
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decarboxylative photoinduced ligand-to-metal charge transfer reaction: synthesis of 2-substituted chroman-4-ones†
Mohsen
Monirialamdari
 a and
Anna
Albrecht
*b
a and
Anna
Albrecht
*b
aInstitute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
bInstitute of General and Ecological Chemistry, Faculty of Chemistry, Lodz University of Technology Żeromskiego 116, 90-924 Łódź, Poland. E-mail: anna.albrecht@p.lodz.pl
First published on 29th December 2023
Abstract
In this manuscript, a photoinduced ligand-to-metal charge transfer (LMCT) approach, employing transition-metal-based photocatalysts, for the efficient alkylation of electron-poor olefin is described. The developed redox-neutral process benefits from mild reaction conditions and involves a wide range of chromone-3-carboxylic acids as well as nucleophiles amenable to selective C–H functionalization leading to the formation of 2-substituted chroman-4-one compounds with potential biological activity.
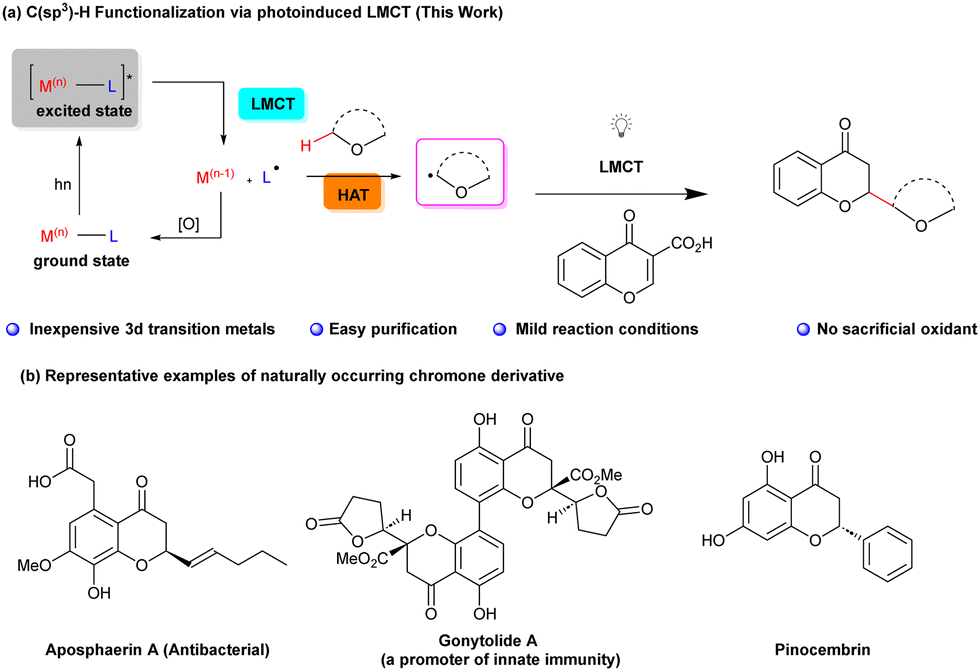
The selective functionalization of aliphatic carbon–hydrogen (C–H) bonds has been a longstanding pursuit in the realm of organic synthesis.1,2 While the functionalization of sp2 and activated sp3 C–H bonds using transition metal catalysts is a well-established field,3 addressing the challenges associated with inert C(sp3)–H functionalization has been proven to be a complex endeavour, marked by issues related to selectivity.4 Recently, considerable efforts in this domain, driven by the application of 4d or 5d transition metal catalysts (mostly Pd, Ru, and Rh), have been made. Remarkably, 3d transition metals, appreciated for their environmental and economic benefits, also contributed significantly to the functionalization of otherwise inert C(sp3)–H bonds.3a,5 Among these, copper (Cu) stands out due to its abundance and versatile redox reactivity, stemming from its array of oxidation states and its ability to coordinate with π-bonds and heteroatoms.3a,5,6
An interesting strategy to C(sp3)–H functionalization is the photoinduced radical-mediated hydrogen atom transfer (HAT) (Scheme 1a). This approach enables the selective introduction of functional groups into molecules by generating alkyl radicals as versatile intermediates, thereby avoiding the need for pre-functionalization or the use of directing groups.7,8 The efficacy of HAT depends on the intricate interplay of electronic and steric factors of both C(sp3)–H bonds within the substrate as well as of the HAT reagent itself.9,10 Cyclic and acyclic ethers constitute particularly interesting groups of reactants in such transformations due to their ability to undergo selective transformation with participation of the electron-rich C(sp3)–H bonds adjacent to the oxygen atom.11
 | ||
| Scheme 1 Cu-catalyzed LMCT enables unactivated C(sp3)–H alkylation. | ||
Interestingly, photoinduced ligand-to-metal charge transfer (LMCT) has been found to promote radical-mediated processes, thus bypassing more traditional photoredox processes.12 It was demonstrated that by direct excitement of high-valency metal–ligand complexes the formation of highly reactive radical species was possible. These key intermediates subsequently participate in the intermolecular HAT processes with hydrocarbons opening exciting possibilities in modern organic synthesis.13,14 In 2019 the Rovis group made a groundbreaking discovery by demonstrating that Co(II)-acetylides possessed the capability to engage in the LMCT process, effectively promoting [2+2+2]-cycloadditions with alkynes.15
