
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From a mercury(II) bis(yldiide) complex to actinide yldiides†
Mike
Jörges
ab,
Alexander J.
Gremillion
ab,
Daniel
Knyszek
 b,
Steven P.
Kelley
a,
Justin R.
Walensky
*a and
Viktoria H.
Gessner
*b
b,
Steven P.
Kelley
a,
Justin R.
Walensky
*a and
Viktoria H.
Gessner
*b
aDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA. E-mail: walenskyj@missouri.edu
bFaculty of Chemistry and Biochemistry, Ruhr-University Bochum, Bochum 44801, Germany. E-mail: viktoria.gessner@rub.de
First published on 20th February 2024
Abstract
The bis(yldiide) mercury complex, (L–Hg–L) [L = C(PPh3)P(S)Ph2], is prepared from the corresponding potassium yldiide and used to access the first substituted yldiide actinide complexes [(C5Me5)2An(L)(Cl)] (An = U, Th) via salt metathesis. Compared to previously reported phosphinocarbene complexes, the complexes exhibit long actinide–carbon distances, which can be explained by the strong polarization of the π-electron density toward carbon.
Actinide–carbon multiple bonding has attracted attention in the past decade due to efforts to better understand actinide–ligand bonding and differences in coordination chemistry and reactivity between actinides and the rest of the periodic table.1 Actinide carbene and carbene-like complexes have been reported with different ligand systems including widely used singlet carbenes2–13 carbodiphosphoranes,14–17 as well as highly nucleophilic methandiides (e.g. in A and B, Fig. 1A),18–22 which exhibited most different bonding situations and reactivities depending on the substituents at the carbon atom and the metal oxidation state.23 In recent years, yldiides (as their alkali metal complexes) have emerged as a further class of di-substituted carbon ligands (Fig. 1B)24 which have particularly been applied for the synthesis of main group complexes leading to seminal contributions in structure, bonding, and reactivity.25–29 Analogous complexes with transition metals are rare,30–33 but have demonstrated the unusual donor strength of the respective phosphonio-carbene/yldiide ligands.34–36 Likewise phosphonio-carbene complexes of the actinides have been reported, but only with the parent phosphoranomethylide ligand (C and D),37–40 