
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeleno-relaxin analogues: effect of internal and external diselenide bonds on the foldability and a fibrosis-related factor of endometriotic stromal cells†
Yuri
Satoh
a,
Yosuke
Ono
b,
Rikana
Takahashi
a,
Hidekazu
Katayama
c,
Michio
Iwaoka
 ad,
Osamu
Yoshino
b and
Kenta
Arai
*ad
ad,
Osamu
Yoshino
b and
Kenta
Arai
*ad
aDepartment of Chemistry, School of Science, Tokai University, Kitakaname, Hiratsuka-shi, Kanagawa 259-1292, Japan. E-mail: k-arai4470@tokai-u.jp; Fax: +81-463-50-2094; Tel: +81-463-58-1211
bDepartment of Obstetrics and Gynecology, University of Yamanashi, 1110 Shimokato, Chuo-shi, Yamanashi 409-3898, Japan
cDepartment of Bioengineering, School of Engineering, Tokai University, Kitakaname, Hiratsuka-shi, Kanagawa 259-1292, Japan
dInstitute of Advanced Biosciences, Tokai University, Kitakaname, Hiratsuka-shi, Kanagawa 259-1292, Japan
First published on 31st May 2024
Abstract
Human relaxin-2 (H2 relaxin) is a peptide hormone of about 6 kDa, first identified as a reproductive hormone involved in vasoregulation during pregnancy. It has recently attracted strong interest because of its diverse functions, including anti-inflammatory, anti-fibrotic, and vasodilatory, and has been suggested as a potential peptide-based drug candidate for a variety of diseases. Mature H2 relaxin is constituted by the A- and B-chains stabilized by two interchain disulfide (SS) bridges and one intrachain SS linkage. In this study, seleno-relaxins, SeRlx-α and SeRlx-β, which are [C11UA,C11UB] and [C10UA,C15UA] variants of H2 relaxin, respectively, were synthesized via a one-pot oxidative chain assembly (folding) from the component A- and B-chains. The substitution of SS bonds in a protein with their analogue, diselenide (SeSe) bonds, has been shown to alter the physical, chemical, and physiological properties of the protein. The surface SeSe bond (U11A–U11B) enhanced the yield of chain assembly while the internal SeSe bond (U10A–U15A) improved the reaction rate of the folding, indicating that these bridges play a major role in controlling the thermodynamics and kinetics, respectively, of the folding mechanism. Furthermore, SeRlx-α and SeRlx-β effectively reduced the expression of a tissue fibrosis-related factor in human endometriotic stromal cells. Thus, the findings of this study indicate that the S-to-Se substitution strategy not only enhances the foldability of relaxin, but also provides new guidance for the development of novel relaxin formulations for endometriosis treatment.
Introduction
Human relaxin-2 (H2 relaxin; ca. 6 kDa), which is mainly secreted by the corpus luteum and placenta during pregnancy, is a member of the insulin superfamily proteins (ISPs) and composed of two polypeptide chains, A- and B-chains (Fig. 1a).1–3 The three-dimensional structure of H2 relaxin is stabilized by two interchain disulfide (SS) bridges (C11A–C11B and C24A–C23B) and one intrachain SS bond (C10A–C15A), with the SS topology being identical to that of insulin, a hypoglycemic hormone (Fig. 1b).4 H2 relaxin exerts its biological effects through activation of the G-protein-coupled receptor, relaxin family peptide receptor 1 (RXFP1). Although H2 relaxin is known as a uterine relaxing factor, H2 relaxin and its primary receptor are ubiquitous in cells of various tissues in both sexes.5,6 Consequently, H2 relaxin shows diverse physiological functions, including anti-inflammatory, anti-fibrotic, and vasodilatory, and is currently undergoing clinical trials for the treatment of a variety of diseases in several countries.7–13 Although a recent clinical trial (Novartis; second phase III)14 found no significant effect of H2 relaxin on the treatment of acute heart failure, there is still a great deal of interest in H2 relaxin's pharmacological applications. Several research institutions, including pharmaceutical companies, have developed a variety of H2 relaxin-related formulation candidates, such as long-acting relaxins,15,16 single-chain relaxin mimics,17–19 and small molecular RXFP1 agonists,20,21 some of which are undergoing clinical trials. Endometriosis, which affects approximately 5–10% of women of childbearing age, induces dysmenorrhea, dyspareunia, and dyschezia, substantially compromising patients’ quality of life.22,23 Recently, Yoshino et al.24 reported that H2 relaxin possesses an inhibitory effect on endometriosis.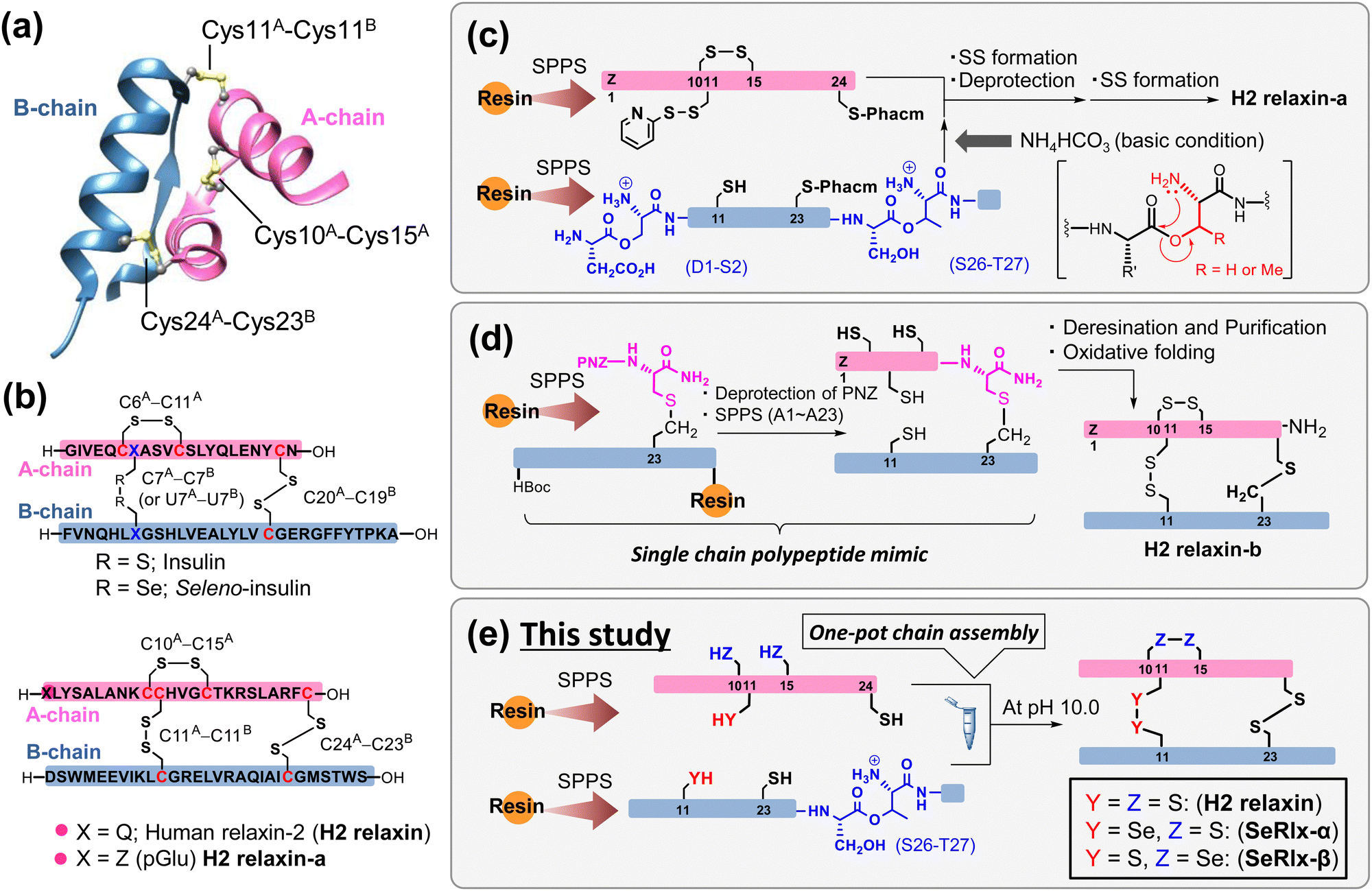 | ||
| Fig. 1 Synthetic strategies for the generation of human relaxin-2 (H2 relaxin) based on solid-phase peptide synthesis (SPPS). (a) Three-dimensional structure of H2 relaxin (PDB code: 6rlx). (b) Primary sequences and disulfide (SS) bond topologies of insulin (top) and relaxin analogues (bottom). (c) Representative chemical synthesis technique for the preparation of H2 relaxin-a via regioselective SS bond formation using orthogonal protection of Cys residues.25 (d) Preparation of a relaxin analogue having SS-like linkage at the C-terminal position via diaminodiacid-assisted single-shot peptide synthesis.26 (e) Preparation of relaxin analogues having SeSe linkage at the internal position of the primary sequence via one-pot oxidative chain assembly. | ||
