
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of dibenzothiophene S-oxides from sulfinate esters†
Yukiko
Kumagai
a,
Akihiro
Kobayashi
ab,
Keisuke
Nakamura
a and
Suguru
Yoshida
 *a
*a
aDepartment of Biological Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan. E-mail: s-yoshida@rs.tus.ac.jp
bLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan
First published on 10th January 2024
Abstract
An efficient method to prepare dibenzothiophene S-oxides is disclosed. Suzuki–Miyaura cross-coupling of 2-bromoaryl sulfinate esters with arylboronic acids selectively at the bromo group followed by electrophilic cyclization of the resulting sulfinate ester moiety provides diverse dibenzothiophene S-oxides. Further transformations including Pummerer-type C–H propargylation and aryne reactions realize to synthesize highly functionalized dibenzothiophene derivatives.
Dibenzothiophenes and their derivatives having S-oxide or S,S-dioxide moieties are of great significance in broad research fields such as pharmaceutical sciences, materials chemistry, and chemical biology (Fig. 1A).1–3 Particularly, dibenzothiophene S-oxides are gaining attention as precursors to generate atomic oxygen triggered by UV irradiation, which can facilitate DNA cleavage, oxidation of adenosine-S′-phosphosulfate kinase, and so on.1 In addition, recent advances in synthetic methods through the activation of S–O bond have allowed us to prepare diverse functionalized dibenzothiophenes from dibenzothiophene S-oxides (Fig. 1B).4 Despite the importance of dibenzothiophene S-oxides, it is not always easy to synthesize highly functionalized dibenzothiophene S-oxides in a conventional manner, which involves thiophene ring formation of biaryls using S8 and subsequent S-oxidation with oxidants such as mCPBA (Fig. 1C).5,6 Herein, we disclose a novel method to prepare dibenzothiophene S-oxides from 2-bromoaryl-substituted sulfinate esters by Br-selective Suzuki–Miyaura coupling followed by intramolecular electrophilic sulfinylation (Fig. 1D). Since we recently found that sulfinate esters can be activated easily by triflic anhydride (Tf2O) realizing S-allylation with allylsilanes, we conceived the synthesis of dibenzothiophene S-oxides from 2-biarylyl sulfinate ester through the electrophilic activation.7,8
 | ||
| Fig. 1 (A) Significant dibenzothiophene derivatives. (B) Transformations of dibenzothiophene S-oxide. (C) Representative synthesis of dibenzothiophene S-oxide. (D) This work. | ||
We succeeded in the synthesis of dibenzothiophene S-oxide 4a through Br-selective coupling and subsequent cyclization by electrophilic activation (Table 1). Treatment of 2-bromobenzenesulfinic acid methyl ester (1a)9 with 4-tolylboronic acid (2a) in the presence of potassium phosphate and a catalytic amount of (amphos)2PdCl2 afforded 2-(4-tolyl)benzenesulfinic acid methyl ester (3a) in an excellent yield. Of note, the coupling proceeded selectively at the bromo group, where palladium-catalyzed sulfoxide formation was not observed.10 Then, reaction conditions were screened for the cyclization of methyl 2-(4-tolyl)benzenesulfinate (3a) by electrophilic activation using acid anhydrides. As a result, we successfully synthesized dibenzothiophene S-oxide 4a in high yield from 3a with Tf2O. Indeed, treatment of 3a with Tf2O in dichloromethane at room temperature followed by the addition of aqueous sodium bicarbonate provided the desired dibenzothiophene S-oxide 4a in high yield, where side-products from further Pummerer-type reactions were not observed (entry 1). The reaction also took place smoothly when using Tf2O with 2,6-di(t-butyl)pyridine as a base (entry 2). In contrast, trifluoroacetic anhydride (TFAA) or acetic anhydride did not activate sulfinate ester 3a (entries 3 and 4). Dibenzothiophene S-oxide 4a was also obtained in moderate yield using triflic acid (TfOH) (entry 5). The yield was slightly improved when the reaction was conducted at 0 °C to realize the synthesis of dibenzothiophene S-oxide 4a in an excellent yield (entry 6). We accomplished the gram-scale synthesis of 4a in good yield (entry 7). No reaction took place at –40 °C (entry 8). While the efficient synthesis of cyclized product 4a failed when the reaction was conducted in toluene, acetonitrile, or nitromethane (entries 9–11), we also succeeded in the facile synthesis of dibenzothiophene S-oxide in diethyl ether at room temperature or 0 °C (entries 12 and 13). The synthesis of 4a in the presence of 2,6-di(t-butyl)pyridine was achieved also in diethyl ether (entry 14). We also prepared dibenzothiophene S-oxide 4a from 1a in high yield through a single silica-gel column chromatography, obviously indicating good practicality of the benzothiophene S-oxide synthesis from sulfinate esters (Fig. 2A).