which was synthesized in the coordination sphere of the metal and was shown to produce short actinide–carbon bonds with some degree of multiple bonding character. Albeit in the meantime various yldiides have been isolated,41–44 no actinide complexes have been prepared from these precursors, so that the impact of the substitution pattern on the bonding situation remains completely unclear.
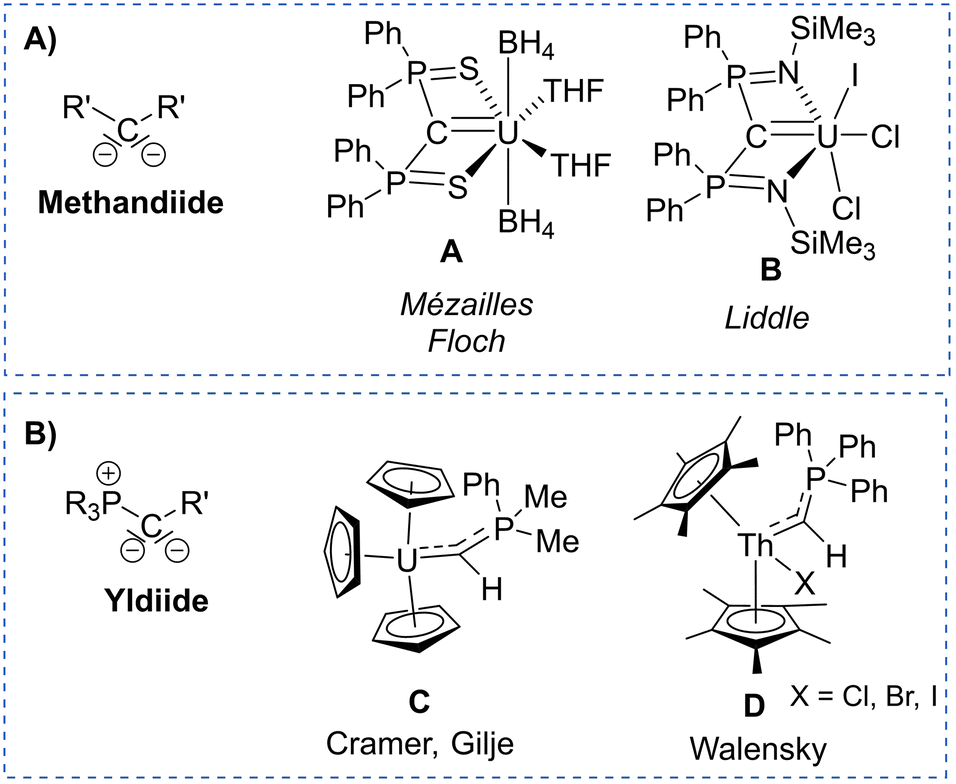 | ||
| Fig. 1 Examples of reported actinide carbene complexes based on (A) methandiide and (B) yldiide ligands. | ||
Previous attempts by our group to close this gap with various transition metal precursors remained unsuccessful, which we attributed to the reducing power of the alkali metal yldiides. Therefore, we turned our attention towards a milder method for introducing yldiide ligands and selected metathesis via a mercury compound as an alternative strategy. Herein, we report the first examples of substituted yldiide actinide complexes, which were prepared through the salt metathesis between the bis(yldiide) mercury complex, [Hg{C(PPh3)(SPPh2)}2], and [(C5Me5)2AnCl2] (Scheme 1).
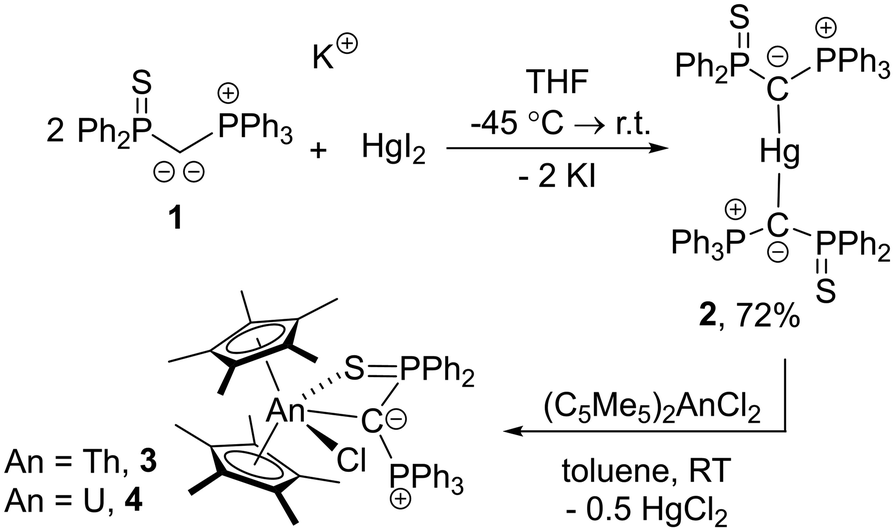 | ||
| Scheme 1 Synthesis of bis(yldiide)mercury, 2, and Th, 3, and U, 4, yldiide complexes. | ||
Due to the intrinsic ability of organomercury compounds to undergo transmetalation under mild conditions, we targeted the synthesis of an yldiide mercury complex. To date, only one mercury yldiide complex has been reported by Niecke and coworkers, but no reactivity studies were presented.45 We selected the thiophosphinoyl-substituted potassium yldiide [K{C(PPh3)(SPPh2)}] (1) previously reported by us as a test system,46 which we anticipated to form more stable complexes due to the potential additional coordination of the sulphur donor (for crystal structure of 1 see the ESI†). Treatment of HgI2 with two equivalents of 1 results in a shift in the 31P{1H} NMR spectrum from −15.8 and 24.2 ppm for the potassium yldiide to 21.8 and 43.6 ppm. The complete consumption of the starting materials and the appearance of a single set of signals suggested the formation of a symmetric bis-yldiide complex, which was confirmed by single crystal X-ray diffraction (sc-XRD) analysis (Fig. 2, top).