Further basic research on relaxins along with rapid drug screening are required for identifying artificial relaxin analogues that could be potent drug candidates. Microbial protein expression systems are the most common method for the preparation of relaxin analogues. In this method, a single-chain relaxin precursor, so-called prorelaxin, is first prepared in which the A- and B-chains are linked via C-peptide. After oxidative folding, C-peptide is removed by protease to obtain the mature relaxin.27–30 However, synthesis of novel relaxins using this method requires the development of corresponding synthetic genes and thus is not suitable for the rapid production of a wide variety of formulations. Alternatively, chemical synthetic technology facilitates the introduction of artificial amino acids and chemical modifications of proteins. However, gaining the SS bonding pattern found in the native ISPs is one of the biggest challenges in the chemical synthesis.31–33 Interchain coupling of A- and B-chains, in which cysteinyl thiol (SH) groups are orthogonally protected, through regioselective deprotection and subsequent SS bond formation is one of the most successful strategies to prepare ISPs despite the fact that multiple reaction and purification steps are required (Fig. 1c).25,34–39
Replacing part of the SS bonds in the native state with a non-reducible surrogate bond is also frequently utilized to improve the oxidative folding efficiency because they play a role as an anchor bridge, simplifying complicated oxidative folding pathways, on which various folding intermediates having non-native SS bonds are involved.40–45 Furthermore, the SS surrogates in ISPs can modify their physical and chemical properties such as thermal stability and resistance against enzymatic degradation.46–51 Recently, Liu et al.26 reported that a single-chain relaxin analogue, in which the SS bond (C24A–C23B) at the C-terminus is replaced by a S–CH2 bond, can be synthesized rapidly with a high yield through a single-shot automated solid-phase peptide synthesis (SPPS), and that the synthetic polypeptide folds into a biologically active state (H2 relaxin-b), also with a good yield (isolated yield = 48.5%) (Fig. 1d). Although the synthesis of an analogue with an S–CH2 bond instead of an interchain SS bond (C11A–C11B) is not technically impossible, the reaction process is substantially more complicated than that for H2 relaxin-b due to the general limitations of SPPS.52
Diselenide (SeSe), an analogue of SS, is also a typical SS bond surrogate.53–56 SeSe linkages formed between selenocysteine (Sec; U) residues in proteins are more thermodynamically stable than SS linkages. In addition, the pKa value of the reduced SeSe (ca. 5.2),57 that is the selenol (SeH) group, is significantly lower than that of the SH group (ca. 8.2).58 Thus, SeH exists almost as a reactive selenolate anion (Se−) even at neutral pH, and hence, the SeSe bond is preferentially formed over the SS bond. In short, the substituted Sec residues can simultaneously modify the kinetics and thermodynamics of oxidative protein folding.59–61
Although the oxidative chain assembly of A- and B-chains of wild-type bovine insulin produces the folded state with a modest yield (39%) even under optimized conditions,62 applying the C-to-U substitution strategy at C7A and C7B in the A- and B-chains, respectively, the [C7UA,C7UB]-insulin variant, seleno-insulin (SeIns),63 was obtained at a higher yield (72%) from the individual chains (Fig. 1b).64 SeIns has higher thermal stability and resistance to degradation by human insulin degrading enzyme than the wild type, resulting in SeIns exhibiting a sustained hypoglycemic effect in diabetic rats.64 These findings suggest that incorporating the C-to-U substitution strategy into ISPs may impact not only their foldability but also their physical and pharmacological characteristics. Later, Metanis et al.65 reported that the replacement of an intrachain SS bond (C6A–C11A) in insulin by a SeSe bond also enhances the efficiency of oxidative chain assembly. Nevertheless, the C-to-U substitution strategy has not been applied to other proteins composed of multiple polypeptide chains. Folding behavior varies greatly, even among ISPs,66 and whether the C-to-U strategy can be applied universally must be examined using a variety of model proteins.
In this study, we demonstrate the complete synthesis of the [C11UA,C11UB]- and [C10UA,C15UA]-variants, namely, seleno-relaxin (SeRlx)-α and -β, respectively (Fig. 1e). Furthermore, the effects of the external SeSe (U11A–U11B) and internal SeSe (U10A–U15A) bonds in the variants on the foldability, structure, and chemical stability are also validated. In addition, we also evaluate the inhibitory effect of SeRlx analogues on the expression of a tissue fibrosis-related factor in human endometriotic stromal cells (ESCs) and show that they could be a potent pharmacological candidate for endometriosis treatment.
Results and discussion