Building upon this pivotal advancement and driven by the hypothesis that other air-stable metal salts could complement HAT-based methodologies in enhancing substrate diversity, selectivity, and reactivity, our research endeavours took a significant leap forward. We initiated an extensive exploration into the potential of various metal halides to participate in LMCT. Our research was also inspired by the pioneering work of Kochi, who demonstrated the utility of cupric chloride in the stoichiometric chlorination of alkanes in acetonitrile.16 Furthermore, Rovis and Tarnovsky demonstrated the utility of various photoactive species capable of undergoing ligand-to-metal charge transfer (LMCT) within Cu(II) chlorocomplexes in acetonitrile.17
Inspired by these research studies, we decided to turn our attention to the potential of photoinduced LMCT using earth-abundant transition-metal-based photocatalysts. Through an exploration of photoinduced HAT and the role of chloride salts, we decided to perform a photoactivation-based site-specific tandem C(sp3)–H functionalization/decarboxylation reaction of chromone-3-carboxylic acids, leading to the formation of 2-substituted chroman-4-ones (Scheme 1a). In this context it is worth noting that chromanones and related compounds (Scheme 1b) constitute privileged structural motifs present in many natural products and exhibit a wide variety of biological activities.18 Selected examples of natural flavanones and chromanones are shown in Scheme 1 featuring notable members like aposphaerin A (recognized for its antibacterial properties)19a and gonytolide A (a promoter of innate immunity).19b–d Furthermore, flavonoid pinocembrin is associated with mitigating the risk of specific chronic diseases.20
Decarboxylative Michael reaction based on the nucleophilic addition to carboxylic-acid-activated olefins followed by the decarboxylation reaction constitute a powerful synthetic tool.21
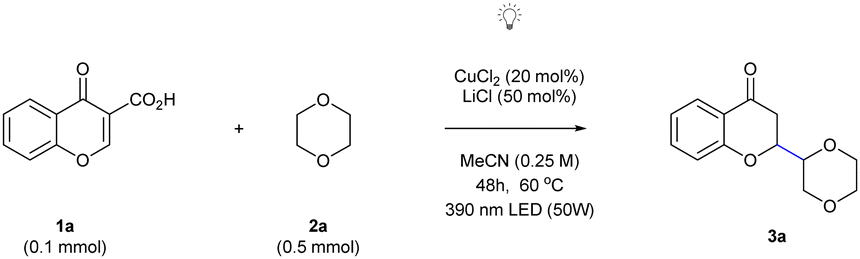
Recently, we described the photocatalytic, doubly decarboxylative Giese reaction applicable to a wide range of carboxylic acids22a and visible-light driven reductive azaarylation of coumarin-3-carboxylic acids,22b thus demonstrating the potential of decarboxylative radical strategies. Initially, the optimization studies were performed using chromone-3-carboxylic acid 1a and 1,4-dioxane 2a as model substrates and it was found that it is possible to realize the devised synthetic strategy in the presence of 20 mol% of anhydrous CuCl2 as the catalyst and 50 mol% of LiCl in acetonitrile, under an argon atmosphere and utilizing 390 nm LEDs (as they excite synthetically useful LMCT absorption bands) at an elevated temperature of 60 °C for 48 hours. The outcome was remarkable yielding the exclusive, regioselective formation of the 2-substituted chroman-4-one 3a with a good yield of 83%. This approach was systematically explored in order to fine-tune the critical parameters, resulting in the conditions necessary for achieving the desired outcome in this study. One notable enhancement was the utilization of a well-sealed crimp vial instead of a screw-capped vial, which led to a substantial increase in the yield, reaching to 91% (Table 1, entry 1). The utilization of 10-fold excess of 2a resulted in a decreased yield of the desired product 3a (Table 1, entry 2). Throughout these optimization studies, the optimal 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a ratio (1.0:5.0 equiv.) was the most favourable for achieving good results. It became evident that these selected parameters encompassed the ideal conditions to enable LMCT within a redox-neutral Giese-type coupling