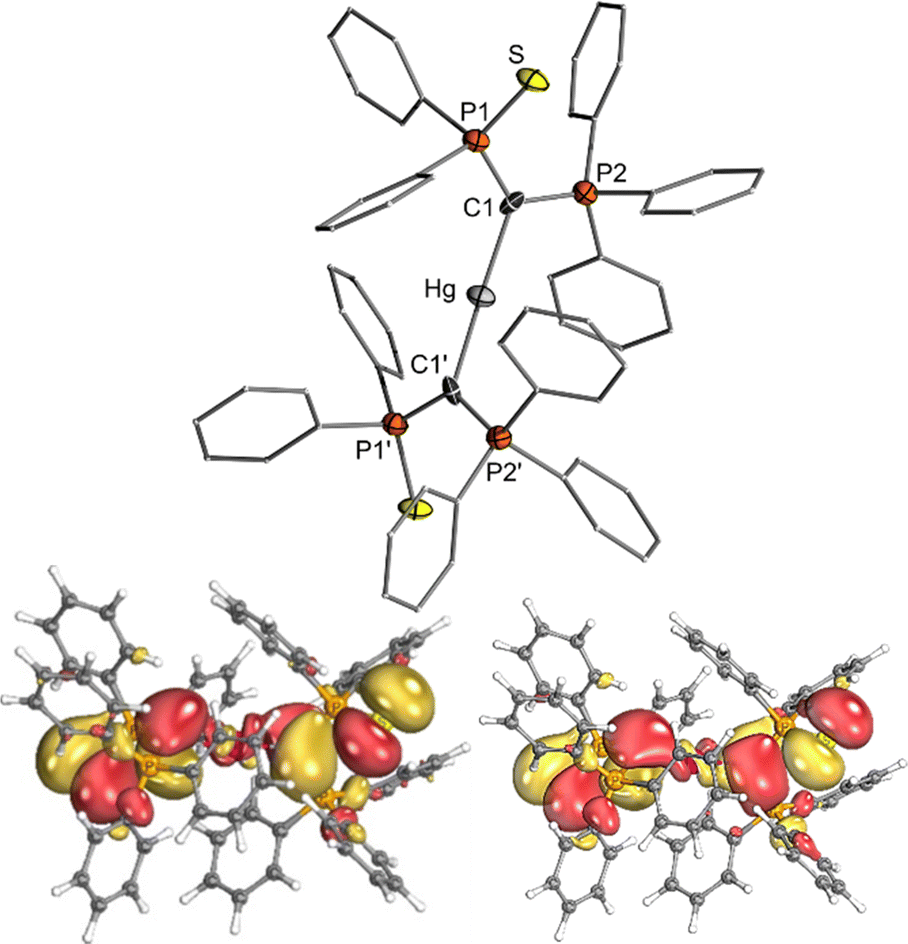 | ||
| Fig. 2 (top) Crystal structure of mercury complex 2. Ellipsoids are drawn at the 50% probability level. Important bond lengths and angles are given in Table 1. (bottom) Representations of the HOMO and HOMO−1 of 2 (isosurface value: 0.2 e Å−3). | ||
2 crystallizes as a C2 symmetric complex with the expected linear geometry around mercury (C–Hg–C angle: 178.3(7)°) and an almost perpendicular arrangement of the two ylide ligands (P1–C1–C1′–P1′: 92.8(9)°) relative to each other. The Hg–C bond lengths amount to 2.096(11) Å and are thus similar to those (2.051(4) Å) reported by Niecke et al. for a bis(yldiide) complex with an ArNP(Ar)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H) ligand (Ar = 2,4,6-tBu3C6H2).45 This distance is in the range of reported Hg–C single bonds,47 thus ruling out any double bond character. This is also reflected by the small Wiberg bond index (WBI) of only 0.46 suggesting a strong ionic contribution to the bonding situation. Nonetheless, the P–C bonds in the ylide ligands of 2 are distinctly elongated compared to those found in the potassium yldiide 1-K indicating a still significant charge transfer from the yldiide ligand to mercury. This increase in bond distances is more pronounced for the C1–P2 bond to the phosphonium group which elongates by more than 0.2 Å, indicating the presence of a single rather than an ylidic bonding due to the reduced negative natural charge at C1. This is further supported by density functional theory (DFT) studies (b3pw91/def2TZVP/MWB60) together with natural bond orbital (NBO) analyses, which revealed a decreased Wiberg bond index of this bond and a surprisingly high negative NBO charge at the ylidic carbon atom (−1.634 e). NBO analysis yields a fully ionic bonding situation with a mercury dication bonded by two anionic yldiide ligands. However, strong second-order perturbation interactions between the σ-symmetric lone pair at carbon and an empty orbital at mercury indicate a strong dative bonding in line with the increased C1–P distances. The π symmetric lone pair is only minimally involved in the Hg–C bonding as is also apparent from the two highest molecular orbitals (HOMO and HOMO−1), which represent the combination of the π symmetric lone pairs at the ylidic carbon atom, with little contribution of the metal center (Fig. 2).
