. 6 Schematics of the methodology used to perform qualitative and quantitative characterization of different chemi- (A)–(C) and physisorbed (D)–(F) CO2 species formed at different gas partial pressures. Reproduced from ref. 121 with permission from ACS, copyright 2023. | ||
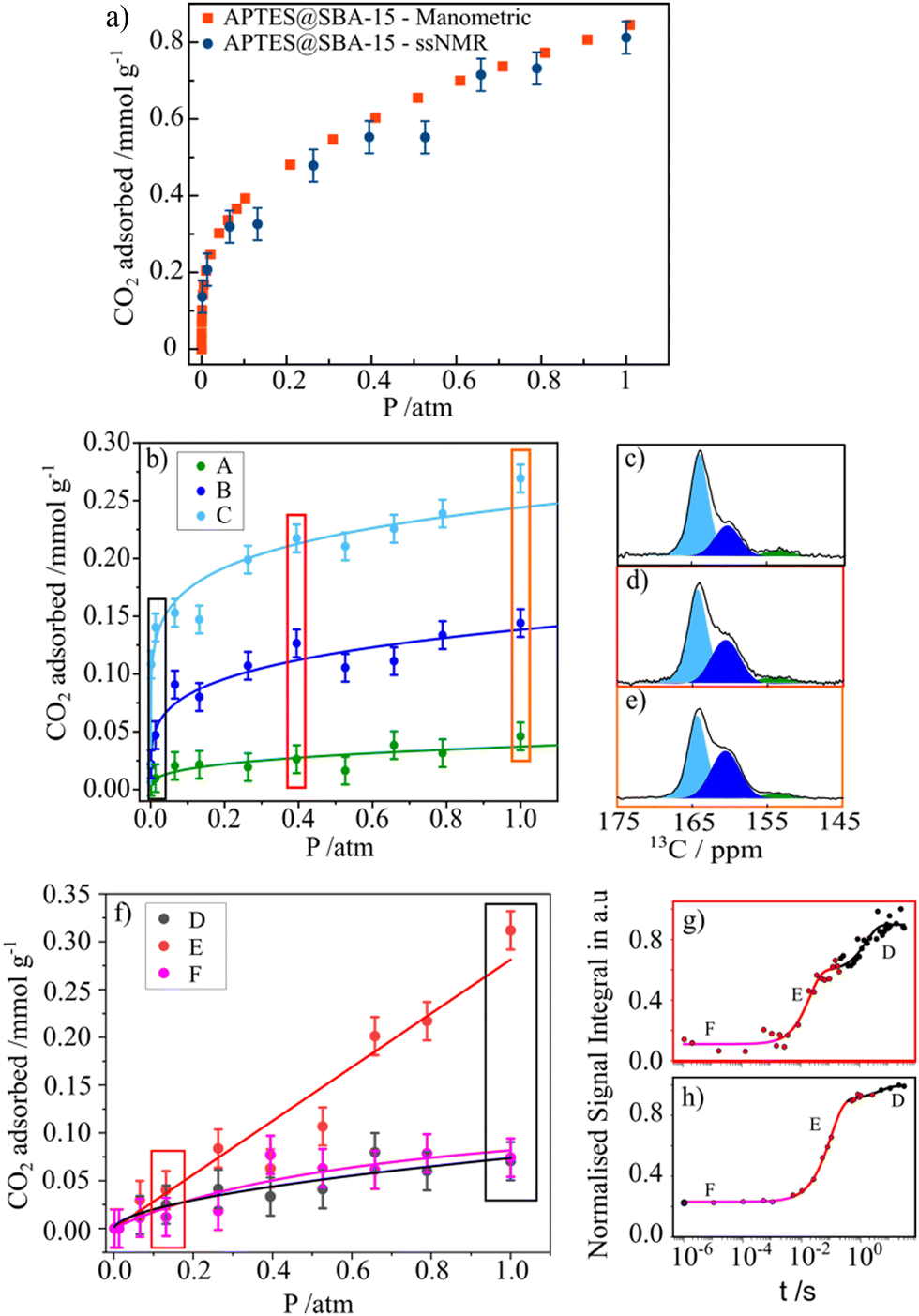 | ||
| Fig. 7 (a) CO2 adsorption isotherms (recorded at 298 K) for APTES@SBA-15 obtained by the manometric (red) and ssNMR (blue) techniques (vertical lines depict the error bars). (b) ssNMR isotherms of the chemisorbed CO2 components (A)–(C) fitted with the Freundlich model. (c)–(e) Selected 13C MAS NMR spectra corresponding to the isotherm data points recorded at 0.01, 0.39 and 1.00 atm, respectively. (f) ssNMR isotherm of the three physisorbed CO2 components (D)–(F) fitted using the Langmuir (E) and (F) and the Freundlich (D) models. (g) and (h) NMR saturation recovery curves of the corresponding isotherm data points recorded at 0.13 and 1.00 atm. Reproduced from ref. 121 with permission from ACS, copyright 2023. | ||
Moreover, applying the ssNMR-assisted approach enabled the collection not only of the full adsorption isotherm, but also of individual isotherms for each chemi- (A, B and C, Fig. 7(b)–(e)) and physisorbed CO2 species (D, E and F, Fig. 7(f)–(h)), which is impossible using conventional adsorption isotherm methods. The obtained isotherms were fitted using Langmuir, Freundlich or Sips models and corresponding parameters (maximum gas uptake, affinity of the gas to the material and heterogeneity of the adsorption sites) were derived from them. This methodology is already being applied by our group to a broader list of materials such as zeolites, carbons and MOFs, thus showing its potential in tailoring more efficient materials for gas adsorption and separation processes.
5. Computational modelling of silica-based sorbents at the meso- and molecular-scale
Computational studies provide insights into the gas–surface interactions in a myriad of materials, including silica-based sorbents. Computational tools have already been applied to (i) assist ssNMR to unravel the mechanisms of CO2 adsorption on AMPS,46,69,71 (ii) understand and predict the CO2 physisorption performance of a variety of PMO materials displaying different chemical functionalities48,122–125 and (iii) simulate dynamic gas sorption measurements on silica-based sorbents.126–129 Apart from silicas, these studies place significant emphasis on investigating other materials such as MOFs, COFs, and zeolites.130,1315.1. Modelling silica-based adsorbents
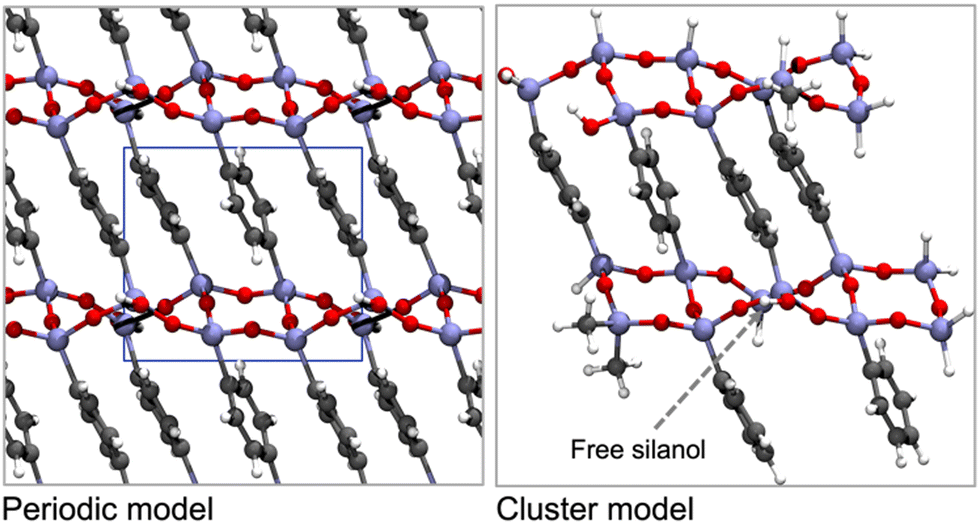 | ||
| Fig. 8 Top views of the periodic model (left, with the unit cell depicted inline) and the selected cluster model (right) for Ph-PMO. The colour code shows H in white, C in grey, O in red, and Si in beige. Schemes are based on the models presented by Lourenço et al.123 | ||
Later, our group selected a model encompassing six- and four-member rings (Fig. 9) of organosilica with T3 to T2 silicon environments in a 2:1 ratio (Tn = RSi(OH)(3−n)(OSi)n, R represents the organic bridge), as this seemed more realistic considering the characterization data from ssNMR obtained by Comotti et al.,132 for studying the interaction of gas molecules on the surface of a variety of PMO sorbents.