First, the component polypeptides (1–5) were synthesized as ingredients for relaxin analogues (Fig. 2). Prior to the synthesis of SeRlx-α and -β, H2 relaxin was synthesized as a reference sample. H2 relaxin A-chain (peptide 1) was prepared by a general 9-fluorenyl methoxycarbonyl (Fmoc)-based SPPS method as described previously.67 Not only is it difficult to assemble the individual peptides in the synthesis of H2 relaxin, but the B-chain is poorly soluble in an acidic solvent.31 Incorporation of a non-native O-acyl isopeptide (O-AIP) unit, which can convert into a native peptide bond under basic conditions, in the sequence of insoluble peptides improves their solubility due to the ionized free amino group under acidic conditions, thereby facilitating its purification process (see Fig. 1c for the principle of the O-AIP strategy).68 Here, the B-chain analogue (peptide 2; isolated yield = 3.5%), in which S26B–T27B was replaced with the corresponding O-AIP unit,69 was obtained through the same SPPS method that was used to synthesize peptide 1 (Fig. 2a). In the synthesis of the B-chain having a phenylacetamidomethyl (Phacm) protecting group on one of the SH groups, two O-AIP units were inserted to achieve sufficient solubilization (Fig. 1c),25 whereas in the B-chain having no protecting group, only one O-AIP unit insertion was sufficient to improve the solubility.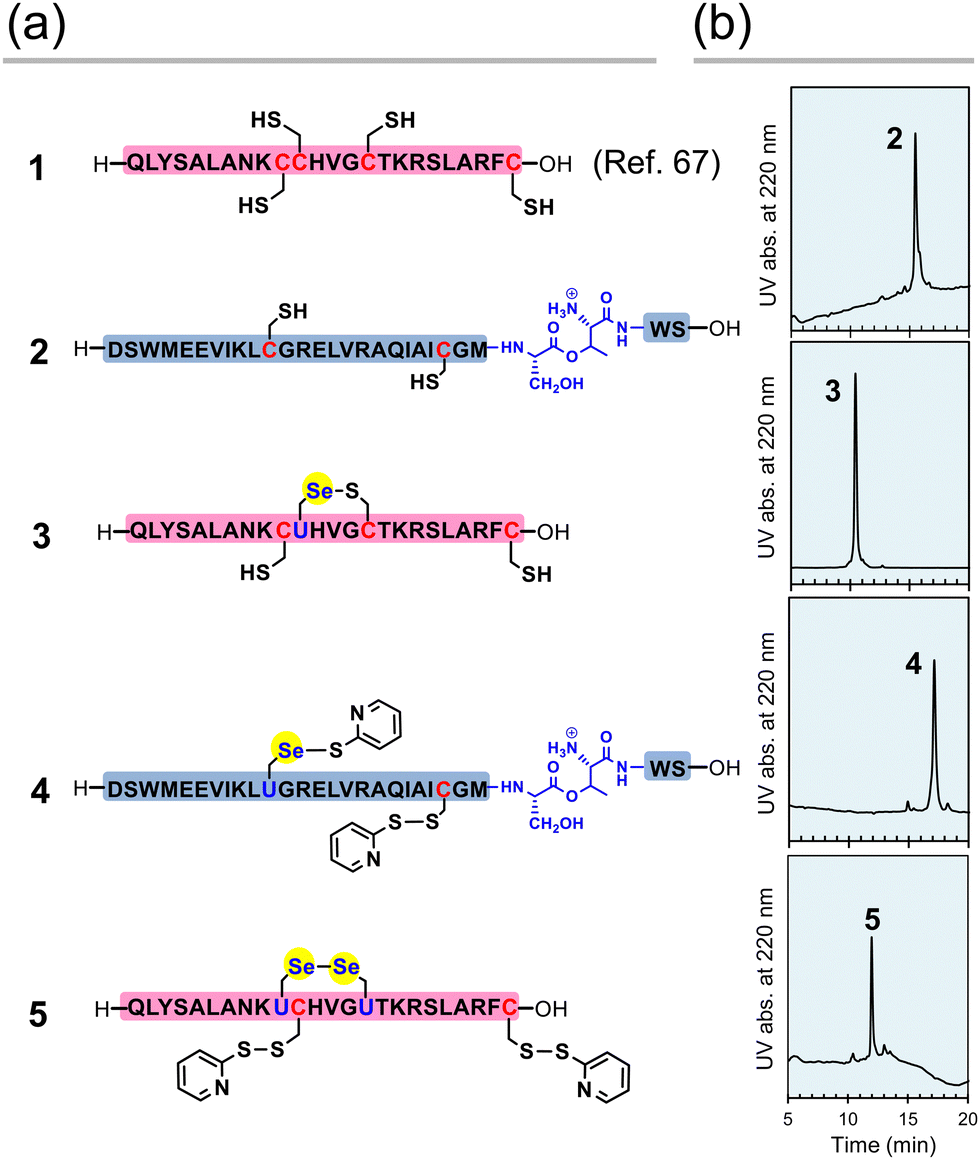 | ||
| Fig. 2 Component peptides of H2 relaxin, SeRlx-α, and SeRlx-β. (a) Primary sequences of synthetic peptides. U represents a selenocysteine (Sec) residue. The position of the Se–S bond in selenopeptide 3 was not identified. (b) HPLC chromatograms of peptides 2–5 obtained after purification. See ESI† for details of the analytical conditions and results of MS analysis of the samples. | ||
As with peptides 1 and 2, selenopeptides 3 and 4 were synthesized by the SPPS method based on the N,N’-dicyclohexylcarbodiimide (DCC)-hydroxybenzotriazole (HOBt) coupling system. Note that for coupling of the Sec derivative (Fmoc-Sec[MPM]-OH),70 diisopropylcarbodiimide (DIC) was used as a condensing agent instead of DCC based on our previous method.64 The resulting selenopeptides attached to the resin were then treated with a trifluoroacetic acid-based cocktail containing 2,2′-dipyridyldisulfide, which is a capping-reagent for SH and SeH groups, to remove protecting groups and the resin. High performance liquid chromatography (HPLC) analysis of the crude peptide preparations of 3 showed that multiple isomers with SeH and SH groups blocked by a pyridylsulfanyl (Pys) group were generated. Since the Pys groups can be easily removed by thiol-based reducing agents in a buffer solution of basic pH, the crude peptide was treated with DL-dithiothreitol (DTTred) at pH 10.0 and 25 °C for 24 h, yielding selenopeptide 3 (isolated yield = 8.1%) as a single isomer presumably having a selenenyl sulfide (Se–S) bond that is thermodynamically more stable than an SS bond (Fig. 2b).71 On the other hand, selenopeptide 4 (isolated yield = 2.9%) with an O-AIP unit at S26B–T27B was obtained as a major product having two Pys groups on the Sec and Cys residues after the de-resination and deprotection (Fig. 2a). Applying a similar protocol as for 4, selenopeptide 5 (isolated yield = 1.3%), which has two Pys groups and possibly one SeSe bond in the molecule, was synthesized as an ingredient for SeRlx-β (Fig. 2a). HPLC analysis showed that all synthetic peptides were obtained in a good purity (Fig. 2b).
Next, oxidative chain assembly was performed using synthetic peptides to obtain relaxin analogues (Fig. 3). The folding pathway from native A- and B-chains to H2 relaxin has been partially revealed by Wade et al. (Fig. 3a).72 To obtain H2 relaxin, peptides 1 and 2 were mixed and incubated in the presence of glutathione (GSH) and its oxidized form (GSSG) under optimized conditions. Since oxidative chain assembly of ISPs efficiently proceeds under basic conditions (pH 10.0),62 the formation of native SS or SeSe-bonding patterns and conversion of the O-AIP into the native peptide bond should simultaneously progress, directly yielding the desired relaxins in a one-pot manner (Fig. 1e). Indeed, it was confirmed that peptide 2 dissolved in a buffer solution at pH 10.0 was rapidly converted to the native B-chain of H2 relaxin (Fig. S1, ESI†). At specific time points, an aliquot of the folding sample solution was taken and the reaction was quenched by the addition of 2-aminoethyl methanethiosulfonate (AEMTS), a capping reagent for reactive SH (and SeH) groups. Reverse phase (RP) HPLC chromatograms of the resulting sample solutions showed that intrachain SS formation of the reduced A-chain (RA; peptide 1) proceeded at a faster rate than the reduced B-chain (RB) (Fig. 3b). After sufficient accumulation of oxidized species (1SSA) of the A chain, the native state (H2 relaxin) was generated, which finally reached a plateau with a reasonable yield (47% based on HPLC) after 48 h (Fig. S2, ESI†). See ESI† for HPLC yield calculations (Fig. S3). This folding behavior is consistent with the folding pathway of H2 relaxin proposed previously (Fig. 3a). The B-chain was apparently consumed stoichiometrically during the folding process of H2 relaxin. However, as the reaction progressed, visible precipitates were gradually generated in the sample solution. This implies that under the applied conditions, together with the correct hetero-dimerization, undesired oligomerization, mainly involving the B-chain, also progressed as a side reaction, which reduces the efficiency of the oxidative chain assembly.