|
|
||||
|---|---|---|---|---|
| Entry | Activator | Solv. | Temp. | Yield (%)a |
| a Isolated yields. b The reaction was conducted using 2,6-di(tert-butyl)pyridine (4.0 equiv.) with Tf2O (2.0 equiv.). c Gram-scale synthesis. See details in the ESI. | ||||
| 1 | Tf2O | CH2Cl2 | rt | 90 |
| 2 | Tf2O | |||
| 2,6-(t-Bu)2pyb | CH2Cl2 | rt | 88 | |
| 3 | (CF3CO)2O | CH2Cl2 | rt | 0 |
| 4 | (CH3CO)2O | CH2Cl2 | rt | 0 |
| 5 | TfOH | CH2Cl2 | rt | 51 |
| 6 | Tf2O | CH2Cl2 | 0 °C | 97 |
| 7c | Tf2O | CH2Cl2 | 0 °C | 83 |
| 8 | Tf2O | CH2Cl2 | –40 °C | 0 |
| 9 | Tf2O | Toluene | rt | 3 |
| 10 | Tf2O | CH3CN | rt | 0 |
| 11 | Tf2O | CH3NO2 | rt | 0 |
| 12 | Tf2O | Et2O | rt | 97 |
| 13 | Tf2O | Et2O | 0 °C | Quant. |
| 14 | Tf2O | |||
| 2,6-(t-Bu)2pyb | Et2O | 0 °C | 88 | |

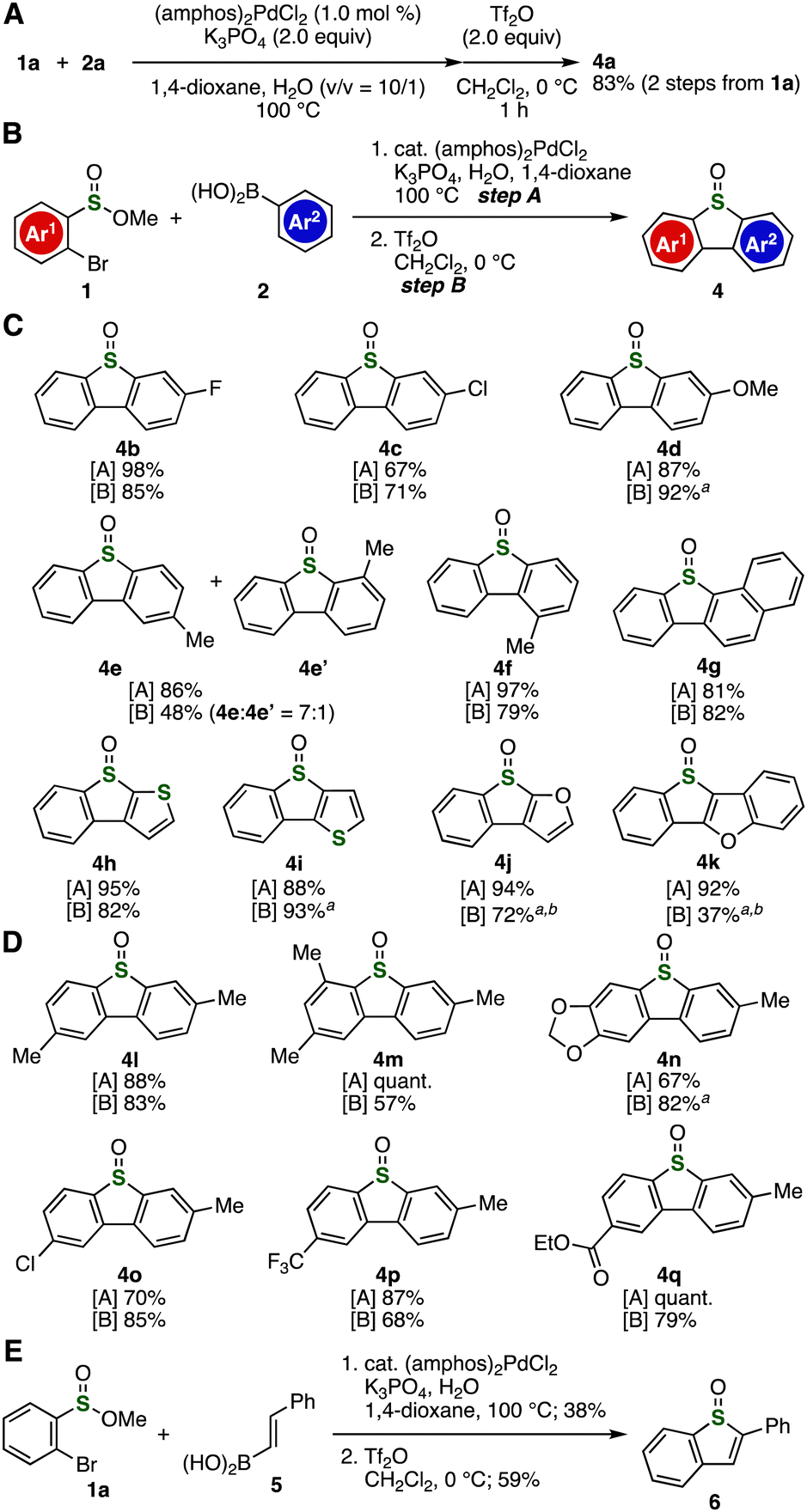 | ||
Fig. 2 (A) Synthesis of 4a with a single silica-gel column chromatography purification. (B) General scheme for synthesizing various dibenzothiophene S-oxides. (C) Results using various arylboronic acids 2 with 1a. (D) Results using various sulfinate esters 1 with 2a. (E) Synthesis of benzothiophene S-oxide 6. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Et2O was used as a solvent instead of CH2Cl2. b Et2O was used as a solvent instead of CH2Cl2. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was conducted in the presence of 2,6-di(tert-butyl)pyridine (4.0 equiv.). See details in the ESI.† amphos = di(tert-butyl)(4-(dimethylamino)phenyl)phosphine. The reaction was conducted in the presence of 2,6-di(tert-butyl)pyridine (4.0 equiv.). See details in the ESI.† amphos = di(tert-butyl)(4-(dimethylamino)phenyl)phosphine. | ||
A wide range of dibenzothiophene S-oxides having diverse functional groups were prepared from sulfinate ester 1a and various arylboronic acids 2 (Fig. 2B and C). Due to the good solubility of dichloromethane for broad aromatic compounds, we chose the conditions using dichloromethane for synthesizing various dibenzothiophene S-oxides. For example, we succeeded in the synthesis of dibenzothiphene S-oxides 4b and 4c in good yields