C(H) ligand (Ar = 2,4,6-tBu3C6H2).45 This distance is in the range of reported Hg–C single bonds,47 thus ruling out any double bond character. This is also reflected by the small Wiberg bond index (WBI) of only 0.46 suggesting a strong ionic contribution to the bonding situation. Nonetheless, the P–C bonds in the ylide ligands of 2 are distinctly elongated compared to those found in the potassium yldiide 1-K indicating a still significant charge transfer from the yldiide ligand to mercury. This increase in bond distances is more pronounced for the C1–P2 bond to the phosphonium group which elongates by more than 0.2 Å, indicating the presence of a single rather than an ylidic bonding due to the reduced negative natural charge at C1. This is further supported by density functional theory (DFT) studies (b3pw91/def2TZVP/MWB60) together with natural bond orbital (NBO) analyses, which revealed a decreased Wiberg bond index of this bond and a surprisingly high negative NBO charge at the ylidic carbon atom (−1.634 e). NBO analysis yields a fully ionic bonding situation with a mercury dication bonded by two anionic yldiide ligands. However, strong second-order perturbation interactions between the σ-symmetric lone pair at carbon and an empty orbital at mercury indicate a strong dative bonding in line with the increased C1–P distances. The π symmetric lone pair is only minimally involved in the Hg–C bonding as is also apparent from the two highest molecular orbitals (HOMO and HOMO−1), which represent the combination of the π symmetric lone pairs at the ylidic carbon atom, with little contribution of the metal center (Fig. 2).
Given the weak C–Hg interaction, we next tested the ability of 2 to form actinide phosphoniocarbene complexes. To our delight, reaction of uranium and thorium precursors [(C5Me5)2AnCl2] (An = Th, U) with half an equivalent of 2 led to clean conversion to new products and yielded the corresponding actinide yldiide complexes [(C5Me5)2An{κ2-(C,S)–C(PPh3)(SPPh2)}(Cl)] (An = Th, 3; U, 4) as colourless and orange solids in 36–38% yield.48 The thorium complex features two doublets in the 31P{1H} NMR spectrum at 20.5 and 9.45 (2JPP = 8.2 Hz) ppm, while the two signals for the paramagnetic uranium complex appear at −47.4 and −506.9 ppm. The signals for the central carbon atom in 3 (as well as in 2) could only be detected for derivatives with a 13C labelled ylidic carbon atom. In the case of the diamagnetic Th(IV) complex 3, this carbenic carbon atom was observed at 42.0 ppm as a doublet of doublets (1JCP = 38.2 Hz, 16.5 Hz) in the 13C{1H} NMR spectrum. This signal appears in a similar range as the multiplet observed for 2 (43.9 ppm; 1JCP = 79.4 Hz, 67.1 Hz), but with markedly lower 1JCP coupling constants. Notably, these 13C signals are down-field shifted compared to yldiide 1-K (27.6 ppm; dd, 80.6 + 77.6 Hz), but clearly high-field shifted compared to carbene complexes with MC double bonds, suggesting a carbanionic character.39,40
The structure of both actinide complexes was unambiguously elucidated by sc-XRD analysis (Fig. 3 and ESI†) and confirms the expected carbene complex formation, with the ligand functioning as a bidentate C,S donor. Interestingly, both complexes feature distinct differences in the metal–carbon bond lengths relative to reported carbene and methandiide complexes, which argue for different bonding situations in these complexes. For example, the Th–C(yldiide) bond length of 2.6220(16) Å is long compared to those reported for Th–C(phosphonio-carbene) complexes (e.g. 2.3235(1)–2.299(6) Å in complexes D38,49,50 or 2.362(2) Å in [{(Me3Si)2N}3Th(CHPPh3)]) or Th–C(methandiide) complexes (2.410(8)–2.489(14) Å).23 Likewise, the U–C(yldiide) bond of 2.555(3) Å is long compared to U–C(methandiide) bonds (2.274(8)–2.393(2) Å),23 but shorter than U(IV)–C(NHC) distances (2.573(5)–2.668(2) Å).7,51–53 In contrast, the An–S bond lengths of 2.8885(4) and 2.8357(8) Å for 3 and 4, respectively, are slightly shorter than the average An–S bond distances in dithiophosphinate complexes (e.g. [An(S2PiPr2)4] with 2.907 Å for Th and 2.848 Å for U),54 suggesting a strong covalent interaction. Comparison of the C–P distances in the yldiide ligands of 3 and 4 with the yldiide 1-K and the mercury complex 2 was revealed to be highly informative regarding the bonding interaction. Both bond lengths in the actinide complexes are in between those found for 1-K and 2, indicating a high negative charge at the C1 carbon atom and hence a more ionic metal–carbon bond.