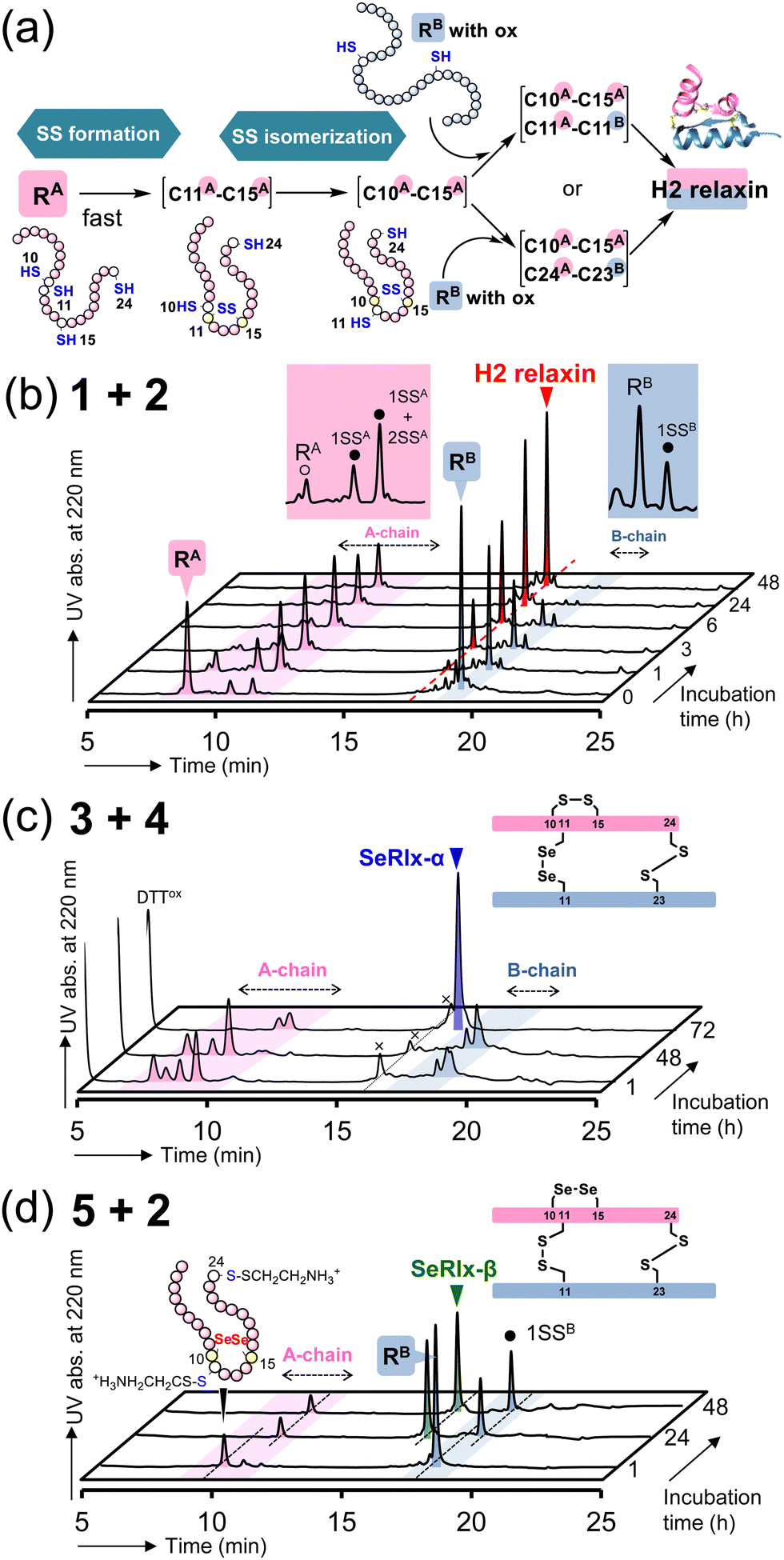 | ||
| Fig. 3 Oxidative chain assembly of relaxin analogues. (a) Double-chain oxidative folding pathway of H2 relaxin proposed by Wade et al.72 (b)–(d) HPLC chromatograms of samples obtained from double-chain oxidative folding of peptides 1 and 2 (b), peptides 3 and 4 (c), and peptides 5 and 2 (d). The symbols 1SSA, 2SSA, and 1SSB in (b) represent folding intermediates having one, two, and one SS bond(s) in the peptides, respectively, as characterized in ref. 67. Reaction conditions: for b, [1]0 = [2]0 = 160 μM, [GSH]0 = 1.0 mM, [GSSG]0 = 0.20 mM, 4 °C, and pH 10.0 in the presence of 0.9 M urea; for c, [3]0 = [4]0 = 160 μM, [DTTred]0 = 4.8 mM, 4 °C and pH 10.0 in the presence of 0.9 M urea; for d, the same conditions as those for c except for the constituent peptides. The reaction was repeated more than four times, and good reproducibility was confirmed. | ||
SeRlx-α was then obtained by the oxidative chain assembly of selenopeptides 3 and 4. Since 4 is protected by Pys groups, the selenopeptides were mixed with DTTred as a reductant and incubated at pH 10.0 and 4 °C under aerobic conditions. It should be noted that the GSH/GSSG system that was applied to the chain assembly of H2 relaxin was not suitable for the folding of SeRlx analogues in terms of folding yield, presumably due to low reducing ability to remove Pys groups (Fig. S4, ESI†). HPLC chromatograms showed that 3 was converted to multiple isomers within 1 h (Fig. 3c). After a steady state for more than 48 h that corresponds to the time required for the process of consumption of the reductant (DTTred), oxidative chain assembly progressed swiftly and SeRlx-α was produced at a high yield (73% based on HPLC) within 72 h (Fig. 3c and S2, ESI†). We recently reported that SeIns with a solvent-exposed SeSe bond (U7A–U7B) has greater foldability than wild-type insulin.64 Thus, these facts would indicate that the thermodynamic stabilization of the SS bond at the surface of the protein molecule is crucial to improve the yield of the oxidative chain assembly of ISPs. Applying a similar protocol, SeRlx-β was prepared through the combination of peptide 2 and selenopeptide 5. Although similar folding behavior to that of SeRlx-α was observed, the initial steady state with consumption of DTTred was noticeably shorter than that for SeRlx-α, and the folded state was generated about 12 h after the initiation of the reaction (Fig. S2, ESI†). While the final yield of SeRlx-β (34% based on HPLC) was lower than that of SeRlx-α, the completion of the reaction took less time (∼24 h) than that for SeRlx-α (Fig. 3d). These facts may suggest that SeRlx-β is more resistant to reducing agents than SeRlx-α (see below for the stability of SeRlx analogues to a reductant [Fig. 4c]). Furthermore, in the case of SeRlx-β, in contrast to the folding of SeRlx-α, the A-chain was observed as a single isomer in the initial folding event. Mass spectroscopic (MS) analysis of this fraction suggested that this intermediate would have thermodynamically more stable SeSe bonds (U11A–U11B), indicating the selective generation of a key intermediate that can couple directly to the B-chain (Fig. 3a). In short, limiting the isomeric species of SS intermediates produced on the folding pathway would be important to accelerate the folding rate. Indeed, when peptides 2 and 5 were mixed under the conditions applied for the chain assembly of H2 relaxin, a small amount of SeRlx-β (6%) was generated within just 10 min, along with misfolded state (Fig. S2 and S4, ESI†). Collectively, these results indicate that external SS (C11A–C11B) and internal SS (C10A–C15A) of H2 relaxin likely play different roles as thermodynamic and kinetic stabilizers, respectively, during the process of oxidative chain assembly.