by the 2-step protocol without damaging fluoro and chloro groups. In the case of synthesis of dibenzothiophene S-oxide 4d from 1a and 4-methoxyphenylboronic acid, the electrophilic cyclization of methyl 2-(4-methoxyphenyl)benzenesulfinate bearing an electron-rich aryl group took place efficiently when using diethyl ether as a solvent.11 We succeeded in the preparation of 2-methyldibenzothiophene S-oxide (4e) along with minor isomer 4e’ from 3-methylboronic acid by efficient Suzuki–Miyaura coupling and following intramolecular arylation, in which cyclization at sterically unhindered site predominantly proceeded. The 2-step protocol using sulfinate ester 1a and bulky 2-methylphenyl boronic acid allowed us to synthesize dibenzothiophene S-oxide 4f in high yields. We successfully synthesized π-extended dibenzothiophene S-oxide 4g, which was reported as a promising dibenzothiophene S-oxide derivative having an extended chromophore to red-shift the absorption spectra for atomic oxygen precursors,12 from 2-naphthylboronic acid through regioselective cyclization, where the regioisomer was not detected. It is worth noting that a variety of heteroarylboronic acids participated in the coupling–cyclization protocol to furnish heteroaromatic ring-fused dibenzothiophene S-oxides 4h–4k. When using 3-thienyl- or 3-furylboronic acid, intramolecular S-arylation proceeded at 2-position selectively to afford dibenzothiophene S-oxide analog 4h or 4j, respectively. The synthesis of furan- or benzo[b]furan-fused benzo[b]thiophene 4j or 4k was realized by the addition of 2,6-di(tert-butyl)pyridine as a base. We also succeeded in the preparation of benzo[b]thiophene S-oxide 6 from sulfinate ester 1a and 2-styrylboronic acid (5) through the intramolecular S-alkenylation (Fig. 2E).