reaction.23 Furthermore, the influence of solvents on the reaction outcome was evaluated and identified that anhydrous acetonitrile is the pivotal solvent for enhancing the efficiency of the proposed coupling of inactivated C(sp3)–H bonds with electron-deficient olefins. Alternative solvents like acetone, DMF, CH2Cl2, and CHCl3 (as indicated in Table 1, entry 3, and details in ESI,† Table S2) exhibited substantially reduced effectiveness, resulting in notably lower yields (65%, 16%, 13%, and 9%, respectively). To increase the redox-neutral transformation, an exogenous chloride source in conjunction with catalytic amounts of CuCl2 was introduced. Application of TMSCl (as denoted in Table 1, entry 4) and TBACl (see ESI,† Table S3) in both cases demonstrated comparable efficiency, affording the desired product with 60% and 53% yields, respectively. Notably, the presence of HCl did not yield any product (see ESI,† Table S3). In the course of further studies, alternative LMCT catalysts such as RuCl3 and FeCl3 as photocatalysts were tested (Table 1, entries 5 and 6). These catalysts delivered product 3a in 79% and 70% yield, respectively, and did not provide a significant improvement over CuCl2. In contrast, TiCl4 and BiCl3 had a dramatically adverse impact on the reaction outcome (ESI,† Table S4). Turning our attention to solvent concentration, we found that CH3CN (0.3 M) (Table 1, entry 7) provided the desired product in 57% yield. Control experiments pointed out the indispensability of CuCl2 and shed light on this transformation (Table 1, entries 8 and 9). Diminished yields were observed when 440 nm irradiation was employed (Table 1, entry 10). The best results were obtained using 5-fold excess of 2a with respect to 1a with application of 20 mol% of CuCl2 as a catalyst, 50 mol% of LiCl in acetonitrile and irradiation of the reaction mixture for 48 h at 60 °C (Table 1, entry 1). Under these conditions 3a was obtained in 91% yield as a separable mixture of two diastereoisomers in a 1:1 ratio (see the ESI,† for the assignment of the relative configuration). With optimized reaction conditions in hand, the scope of this photocatalytic copper-mediated Giese-type coupling reaction with regard to both substrates was examined (Scheme 2). In the first part of the scope, various chromone-3-carboxylic acids 1, encompassing a range of electron-donating and electron-withdrawing functional groups, were evaluated as potential coupling partners (Scheme 2). Chromone-3-carboxylic acids (1a–1e) featuring electron-donating groups on the aromatic ring displayed remarkable compatibility with these photoinduced conditions, yielding separable diastereomeric mixtures of desired products 3a–3e with high efficiency. Similarly, chromone-3-carboxylic acids (1f–h) containing electron-withdrawing groups smoothly underwent the reaction under photoinduced LMCT conditions, affording the desired products 3f–3h as separable diastereomeric mixtures with commendable yields (ranging from 48% to 34%). Furthermore, chromone-3-carboxylic acids bearing two substituents, such as 6,8-dichloride 1i were obtained as single diastereomers, and those featuring opposite electronic effects (3j, obtained as a separable mixture of diastereomers) successfully furnished the corresponding products 3i and 3j in moderate yields (40% and 39% yields, respectively). Notably, hydroxy-substituted chromone-3-carboxylic acids 1k on the position 6 of the aromatic ring gave desired product in 65% yield under the photoinduced LMCT strategy. It is also worth noting that coumarin-3-carboxylic acid 1l is applicable in the developed methodology to give 3l.