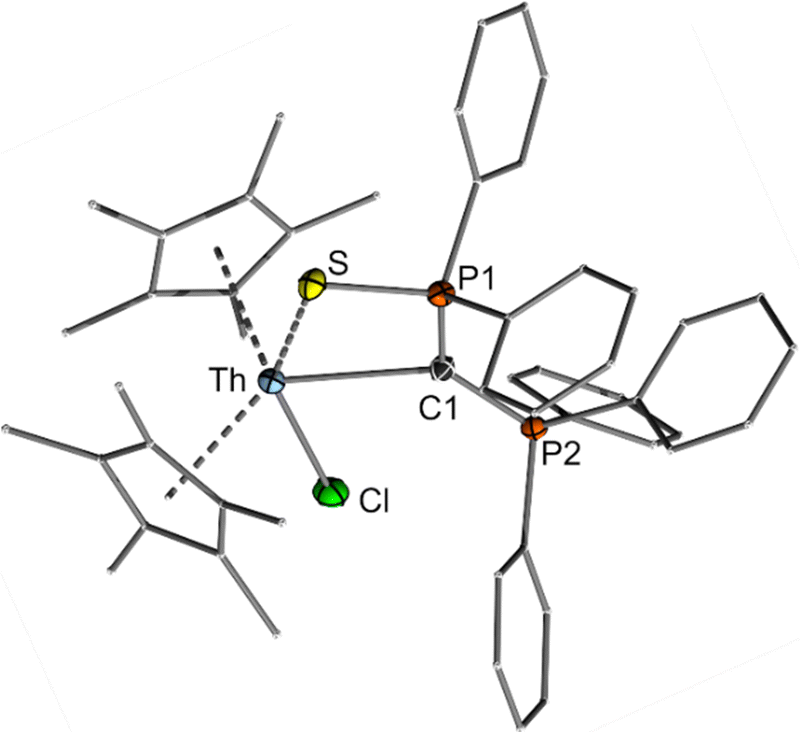 | ||
| Fig. 3 Crystal structure of the thorium complex 3. The uranium complex is isostructural (Fig. S24, ESI†). Important bond lengths and angles are given in Table 1. | ||
DFT calculations (see the ESI† for details and a more detailed discussion of the bonding situation in compounds 1–4) were performed to obtain further insights into the bonding situation of 3 and 4. Both complexes feature low Wiberg bond indices for the metal carbon bond, arguing for a single rather than a double bond, in line with the long metal carbon distances observed in the crystal structures (Table 1). This single bond character is further confirmed by atoms in molecules (AIM) calculations,55 which in all cases yielded bond critical points (BCP) as a decisive criterion for the bonding interaction, but with ellipticity values close to zero as a result of the cylindrical symmetry of the M–C single bond. Natural bond orbital (NBO)56 analysis again resulted in an extreme bonding picture with two lone pairs remaining at the C1 carbon atom. The covalent bonding is solely expressed by strong second-order perturbation interactions between the σ-symmetric lone pair at carbon and empty metal orbitals. The π-symmetric lone-pair at carbon shows again no or only a minimal interaction with metal orbitals. Overall, the bonding analysis argues for the presence of a M–C single bond in both complexes with a high ionic contribution. Accordingly, a high negative charge (approx. −1.4 for both actinide complexes) remains at the ylidic carbon centre. However, comparison of the overall charge of the yldiide ligand in the mercury complex 2 and the actinide complexes 3 and 4 revealed a significant charge transfer in the course of the transmetalation process from −0.625 e in 2 to almost zero in the thorium and the uranium complex. This net charge transfer mostly arises from the sulphur rather than from the carbon donor and is presumably the driving force of the transmetalation process. The calculated high ionic bonding character is commensurate with the long Th–C bond and upfield ylidic carbon resonances found in the solid-state structure and NMR spectrum, respectively. Hence, the trend of more downfield shifts in the 13C NMR spectra equating to increased covalent character holds in this case.50,51
| 1-K | 1 2Hg (2) | Th complex 3 | U complex 4 | |
|---|---|---|---|---|
| d(P(S)–C) [Å] | 1.664(3) | 1.750(11) | 1.706(2) | 1.709(4) |
| d(PPPh3–C) [Å] | 1.607(3) | 1.839(13) | 1.699(2) | 1.704(4) |
|
d(PS) [Å] |