Various sulfinate esters served in synthesizing dibenzothiophene S-oxides having various electron-donating or electron-withdrawing functional groups (Fig. 2B and D). Indeed, 2,7-dimethyl- and 2,4,7-trimethyl-substituted unsymmetric dibenzothiophene S-oxides 4l and 4m were prepared efficiently from 4-methyl- and 4,6-dimethyl-substituted 2-bromobenzenesulfinic acid esters, respectively. The synthesis of dibenzothiophene S-oxide 4n bearing a methylenedioxy moiety was achieved efficiently through cross-coupling and cyclization in diethyl ether. We also accomplished the preparation of dibenzothiophene S-oxides 4o–4q leaving electron-withdrawing chloro, trifluoromethyl, and ethoxycarbonyl groups untouched. These results obviously indicated that Br-selective coupling and following cyclization were not affected by a broad range of electron-donating or -withdrawing functional groups.
To gain insight into the reaction mechanism of the electrophilic cyclization of sulfinate esters, we then isolated sulfuran intermediate 7 by changing the protocol (Fig. 3A). Indeed, after treatment of sulfinate ester 3a with Tf2O, the resulting mixture was quenched with solid sodium bicarbonate instead of aqueous sodium bicarbonate. Then, filtrating the mixture to remove sodium bicarbonate and washing the resulting filtrate with n-hexane furnished sulfuran 7 quantitatively. We achieved the synthesis of dibenzothiophene S-oxide 4a with the hydrolysis of sulfuran 7 with aqueous sodium bicarbonate. Based on this result, we proposed a plausible reaction mechanism for the dibenzothiophene S-oxide synthesis (Fig. 3B). First, Tf2O activates S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety to form sulfuran intermediate I. Second, electrophilic aromatic substitution at the electrophilic sulfur provides sulfuran 7. Finally, hydrolysis of sulfuran 7 with aqueous sodium bicarbonate furnishes dibenzothiophene S-oxide 4avia the nucleophilic attack of water or bicarbonate anion to the sulfur atom. When sulfoxide 8 was treated with Tf2O followed by the addition of sodium bicarbonate, dibenzothiophene 9a was obtained in good yield through the demethylative hydrolysis of sulfonium intermediate III, where sulfoxide 4a was not obtained (Fig. 3C).13 This result shows that the difference between sulfinate esters and sulfoxides in the electrophilic cyclization lies in the reaction sites in the hydrolysis.