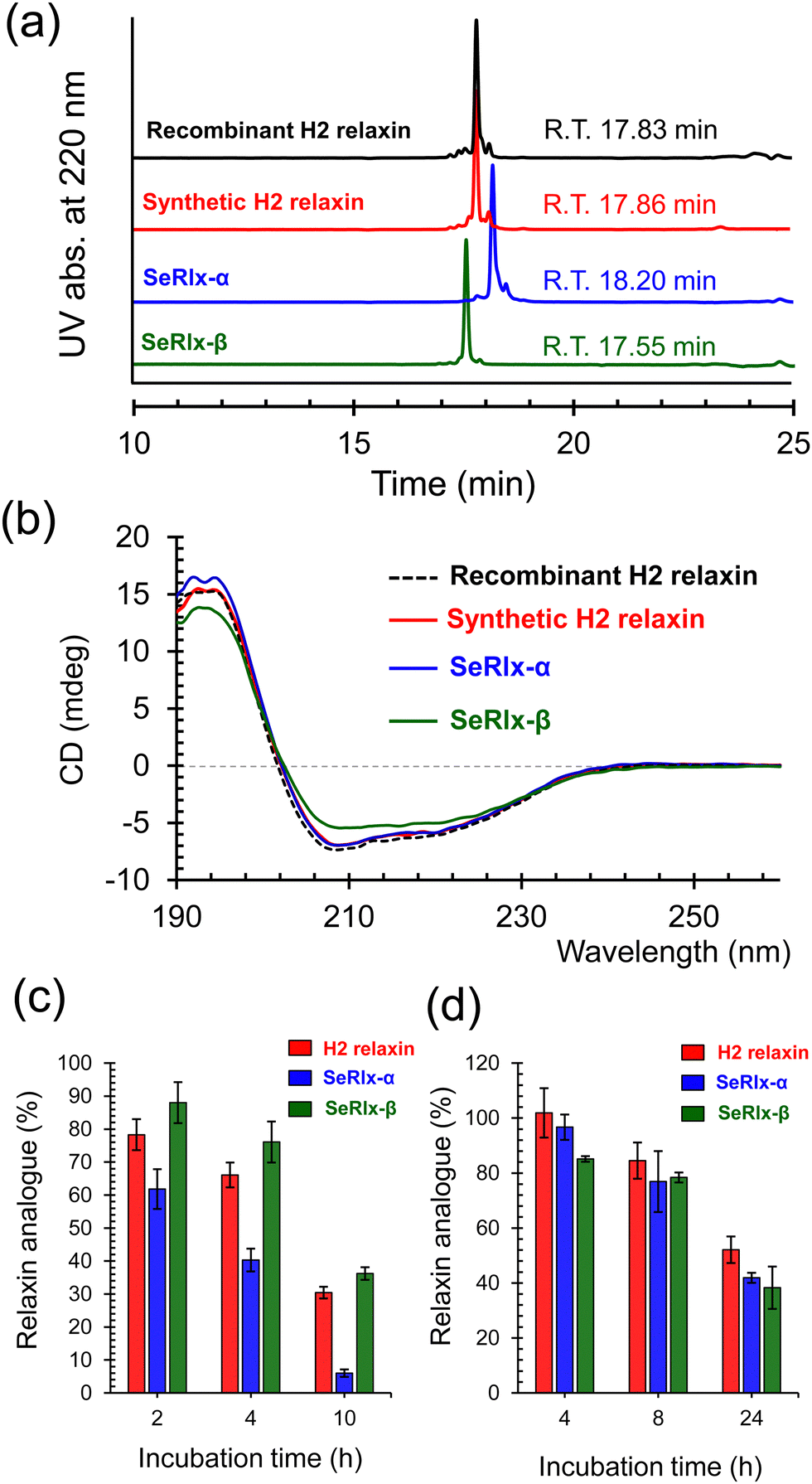 | ||
| Fig. 4 Characterization of the structure and chemical stability of relaxin analogues. (a) Comparison of the retention time of purified relaxin analogues in RP HPLC analysis. (b) CD spectra of relaxin analogues in 10 mM Tris–HCl buffer solution at pH 7.5 and 25 °C. The spectra were measured at the concentration of 10 μM using a quartz cell (path length = 1 mm). The measurement was repeated three times and good reproducibility was confirmed. Typical spectra are shown in the panel. (c) Reductive unfolding of relaxins by reduced glutathione (GSH). Reaction conditions: [relaxins]0 = 30 μM and [GSH]0 = 1.0 mM in 100 mM Tris–HCl buffer solution at pH 7.5 and 37 °C. Data are shown as mean ± standard error of the mean (SEM) (n = 3). (d) Stability assay of relaxins in human serum. Reaction conditions: [relaxins]0 = 10 μM in a pooled human serum at 37 °C. Data are shown as mean ± SEM (n = 3). | ||
HPLC chromatograms of the isolated relaxin analogues showed that all products were obtained with high purities with similar retention times (Fig. 4a). In addition, treatment of the isolated relaxins with trypsin produced appropriate peptide fragments that were derived from the folded states having the correct SS- or SeSe-bonding patterns (Fig. S5 and Table S1, ESI†). To further characterize the structure of the relaxins, circular dichroism (CD) spectroscopic analysis was performed (Fig. 4b). The CD spectrum of SeRlx-α showed a typical spectral profile, as observed for α-helical proteins, and was identical to that of H2 relaxin. On the other hand, the CD spectrum of SeRlx-β indicated that its α-helical content was slightly lower than that of other analogues. Nevertheless, evaluation of the affinity of relaxins with a specific antibody by enzyme-linked immunosorbent assay (ELISA) showed that SeRlx analogues have similar affinity like that of H2 relaxin, indicating that the overall structure of SeRlx is comparable to that of the wild type protein (Fig. S6, ESI†).
Next, the chemical stabilities of the relaxins were assessed. First, to investigate the rate of degradation under reducing conditions, synthetic relaxins were treated with GSH, which is the most ubiquitous reducing agent in vivo. After a certain time, an aliquot of the sample was analyzed by RP HPLC (Fig. S7, ESI†) to estimate the residual relaxin analogue (Fig. 4c). The reductive degradation rate of SeRlx-β was similar to or slightly slower than that of H2 relaxin, whereas SeRlx-α was reduced more rapidly. Although SeSe bonds are thermodynamically more stable than SS bonds, they are kinetically more unstable,73,74 suggesting that the solvent-exposed SeSe (U11A–U11B) bond in SeRlx-α was more rapidly cleaved by GSH, causing cooperative unfolding. Next, the synthetic analogues were incubated with human serum, and their residual amounts after specific time points were estimated by HPLC (Fig. S8, ESI†). All the relaxin analogues degraded at approximately the same rate (Fig. 4d). While the instability of peptide hormones and enzymes in the blood is one of the main concerns when considering their pharmaceutical applications, the result that C-to-U substitution does not affect their stability in serum and the fact that H2 relaxin is already under clinical trials would be advantageous for pharmacological applications of SeRlx analogues.
Finally, the potential therapeutic application of the SeRlx analogues for endometriosis was examined. Leucine-rich repeat-containing G-protein-coupled receptor-7 (LGR-7),75,76 a primary receptor for H2 relaxin, is expressed in ESCs obtained from patients with endometriosis.24 Previously, Yoshino and coworkers found H2 relaxin to be a potential suppressor for endometriosis,24 and hence, it would be intriguing to explore whether or not SeRlx analogues also show a similar effect. We evaluated the effect of relaxins on ESCs, focusing on the expression of a tissue fibrosis-related factor, namely, plasminogen activator inhibitor-1 (PAI-1) (Fig. 5).