2.018(1) | 1.994(4) | 2.032(1) | 2.031(1) |
| d(C–M) [Å] | — | 2.096(11) | 2.622(2) | 2.555(3) |
| d(S–M) [Å] | 3.157(1) | — | 2.888(1) | 2.836(1) |
| P–C–P [°] | 141.3(2) | 120.5(7) | 123.7(1) | 123.4(2) |
| WBI (C–M) | 0.0180 | 0.4607 | 0.605 | 0.7229 |
| WBI (C–P(S)) | 1.1775 | 0.9855 | 1.077 | 1.0857 |
| WBI (C–PPPh3) | 1.3865 | 1.1642 | 1.1467 | 1.15 |
| WBI (PS) |
1.1605 | 1.3118 | 1.0077 | 1.0464 |
| q(C1) [e] | −1.556 | −1.634 | −1.488 | −1.421 |
| q(PPPh3) [e] | 1.560 | 1.624 | 1.652 | 1.651 |
| q(P(S)) [e] | 1.395 | 1.383 | 1.515 | 1.492 |
| q(M) [e] | 0.876 | 1.249 | 0.755 | 0.575 |
| q(S) [e] | −0.789 | −0.661 | −0.388 | −0.389 |
| Σq(ligand) | −0.954 | −0.625 | −0.007 | −0.011 |
| ρ(BCP–MC) | 0.014 | 0.126 | 0.062 | 0.069 |
| ε(BCP–MC) | 0.039 | 0.041 | 0.018 | 0.041 |
In conclusion, we report the first synthesis of yldiide actinide complexes via salt metathesis with a bis(yldiide) mercury complex, which showed promising capabilities as a yldiide transfer reagent also for broader applications. The obtained thorium and uranium complexes exhibit long metal carbon distances as a consequence of a highly ionic bonding situation. DFT studies confirm that only little electron density is shifted from the yldiide towards the metal centre resulting in a M–C single bond with high ionic contribution. This contrasts previously reported phosphino carbene actinide complexes, which implies that yldiides depending on the substituents at the carbon may form Schrock-type carbene, but also ionic bonding situations similar to methandiide ligands.
This project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC-2033 – 390677874 – RESOLV and through research grant (DA 1402/6-1). J. R. W. gratefully acknowledges the Alexander von Humboldt Foundation for the award of a research fellowship and the Department of Energy, Office of Basic Energy Sciences, Heavy Element Program under Award DE-SC0021273. M. J. and A. J. G. thank the RUB research school for travel grants.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- J. Warren, J. Oldham, S. M. Oldham, B. L. Scott, K. D. Abney, W. H. Smith and D. A. Costa, Chem. Commun., 2001, 1348–1349 Search PubMed.
- H. Nakai, X. Hu, L. N. Zakharov, A. L. Rheingold and K. Meyer, Inorg. Chem., 2004, 43, 855–857 CrossRef CAS PubMed.
- T. Mehdoui, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Chem. Commun., 2005, 2860–2862 RSC.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS.
- D. Pugh, J. A. Wright, S. Freeman and A. A. Danopoulos, Dalton Trans., 2006, 775–782 RSC.