O moiety to form sulfuran intermediate I. Second, electrophilic aromatic substitution at the electrophilic sulfur provides sulfuran 7. Finally, hydrolysis of sulfuran 7 with aqueous sodium bicarbonate furnishes dibenzothiophene S-oxide 4avia the nucleophilic attack of water or bicarbonate anion to the sulfur atom. When sulfoxide 8 was treated with Tf2O followed by the addition of sodium bicarbonate, dibenzothiophene 9a was obtained in good yield through the demethylative hydrolysis of sulfonium intermediate III, where sulfoxide 4a was not obtained (Fig. 3C).13 This result shows that the difference between sulfinate esters and sulfoxides in the electrophilic cyclization lies in the reaction sites in the hydrolysis.
 | ||
| Fig. 3 (A) Stepwise synthesis of 4avia isolation of 7. (B) Plausible mechanism for the synthesis of 4a from 3a. (C) Electrophilic cyclization of sulfoxide 8. | ||
Good transformability of dibenzothiophene S-oxide enabled us to synthesize a wide variety of dibenzothiophene derivatives (Fig. 4A). For instance, S-oxidation of 4a with mCPBA afforded dibenzothiophene S,S-dioxide 10a in high yield.14 Rhodium-catalyzed imidation of 4a with trifluoroacetamide took place efficiently in the presence of iodobenzene diacetate and magnesium oxide to provide sulfoximine 11.15 Reduction of sulfoxide 4a with TFAA and sodium iodide proceeded smoothly to furnish dibenzothiophene 9a in high yield.16 We also accomplished the preparation of thiophene- and furan-fused benzothiophenes 9b and 9c by the reduction of sulfoxides 4i and 4j. To our surprise, we found that the decomposition of furan-fused benzothiophene 9c took place gradually in CDCl3 at room temperature.17
 | ||
| Fig. 4 (A) Transformations of 4a, 4i, and 4j. (B) Oxidation of 9b. (C) Oxidation of 9c. | ||
We clarified that it is not easy to achieve the efficient synthesis of sulfoxides 4i and 4j by the conventional oxidation with mCPBA (Fig. 4B and C).5 Indeed, the oxidation of thienobenzothiophene 9b with mCPBA (0.7 or 1.4 equiv.) afforded a mixture of sulfoxide 4i and sulfone 10b, clearly showing the problematic reactivity of sulfoxides under the oxidation conditions leading to sulfones (Fig. 4B). Furthermore, mCPBA oxidation of furanobenzothiophene 4j provided a complex mixture of products (Fig. 4C). These results obviously indicate an advantage of the newly developed method to synthesize dibenzothiophene S-oxides.
Further transformations of dibenzothiophene S-oxides realized a wide variety of highly functionalized dibenzothiophene derivatives (Fig. 5). An extended Pummerer-type propargylation of dibenzothiophene S-oxide 4a with silane 12 took place efficiently to provide 13 and 13′ in high combined yield, where C–C formation at the electron-rich but bulky site occurred smoothly to furnish 13 as a major product (Fig. 5A).4a It is worth noting that the good functional group tolerance allowed us to prepare o-silylaryl triflate 4r efficiently by the coupling–cyclization approach without damaging reactive silyl and triflyloxy groups (Fig. 5B). This result shows a clear advantage over other dibenzothiophene S-oxide synthesis with nucleophilic carbanions or heating under basic conditions.6 Then, regioselective N-arylation of aniline with aryne IV took place smoothly to afford adduct 14 in moderate yield without forming the regioisomer, in which the sulfoxide moiety remained unreacted.18–20 Moreover, we accomplished the synthesis of triazole-fused dibenzothiophene S-oxide 15 by regioselective cycloaddition of aryne IV with benzyl azide. Since o-silylaryl triflate moiety serves to synthesize highly substituted arenes through aryne intermediates by a wide variety of transformations, a broad range of dibenzothiophene derivatives will be prepared by diverse aryne reactions.