:2a ratio (1.0:5.0 equiv.) was the most favourable for achieving good results. It became evident that these selected parameters encompassed the ideal conditions to enable LMCT within a redox-neutral Giese-type coupling reaction.23 Furthermore, the influence of solvents on the reaction outcome was evaluated and identified that anhydrous acetonitrile is the pivotal solvent for enhancing the efficiency of the proposed coupling of inactivated C(sp3)–H bonds with electron-deficient olefins. Alternative solvents like acetone, DMF, CH2Cl2, and CHCl3 (as indicated in Table 1, entry 3, and details in ESI,† Table S2) exhibited substantially reduced effectiveness, resulting in notably lower yields (65%, 16%, 13%, and 9%, respectively). To increase the redox-neutral transformation, an exogenous chloride source in conjunction with catalytic amounts of CuCl2 was introduced. Application of TMSCl (as denoted in Table 1, entry 4) and TBACl (see ESI,† Table S3) in both cases demonstrated comparable efficiency, affording the desired product with 60% and 53% yields, respectively. Notably, the presence of HCl did not yield any product (see ESI,† Table S3). In the course of further studies, alternative LMCT catalysts such as RuCl3 and FeCl3 as photocatalysts were tested (Table 1, entries 5 and 6). These catalysts delivered product 3a in 79% and 70% yield, respectively, and did not provide a significant improvement over CuCl2. In contrast, TiCl4 and BiCl3 had a dramatically adverse impact on the reaction outcome (ESI,† Table S4). Turning our attention to solvent concentration, we found that CH3CN (0.3 M) (Table 1, entry 7) provided the desired product in 57% yield. Control experiments pointed out the indispensability of CuCl2 and shed light on this transformation (Table 1, entries 8 and 9). Diminished yields were observed when 440 nm irradiation was employed (Table 1, entry 10). The best results were obtained using 5-fold excess of 2a with respect to 1a with application of 20 mol% of CuCl2 as a catalyst, 50 mol% of LiCl in acetonitrile and irradiation of the reaction mixture for 48 h at 60 °C (Table 1, entry 1). Under these conditions 3a was obtained in 91% yield as a separable mixture of two diastereoisomers in a 1:1 ratio (see the ESI,† for the assignment of the relative configuration). With optimized reaction conditions in hand, the scope of this photocatalytic copper-mediated Giese-type coupling reaction with regard to both substrates was examined (Scheme 2). In the first part of the scope, various chromone-3-carboxylic acids 1, encompassing a range of electron-donating and electron-withdrawing functional groups, were evaluated as potential coupling partners (Scheme 2). Chromone-3-carboxylic acids (1a–1e) featuring electron-donating groups on the aromatic ring displayed remarkable compatibility with these photoinduced conditions, yielding separable diastereomeric mixtures of desired products 3a–3e with high efficiency. Similarly, chromone-3-carboxylic acids (1f–h) containing electron-withdrawing groups smoothly underwent the reaction under photoinduced LMCT conditions, affording the desired products 3f–3h as separable diastereomeric mixtures with commendable yields (ranging from 48% to 34%). Furthermore, chromone-3-carboxylic acids bearing two substituents, such as 6,8-dichloride 1i were obtained as single diastereomers, and those featuring opposite electronic effects (3j, obtained as a separable mixture of diastereomers) successfully furnished the corresponding products 3i and 3j in moderate yields (40% and 39% yields, respectively). Notably, hydroxy-substituted chromone-3-carboxylic acids 1k on the position 6 of the aromatic ring gave desired product in 65% yield under the photoinduced LMCT strategy. It is also worth noting that coumarin-3-carboxylic acid 1l is applicable in the developed methodology to give 3l.