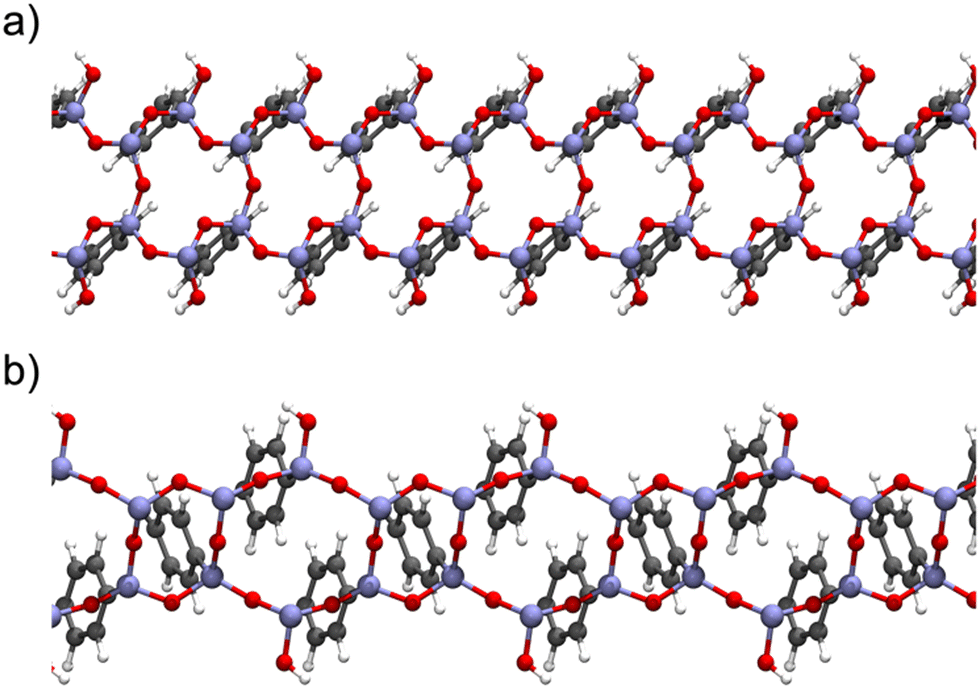 | ||
| Fig. 9 Lateral view of the Ph-PMO periodic models (a) encompassing six- and four-member rings of organosilica with T3 to T2 silicon environments in a 2:1 ratio and (b) corresponding to a regular sequence of six-member silica ring with alternate T3 and T2 species in a 1:1 ratio. Schemes are based on the models presented by Martinez and Pacchioni.122 | ||
Cluster models are more affordable than periodic models, and their validation is fundamental to the computational study of the interaction of different gas molecules at the surface of PMO structures. Our group observed that both types of models,48,125 were accurate in the determination of CO2 and CH4 adsorption performances on Ph-PMO sorbents, as the obtained data agrees with the experimental adsorption measurements, regardless of the chosen DFT exchange–correlation functional (Table 3 and Fig. 8).
| Gas | Modela | DFT method | E ads (kJ mol−1) |
b ratio |
|---|---|---|---|---|
| a PW/PAW and GTO stand for plane-wave/projected-augmented wave and Gaussian type orbitals, respectively. b Ratio between the calculated adsorption energies of CO2 and CH4. c Experimental isosteric heat of adsorption. d From adsorption isotherms (temperatures, pressure range and uncertainty not specified) in ref. 132. e Ratio between the Henry's constant for CO2 and for CH4 determined in ref. 48. f Second most stable structure for the PBE-D2 method. g From adsorption isotherms at T = 285 K, 298 K and 314 K and pressures of 0.01 to 0.1 MPa determined in ref. 133. | ||||
| CO2 | Periodic PW/PAW | PBE-D2 | −21.7 | 2.24 |
| Cluster GTO | PBE-D2 | −25.8 | 1.79 | |
| M06-2X | −25.1 | 2.46 | ||
| Experimentalcd | −19 | 3.2e | ||
| CH4 | Periodic PW/PAW | PBE-D2 | −13.1 | |
| −10.1f | ||||
| Cluster GTO | PBE-D2 | −14.4 | ||
| M06-2X | −10.2 | |||
| Experimentalcg | −12.6 ± 0.8 | |||
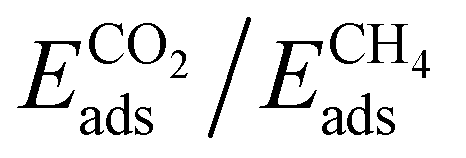
Although the cluster model represents a small fraction of the silica wall surface structure, its validation allows studying the adsorption of diatomic (e.g., CO, H2, N2, O2, and NO), triatomic (e.g., CO2, H2O, H2S, and SO2) and tetratomic (SO3 and NH3) gas molecules onto the PhPMO pore wall surface at lower computational cost compared with the analogous periodic system.123 The CO2 and CH4 adsorption behaviour of other PMO sorbents containing different organic moieties, such as biphenylene (Bph-), pyridine (Py-) and bipyridine (Bpy-) bridges and their mixtures (Ph/Py- and Bph/Bpy-moieties), was also assessed in previous studies.125 Building these cluster models presents a significant challenge, requiring consideration of the molecular-scale periodicity determined by XRD analysis of experimentally prepared PMO materials. The periodicity varies depending on the size of the target organic moiety. A convenient approach to build the models involves using the existing and optimized Ph-PMO model as a starting point. The next step is to replace the phenylene moieties of the material with desired organic bridges while considering its molecular-scale periodicity, followed by geometry optimization (DFT energy minimisation).125
These challenges have, over the years, been tackled with the development of more realistic models for mesoporous silicas, ranging from reconstruction to mimetic models. The former models are custom-designed to replicate structural properties observed in experimental studies, while the latter models aim at simulating the material synthesis process using molecular simulation techniques (Fig. 10).126
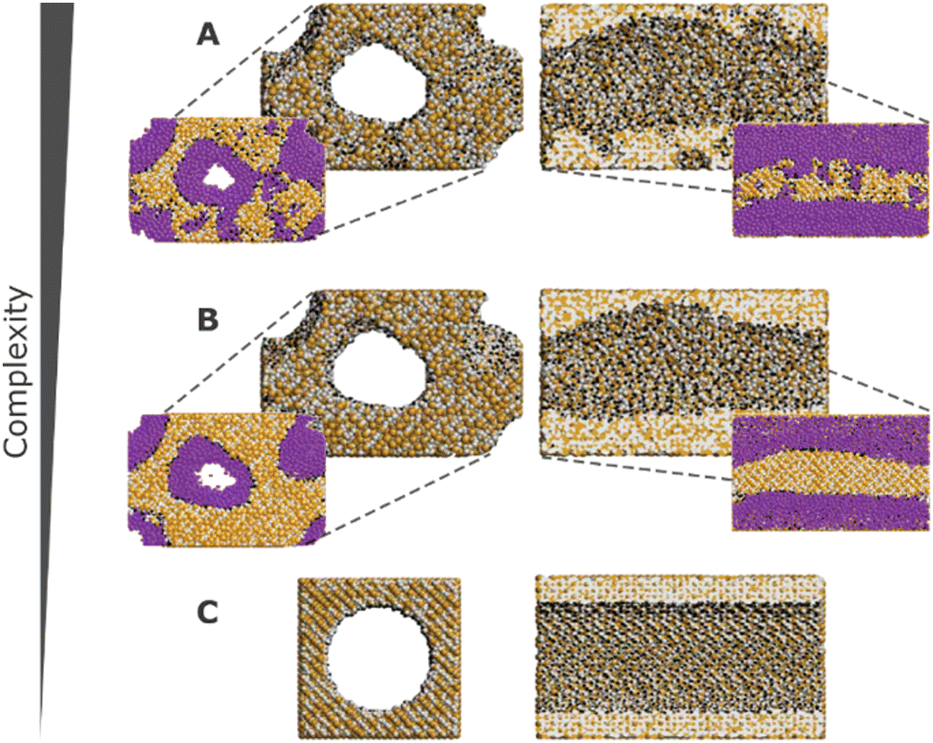 | ||
| Fig. 10 Front and cross section views of different models with decreasing complexity. Silicon, oxygen, and hydrogen atoms are represented in tan, grey, and black, respectively. Argon (represented in purple) adsorption snapshots for a relative pressure of P/P0 = 0.67 are also depicted for models A and B. Model A possesses meso- and microporosity, while model B exhibits no microporosity, and model C is a simplified cylindrical pore model. Adapted from ref. 126 with permission from ACS, copyright 2009. | ||
In computer simulations, the complexity of the system must be tailored to suit each specific problem, including the computational cost. This can range from more realistic structural models that incorporate both mesopores and micropores with appropriate pore roughness, to simpler cylindrical pores created from crystalline or pre-amorphized silica blocks. The modelling of such systems can be accomplished through multiple pre-processing approaches, including both specialized software and programming-based approaches. For instance, widely recognized molecular builders like Moltemplate and PACKMOL, although primarily used for biological and soft-condensed matter systems, can also be adapted for modelling silica materials. While these tools were originally developed for different applications, they offer versatile functionalities that can be harnessed to construct and arrange silica structures in a systematic manner. Another valuable tool for molecular modelling of silica systems is VMD134 (Visual Molecular Dynamics). VMD packages, like InorganicBuilder, offer user-friendly interfaces and tools specifically designed for modelling inorganic materials. These packages provide a convenient way to construct and manipulate silica structures, making them accessible to researchers with varying levels of expertise. Alternatively, for more advanced modelling and simulation techniques, researchers can leverage their programming skills. They can opt to use programming languages like Python and take advantage of specialized packages such as PoreMS135 or MoSDeF (Molecular Simulation Design Framework),136 which provide functionalities tailored to this type of systems. These programming-based approaches offer greater flexibility and customization options, enabling the implementation of sophisticated algorithms and to perform complex simulations tailored to specific research objectives.