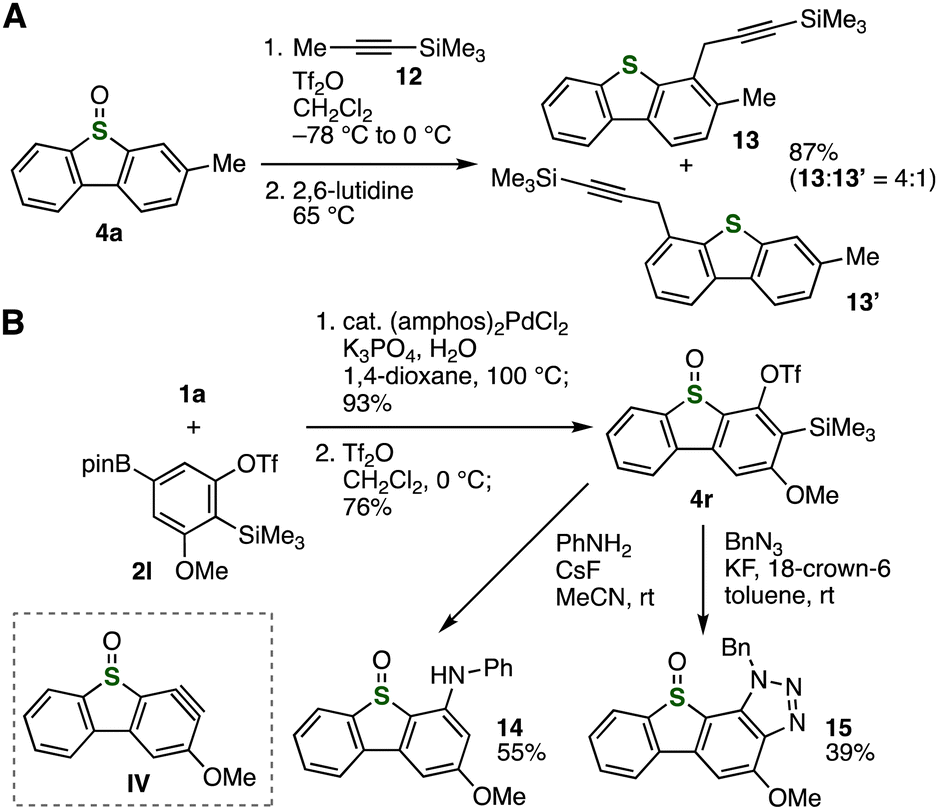 | ||
| Fig. 5 (A) C–H propargylation using 4a. (B) Synthesis and transformations of o-silylaryl triflate 4r. | ||
In summary, we have developed an efficient method to prepare dibenzothiophene S-oxides from 2-bromoaryl sulfinate esters and arylboronic acids in 2 steps. Diverse dibenzothiophene S-oxides having various functionalities were synthesized by Br-selective cross-coupling and subsequent cyclization with Tf2O without side reactions through the activation of sulfoxides. The good transformability of dibenzothiophene S-oxides allowed us to access novel heterocyclic molecules such as highly ring-fused thiophenes. Further studies including applications for the construction of new organosulfur skeletons by transformations of dibenzothiophene S-oxides are ongoing in our laboratory.
The authors thank Central Glass Co., Ltd. for providing Tf2O. This work was supported by JSPS KAKENHI Grant Number JP22H02086 (S. Y.); The Uehara Memorial Foundation (S. Y.); Tokuyama Science Foundation (S. Y.); The Ube Foundation (S. Y.); and Inamori Research Grants (S. Y.)
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Zhang, G. E. Ravilious, L. M. Hicks, J. M. Jez and R. D. McCulla, J. Am. Chem. Soc., 2012, 134, 16979 CrossRef CAS PubMed.