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | dr | Yield (%) |
| All reactions were performed in a crimp vial using 1a (0.1 mmol), 2a (0.5 mmol), CuCl2 (20 mol%), LiCl (50 mol%), anhydrous MeCN [0.25 M], 50 W blue LED irradiation (390 nm), and a 7 cm distance from the vial at 60 °C for 48 h. TMSCl = trimethylsilyl chloride; yields of isolated product 3a are reported after chromatographic purification. dr – diastereoisomeric ratio | |||
| 1 | None | 1:1 |
91 |
| 2 | 10.0 (equiv.) 2a | 1:1 |
21 |
| 3 | Acetone | 1:1 |
65 |
| 4 | TMSCl | 1:1 |
60 |
| 5 | RuCl3 | 1:1 |
79 |
| 6 | FeCl3 | 1:1 |
70 |
| 7 | CH3CN (0.3 M) | 1:1 |
57 |
| 8 | 0% CuCl2 | n.d. | <5 |
| 9 | In the dark | n.d. | <5 |
| 10 | Kessil (440 nm) | 1:1 |
37 |
 | ||
| Scheme 2 Scope of the reaction. | ||
In the second part of the scope research, we embarked on an extensive exploration of nucleophiles amenable to selective C–H functionalization. Unactivated cyclic alkanes, including cyclooctane, cyclohexane, and cyclopentane (2m–2o), emerged as effective coupling partners, showcasing the versatility of this method. Buoyed by these promising results, we turned our attention to various ether substrates for the C–H functionalization reaction. Gratifyingly, not only did 1,4-dioxane exhibit compatibility, but other cyclic and acyclic ethers such as tetrahydrofuran, tetrahydropyran, diethyl ether, ethyl acetate and γ-butyrolactone also proved to be amenable, yielding the corresponding ether-derived chromanones 3p–3t in moderate to good yields (ranging from 57% to 74%). In almost all cases, no discernible diastereoselectivity was observed, resulting in a 1:1 mixture of products. While the diastereomers of 3p could be separated through silica gel chromatography, the diastereomers of products 3q–3s remained inseparable. Subsequently, the products 3a and 3a′ were subjected to chemo- and diastereoselective reductions of the carbonyl group (Scheme 3). Depending on the conditions employed the reaction afforded alcohol 4a or dihydrobenzopyran 5a in 83 and 95% yields, with complete diastereoselectivity.
 | ||
| Scheme 3 Chemo- and diastereoselective reduction of 3a and 3a′. | ||
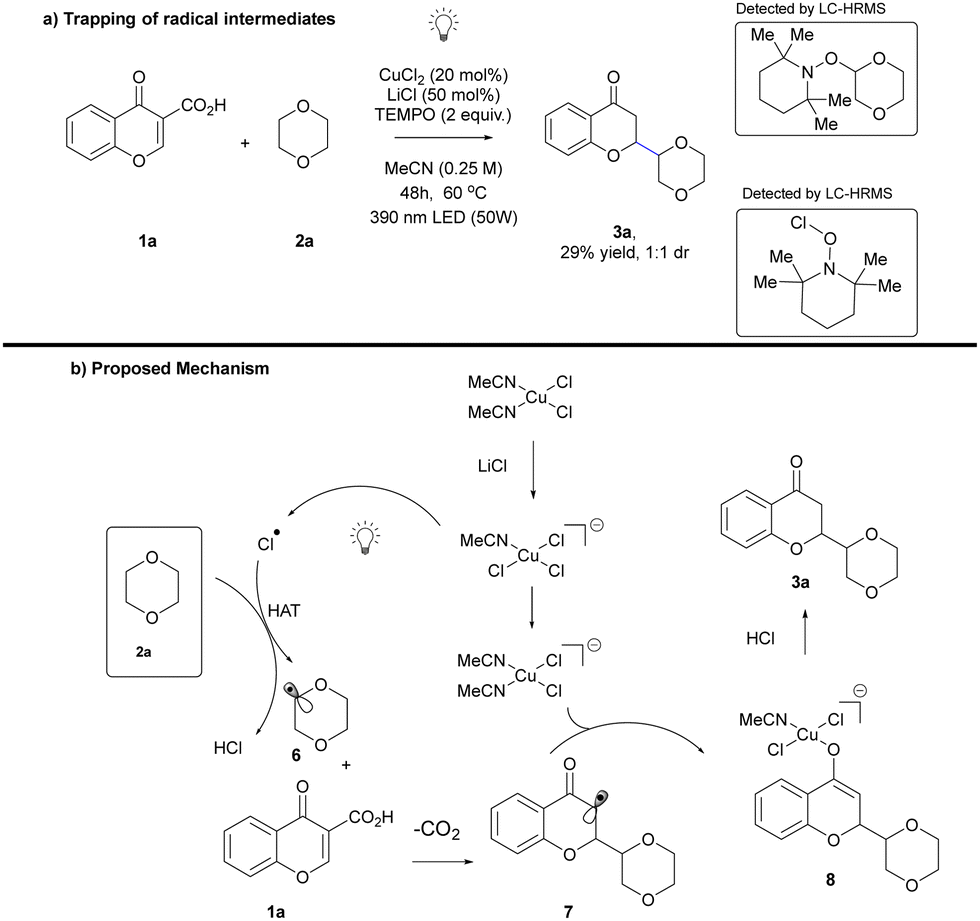
Furthermore, to emphasize the synthetic utility of this methodology, the scaled up protocol to a 1 mmol scale was successfully developed (see ESI,† Table S6). To gain deeper insights into the potential reaction pathway, a series of control experiments were conducted. The addition of 2.0 equivalents of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), a widely recognized radical scavenger, to the standard reaction mixture resulted in a noticeable reduction in the yield of the desired reaction pathway (Scheme 4a). Electrospray ionization-high-resolution mass spectrometry further revealed that both chlorine and aliphatic radicals were effectively trapped by TEMPO (see the ESI† for details). These compelling observations strongly indicate the involvement of radical intermediates within the reaction mechanism, substantiating our proposed pathway under the photoinduced ligand-to-metal charge transfer (LMCT) conditions. Based on these results, the mechanism of developed transformation was proposed. The process is initiated by coordination of acetonitrile to copper chloride and ligand transfer from lithium chloride. Irradiation