- P. L. Arnold, A. J. Blake and C. Wilson, Chem. – Eur. J., 2005, 11, 6095–6099 CrossRef CAS PubMed.
- S. A. Mungur, S. T. Liddle, C. Wilson, M. J. Sarsfield and P. L. Arnold, Chem. Commun., 2004, 2738–2739 RSC.
- P. L. Arnold, I. J. Casely, Z. R. Turner and C. D. Carmichael, Chem. – Eur. J., 2008, 14, 10415–10422 CrossRef CAS PubMed.
- (a) J. A. Seed, L. Vondung, R. W. Adams, A. J. Wooles, E. Lu and S. T. Liddle, Organometallics, 2022, 41, 1353–1363 CrossRef CAS; (b) E. Lu, J. T. Boronski, M. Gregson, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 5506 CrossRef CAS.
- J. A. Seed, M. Gregson, F. Tuna, N. F. Chilton, A. J. Wooles, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2017, 56, 11534–11538 CrossRef CAS.
- A. K. Maity, R. J. Ward, D. M. R. Y. P. Rupasinghe, M. Zeller, J. R. Walensky and S. C. Bart, Organometallics, 2020, 39, 783 CrossRef CAS.
- J. F. DeJesus, R. W. F. Kerr, D. A. Penchoff, X. B. Carroll, C. C. Peterson, P. L. Arnold and D. M. Jenkins, Chem. Sci., 2021, 12, 7882–7887 RSC.
- W. Fang, S. Pan, W. Su, L. Zhao, G. Frenking and C. Zhu, CCS Chem., 2021, 3, 2324 Search PubMed.
- W. Su, S. Pan, S. Wang, L. Zhao, G. Frenking and C. Zhu, Nat. Commun., 2018, 9, 4997 CrossRef PubMed.
- M. Gregson, E. Lu, D. P. Mills, F. Tuna, E. J. L. McInnes, C. Hennig, A. C. Scheinost, J. McMaster, W. Lewis, A. J. Blake, A. Kerridge and S. T. Liddle, Nat. Commun., 2017, 8, 14137 CrossRef CAS PubMed.
- W. Su, Y. Ma, L. Xiang, J. Wang, S. Wang, L. Zhao, G. Frenking and Q. Ye, Chem. – Eur. J., 2021, 27, 10006–10011 CrossRef CAS PubMed.
- T. Cantat, T. Arliguie, A. Noël, P. Thuéry, M. Ephritikhine, P. L. Floch and N. Mézailles, J. Am. Chem. Soc., 2009, 131, 963–972 CrossRef CAS.
- J.-C. Tourneux, J.-C. Berthet, T. Cantat, P. Thuéry, N. Mézailles, P. Le Floch and M. Ephritikhine, Organometallics, 2011, 30, 2957–2971 CrossRef CAS.
- J.-C. Tourneux, J.-C. Berthet, T. Cantat, P. Thuéry, N. Mézailles and M. Ephritikhine, J. Am. Chem. Soc., 2011, 133, 6162–6165 CrossRef CAS PubMed.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 2383–2386 CrossRef CAS.
- O. J. Cooper, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 39, 5074–5076 RSC.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Eur. J., 2013, 19, 7071–7083 CrossRef CAS PubMed.
- L. T. Scharf and V. H. Gessner, Inorg. Chem., 2017, 56, 8599–8607 CrossRef CAS.
- A. Sarbajna, V. S. V. S. N. Swamy and V. H. Gessner, Chem. Sci., 2021, 12, 2016–2024 RSC.
- T. Scherpf, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2017, 56, 3275–3279 CrossRef CAS.
- C. Mohapatra, L. T. Scharf, T. Scherpf, B. Mallick, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 7459–7463 CrossRef CAS PubMed.
- C. Mohapatra, H. Darmandeh, H. Steinert, B. Mallick, K.-S. Feichtner and V. H. Gessner, Chem. – Eur. J., 2020, 26, 15145 CrossRef CAS PubMed.
- T. Stalder, F. Krischer, H. Steinert, P. Neigenfind and V. H. Gessner, Chem. – Eur. J., 2022, 28, e202104074 CrossRef CAS.
- W. C. Kaska, D. K. Mitchell, R. F. Reichelderfer and W. D. Korte, J. Am. Chem. Soc., 1974, 96, 2847–2854 CrossRef CAS.