- G. C. M. Smith, A. J. Slade, C. J. Richardson and B. W. Durkacz, WO2006/126010 A2, 2006.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135 RSC.
- (a) J. A. Fernańdez-Salas, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2016, 138, 790 CrossRef PubMed; (b) X. Wang, X. Xun, H. Song, Y. Liu and Q. Wang, Org. Lett., 2022, 24, 4580 CrossRef CAS PubMed; (c) X. Li, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2021, 60, 6943 CrossRef CAS PubMed.
- (a) D. Villemin and X. Vlieghe, Sulfur Lett., 1998, 21, 199 CAS; (b) M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163 CrossRef CAS.
- For other dibenzothiophene S-oxide synthesis, see: (a) T. Wesch, A. Berthelot-Bréhier, F. R. Leroux and F. Colobert, Org. Lett., 2013, 15, 2490 CrossRef CAS PubMed; (b) R. Wada, S. Kaga, Y. Kawai, K. Futamura, T. Murai and F. Shibahara, Tetrahedron, 2021, 83, 131978 CrossRef CAS.
- (a) A. Kobayashi, T. Matsuzawa, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 5429 RSC; (b) A. Kobayashi, T. Matsuzawa, T. Hosoya and S. Yoshida, Chem. Lett., 2020, 49, 813 CrossRef CAS.
- For arylation of sulfinate esters using AlCl3, see: F. Yuste, A. H. Linares, V. M. Mastranzo, B. Ortiz, R. Sánchez-Obregón, A. Fraile and J. L. G. Ruano, J. Org. Chem., 2011, 76, 4635 CrossRef CAS PubMed . In this report, it is not easy to perform ortho-sulfinylation of arenes.
- K. Nakamura, Y. Kumagai, A. Kobayashi and S. Yoshida, Org. Biomol. Chem., 2023, 21, 6886 RSC.
- M. Suzuki, K. Kanemoto, Y. Nakamura, T. Hosoya and S. Yoshida, Org. Lett., 2021, 23, 3793 CrossRef CAS.
- When the reactions were performed in CH2Cl2 at 0 °C, dibenzothiophene S-oxides 4d, 4i, 4j, 4k, and 4n were obtained in 28, 76, 25, 0, and 0% yield, respectively.
- A. Isor, B. V. Chartier, M. Abo, E. R. Currens, E. Weerapana and R. D. McCulla, RSC Chem. Biol., 2021, 2, 577 RSC.
- D. Vasu, J. N. Hausmann, H. Saito, T. Yanagi, H. Yorimitsu and A. Osuka, Asian J. Org. Chem., 2017, 6, 1390 CrossRef CAS.
- H. Hussain, A. Al-Harrasi, V. R. Green, I. Ahmed, G. Abbas and N. U. Rehman, RSC Adv., 2014, 4, 12882 RSC.
- H. Okamura and C. Bolm, Org. Lett., 2004, 6, 1305 CrossRef CAS PubMed.
- M. de Greef and S. Z. Zard, Tetrahedron, 2004, 60, 7781 CrossRef CAS.
- When a time-course monitoring experiment of furan-fused benzothiophene 9c in CDCl3 at room temperature was conducted, 85% of 9c was decomposed after 7 days.
- Y. Nakamura, Y. Miyata, K. Uchida, S. Yoshida and T. Hosoya, Org. Lett., 2019, 21, 5252 CrossRef CAS PubMed.
- (a) T. Matsuzawa, K. Uchida, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 5521 CrossRef CAS; (b) S. Yoshida, H. Nakajima, K. Uchida, T. Yano, M. Kondo, T. Matsushita and T. Hosoya, Chem. Lett., 2017, 46, 77 CrossRef CAS.
- S. Yoshida, T. Kuribara, T. Morita, T. Matsuzawa, K. Morimoto, T. Kobayashi and T. Hosoya, RSC Adv., 2018, 8, 21754 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: https://doi.org/10.1039/d3cc05703h |
| This journal is © The Royal Society of Chemistry 2024 |