of the complex obtained initiates ligand-to-metal charge transfer resulting in the homolysis of the Cu–Cl bond and chlorine radical formation. It acts as a powerful hydrogen atom transfer reagent, which undergoes a reaction with 1,4-dioxane to give radical 6 and HCl. The newly formed alkyl radical 6 undergoes Giese reaction with chromone-3-carboxylic acid 1a followed by decarboxylation. Subsequent newly generated more stable radical 7 takes part in the metallization tautomerization process to give corresponding enolate that undergoes photodemetallation that terminates the reaction affording 3 as target product (Scheme 4b).
 | ||
| Scheme 4 Mechanistic investigations. | ||
In summary, our study successfully showcases a photoinduced LMCT approach for achieving site-specific functionalization of carboxylic acid-activated olefins, ultimately leading to the formation of new C–C σ-bonds. This redox-neutral, photocatalytic alkylation of nonactivated C(sp3)–H bonds under mild conditions represents a synthetically practical and operationally feasible route for the synthesis of 2-substituted chroman-4-ones. The versatility of this reaction was underscored by its successful application to various electron-rich and electron-deficient substrates.
This contribution has been completed while the first author (MM) was the Doctoral Candidate in the Interdisciplinary Doctoral School of TUL, Poland. This work was financially supported by Young Scientists’ Fund at the Faculty of Chemistry, Lodz University of Technology W-3D/FMN/8G/2022 (MM).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed
.
- J. C. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed
-
(a) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2018, 119, 2192–2452 CrossRef PubMed
- R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654–2672 CrossRef CAS PubMed
- M. Neetha, S. Saranya, N. Ann Harry and G. Anilkumar, ChemistrySelect, 2020, 5, 736–753 CrossRef CAS
- R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2021, 122, 1875–1924 CrossRef PubMed
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed
-
(a) J. M. Tedder, Tetrahedron, 1982, 38, 313–329 CrossRef CAS
- M. Bietti, Angew. Chem., Int. Ed., 2018, 57, 16618–16637 CrossRef CAS PubMed
- A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed
- H.-P. Deng, X.-Z. Fan, Z.-H. Chen, Q.-H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef CAS PubMed
- B. D. Ravetz, J. Y. Wang, K. E. Ruhl and T. Rovis, ACS Catal., 2018, 9, 200–204 CrossRef
- J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 2121–2127 CrossRef CAS
-
(a) S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed
-
(a) S. T. Saengchantara and T. W. Wallace, Nat. Prod. Rep., 1986, 3, 465–475 RSC
-
(a) P. N. Moquist, T. Kodama and S. E. Schaus, Angew. Chem., Int. Ed., 2010, 49, 7096 CrossRef CAS PubMed
- D. Genovese, C. Conti, P. Tomao, N. Desideri, M. L. Stein, S. Catone and L. Fiore, Antiviral Res., 1995, 27, 123–136 CrossRef CAS PubMed
- For reviews on decarboxylative strategies, see:
(a) Z.-L. Wang, Adv. Synth. Catal., 2013, 355, 2745 CrossRef CAS
-
(a) M. Moczulski, E. Kowalska, E. Kuśmierek, Ł. Albrecht and A. Albrecht, RSC Adv., 2021, 11, 27782–27786 RSC
-
(a) B. Giese, J. A. Gonzalez-Gomez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05331h |
| This journal is © The Royal Society of Chemistry 2024 |