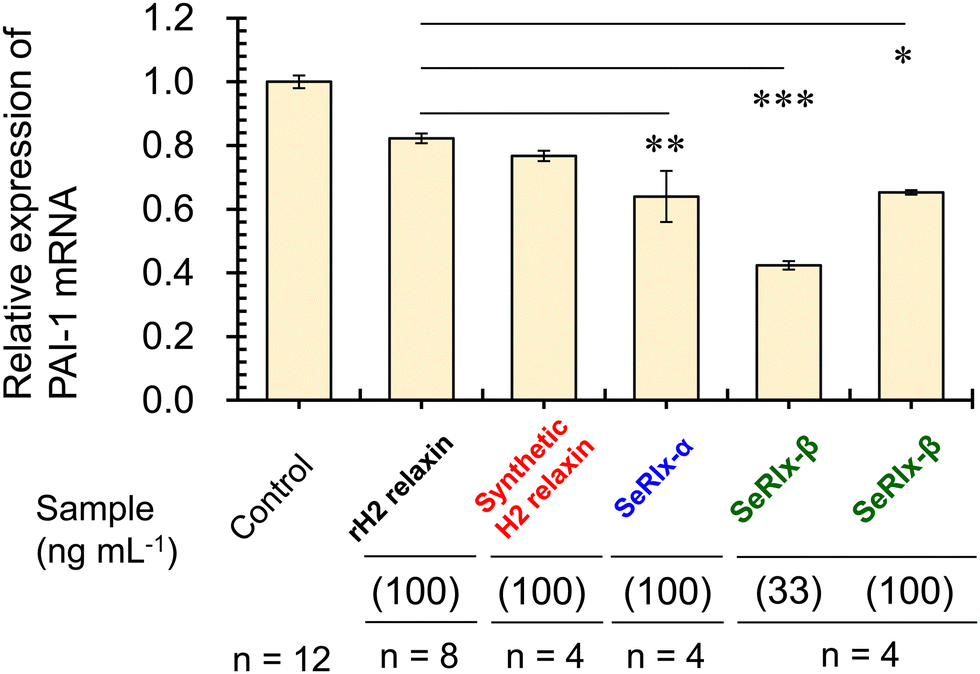 | ||
| Fig. 5 The effect of relaxins on endometriotic stromal cells (ESCs). ESCs were incubated with relaxins (33 or 100 ng mL−1) for 8 h, and quantitative PCR was performed to measure the mRNA expression level of PAI-1 in the control and relaxin-treated group. In this assay, primary cultured cells were used. All data were normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels to show the relative abundance. rH2 relaxin represents recombinant H2 relaxin. Experiments were performed more than four times, and data are shown as the mean ± SEM relative to an adjusted value of 1.0 for the mean value of the control. The symbols *, **, and *** represent p < 0.05, p < 0.01, and p < 0.001, respectively. | ||
PAI-1 plays an important role in coagulation-fibrinolysis and fibrosis, and a high level of PAI-1 in the intraperitoneal fluid of patients with endometriosis has been suggested to contribute to the development of peritoneal lesions.77,78 As a preliminary experiment, ESCs, which were purified from surgical specimens and cultured as described previously,24 were stimulated with SeRlx-β (33 ng mL−1) for 8 h, and the expression level of PAI-1 mRNA was estimated by quantitative polymerase chain reaction (qPCR). It was observed that SeRlx-β significantly reduced PAI-1 mRNA expression in ESCs (Fig. 5). Besides, we previously reported that stimulation of ESCs with recombinant H2 relaxin at 100 ng mL−1 significantly reduced PAI-1 mRNA expression.24 Therefore, to compare the effect of SeRlx analogues and H2 relaxin on the PAI-1 mRNA expression, ESCs were stimulated at the same concentrations. Importantly, we found that SeRlx analogues (100 ng mL−1) decreased the PAI-1 mRNA expression in ESCs more effectively than H2 relaxin (Fig. 5). Since the SeSe bond has a higher polarizability than the SS bond, the external surface SeSe bond in SeRlx-α may enhance intermolecular interactions to facilely associate with LGR-7, resulting in high suppression of PAI-1 mRNA expression. The fact that a solvent-exposed SeSe enhances inter-protein interactions has been suggested by our recent study on the oligomerizing behavior of SeIns, which also has an external SeSe bond.64 On the other hand, SeRlx-β's high suppressing ability may be attributed to fine conformational changes induced by the substitution of SS by SeSe, as shown in the CD spectral analysis of relaxins (Fig. 4b), which enhances the protein's ability to bind with LGR-7. These results imply that SeRlx analogues may have a high therapeutic potential for the treatment of endometriosis in terms of suppressing PAI-1 production. In light of the fact that lower concentrations of SeRlx-β suppress PAI-1 production more effectively, detailed investigation of the dose dependence of SeRlx analogues on their efficacy is warranted.
Conclusions
In conclusion, the [C11UA,C11UB] and [C10UA,C15UA] variants of H2 relaxin, namely, SeRlx-α and SeRlx-β, respectively, were successfully synthesized from the individual component peptides via one-pot oxidative chain assembly. The external SeSe bond (U11A–U11B) enhanced the folding yield while the internal SeSe bond (U10A–U15A) improved the folding rate, indicating that C11A–C11B and C10A–C15A in H2 relaxin govern the thermodynamics and kinetics of the oxidative folding, respectively. Furthermore, treatment of ESCs with the SeRlx analogues effectively suppressed the mRNA expression of PAI-1, a tissue fibrosis factor, by up to 40%. Importantly, the effect of SeRlx analogues was significantly higher than that of H2 relaxin. These results suggest that the C-to-U substitution strategy not only enhances the foldability of relaxin, but also provides new guidance for the development of novel relaxin formulations for endometriosis treatment. Besides, Sec residues can be artificially introduced into a protein by genetic engineering technology.79,80 This is a notable advantage of the C-to-U substitution strategy over other strategies utilizing non-reducible SS surrogates such as thioether linkages,26,48 and may imply the possibility of mass production of SeRlx analogues by microbial expression systems.Structural and physicochemical studies on the complex formation of SeRlx with LGR-7 should be crucial in further enhancing the capability of SeRlx to suppress PAI-1 expression. To practically apply SeRlx as a pharmaceutical formulation, it is also necessary to comprehensively examine the inhibitory effects of SeRlx on the expression of other endometriosis-related factors, as well as to investigate its pharmacodynamics and pharmacokinetics using disease-model animals. Although we have previously shown that SeIns has no short-term toxicity in rats, the long- and medium-term toxicities of artificial selenoproteins have not been evaluated. Therefore, careful safety evaluation in preclinical studies would also be necessary for the application of SeRlx as a selenopeptide formulation.
Author contributions
K. A. developed the idea of this project and initiated it. K. A. and Y. S. designed and conducted the experiments. R. T. and M. I. prepared essential materials used in the peptide synthesis, such as the O-AIP unit and Sec derivative. H. K. cooperatively conducted the SPPS with Y. S. O. Y. and Y. O. assessed the biological functions of relaxins by using ESCs. K. A. prepared the manuscript, which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Japan Society for the Promotion of Science (JSPS) [KAKENHI: grant number 23K04933 (to K. A.)]; Promotion and Mutual Aid Corporation for Private Schools of Japan (PMAC) [Science Research Promotion Fund (to K. A., M. I., H. K., O. Y.)]; and Research and Study Project of Tokai University, Educational System General Research Organization (to K. A., M. I., H. K., O. Y.).Notes and references
- A. H. MacLennan, Aust. N. Z. J. Obstet. Gynaecol., 1981, 21, 195–202 CrossRef CAS PubMed.
- L. T. Goldsmith, G. Weiss and B. G. Steinetz, Endocrinol. Metab. Clin. North Am., 1995, 24, 171–186 CrossRef CAS.
- L. T. Goldsmith and G. Weiss, Ann. N. Y. Acad. Sci., 2009, 1160, 130–135 CrossRef CAS PubMed.
- N. A. Patil, K. J. Rosengren, F. Separovic, J. D. Wade, R. A. D. Bathgate and M. A. Hossain, Br. J. Pharmacol., 2017, 174, 950–961 CrossRef CAS PubMed.
- O. D. Sherwood, Endocr. Rev., 2004, 25, 205–234 CrossRef CAS PubMed.
- R. A. D. Bathgate, M. L. Halls, E. T. van der Westhuizen, G. E. Callander, M. Kocan and R. J. Summers, Physiol. Rev., 2013, 93, 405–480 CrossRef CAS PubMed.
- S. L. Teichman, E. Unemori, J. R. Teerlink, G. Cotter and M. Metra, Curr. Heart Failure Rep., 2010, 7, 75–82 CrossRef CAS PubMed.
- C. Sassoli, S. Nistri, F. Chellini and D. Bani, Curr. Mol. Med., 2022, 22, 196–208 CrossRef CAS PubMed.
- T. Devarakonda and F. N. Salloum, Trends Endocrinol. Metab., 2018, 29, 338–348 CrossRef CAS PubMed.
- H. H. Ng, C. H. Leo, L. J. Parry and R. H. Ritchie, Front. Pharmacol., 2018, 9, 501 CrossRef PubMed.
- C. H. Leo, M. Jelinic, H. H. Ng, L. J. Parry and M. Tare, Curr. Opin. Pharmacol., 2019, 45, 42–48 CrossRef CAS PubMed.
- C. S. Samuel and R. G. Bennett, Biochem. Pharmacol., 2022, 197, 114884 CrossRef CAS PubMed.
- N. Almeida-Pinto, T. B. Dschietzig, C. Brás-Silva and R. Adão, Clin. Res. Cardiol., 2023 DOI:10.1007/s00392-023-02305-1.
- Top, heart-failure contender serelaxin flops, Nat. Biotechnol., 2017, 35, 297 Search PubMed.