In certain instances, the utilization of a complex model becomes necessary to ensure the accurate simulation of the desired properties. Recognizing this, for a more precise and comprehensive modelling of the system, it is crucial to employ a synergistic combination of various molecular simulation techniques. By deepening our understanding and effectively integrating these techniques, we can achieve a more refined representation of the system, enabling us to capture intricate details and accurately predict the properties of interest. Bhattacharya et al. used coarse-grained simulations of surfactants to obtain a model of the main physical features of the pore.126 It is noteworthy that the mentioned methodologies are most widely described in the literature, as some novel modelling methods like self-assembly of silica-based mesoporous materials or modelling materials using reactive force fields are starting to emerge.137
A proper characterization of the model must be performed, to confirm its validity. This can be achieved by comparing structural properties like the pore size distribution, specific surface area, framework density, surface hydroxyl group density, and many others, with experimental values. In contrast to periodic crystalline materials, these silica models must also ensure a suitable degree of amorphization, which can be obtained using quenching methods like temperature increase through molecular dynamics canonical ensemble simulation.138 Recently, machine-learning driven molecular dynamics has also been employed in the simulation of accurate amorphous silicon materials.137
The simulation of gas adsorption solely on bare silica surfaces has limited relevance. As a result, these materials are predominantly simulated as a support medium for organic molecules, particularly amines. Hence, the models undergo subsequent functionalization procedures prior to gas adsorption studies, as described in the following section.
Computational tools are thus an environmentally friendly way to predict if different sorbents could perform better without the need to synthesize them in the lab. Up to now, to design the functionalized sorbent models, the desired functional group replaces a selected hydrogen atom. Typically, this is made on all possible functionalization sites, as the distribution of the functional group on the material is mostly unknown, generating several geometries that must be optimized and studied. Our group performed this type of model building on both cluster PMS46,69,71 and periodic PMO models.48,124 In the case of PMO sorbents, functionalization is easier to predict, as the crystal-like structure and fewer possible defects are an advantage in analysing data from characterization techniques like PXRD, elemental analysis and ssNMR.124,125
However, for modelling specific structures, certain structural parameters may need to be fixed at non-optimal values, which can result in vibrational modes with associated imaginary wavenumbers. Cluster size can vary depending on the number of amine chains and surface silanols, with small clusters composed of a single amine chain and surface silanol (Fig. 11(a)), medium clusters with two amine chains and a surface silanol (Fig. 11(b)), and large clusters made up of one amine chain and five surface silanols (Fig. 11(c)). For the case of an isolated amine, a (CH3)3SiCH2CH2CH2NH2 model can be applied, as it is unable to form hydrogen bonds with the silica framework (Fig. 11(d)).71 In this model, terminal OH moieties are replaced by non-interacting methyl groups. While functionalization tasks have traditionally been accomplished through manual manipulation of the model, the advent of software packages mentioned in the previous section has significantly simplified this process. These software packages provide a user-friendly interface that enables the swift and convenient introduction of different functional groups in varying proportions, facilitating easy customization of the models.
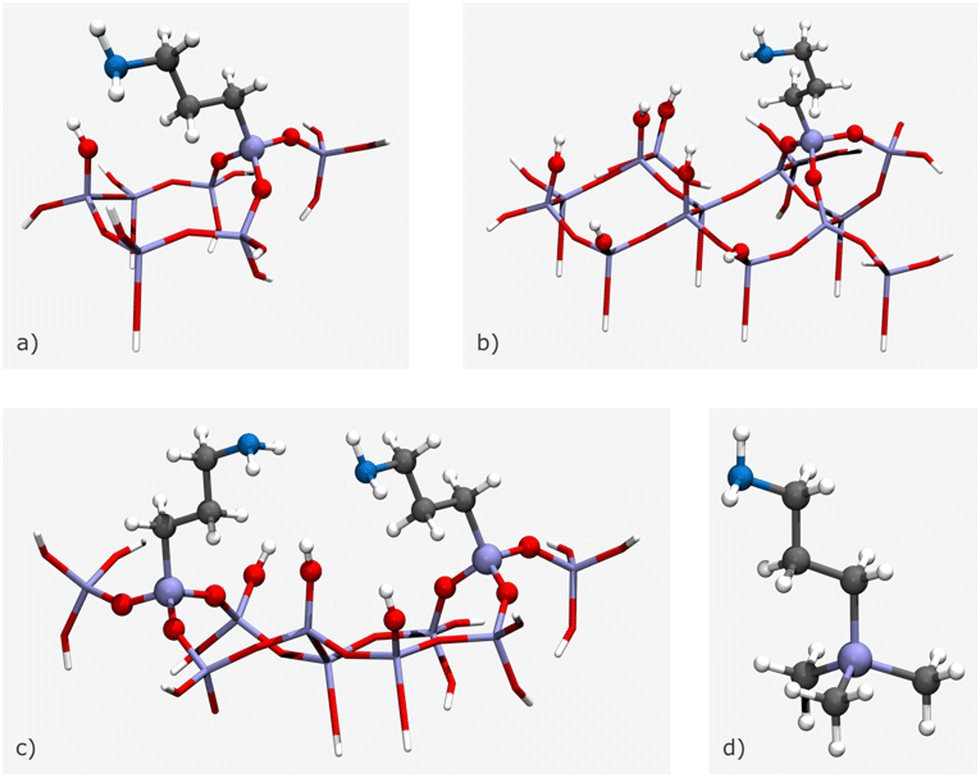 | ||
| Fig. 11 3D illustrations of the different size clusters: (a) small cluster, (b) medium cluster, (c) large cluster, and (d) isolated amine. Stick and ball-and-stick representations indicate frozen and fully optimised atoms, respectively. The colour code is as follows: white for H, dark grey for C, blue for N, red for O, and purple for Si. The schemes are based on the models presented by Mafra et al.69 | ||
For instance, to graft silylpropylamines onto the clusters, optimization is employed to bind three surface OH groups by each alkylamine. Subsequent optimizations involve the relaxation of various species, such as the alkyl chain (along with its respective functional group at the end), water or CO2 molecules (when present), the SiO3 moieties binding the alkylamines, and the surface OH groups.71 The remaining Si and O atoms are held constant at their crystallographic positions during these optimizations. The absence of imaginary values in the phonon mode frequencies pertaining to the atoms optimized in the various structural models confirms that these structures are true minima on their potential energy surfaces.