- X. Li, A. Wang, L. Wang, H. Sun, K. Harms and J. Sundermeyer, Organometallics, 2007, 26, 1411–1413 CrossRef CAS.
- (a) X. Li, M. Schopf, J. Stephan, K. Harms and J. Sundermeyer, Organometallics, 2002, 21, 2356–2358 CrossRef CAS; (b) S. Yogendra, T. Weyhermüller, A. W. Hahn and S. DeBeer, Inorg. Chem., 2019, 58, 9358 CrossRef CAS PubMed.
- A. Y. Khalimon, E. M. Leitao and W. E. Piers, Organometallics, 2012, 31, 5634–5637 CrossRef CAS.
- R. Zurawinski, C. Lepetit, Y. Canac, M. Mikolajczyk and R. Chauvin, Inorg. Chem., 2009, 48, 2147–2155 CrossRef CAS PubMed.
- S. Lapointe, P. Duari and V. H. Gessner, Chem. Sci., 2023, 14, 3816–3825 RSC.
- R. Taakili, C. Barthes, C. Lepetit, C. Duhayon, D. A. Valyaev and Y. Canac, Inorg. Chem., 2021, 60, 12116–12128 CrossRef CAS PubMed.
- R. E. Cramer, R. B. Maynard, J. C. Paw and J. W. Gilje, J. Am. Chem. Soc., 1981, 103, 3589–3590 CrossRef CAS.
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 6894–6897 CrossRef CAS PubMed.
- P. Rungthanaphatsophon, A. Bathelier, L. Castro, A. C. Behrle, C. L. Barnes, L. Maron and J. R. Walensky, Angew. Chem., Int. Ed., 2017, 56, 12925–12929 CrossRef CAS PubMed.
- W. Mao, L. Xiang, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2017, 139, 17759–17762 CrossRef CAS PubMed.
- T. Scherpf, R. Wirth, S. Molitor, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2015, 54, 8542–8546 CrossRef CAS PubMed.
- C. Schwarz, L. T. Scharf, T. Scherpf, J. Weismann and V. H. Gessner, Chem. – Eur. J., 2019, 25, 2793–2802 CrossRef CAS PubMed.
- H. Darmandeh, T. Scherpf, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Z. Anorg. Allg. Chem., 2020, 646, 835–841 CrossRef CAS PubMed.
- M. Jörges, R. M. Gauld, H. Steinert, L. Kelling, V. S. V. S. N. Swamy, A. Kroll, B. Mallick and V. H. Gessner, Chem. – Eur. J., 2023, 29, e202300504 CrossRef.
- T. Baumgartner, B. Schinkels, D. Gudat, M. Nieger and E. Niecke, J. Am. Chem. Soc., 1997, 119, 12410–12411 CrossRef CAS.
- M. Jörges, F. Krischer and V. H. Gessner, Science, 2022, 378, 1331–1336 CrossRef PubMed.
- J. G. Melnick and G. Parkin, Science, 2007, 317, 225–227 CrossRef CAS PubMed.
- The Th complex contained residual toluene, which was difficult to remove without decomposition. Therefore, the actual yield of the complex is slightly lower (see ESI†).
- P. Rungthanaphatsophon, P. Huang and J. R. Walensky, Organometallics, 2018, 37, 1884–1891 CrossRef CAS.
- D. E. Smiles, G. Wu, P. Hrobárik and T. W. Hayton, Organometallics, 2017, 36, 4519–4524 CrossRef CAS.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS.
- D. Pugh, J. A. Wright, S. Freeman and A. A. Danopoulos, Dalton Trans., 2006, 775–782 RSC.
- J. F. DeJesus, R. W. F. Kerr, D. A. Penchoff, X. B. Carroll, C. C. Peterson, P. L. Arnold and D. M. Jenkins, Chem. Sci., 2021, 12, 7882–7887 RSC.
- A. C. Behrle, A. Kerridge and J. R. Walensky, Inorg. Chem., 2015, 54, 11625–11636 CrossRef CAS PubMed.
- R. F. A. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic, crystallographic, and computational details are given in the ESI. CCDC 2307044–2307048. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05553a |
| This journal is © The Royal Society of Chemistry 2024 |