- A. Muppidi, S. J. Lee, C.-H. Hsu, H. Zou, C. Lee, E. Pflimlin, M. Mahankali, P. Yang, E. Chao, I. Ahmad, A. Crameri, D. Wang, A. Woods and W. Shen, Bioconjugate Chem., 2019, 30, 83–89 CrossRef CAS PubMed.
- P. Verdino, S. L. Lee, F. N. Cooper, S. R. Cottle, P. F. Grealish, C. C. Hu, C. M. Meyer, J. Lin, V. Copeland, G. Porter, R. L. Schroeder, T. D. Thompson, L. L. Porras, A. Dey, H. Y. Zhang, E. C. Beebe, S. J. Matkovich, T. Coskun, A. M. Balciunas, A. Ferrante, R. Siegel, L. Malherbe, N. Bivi, C. D. Paavola, R. J. Hansen, M. M. Abernathy, S. O. Nwosu, M. C. Carr, J. G. Heuer and X. Wang, Br. J. Pharmacol., 2023, 180, 1965–1980 CrossRef CAS PubMed.
- K. Hojo, M. A. Hossain, J. Tailhades, F. Shabanpoor, L. L. L. Wong, E. E. K. Ong-Pålsson, H. E. Kastman, S. Ma, A. L. Gundlach, K. J. Rosengren, J. D. Wade and R. A. D. Bathgate, J. Med. Chem., 2016, 59, 7445–7456 CrossRef CAS PubMed.
- M. A. Hossain, M. Kocan, S. T. Yao, S. G. Royce, V. B. Nair, C. Siwek, N. A. Patil, I. P. Harrison, K. J. Rosengren, S. Selemidis, R. J. Summers, J. D. Wade, R. A. D. Bathgate and C. S. Samuel, Chem. Sci., 2016, 7, 3805–3819 RSC.
- S. Illiano, B. Poirier, C. Minoletti, O. Pasquier, L. Riva, X. Chenede, I. Menguy, M. Guillotel, P. Prigent, S. Le Claire, F. Gillot, G. Thill, F. Lo Presti, A. Corbier, J.-C. Le Bail, P. Grailhe, E. Monteagudo, R. Ingenito, E. Bianchi, C. Philippo, O. Duclos, S. Mallart, R. Bathgate and P. Janiak, Sci. Rep., 2022, 12, 20435 CrossRef CAS PubMed.
- K. L. Granberg, S. Sakamaki, R. Fuchigami, Y. Niwa, M. Fujio, H. Kato, F. Bergström, N. Larsson, M. Persson, I. C. Villar, T. Fujita, E. Sugikawa, M. Althage, N. Yano, Y. Yokoyama, J. Kimura, M. Lal and H. Mochida, J. Med. Chem., 2024, 67, 4442–4462 CrossRef CAS PubMed.
- K. L. Granberg, S. Sakamaki, N. Larsson, F. Bergström, R. Fuchigami, Y. Niwa, E. Ryberg, A. Backmark, H. Kato, S. Miyazaki, K. Iguchi, T. Sakamoto, M. Persson, A. Idei, L. Prieto Garcia, I. C. Villar, H. Gradén, G. Bergonzini, T. Arvidsson, T. Fujita, M. Althage, J. Ulander, J. Kimura, H. Yoneda, O. Fjellström, H. Mochida and M. Lal, J. Med. Chem., 2024, 67, 4419–4441 CrossRef CAS PubMed.
- K. E. Nnoaham, L. Hummelshoj, P. Webster, T. d’Hooghe, F. de Cicco Nardone, C. de Cicco Nardone, C. Jenkinson, S. H. Kennedy and K. T. Zondervan, Fertil. Steril., 2011, 96(366–373), e8 Search PubMed.
- K. T. Zondervan, C. M. Becker, K. Koga, S. A. Missmer, R. N. Taylor and P. Viganò, Nat. Rev. Dis. Primers, 2018, 4, 1–25 Search PubMed.
- O. Yoshino, Y. Ono, M. Honda, K. Hattori, E. Sato, T. Hiraoka, M. Ito, M. Kobayashi, K. Arai, H. Katayama, H. Tsuchida, K. Yamada-Nomoto, S. Iwahata, Y. Fukushi, S. Wada, H. Iwase, K. Koga, Y. Osuga, M. Iwaoka and N. Unno, Biomedicines, 2020, 8, 467 CrossRef CAS PubMed.
- X. Yang, V. Gelfanov, F. Liu and R. DiMarchi, Org. Lett., 2016, 18, 5516–5519 CrossRef CAS PubMed.
- R. Zhao, P. Shi, J. Cui, C. Shi, X.-X. Wei, J. Luo, Z. Xia, W.-W. Shi, Y. Zhou, J. Tang, C. Tian, M. Meininghaus, D. Bierer, J. Shi, Y.-M. Li and L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202216365 CrossRef CAS PubMed.
- G. K. Reddy, S. Gunwar, C. B. Green, D. T. W. Fei, A. B. Chen and S. C. M. Kwok, Arch. Biochem. Biophys., 1992, 294, 579–585 CrossRef CAS PubMed.
- S. Yang, H. Heyn, Y. Z. Zhang, E. E. Bullesbach and C. Schwabe, Arch. Biochem. Biophys., 1993, 300, 734–737 CrossRef CAS PubMed.
- D. Cimini, K. D. Corte, R. Finamore, L. Andreozzi, A. Stellavato, A. V. A. Pirozzi, F. Ferrara, R. Formisano, M. De Rosa, M. Chino, L. Lista, A. Lombardi, V. Pavone and C. Schiraldi, BMC Biotechnol., 2017, 17, 4 CrossRef CAS PubMed.
- R. Vandlen, J. Winslow, B. Moffat and E. Rinderknecht, in Progress in relaxin research, ed. A. H. MacLennan, G. W. Tregear and G. D. Bryant-Greenwood, Global Publications Services, Singapore, 1995, pp. 59–72 Search PubMed.
- M. A. Hossain and J. D. Wade, Curr. Opin. Chem. Biol., 2014, 22, 47–55 CrossRef CAS PubMed.
- F. Liu, A. N. Zaykov, J. J. Levy, R. D. DiMarchi and J. P. Mayer, J. Pept. Sci., 2016, 22, 260–270 CrossRef CAS PubMed.
- J. A. Karas, J. D. Wade and M. A. Hossain, Chem. Rev., 2021, 121, 4531–4560 CrossRef CAS PubMed.
- E. E. Büllesbach and C. Schwabe, J. Biol. Chem., 1991, 266, 10754–10761 CrossRef.
- K. Akaji, K. Fujino, T. Tatsumi and Y. Kiso, J. Am. Chem. Soc., 1993, 115, 11384–11392 CrossRef CAS.
- M. A. Hossain, A. Belgi, F. Lin, S. Zhang, F. Shabanpoor, L. Chan, C. Belyea, H.-T. Truong, A. R. Blair, S. Andrikopoulos, G. W. Tregear and J. D. Wade, Bioconjugate Chem., 2009, 20, 1390–1396 CrossRef CAS PubMed.
- J. A. Karas, D. B. Scanlon, B. E. Forbes, I. Vetter, R. J. Lewis, J. Gardiner, F. Separovic, J. D. Wade and M. A. Hossain, Chem. – Eur. J., 2014, 20, 9549–9552 CrossRef CAS PubMed.
- F. Liu, E. Y. Luo, D. B. Flora and A. R. Mezo, Angew. Chem., Int. Ed., 2014, 53, 3983–3987 CrossRef CAS PubMed.
- N. A. Patil, J. A. Karas, J. D. Wade, M. A. Hossain and J. Tailhades, Chem. – Eur. J., 2019, 25, 8599–8603 CrossRef CAS PubMed.
- A. Gori, P. Gagni and S. Rinaldi, Chem. – Eur. J., 2017, 23, 14987–14995 CrossRef CAS PubMed.
- R. Zhao, P. Shi, J. Chen, S. Sun, J. Chen, J. Cui, F. Wu, G. Fang, C. Tian, J. Shi, D. Bierer, L. Liu and Y.-M. Li, Chem. Sci., 2020, 11, 7927–7932 RSC.