Additionally, for constructing more intricate functionalized models, pre-processing simulations can be employed. Techniques like Configurational Bias Monte Carlo (CBMC) offer a controlled approach to grow chains, enhancing the precision of surface functionalization. These methods allow researchers to achieve a more refined and tailored functionalization of the surface, expanding the possibilities for studying complex systems in depth.139
5.2. Modelling NMR parameters
The development of software dedicated to modelling ssNMR parameters has gained significant traction in recent years. Prominent among these software packages are SIMPSON, SpinDynamica, SPINEVOLUTION, Spinach, and EXPRESS. These platforms offer a versatile suite of simulation capabilities, empowering users to perform different experiments across various spin systems within a virtual NMR spectrometer.28In the context of investigating the complexities inherent to porous adsorbent materials, researchers frequently integrate ssNMR data with theoretical methodologies.50,71 Central to this synergistic strategy is the combination of NMR lineshape simulations, DFT calculations, and molecular dynamics (MD) simulations, collectively fostering a holistic comprehension of these materials.71,140
DFT calculations, which are rooted in the fundamental principles of electronic structure determination, can be used to compute tensors related to various NMR phenomena, such as CS, quadrupolar coupling, hyperfine interactions, and binding energies. These tensors can then be compared with experimental data to provide insights into the nature of adsorption sites.
5.3. Modelling pore surface interactions with adsorbates
In addition to the chemisorption process, CO2 molecules can also interact with the amine functionalities without establishing chemical bond(s). In this case, the interaction between the adsorbate molecules and the surface of the material occurs by a physisorption process mediated by van der Waals forces.
Our group studied both processes by combining DFT and ssNMR studies.46,71 DFT studies were relevant to distinguish physi- and chemisorption by simulating the CO2 physisorption on the surface of three amine-modified silica sorbents (SBA-15 modified with APTES, TMMAP, and N-3) and comparing the experimental 13C and 15N NMR CS with those obtained computationally (see Section 3 for further details). This approach was also used to verify that physisorbed CO2 can interact with the chemisorbed species, giving a possible explanation for the high CO2 adsorption capacity and selectivity (on CO2/CH4 gas mixtures) observed even beyond the saturation of amines.46
In the case of PMO sorbents, DFT studies confirmed that CO2 is preferentially physisorbed on the PMO walls independently of the type of chemical functionalities introduced, being advantageous for adsorbent regeneration.43,94–97 The CO2 preferential adsorption on PMO materials without N groups, like Ph-48,122–125 and Bph-125PMO sorbents, occurs close to the isolated T2 type silanol species. The preferential adsorption sites seem to be the same on PMO sorbents with Py-, Bpy-, and Bph-moieties, or by mixtures of Ph/Py- and Bph/Bpy-species,97 but with the later sorbent showing enhanced CO2 adsorption.
The introduction of different types and amounts of amino groups on the PhPMO sorbent increased the CO2 adsorption energy (obtained from DFT calculations using periodic PMO models) and Henry constant for CO2 (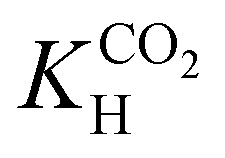 ), showing a good agreement between calculated and experimental data.48 In this study, we also demonstrated that the modification of the PMO sorbents with alkyl amines (APTMS@PhPMO) improved the CO2 adsorption more than when aromatic amines (–NH2) were introduced into the material. Interestingly, the CO2 sorption affinity seemed to be more controlled by the amine type than by the nitrogen quantity in the PMO channels. Experimental and DFT studies displayed the same adsorption–separation trend, allowing the extension of the computational approach to study the CO2 adsorption–separation performance on other modified Ph-PMO sorbents, e.g., containing –NO2, –NH-i-Pr, –CH2NH2, and –SO3H groups instead of aromatic amines. The PMO modified with CH2NH2 groups bonded to the phenylene moiety enhanced CO2 adsorption capacity.48
), showing a good agreement between calculated and experimental data.48 In this study, we also demonstrated that the modification of the PMO sorbents with alkyl amines (APTMS@PhPMO) improved the CO2 adsorption more than when aromatic amines (–NH2) were introduced into the material. Interestingly, the CO2 sorption affinity seemed to be more controlled by the amine type than by the nitrogen quantity in the PMO channels. Experimental and DFT studies displayed the same adsorption–separation trend, allowing the extension of the computational approach to study the CO2 adsorption–separation performance on other modified Ph-PMO sorbents, e.g., containing –NO2, –NH-i-Pr, –CH2NH2, and –SO3H groups instead of aromatic amines. The PMO modified with CH2NH2 groups bonded to the phenylene moiety enhanced CO2 adsorption capacity.48
Classical MD simulations are frequently used to model the motion and behaviour of CO2 molecules within porous materials, providing insights into diffusion, adsorption kinetics, and the dynamics of the sorption process.144 Another commonly utilized technique is grand canonical Monte Carlo (GCMC) simulation, which enables the prediction of adsorption isotherms and the determination of adsorption capacities at different temperatures and pressures, as depicted in Fig. 12.144 Advanced methods, such as replica exchange145 and umbrella sampling,146 can also be applied to explore the free energy landscape and overcome energy barriers associated with CO2 physisorption.
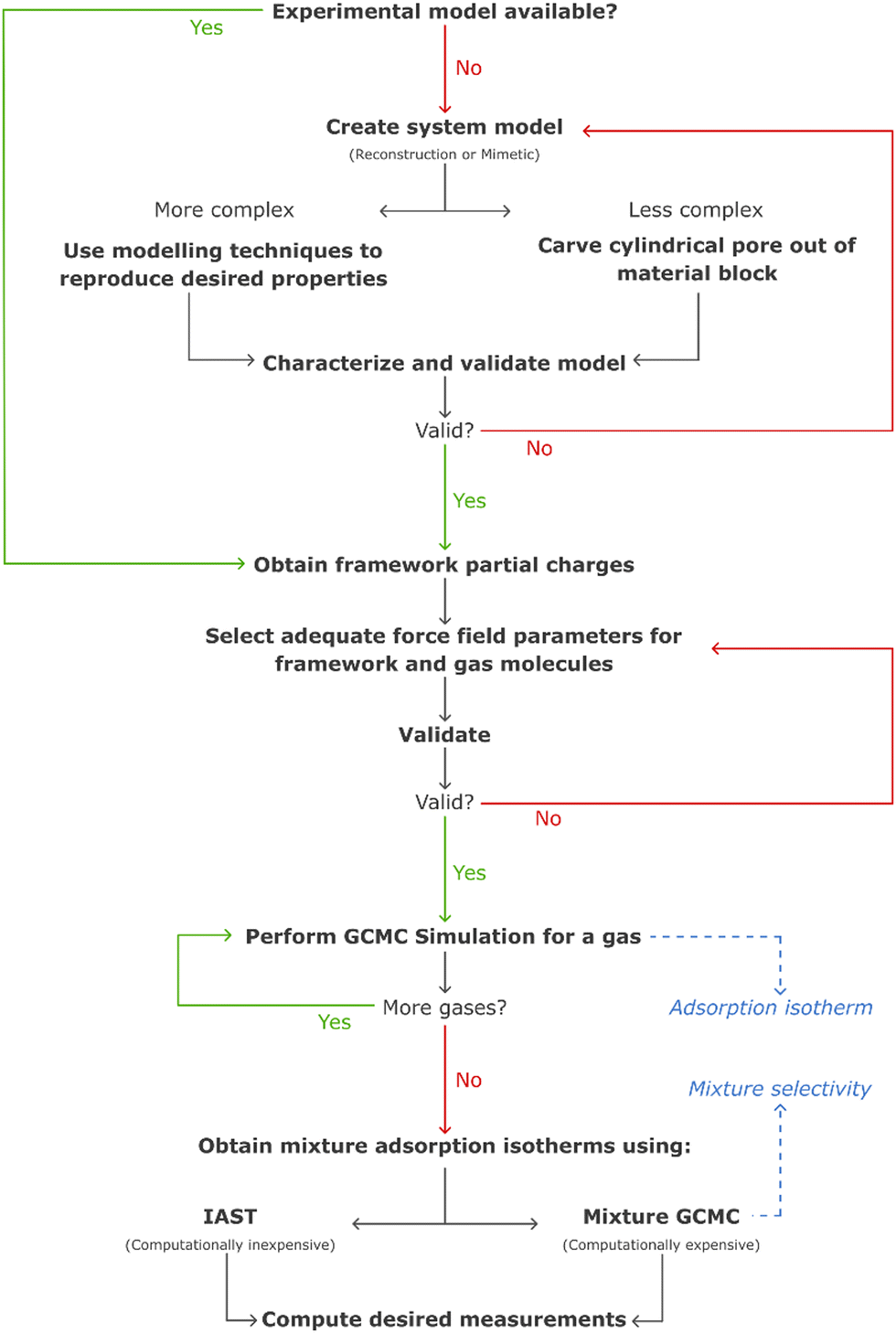 | ||
| Fig. 12 Flowchart illustrating a possible methodology used for gas adsorption simulations in porous frameworks (GCMC: grand canonical Monte Carlo; IAST: ideal adsorbed solution theory147). | ||
Studies have suggested that bare amorphous mesoporous silicas, like SBA-15 and MCM-41, exhibit larger CO2 adsorption capacities when compared with N2 and CH4, with the latter adsorbing to a larger extent as a result of stronger interactions with solids.129,137 Generally, CO2 is preferentially adsorbed over N2 at all temperatures, given the stronger dispersion interactions and larger quadrupole moment.137 In binary gas adsorption studies of CO2 and H2O, the material's lipophilicity affects the amount of water that gets adsorbed on the pore surface. Hydrophilic materials tend to adsorb more water, reducing adsorption and increasing diffusion of other gases. For hydrophobic surfaces, water molecules clump in the middle of the pore, which reduces CO2 diffusion and increases its adsorption.148 O2 in CO2/N2/O2 ternary mixtures has no effect on the selectivity of CO2 towards N2, and this is also observed in quaternary mixtures with additional H2O.128 Nonetheless, the presence of other gas molecules, even in small partial pressures, can significantly reduce CO2 adsorption if these molecules have preferential adsorption in the material's micropores.148 A possible workflow to perform computational studies on silica materials is presented in Fig. 12, encompassing the entire process from modelling to simulating gas mixtures.
In the case of PMO adsorbents, both experimental findings and computational results (using a DFT approach, see Table 3) unveiled their tendency to selectively adsorb CO2 rather than CH4. This feature positions them as prospective materials for separating these two gases.48,122–125 The incorporation of amino groups further enhanced the CO2/CH4 selectivity, with a more pronounced effect observed when using alkylamines.48 An M06-2X/cluster approach was employed to study the gas–host interactions between the additional components of the flue gas mixture and the PhPMO.123 Determining the adsorption energy of these gases plays a pivotal role in anticipating the uptake of CO, H2, N2, O2, NO, H2O, H2S, SO2, SO3, and NH3 by this material, eliminating the need for extensive Ph-PMO synthesis and numerous adsorption experiments. The preferential adsorption location for all these gases, also including CO2 and CH4, is near the silanols for this material. The energetic data reveal the preference of PhPMO for adsorbing CO2 over CO, CH4, N2, and H2, while NH3, H2O, SO2, SO3, and H2S exhibit higher adsorption energies than CO2, indicating their preferential adsorption over CO2.
6. Conclusion and perspectives
Technical and experimental advancements in the field of ssNMR and computational methods have enabled gaining insights into the molecular-level structure of porous surfaces, silica-based adsorbents. The progress made in studying host–gas interactions in the field of CO2 adsorption and separation has been greatly influenced by surface enhanced innovative techniques applied to sample preparation in controlled environments and the use of isotopically enriched gases and/or materials. The sample preparation can sometimes be difficult and hamper the obtained results (rigorous control of temperature, pressure and moisture levels is crucial) and good knowledge about the material textural properties (amount and type of functionalities, sample crystallinity and porosity, etc.) is important to correctly determine CO2 speciation. By combining computational tools, adsorption and ssNMR data, new chemisorbed and physisorbed CO2 species have been identified by controlling the former variables and working under controlled and reproducible conditions. Progressing beyond the state of the art requires getting closer to realistic gas adsorption conditions. In real world applications, the adsorption process is dynamic encompassing multiple gases flowing through the adsorbent material. In situ or operando NMR methods can be adapted to the study of adsorbents under real-time gas adsorption. Developing such methods is difficult for ssNMR applications as it requires a gas flowing through a spinning sample holder involving costly custom-made probe modifications. To circumvent this inconvenience, the use of 3D printing has become a popular and cost-effective way to fabricate components for NMR applications.149–152 Creating printed ssNMR components in-house will boost custom probe modifications, leading to new concepts and designs that can be tailored to specific applications. In the field of CO2 capture, we anticipate that 3D printing will play a crucial role, for instance in the adaptation of standard NMR probes for in situ flow MAS spectroscopy, in developing components for high-pressure rotor apparatus, and creating new gas-flow stator concepts (e.g., breakthrough-style systems for studying gas separation).As for computational methods, the integration of chemical information with artificial intelligence, through machine learning algorithms using, for instance, neural network prediction approaches will quickly become ubiquitous in structural determination in disordered systems as they offer considerable speed-up compared to traditional force fields, enabling simulations over longer time scales, typically ranging from nanoseconds to microseconds or more. MLPs are particularly useful for studying complex processes and large systems, as they can capture the underlying physics with reduced computational costs.143,153
As an example, Choudhary et al.154 have recently performed graph neural network predictions of the CO2 adsorption properties of hypothetical MOF materials, developing high accuracy and fast models for pre-screening applications. These tools offer new opportunities for predicting the structure of surface species in confined spaces and for optimizing distinct material properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI: 10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI: 10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI: 10.54499/LA/P/0006/2020), financed from national funds through the FCT/MCTES (PIDDAC). We also acknowledge the funding from project PTDC/QUI-QFI/28747/2017 (GAS2MAT-DNPSENS-POCI-01-0145-FEDER-028), financed through FCT/MEC and cofinanced by FEDER under the PT2020 Partnership Agreement. The NMR spectrometers are part of the National NMR Network (PTNMR) and are partially supported by Infrastructure Project 022161 (cofinanced by FEDER through COMPETE 2020, POCI and PORL and FCT through PIDDAC). This work has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement 865974). FCT is also acknowledged by R. V., M. I. and M. A. O. L. for a Junior Researcher Position (CEECIND/02127/2017, CEECIND/00546/2018 and CEECIND/01158/2021 (DOI: 10.54499/2021.01158.CEECIND/CP1659/CT0022), respectively) and M. S. for an Assistant Research Position CEECIND/00056/2020 (DOI 10.54499/2020.00056.CEECIND/CP1589/CT0005). M. A. O. L. further acknowledges funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 101090287. MI also acknowledges Spanish Ministry of Science, Innovation and Universities for the “Beatriz Galindo” Scholarship (MU-23-BG22/00145).References
- Sixth Assessment Report, Intergovernmental Panel on Climate Change (IPCC AR6 SYR), 2023.
- IPCC, IPCC Special Report on Carbon Dioxide Capture and Storage, Cambridge University Press, Cambridge, United Kingdom and NY, USA, 2005, pp. 297–348.
- IEA – International Energy Agency, Global Energy Review: CO2 Emissions in 2021, 2022.
- Y. Deng, J. Li, Y. Miao and D. Izikowitz, Energy Rep., 2021, 7, 3506–3516 CrossRef.
- J. Park, S. Y. Cho, M. Jung, K. Lee, Y.-C. Nah, N. F. Attia and H. Oh, J. CO2 Util., 2021, 45, 101404 CrossRef CAS.