- R. Mousa, S. Lansky, G. Shoham and N. Metanis, Chem. Sci., 2018, 9, 4814–4820 RSC.
- N. Zheng, S. B. Christensen, C. Dowell, L. Purushottam, J. J. Skalicky, J. M. McIntosh and D. H.-C. Chou, J. Med. Chem., 2021, 64, 9513–9524 CrossRef CAS PubMed.
- Y.-K. Qi, Q. Qu, D. Bierer and L. Liu, Chem. – Asian J., 2020, 15, 2793–2802 CrossRef CAS PubMed.
- Y. Guo, D.-M. Sun, F.-L. Wang, Y. He, L. Liu and C.-L. Tian, Angew. Chem., Int. Ed., 2015, 54, 14276–14281 CrossRef CAS PubMed.
- M. A. Hossain, K. J. Rosengren, S. Zhang, R. A. D. Bathgate, G. W. Tregear, B. J. van Lierop, A. J. Robinson and J. D. Wade, Org. Biomol. Chem., 2009, 7, 1547–1553 RSC.
- M. A. Hossain, L. M. Haugaard-Kedström, K. J. Rosengren, R. A. D. Bathgate and J. D. Wade, Org. Biomol. Chem., 2015, 13, 10895–10903 RSC.
- J. A. Karas, N. A. Patil, J. Tailhades, M.-A. Sani, D. B. Scanlon, B. E. Forbes, J. Gardiner, F. Separovic, J. D. Wade and M. A. Hossain, Angew. Chem., Int. Ed., 2016, 55, 14743–14747 CrossRef CAS PubMed.
- B. van Lierop, S. C. Ong, A. Belgi, C. Delaine, S. Andrikopoulos, N. L. Haworth, J. G. Menting, M. C. Lawrence, A. J. Robinson and B. E. Forbes, Sci. Rep., 2017, 7, 17239 CrossRef PubMed.
- S. C. Ong, A. Belgi, B. van Lierop, C. Delaine, S. Andrikopoulos, C. A. MacRaild, R. S. Norton, N. L. Haworth, A. J. Robinson and B. E. Forbes, J. Biol. Chem., 2018, 293, 11928–11943 CrossRef CAS PubMed.
- N. Zheng, P. Karra, M. A. VandenBerg, J. H. Kim, M. J. Webber, W. L. Holland and D. H.-C. Chou, J. Med. Chem., 2019, 62, 11437–11443 CrossRef CAS PubMed.
- R. Zhao, P. Shi, X.-X. Wei, Z. Xia, C. Shi and J. Shi, Org. Lett., 2023, 25, 6544–6548 CrossRef CAS PubMed.
- R. Mousa, R. Notis Dardashti and N. Metanis, Angew. Chem., Int. Ed., 2017, 56, 15818–15827 CrossRef CAS PubMed.
- L. Moroder and H.-J. Musiol, J. Pept. Sci., 2020, 26, e3232 CrossRef CAS PubMed.
- K. Arai and M. Iwaoka, Molecules, 2021, 26, 195 CrossRef CAS PubMed.
- K. Arai and R. Mikami, Metallomics Res., 2022, 2, 1–17 Search PubMed.
- R. E. Huber and R. S. Criddle, Arch. Biochem. Biophys., 1967, 122, 164–173 CrossRef CAS PubMed.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, J. Am. Chem. Soc., 2004, 126, 10508–10509 CrossRef CAS PubMed.
- N. Metanis and D. Hilvert, Angew. Chem., Int. Ed., 2012, 51, 5585–5588 CrossRef CAS PubMed.
- N. Metanis and D. Hilvert, Chem. Sci., 2014, 6, 322–325 RSC.
- R. Mousa, T. Hidmi, S. Pomyalov, S. Lansky, L. Khouri, D. E. Shalev, G. Shoham and N. Metanis, Commun. Chem., 2021, 4, 1–9 CrossRef PubMed.
- K. Arai, T. Takei, R. Shinozaki, M. Noguchi, S. Fujisawa, H. Katayama, L. Moroder, S. Ando, M. Okumura, K. Inaba, H. Hojo and M. Iwaoka, Commun. Chem., 2018, 1, 1–11 CrossRef CAS.
- K. Arai, T. Takei, M. Okumura, S. Watanabe, Y. Amagai, Y. Asahina, L. Moroder, H. Hojo, K. Inaba and M. Iwaoka, Angew. Chem., Int. Ed., 2017, 56, 5522–5526 CrossRef CAS PubMed.
- K. Arai, M. Okumura, Y.-H. Lee, H. Katayama, K. Mizutani, Y. Lin, S.-Y. Park, K. Sawada, M. Toyoda, H. Hojo, K. Inaba and M. Iwaoka, Commun. Chem., 2023, 6, 1–10 CrossRef PubMed.
- O. Weil-Ktorza, N. Rege, S. Lansky, D. E. Shalev, G. Shoham, M. A. Weiss and N. Metanis, Chem. – Eur. J., 2019, 25, 8513–8521 CrossRef CAS PubMed.
- Y.-M. Feng, in Folding of Disulfide Proteins, ed. R. J. Y. Chang and S. Ventura, Springer, New York, NY, 2011, pp. 63–89 Search PubMed.
- K. Arai, M. Noguchi, B. G. Singh, K. I. Priyadarsini, K. Fujio, Y. Kubo, K. Takayama, S. Ando and M. Iwaoka, FEBS Open Bio, 2013, 3, 55–64 CrossRef CAS PubMed.
- Y. Sohma, M. Sasaki, Y. Hayashi, T. Kimura and Y. Kiso, Chem. Commun., 2004, 124–125 RSC.
- T. Yoshiya, A. Taniguchi, Y. Sohma, F. Fukao, S. Nakamura, N. Abe, N. Ito, M. Skwarczynski, T. Kimura, Y. Hayashi and Y. Kiso, Org. Biomol. Chem., 2007, 5, 1720–1730 RSC.
- S. Shimodaira and M. Iwaoka, ARKIVOC, 2016, 2017, 260–271 Search PubMed.
- D. Besse, F. Siedler, T. Diercks, H. Kessler and L. Moroder, Angew. Chem., Int. Ed. Engl., 1997, 36, 883–885 CrossRef CAS.
- J.-G. Tang, Z.-H. Wang, G. W. Tregear and J. D. Wade, Biochemistry, 2003, 42, 2731–2739 CrossRef CAS PubMed.
- N. Metanis, E. Keinan and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 16684–16691 CrossRef CAS PubMed.
- D. Steinmann, T. Nauser and W. H. Koppenol, J. Org. Chem., 2010, 75, 6696–6699 CrossRef CAS PubMed.
- S. Y. Hsu, M. Kudo, T. Chen, K. Nakabayashi, A. Bhalla, P. J. van der Spek, M. van Duin and A. J. Hsueh, Mol. Endocrinol., 2000, 14, 1257–1271 CrossRef CAS PubMed.
- S. Y. Hsu, K. Nakabayashi, S. Nishi, J. Kumagai, M. Kudo, O. D. Sherwood and A. J. W. Hsueh, Science, 2002, 295, 671–674 CrossRef CAS PubMed.
- C. Bruse, A. Bergqvist, K. Carlström, A. Fianu-Jonasson, I. Lecander and B. Åstedt, Fertil. Steril., 1998, 70, 821–826 CrossRef CAS PubMed.
- J. Gilabert-Estelles, R. Castello, J. Gilabert, L. A. Ramon, F. Espana, A. Romeu and A. Estelles, Front. Biosci., 2005, 10, 1162–1176 CrossRef CAS PubMed.
- M. J. Bröcker, J. M. L. Ho, G. M. Church, D. Söll and P. O’Donoghue, Angew. Chem., Int. Ed., 2014, 53, 319–323 CrossRef PubMed.
- J. Liu, Q. Chen and S. Rozovsky, J. Am. Chem. Soc., 2017, 139, 3430–3437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00095a |
| This journal is © The Royal Society of Chemistry 2024 |