- X. Li, Y. Pei, T. Ajmal, K.-J. Rana, A. Aitouche, R. Mobasheri and Z. Peng, Fuel, 2021, 302, 121162 CrossRef CAS.
- R. A. Azpiri Solares, D. Soares dos Santos, A. Ingram and J. Wood, Fuel, 2019, 253, 1130–1139 CrossRef CAS.
- L. Lin, Y. Meng, T. Ju, S. Han, F. Meng, J. Li, Y. Du, M. Song, T. Lan and J. Jiang, J. Environ. Manage., 2023, 325, 116438 CrossRef CAS PubMed.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467–4475 CrossRef CAS PubMed.
- A. Kiani, K. Jiang and P. Feron, Front. Energy Res., 2020, 8, 1–13 CrossRef.
- M. Wang, A. Lawal, P. Stephenson, J. Sidders and C. Ramshaw, Chem. Eng. Res. Des., 2011, 89, 1609–1624 CrossRef CAS.
- K. Li, J. Jiang, F. Yan, S. Tian and X. Chen, Appl. Energy, 2014, 136, 750–755 CrossRef CAS.
- D. W. Keith, G. Holmes, D. St. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- N. R. Stuckert and R. T. Yang, Environ. Sci. Technol., 2011, 45, 10257–10264 CrossRef PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- W. Chaikittisilp, R. Khunsupat, T. T. Chen and C. W. Jones, Ind. Eng. Chem. Res., 2011, 50, 14203–14210 CrossRef CAS.
- P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- Y. Belmabkhout, R. Serna-Guerrero and A. Sayari, Chem. Eng. Sci., 2010, 65, 3695–3698 CrossRef CAS.
- A. Sayari, Q. Liu and P. Mishra, ChemSusChem, 2016, 9, 2796–2803 CrossRef CAS PubMed.
- C. Gebald, J. A. Wurzbacher, P. Tingaut, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2011, 45, 9101–9108 CrossRef CAS PubMed.
- C. Gebald, J. A. Wurzbacher, A. Borgschulte, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2014, 48, 2497–2504 CrossRef CAS PubMed.
- S. Choi, T. Watanabe, T.-H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- J. Wang, M. Wang, W. Li, W. Qiao, D. Long and L. Ling, AIChE J., 2015, 61, 972–980 CrossRef CAS.
- Y. Kong, X. Shen, S. Cui and M. Fan, Green Chem., 2015, 17, 3436–3445 RSC.
- D. Pereira, R. Fonseca, I. Marin-Montesinos, M. Sardo and L. Mafra, Curr. Opin. Colloid Interface Sci., 2023, 64, 101690 CrossRef CAS.
- C. Chen, S. Zhang, K. H. Row and W.-S. Ahn, J. Energy Chem., 2017, 26, 868–880 CrossRef CAS.
- X. (Eric) Hu, L. Liu, X. Luo, G. Xiao, E. Shiko, R. Zhang, X. Fan, Y. Zhou, Y. Liu, Z. Zeng and C. Li, Appl. Energy, 2020, 260, 114244 CrossRef.
- P. A. Saenz Cavazos, E. Hunter-Sellars, P. Iacomi, S. R. McIntyre, D. Danaci and D. R. Williams, Front. Energy Res., 2023, 11, 1–22 Search PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- K. Cassiers, T. Linssen, M. Mathieu, M. Benjelloun, K. Schrijnemakers, P. van der Voort, P. Cool and E. F. Vansant, Chem. Mater., 2002, 14, 2317–2324 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1979, 1998(279), 548–552 Search PubMed.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CrossRef CAS.
- F. de Clippel, M. Dusselier, S. Van de Vyver, L. Peng, P. A. Jacobs and B. F. Sels, Green Chem., 2013, 15, 1398 RSC.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- B. Karimi, N. Ganji, O. Pourshiani and W. R. Thiel, Prog. Mater. Sci., 2022, 125, 100896 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- Y. Tang and K. Landskron, J. Phys. Chem. C, 2010, 114, 2494–2498 CrossRef CAS.
- R. Sanz, G. Calleja, A. Arencibia and E. S. Sanz-Pérez, Microporous Mesoporous Mater., 2015, 209, 165–171 CrossRef CAS.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, Chem. Eng. J., 2018, 336, 612–621 CrossRef CAS.
- K. Sim, N. Lee, J. Kim, E.-B. Cho, C. Gunathilake and M. Jaroniec, ACS Appl. Mater. Interfaces, 2015, 7, 6792–6802 CrossRef CAS.
- M. A. O. Lourenço, C. Siquet, M. Sardo, L. Mafra, J. Pires, M. Jorge, M. L. Pinto, P. Ferreira and J. R. B. Gomes, J. Phys. Chem. C, 2016, 120, 3863–3875 CrossRef.
- E. De Canck, I. Ascoop, A. Sayari and P. Van Der Voort, Phys. Chem. Chem. Phys., 2013, 15, 9792 RSC.
- T. Čendak, L. Sequeira, M. Sardo, A. Valente, M. L. Pinto and L. Mafra, Chem. – Eur. J., 2018, 24, 10136–10145 CrossRef.
- M. Sardo, R. Afonso, J. Juźków, M. Pacheco, M. Bordonhos, M. L. Pinto, J. R. B. Gomes and L. Mafra, J. Mater. Chem. A, 2021, 9, 5542–5555 RSC.
- W. Li, Y. Cheng, D. Yang, Y. Liu and B. Han, Macromol. Rapid Commun., 2023, 44, 1–16 CrossRef CAS PubMed.
- S. Noro and T. Nakamura, NPG Asia Mater., 2017, 9, e433 CrossRef.
- D.-S. Zhang, Z. Chang, Y.-F. Li, Z.-Y. Jiang, Z.-H. Xuan, Y.-H. Zhang, J.-R. Li, Q. Chen, T.-L. Hu and X.-H. Bu, Sci. Rep., 2013, 3, 3312 CrossRef PubMed.
- G. Wang, K. Leus, H. S. Jena, C. Krishnaraj, S. Zhao, H. Depauw, N. Tahir, Y.-Y. Liu and P. Van Der Voort, J. Mater. Chem. A, 2018, 6, 6370–6375 RSC.
- A. S. L. Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102–103, 120–195 CrossRef PubMed.
- A. G. M. Rankin, J. Trébosc, F. Pourpoint, J.-P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2019, 101, 116–143 CrossRef CAS PubMed.
- D. L. Silverio, H. A. van Kalkeren, T.-C. Ong, M. Baudin, M. Yulikov, L. Veyre, P. Berruyer, S. Chaudhari, D. Gajan, D. Baudouin, M. Cavaillès, B. Vuichoud, A. Bornet, G. Jeschke, G. Bodenhausen, A. Lesage, L. Emsley, S. Jannin, C. Thieuleux and C. Copéret, Helv. Chim. Acta, 2017, 100, e1700101 CrossRef.
- M. de Oliveira, K. Herr, M. Brodrecht, N. B. Haro-Mares, T. Wissel, V. Klimavicius, H. Breitzke, T. Gutmann and G. Buntkowsky, Phys. Chem. Chem. Phys., 2021, 23, 12559–12568 RSC.
- W. R. Grüning, H. Bieringer, M. Schwarzwälder, D. Gajan, A. Bornet, B. Vuichoud, J. Milani, D. Baudouin, L. Veyre, A. Lesage, S. Jannin, G. Bodenhausen, C. Thieuleux and C. Copéret, Helv. Chim. Acta, 2017, 100, e1600122 CrossRef.
- D. Bernin and N. Hedin, Curr. Opin. Colloid Interface Sci., 2018, 33, 53–62 CrossRef CAS.
- A. C. Forse, C. Merlet, C. P. Grey and J. M. Griffin, Prog. Nucl. Magn. Reson. Spectrosc., 2021, 124–125, 57–84 CrossRef CAS PubMed.
- X. E. Hu, Q. Yu, F. Barzagli, C. Li, M. Fan, K. A. M. Gasem, X. Zhang, E. Shiko, M. Tian, X. Luo, Z. Zeng, Y. Liu and R. Zhang, ACS Sustain, Chem. Eng., 2020, 8, 6173–6193 CAS.
- Y. Xiao, S. Li, J. Xu and F. Deng, Curr. Opin. Colloid Interface Sci., 2022, 61, 101633 CrossRef CAS.
- X. Zhu, W. Xie, J. Wu, Y. Miao, C. Xiang, C. Chen, B. Ge, Z. Gan, F. Yang, M. Zhang, D. O’Hare, J. Li, T. Ge and R. Wang, Chem. Soc. Rev., 2022, 51, 6574–6651 RSC.
- J. Timm and R. Marschall, Chem. Ing. Tech., 2022, 94, 101–110 CrossRef CAS.
- H. R. N. B. Enninful, D. Enke and R. Valiullin, Materials, 2022, 15, 2350 CrossRef CAS PubMed.
- S. M. Pugh and A. C. Forse, J. Magn. Reson., 2023, 346, 107343 CrossRef CAS PubMed.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, J. Am. Chem. Soc., 2017, 139, 389–408 CrossRef CAS PubMed.
- J. J. Lee, C.-H. Chen, D. Shimon, S. E. Hayes, C. Sievers and C. W. Jones, J. Phys. Chem. C, 2017, 121, 23480–23487 CrossRef CAS.
- R. Afonso, M. Sardo, L. Mafra and J. R. B. B. Gomes, Environ. Sci. Technol., 2019, 53, 2758–2767 CrossRef CAS PubMed.
- M. L. Pinto, L. L. Mafra, J. M. Guil, J. Pires and J. Rocha, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- R. Vieira, I. Marin-Montesinos, J. Pereira, R. Fonseca, M. Ilkaeva, M. Sardo and L. Mafra, J. Phys. Chem. C, 2021, 125, 14797–14806 CrossRef CAS PubMed.
- R. Fonseca, R. Vieira, M. Sardo, I. Marin-Montesinos and L. Mafra, J. Phys. Chem. C, 2022, 126, 12582–12591 CrossRef CAS PubMed.
- R. W. Flaig, T. M. O. Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- D. Shimon, C.-H. Chen, J. J. Lee, S. A. Didas, C. Sievers, C. W. Jones and S. E. Hayes, Environ. Sci. Technol., 2018, 52, 1488–1495 CrossRef CAS PubMed.
- A. H. Berge, S. M. Pugh, M. I. M. Short, C. Kaur, Z. Lu, J.-H. Lee, C. J. Pickard, A. Sayari and A. C. Forse, Nat. Commun., 2022, 13, 7763 CrossRef CAS PubMed.
- J. M. Kolle, M. Fayaz and A. Sayari, Chem. Rev., 2021, 121, 7280–7345 CrossRef CAS PubMed.
- A. E. Szego, A. Jaworski and N. Hedin, Mater. Adv., 2021, 2, 448–454 RSC.
- C.-H. Chen, E. L. Sesti, J. J. Lee, F. Mentink-Vigier, C. Sievers, C. W. Jones and S. E. Hayes, J. Phys. Chem. C, 2021, 125, 16759–16765 CrossRef CAS.
- M. Pacheco, M. Bordonhos, M. Sardo, R. Afonso, J. R. B. Gomes, L. Mafra and M. L. Pinto, Chem. Eng. J., 2022, 443, 136271 CrossRef CAS.
- C. Xu, Z. Bacsik and N. Hedin, J. Mater. Chem. A, 2015, 3, 16229–16234 RSC.
- A. Sayari, Y. Belmabkhout and R. Serna-Guerrero, Chem. Eng. J., 2011, 171, 760–774 CrossRef CAS.
- J. Wang, R. Fu, S. Wen, P. Ning, M. H. Helal, M. A. Salem, B. Bin Xu, Z. M. El-Bahy, M. Huang, Z. Guo, L. Huang and Q. Wang, Adv. Compos. Hybrid Mater., 2022, 5, 2721–2759 CrossRef CAS.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- H. J. Choi, J. G. Min, S. H. Ahn, J. Shin, S. B. Hong, S. Radhakrishnan, C. V. Chandran, R. G. Bell, E. Breynaert and C. E. A. Kirschhock, Mater. Horiz., 2020, 7, 1528–1532 RSC.
- S. Karimi, D. Korelskiy, Y. Mortazavi, A. A. Khodadadi, K. Sardari, M. Esmaeili, O. N. Antzutkin, F. U. Shah and J. Hedlund, J. Membr. Sci., 2016, 520, 574–582 CrossRef CAS.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Snape, T. C. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028–9035 CrossRef CAS PubMed.
- A. B. Jasso-Salcedo, X. Wang, Z. Bacsik and N. Hedin, Inorg. Chim. Acta, 2021, 525, 120443 CrossRef CAS.
- Q. Jia, E. Lasseuguette, M. M. Lozinska, M.-C. Ferrari and P. A. Wright, ACS Appl. Mater. Interfaces, 2022, 14, 46615–46626 CrossRef CAS PubMed.
- N. Popp, T. Homburg, N. Stock and J. Senker, J. Mater. Chem. A, 2015, 3, 18492–18504 RSC.
- P. Amoatey, H. Omidvarborna, M. S. Baawain and A. Al-Mamun, Process Saf. Environ. Prot., 2019, 123, 215–228 CrossRef CAS.
- J. H. Carter, X. Han, F. Y. Moreau, I. da Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2018, 140, 15564–15567 CrossRef CAS PubMed.
- I. J. Uyanga and R. O. Idem, Ind. Eng. Chem. Res., 2007, 46, 2558–2566 CrossRef CAS.
- F. Rezaei and C. W. Jones, Ind. Eng. Chem. Res., 2013, 52, 12192–12201 CrossRef CAS.
- F. Rezaei and C. W. Jones, Ind. Eng. Chem. Res., 2014, 53, 12103–12110 CrossRef CAS.
- P. Rovó, Solid State Nucl. Magn. Reson., 2020, 108, 101665 CrossRef PubMed.
- P. Schanda and M. Ernst, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 96, 1–46 CrossRef CAS PubMed.
- A. Krushelnitsky, T. Zinkevich, B. Reif and K. Saalwächter, J. Magn. Reson., 2014, 248, 8–12 CrossRef CAS PubMed.
- A. Krushelnitsky, T. Zinkevich, D. Reichert, V. Chevelkov and B. Reif, J. Am. Chem. Soc., 2010, 132, 11850–11853 CrossRef CAS PubMed.
- V. J. Witherspoon, J. Xu and J. A. Reimer, Chem. Rev., 2018, 118, 10033–10048 CrossRef CAS PubMed.
- H. J. Moon, J.-M. Carrillo, J. Leisen, B. G. Sumpter, N. C. Osti, M. Tyagi and C. W. Jones, J. Am. Chem. Soc., 2022, 144, 11664–11675 CrossRef CAS PubMed.
- J. K. Moore, M. A. Sakwa-Novak, W. Chaikittisilp, A. K. Mehta, M. S. Conradi, C. W. Jones and S. E. Hayes, Environ. Sci. Technol., 2015, 49, 13684–13691 CrossRef CAS PubMed.
- A. F. Ciftja, A. Hartono and H. F. Svendsen, Int. J. Greenhouse Gas Control, 2013, 16, 224–232 CrossRef CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- R. K. Wangsness and F. Bloch, Phys. Rev., 1953, 89, 728–739 CrossRef CAS.
- A. G. Redfield, IBM J. Res. Dev., 1957, 1, 19–31 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- F. Stallmach, A.-K. Pusch, T. Splith, C. Horch and S. Merker, Microporous Mesoporous Mater., 2015, 205, 36–39 CrossRef CAS.
- H. Thern, C. Horch, F. Stallmach, B. Li, A. Mezzatesta, H. Zhang and R. Arro, Microporous Mesoporous Mater., 2018, 269, 21–25 CrossRef CAS.
- C. Horch, S. Schlayer and F. Stallmach, J. Magn. Reson., 2014, 240, 24–33 CrossRef CAS PubMed.
- M. Bonnet, D. Courtier-Murias, P. Faure, S. Rodts and S. Care, Holzforschung, 2017, 71, 481–490 CrossRef CAS.
- Y. Kawata, Y. Adachi, S. Haga, J. Fukutomi, H. Imai, A. Kimura and H. Fujiwara, Anal. Sci., 2007, 23, 1397–1402 CrossRef CAS PubMed.
- H. Fujiwara, H. Imai, Y. Adachi and A. Kimura, Anal. Sci., 2021, 37, 1803–1810 CrossRef CAS PubMed.
- J. Marreiros, R. de Oliveira-Silva, P. Iacomi, P. L. Llewellyn, R. Ameloot and D. Sakellariou, J. Am. Chem. Soc., 2021, 143, 8249–8254 CrossRef CAS PubMed.
- Y. An, A. Kleinhammes, P. Doyle, E.-Y. Chen, Y. Song, A. J. Morris, B. Gibbons, M. Cai, J. K. Johnson, P. B. Shukla, M. N. Vo, X. Wei, C. E. Wilmer, J. P. Ruffley, L. Huang, T. M. Tovar, J. J. Mahle, C. J. Karwacki and Y. Wu, J. Phys. Chem. Lett., 2021, 12, 892–899 CrossRef CAS PubMed.
- K. Yang, P. R. J. Conolly, L. Liu, X. Yang, N. Robinson, M. Li, M. Mahmoud, A. El-Husseiny, M. Verrall, E. F. May and M. L. Johns, J. Nat. Gas Sci. Eng., 2022, 108, 104847 CrossRef CAS.
- K. Yang, E. Sadeghi Pouya, L. Liu, M. Li, X. Yang, N. Robinson, E. F. May and M. L. Johns, ChemPhysChem, 2022, 23, e202100794 CrossRef CAS PubMed.
- M. Sin, N. Kavoosi, M. Rauche, J. Pallmann, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Langmuir, 2019, 35, 3162–3170 CrossRef CAS PubMed.
- D. Wisser and M. Hartmann, Adv. Mater. Interfaces, 2021, 8, 2001266 CrossRef.
- M. Ilkaeva, R. Vieira, J. M. P. Pereira, M. Sardo, I. Marin-Montesinos and L. Mafra, J. Am. Chem. Soc., 2023, 145, 8764–8769 CrossRef CAS PubMed.
- U. Martinez and G. Pacchioni, Microporous Mesoporous Mater., 2010, 129, 62–67 CrossRef CAS.
- M. A. O. Lourenço, P. Ferreira and J. R. B. Gomes, Phys. Chem. Chem. Phys., 2018, 20, 16686–16694 RSC.
- M. A. O. Lourenço, C. Siquet, J. Santos, M. Jorge, J. R. B. Gomes and P. Ferreira, J. Phys. Chem. C, 2016, 120, 14236–14245 CrossRef.
- M. A. O. Lourenço, P. Ferreira and J. R. B. Gomes, Mater. Today Commun., 2021, 26, 102088 CrossRef.
- S. Bhattacharya, B. Coasne, F. R. Hung and K. E. Gubbins, Langmuir, 2009, 25, 5802–5813 CrossRef CAS PubMed.
- A. A. Sizova, V. V. Sizov and E. N. Brodskaya, Colloids Surf., A, 2017, 524, 87–95 CrossRef CAS.
- K. Kumar and A. Kumar, J. Phys. Chem. C, 2018, 122, 8216–8227 CrossRef CAS.
- K. Kumar and A. Kumar, Korean J. Chem. Eng., 2018, 35, 535–547 CrossRef CAS.
- B. Liu and B. Smit, Langmuir, 2009, 25, 5918–5926 CrossRef CAS PubMed.
- Y. Liu, D. Liu, Q. Yang, C. Zhong and J. Mi, Ind. Eng. Chem. Res., 2010, 49, 2902–2906 CrossRef CAS.
- A. Comotti, S. Bracco, P. Valsesia, L. Ferretti and P. Sozzani, J. Am. Chem. Soc., 2007, 129, 8566–8576 CrossRef CAS PubMed.
- G. N. Kalantzopoulos, M. K. Antoniou, A. Enotiadis, K. Dimos, E. Maccallini, A. Policicchio, E. Colavita and R. G. Agostino, J. Mater. Chem. A, 2016, 4, 9275–9285 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- H. Kraus, J. Rybka, A. Höltzel, N. Trebel, U. Tallarek and N. Hansen, Mol. Simul., 2021, 47, 306–316 CrossRef CAS.
- P. T. Cummings, C. MCabe, C. R. Iacovella, A. Ledeczi, E. Jankowski, A. Jayaraman, J. C. Palmer, E. J. Maginn, S. C. Glotzer, J. A. Anderson, J. Ilja Siepmann, J. Potoff, R. A. Matsumoto, J. B. Gilmer, R. S. DeFever, R. Singh and B. Crawford, AIChE J., 2021, 67, e17206 CrossRef CAS.
- V. L. Deringer, N. Bernstein, A. P. Bartók, M. J. Cliffe, R. N. Kerber, L. E. Marbella, C. P. Grey, S. R. Elliott and G. Csányi, J. Phys. Chem. Lett., 2018, 9, 2879–2885 CrossRef CAS PubMed.
- C. D. Williams, K. P. Travis, N. A. Burton and J. H. Harding, Microporous Mesoporous Mater., 2016, 228, 215–223 CrossRef CAS.
- J. I. Siepmann and D. Frenkel, Mol. Phys., 1992, 75, 59–70 CrossRef CAS.
- A. C. Forse, M. I. Gonzalez, R. L. Siegelman, V. J. Witherspoon, S. Jawahery, R. Mercado, P. J. Milner, J. D. Martell, B. Smit, B. Blümich, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 1663–1673 CrossRef CAS PubMed.
- H. D. M. Pham and R. Z. Khaliullin, J. Phys. Chem. C, 2021, 125, 24719–24727 CrossRef CAS.
- A. C. T. van Duin, A. Strachan, S. Stewman, Q. Zhang, X. Xu and W. A. Goddard, J. Phys. Chem. A, 2003, 107, 3803–3811 CrossRef CAS.
- S. Vandenhaute, M. Cools-Ceuppens, S. DeKeyser, T. Verstraelen and V. Van Speybroeck, Comput. Mater., 2023, 9, 19 CrossRef.
- H. Zeng, Y. Liu and H. Liu, Mol. Simul., 2018, 44, 1244–1251 CrossRef CAS.
- G. A. Ross, H. E. Bruce Macdonald, C. Cave-Ayland, A. I. Cabedo Martinez and J. W. Essex, J. Chem. Theory Comput., 2017, 13, 6373–6381 CrossRef CAS PubMed.
- A. Rahbari, R. Hens, O. A. Moultos, D. Dubbeldam and T. J. H. Vlugt, J. Chem. Theory Comput., 2020, 16, 1757–1767 CrossRef CAS PubMed.
- S. Furmaniak, S. Koter, A. P. Terzyk, P. A. Gauden, P. Kowalczyk and G. Rychlicki, Phys. Chem. Chem. Phys., 2015, 17, 7232–7247 RSC.
- A. A. Sizova, V. V. Sizov and E. N. Brodskaya, Colloids Surf., A, 2015, 474, 76–84 CrossRef CAS.
- D. Pereira, M. Sardo, I. Marín-Montesinos and L. Mafra, Anal. Chem., 2023, 95, 10384–10389 CrossRef CAS PubMed.
- J. L. Uribe, M. D. Jimenez, J. I. Kelz, J. Liang and R. W. Martin, J. Magn. Reson. Open, 2024, 18, 100142 CrossRef PubMed.
- D. Banks, B. Michael, N. Golota and R. G. Griffin, J. Magn. Reson., 2022, 335, 107126 CrossRef CAS PubMed.
- K. Xu, O. Pecher, M. Braun and J. Schmedt auf der Günne, J. Magn. Reson., 2021, 333, 107096 CrossRef CAS PubMed.
- P. Friederich, F. Häse, J. Proppe and A. Aspuru-Guzik, Nat. Mater., 2021, 20, 750–761 CrossRef CAS PubMed.
- K. Choudhary, T. Yildirim, D. W. Siderius, A. G. Kusne, A. McDannald and D. L. Ortiz-Montalvo, Comput. Mater. Sci., 2022, 210, 111388 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |