DOI:
10.1039/D4BM01383B
(Paper)
Biomater. Sci., 2025,
13, 324-329
Thermoresponsive degradable hydrogels with renewable surfaces for protein removal†
Received
17th October 2024
, Accepted 15th November 2024
First published on 16th November 2024
Abstract
Most biological materials used in the body undergo protein adsorption, which alters their biological functions. Previously, we introduced surface-degradable hydrogels as adsorbed protein-removing surfaces. However, only a few surface renewals were possible because of the hydrophilic nature of the hydrogels, which accelerated their degradation. In this research, we introduced thermoresponsive properties of hydrogels for limited degradation for protein removal. Hydrogels were synthesized by the radical polymerization of N-isopropylacrylamide (NIPAAm), 2-methylene-1,3-dioxepane, and poly(ethylene glycol) monomethacrylate (PEGMA). The synthesized hydrogels demonstrated thermoresponsive behavior derived from poly(NIPAAm). At 10 °C, the hydrogels swelled and exhibited bulk degradation. After 2 h, the prepared hydrogels were degraded completely. However, at 37 °C, the hydrogels shrunk and showed surface degradation. After 7 h of degradation, the swelling ratio of the hydrogels changed marginally. The proteins adsorbed on the hydrogel surfaces were removed via surface degradation. However, the fluorescence intensity of adsorbed proteins increased on the hydrogel surfaces without degradable functions. In addition, the fluorescence intensity of adsorbed proteins increased in the hydrogels without PEG graft chains, suggesting that the prepared thermoresponsive hydrogels with PEG chains could be used as potential biomaterial surface coating materials, exhibiting regenerative low-fouling ability.
1. Introduction
For materials used in the living body, the surface design of materials is important, since adsorption of proteins to the surface leads to thrombus formation over time when in contact with blood, and an undesirable tissue response may occur when used in the tissue.1 Protein adsorption suppression is a major issue in the preparation of biomaterial surfaces for use in the living body. Much research has been reported for the suppression of protein adsorption onto biomaterial surfaces by using hydrophilic polymer coatings, such as poly(ethylene glycol) (PEG),2–4 poly(vinylpyrrolidone),5,6 poly(N,N-dimethylacrylamide),7 poly(2-methacryloyloxyethyl phosphorylcholine),8,9 carboxybetaine, and sulfobetaine polymers,10–12 which have already been applied to biomaterial devices. Material surfaces modified with these polymers show low fouling due to the formation of a hydration layer, which efficiently suppresses protein adsorption. Recently, protein removal surfaces have been reported, such as the surface cleaning of contact lenses using lysozymes13 and membrane surfaces that show self-cleaning of adsorbed proteins using modified enzymes.14 These materials clean their surfaces through the enzymatic degradation of adsorbed proteins.
Thus, biomaterial surface cleaning or the removal of adsorbed proteins is important; however, it is generally difficult to construct surfaces that clean regardless of the type of adsorbed protein. Dynamic self-renewing surfaces made of degradable polymers have been used as coating materials for marine-based material surfaces.15,16 The material surfaces exhibit removal properties for marine microorganisms via surface hydrolysis. Hydrophobic materials are often used for surface degradation because they can prevent the penetration of enzymes and/or water inside the materials. However, hydrophobic materials have various drawbacks for use in medical devices, such as adsorption of nonspecific proteins and aggregation of degraded materials. Therefore, the introduction of surface-cleaning properties into biomaterials requires the maintenance of protein removal and the release of hydrophilic oligomers as byproducts after degradation and surface renewal within the lifetime of the material.
We previously reported that poly(2-methylene-1,3-dioxepane (MDO)-co-2-hydroxyethylacrylate (HEA))-g-PEG (poly(MDO-co-HEA)-g-PEG) hydrogels exhibit protein removal properties via hydrolysis of the ester group in the gel surface vicinity.17 This property is realized by the thermoresponsive swelling–shrinking behavior of the base hydrogels. While the adsorbed proteins were removed from the prepared hydrogels, bulk decomposition of the gels occurred over time when the gels were subjected to repetitive protein removal. Poly(MDO-co-HEA)-based hydrogels exhibited liquid–liquid phase separation-type thermoresponsive behavior with a relatively high water content.17,18 The degradable polyester-based particles, PMDO-g-PEG, degrade from the particle surface.19–22 These particles are formed by hydrophobic interactions and decomposed from the particle surfaces by hydrolysis, reducing the particle size. This was due to the aggregation of hydrophobic polymers which suppresses the penetration of water molecules into the particle core, inhibiting bulk degradation. These reports suggest that aggregation of the polymer network is important for controlling the surface degradation of hydrogels. Therefore, we focused on liquid–solid phase separation-type thermoresponsive properties to control the surface degradation of the synthesized hydrogels.
In this paper, thermoresponsive degradable poly(NIPAAm)-based hydrogels are reported, because poly(N-isopropylacrylamide) (PNIPAAm) is a well-known liquid–solid phase separation-type thermoresponsive polymer above the lower critical solution temperature (LCST) and often used as a biomaterial.23–27 The prepared hydrogels showed thermoresponsive swelling–deswelling changes and surface degradable properties in the shrunken state at a physiological temperature of 37 °C. In addition, the adsorbed proteins on the hydrogels were removed through surface degradation (Fig. 1).
 |
| | Fig. 1 Schematic illustration of hydrogels that remove proteins via surface degradation at 37 °C. | |
2. Experimental
2.1 Materials
Dimethyl sulfoxide (DMSO), N,N′-methylenebis(acrylamide) (MBAAm), 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile), N-isopropylacrylamide (NIPAAm), and fluorescein-4-isothiocyanate were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Hexane was purchased from Kanto Chemical (Tokyo, Japan). NIPAAm was purified via recrystallization in hexane. Fibrinogen fraction I type I-S, bovine serum albumin-fluorescein isothiocyanate conjugate (FITC-BSA) and poly(ethylene glycol) monomethacrylate (PEGMA) (MW 2000) were purchased from Sigma-Aldrich (MO, USA). DMSO was purified by vacuum distillation before use (95.0 °C, 0.5 kPa). MDO was synthesized as described in previous studies.28–30 FITC-fibrinogen was prepared by conjugating fibrinogen with fluorescein isothiocyanate in carbonate buffer (pH 8.5) for 24 h at 25 °C, according to a previous study.17
2.2 Synthesis of thermoresponsive degradable hydrogels
Thermoresponsive degradable hydrogels were synthesized through radical polymerization of NIPAAm, MDO and PEGMA in the presence of 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (2 mol% to monomer) and MBAAm (3 mol% to monomer) in DMSO (total monomer conc. of 3.0 mol L−1) (Scheme 1a) using a procedure similar to that reported previously.17 The monomer feed ratio of hydrogel preparation is summarized in Table 1. Briefly, a monomer containing solution was degassed by N2 gas bubbling for 10 min. The degassed monomer containing solution was injected into the gap of 0.5 mm thick poly(dimethylsiloxane) spacer sandwiched two glass plates. Polymerization was performed at 25 °C for 24 h. After gelation, the polymerized hydrogels were dialyzed against methanol for 3 d and distilled water for 2 d. The purified poly(NIPAAm-co-MDO)-g-PEG hydrogels were molded into disk shapes (diameter: 1.0 cm, thickness: 0.5 mm) at 10 °C (Scheme 1b).
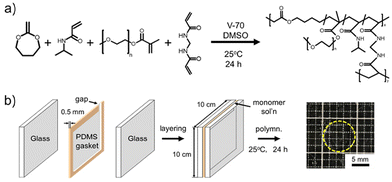 |
| | Scheme 1 Preparation of the poly(NIPAAm-co-MDO)-g-PEG hydrogels. (a) The reaction scheme and chemical structure of the poly(NIPAAm-co-MDO)-g-PEG hydrogels. (b) Preparation method of the poly(NIPAAm-co-MDO)-g-PEG hydrogels. | |
Table 1 Monomer feed ratio for the preparation of poly(NIPAAm-co-MDO)-g-PEG hydrogels
| Sample |
Monomer feed ratio (mol mol−1) |
MDO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NIPAAm :NIPAAm |
(MDO + NIPAAm):PEGMA |
| 1 |
3:7 |
100:1 |
| 2 |
3:7 |
Without PEGMA |
| 3 |
0:10 |
100:1 |
2.3 Thermoresponsive properties of hydrogels
The thermoresponsive properties of the poly(NIPAAm-co-MDO)-g-PEG hydrogels were analyzed by measuring their swelling ratios as a function of temperature. Initially, the hydrogel discs were soaked in distilled water at 50 °C for 24 h to attain an equilibrium state. After reaching the equilibrium state, the hydrogels were soaked in distilled water of each temperature (5–50 °C) for 24 h. Subsequently, the hydrogels were lyophilized overnight to obtain dry gels. After lyophilization, the swelling ratio of the hydrogels was calculated from the weight of the swollen gels (Ws) and dry gels (Wd) using the following equation:| | | Swelling ratio [−] = (Ws − Wd)/Wd. | (1) |
2.4 Degradable properties of hydrogels
The degradable properties of the swollen hydrogels and shrunken hydrogels were analyzed via alkaline hydrolysis in NaOH (1.0 mmol L−1, pH 11.2 (actual measurements)) as an accelerated test at either 10 or 37 °C, respectively. The degradation behavior of the tested hydrogels was estimated based on the increase in the swelling ratio, the ATR-FTIR spectra of the hydrogel surfaces were obtained after freeze-drying, and the 1H nuclear magnetic resonance (NMR) spectra of the supernatant solution were obtained using an AVANCE Neo 400 NMR spectrometer (Bruker, MA, USA).
2.5 Protein removal ability of hydrogels by degradation of hydrogel surfaces
The protein removal ability was analyzed by comparing the fluorescence intensities of the adsorbed proteins on the hydrogels before and after degradation. The prepared hydrogel discs were soaked in phosphate-buffered saline (PBS, 150 mmol L−1) at 37 °C for 24 h to reach an equilibrium state. After 24 h, the hydrogels were soaked in PBS containing FITC-BSA or FITC-fibrinogen (FITC-FBN) (100 μg mL−1 in PBS, pH 7.4) at 37 °C for 1 h, respectively. The hydrogels were washed by gentle agitation in PBS followed by immersing in NaOH solution (1.0 mmol L−1, pH 11.2) at 37 °C for 1 h. The hydrogels were washed again using PBS. This series of methods were repeated five times to evaluate the surface cleaning behavior by degradation due to protein removal. Protein removal properties were evaluated using the signal-to-noise ratio (SN ratio). The SN ratio was determined as the quotient in the fluorescence intensity of the protein adsorbed hydrogel surface (Fh) and the fluorescence intensity of the sliding glass (Fg), calculated using ImageJ software ver. 1.54 (National Institutes of Health, USA) with the following equation:
3. Results and discussion
Hydrogels were prepared through radical polymerization of NIPAAm, MDO, PEGMA and MBAAm as a crosslinker in DMSO (Scheme 1a). Their thermoresponsive behavior was measured in ultrapure water. In Fig. 2a, the prepared hydrogels (sample 1 in Table 1) swelled at 10 °C. At 50 °C, the prepared hydrogels shrunk, and the swelling ratio decreased from 20.4 at 5 °C to 1.3 at 50 °C (Fig. 2b), indicating that the hydrogels showed thermoresponsive properties. The volume phase transition temperature of the gel was 27.9 °C. It is well known that PNIPAAm shows LCST-type thermoresponsive behavior at approximately 32 °C due to hydration and dehydration changes.24–26 The main chains of the hydrogels consisted of PNIPAAm segments, and thermoresponsive hydrogels were obtained.
 |
| | Fig. 2 Thermoresponsive properties of the prepared hydrogels. (a) Image of the thermoresponsive behavior of the hydrogels (sample 1 in Table 1). All scale bars are 5 mm. (b) Thermoresponsive behavior in the swelling ratio of the hydrogels (sample 1 in Table 1). Data are expressed as the mean of 3 samples with S.D. | |
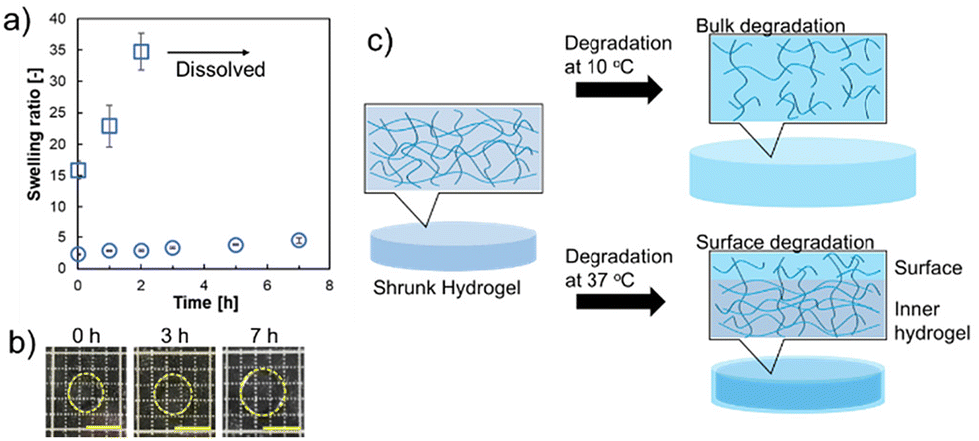
Next, the degradation abilities of the prepared hydrogels were measured. The degradation abilities were analyzed under alkaline conditions. Fig. 3a shows the change in the degradation time-dependent swelling ratio of the hydrogels (sample 1 in Table 1) in NaOH solution at 10 °C and 37 °C. At 37 °C, the swelling ratio showed a slow increase from 2.2 to 4.8 and maintained a shrunken state after 7 h (Fig. 3b). However, upon degradation at 10 °C, the swelling ratio was substantially increased from 16 to 34, and after 2 h of incubation in NaOH solution, the hydrogels showed complete degradation and dissolved. The slow degradation at 37 °C indicated that surface degradation of the hydrogels would preferentially occur in the shrunken state, rather than degradation occurring both at the surface and inner part of the hydrogels as can be seen at 10 °C; an increase in the swelling ratio of the hydrogels was confirmed within 2 h. These results suggest that degradation in the contracted state may increase the swelling of the gel surface and decrease the polymer density on the surface (Fig. 3c). In our previous reports, thermoresponsive degradable poly(HEA-co-MDO)-g-PEG hydrogels also showed suppression of degradation at 37 °C in a shrunken state.17,19 However, after degradation at 37 °C for 7 h, the swelling ratio increased 6-fold (from 3 to 18). The poly(HEA-co-MDO)-g-PEG hydrogels show liquid–liquid phase separation-type thermoresponsive behavior, which indicates a relatively high water content, even at higher temperatures. In contrast, PNIPAAm exhibits liquid–solid phase separation-type thermoresponsive behavior.23–25 It is suggested that the type of thermoresponsive behavior is responsible for suppressing bulk degradation by hydrolysis effectively at 37 °C in which the hydrogels are in a shrunken state due to the thermoresponsive properties of PNIPAAm chains.
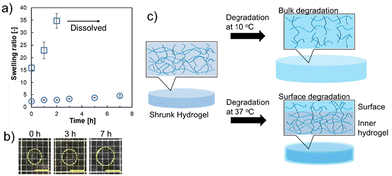 |
| | Fig. 3 Degradable properties of the prepared hydrogels (sample 1) under alkaline conditions. (a) Changes in the degradation time-dependent swelling ratios at 37 and 10 °C. Circle plot: degradation at 37 °C. Square plot: degradation at 10 °C. Data are expressed as the mean of 3 samples with S.D. (b) Image of degradable behavior at 37 °C. Scale bars: 5 mm. (c) Schematic illustration of the degradation behavior of the hydrogels at different temperatures. | |
The surface properties of the hydrogels before and after degradation were analyzed by 1H NMR and ATR-FTIR measurements according to a previous study.18 The supernatant solutions were obtained at degradation times of 2 and 5 h and their 1H NMR spectra were obtained to determine the degraded entities in NaOD (1.0 mmol L−1) containing D2O (Fig. 4a). The peak of PEG was observed at 3.75 ppm after 2 h of degradation, and the intensities of the peaks of PEG and the corresponding degraded products of the hydrophilic oligomers increased after 5 h of degradation. These results indicated that the PEG chains cleaved by surface degradation were released from the hydrogels to the external fluid, and the peak derived from PEG intensified with degradation time. Moreover, the hydrogel surfaces during degradation were analyzed using ATR-FTIR (Fig. 4b). The control (neat) hydrogels soaked in NaOH solution for 5 and 7 h were analyzed after freeze-drying. Before hydrogel degradation, the peaks of ether (–C–O–C–), ester (–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–), and amide (C(O)–NH–) groups derived from the corresponding monomer (MDO, PEG, and NIPAAm, respectively) units in the hydrogel chains were observed at 1750, 1150, 1550, and 1650 cm−1, respectively. In addition, these ATR-FTIR peaks were observed even after surface degradation for at least 7 h. The obtained results indicated that the prepared hydrogels have almost the same surface compositions even after degradation, suggesting that the hydrogels show surface renewable ability via re-orientation of PEG chains on the surfaces by degradation. This is in good agreement with the previous studies on the degradation properties of poly(HEA-co-MDO)-g-PEG hydrogels.17 The change in the swelling ratio of the hydrogels during degradation for at least 5 h was mostly constant (Fig. 3a), and the surface characteristics remained mostly unchanged during degradation (Fig. 4a and b), which suggested degradation of the hydrogel surface. In addition, the thermoresponsive properties of the hydrogel degradation products were evaluated based on temperature-dependent transmittance changes (Fig. S1†). The blue line is the transmittance change of neat PNIPAAm, and the LCST appeared at approximately 32 °C. The orange line represents the hydrogel degradation byproducts. The transmittance of the byproducts decreased above 38 °C, indicating that the products were hydrophilic at 37 °C.
O)–O–), and amide (C(O)–NH–) groups derived from the corresponding monomer (MDO, PEG, and NIPAAm, respectively) units in the hydrogel chains were observed at 1750, 1150, 1550, and 1650 cm−1, respectively. In addition, these ATR-FTIR peaks were observed even after surface degradation for at least 7 h. The obtained results indicated that the prepared hydrogels have almost the same surface compositions even after degradation, suggesting that the hydrogels show surface renewable ability via re-orientation of PEG chains on the surfaces by degradation. This is in good agreement with the previous studies on the degradation properties of poly(HEA-co-MDO)-g-PEG hydrogels.17 The change in the swelling ratio of the hydrogels during degradation for at least 5 h was mostly constant (Fig. 3a), and the surface characteristics remained mostly unchanged during degradation (Fig. 4a and b), which suggested degradation of the hydrogel surface. In addition, the thermoresponsive properties of the hydrogel degradation products were evaluated based on temperature-dependent transmittance changes (Fig. S1†). The blue line is the transmittance change of neat PNIPAAm, and the LCST appeared at approximately 32 °C. The orange line represents the hydrogel degradation byproducts. The transmittance of the byproducts decreased above 38 °C, indicating that the products were hydrophilic at 37 °C.
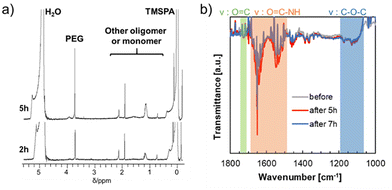 |
| | Fig. 4 Degradable properties of the hydrogel surfaces and supernatants recovered during degradation (sample 1 in Table 1). (a) 1H NMR spectra of the supernatants of degraded hydrogels after degradation for 2 and 5 h. (b) ATR-FTIR spectra of the hydrogel surfaces before and after degradation for 5 and 7 h at 37 °C after drying. | |
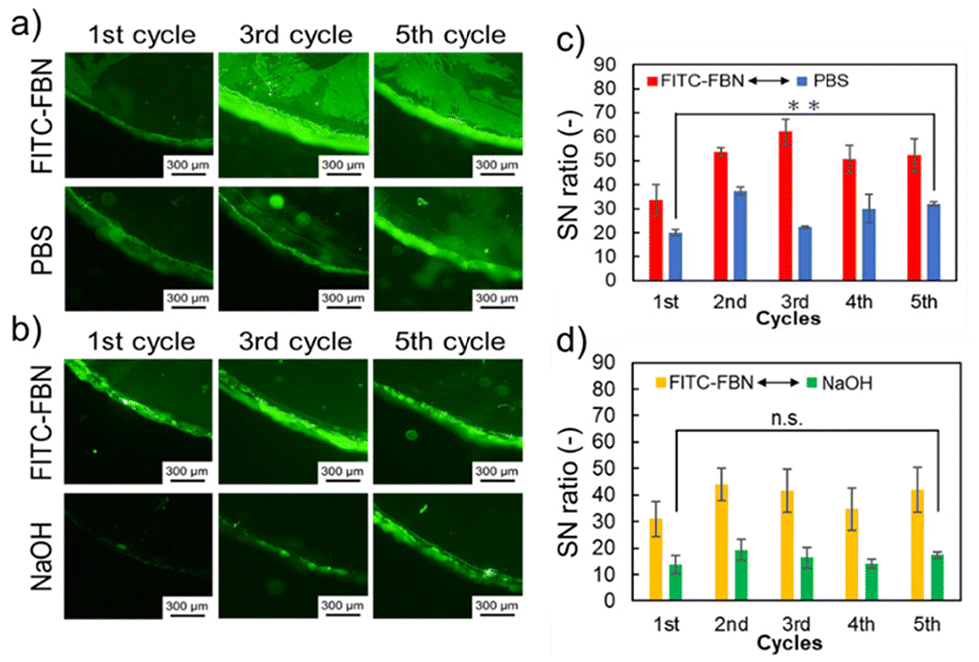
Finally, protein adsorption on the hydrogels and surface renewability of the hydrogels were analyzed using fluorescence microscopy observation. The renewability of the hydrogel surfaces was investigated by measurements of protein adsorption and removal on hydrogel surfaces, repetitively. First, the prepared hydrogels were soaked in either FITC-BSA or FITC-FBN solution for 1 h, followed by agitation and PBS washing at 37 °C. After washing, the protein-adsorbed hydrogels were soaked in NaOH or PBS solution at 37 °C for 1 h. The adsorbed amount of BSA and FBN in the first cycle was 0.76 ± 0.17 μg cm−2 and 0.89 ± 0.06 μg cm−2, respectively. Fluorescence microscopy observation of the FITC-BSA-adsorbed hydrogels was performed at each step of the 5 cycles (Fig. 5). At the first cycle, the fluorescence signal of FITC-BSA was observed on both the hydrogels (Fig. 5a and b; top-left images), which increased with increasing cycle number. After immersion in PBS, the fluorescence from FITC-BSA decreased but remained in each cycle image (Fig. 5a; bottom). As shown in Fig. 5c, the SN ratio increased with increasing cycle numbers because the amount of adsorbed proteins on the hydrogels increased by repetitive immersion in the FITC-BSA solution. In contrast, fluorescence from FITC-BSA was not observed after washing the hydrogels with NaOH solution (Fig. 5b; bottom). In addition, the SN ratio remained unchanged despite the increased number of degradation cycles, and the fluorescence intensities of the hydrogels subjected to surface degradation decreased to equivalent values to those of the sliding glass (Fig. 5d). As the hydrogel surface degraded, the adsorbed proteins were removed and at the same time the byproduct oligomers were released (Fig. 5e). These results indicated that protein removal was effectively achieved, along with surface degradation, at least five times. The protein removal properties of the hydrogels were investigated in detail (Fig. S2†). The same cycle test was performed using a non-degradable thermoresponsive hydrogel, poly(NIPAAm)-g-PEG hydrogel (sample 3 in Table 1), without MDO during polymerization. The prepared hydrogels were soaked in an FITC-BSA solution under the same conditions. Subsequently, the protein-adsorbed hydrogels were soaked in NaOH solution. The SN ratio of the hydrogels increased in the 3rd cycle. This indicated that surface degradation of the hydrogels was necessary for protein removability.
 |
| | Fig. 5 Evaluation of protein removal ability (sample 1 in Table 1). (a) Fluorescence images of the hydrogel surface adsorbed with FITC-BSA after each cycle and after soaking with PBS after FITC-BSA adsorption (bottom). (b) Fluorescence images of the hydrogel surface adsorbed with FITC-BSA after each cycle (top) followed by hydrolysis in NaOH solution (bottom). All scale bars: 300 μm. (c) SN ratio of adsorbed FITC-BSA on the hydrogel surface. Red bars: SN ratio of the hydrogel immersed in PBS solution containing FITC-BSA in each cycle. Blue bars: SN ratio of the hydrogel immersed in PBS in each cycle after 1 h of soaking in PBS solution containing FITC-BSA. Data are expressed as the mean ± SD (n = 3, *: p < 0.05). (d) SN ratio of adsorbed FITC-BSA on the hydrogel surface. Yellow bars: SN ratio of the hydrogel immersed in PBS solution containing FITC-BSA in each cycle. Green bars: SN ratio of the hydrogel immersed in NaOH solution in each cycle after 1 h of soaking in PBS solution containing FITC-BSA. Data are expressed as the mean ± SD (n = 3), n.s.: not significant. (e) Schematic illustration of the surface renewal properties of the hydrogels. | |
Fig. 6 summarizes the FITC-FBN removal results. In the fluorescence images, the same tendency as that in the case of FITC-BSA was observed (Fig. 6a and b). Although the fluorescence intensity increased compared to that of BSA, FITC-FBN was effectively removed by surface degradation (Fig. 6c and d). These results suggest that protein removal by degradation is effective regardless of the protein type. We then analyzed the effect of the PEG chains on protein adsorption onto the hydrogel surfaces. The same cycle test was performed using the poly(MDO-co-NIPAAm) hydrogels (sample 2 in Table 1) (Fig. S3a and b†). The prepared hydrogels were soaked in either FITC-BSA or FITC-FBN solution for 1 h. Subsequently, the protein-adsorbed hydrogels were soaked in PBS or NaOH solution. The initial amount of BSA adsorbed on the poly(MDO-co-NIPAAm) hydrogels without PEG was lower than that on the hydrogels with PEG graft chains. The swelling ratio of the poly(MDO-co-NIPAAm) hydrogels without the PEG chain was 0.25 at 37 °C which is lower compared to that of the poly(NIPAAm-co-MDO)-g-PEG hydrogels with the PEG chain: 2.2 at 37 °C, as seen in Fig. 3a. This difference could be due to the presence of PEG chains inside the hydrogels that create hydration domains within the hydrogels, leading to the migration of some BSA molecules inside the poly(NIPAAm-co-MDO)-g-PEG hydrogels. However, the SN ratio of the degraded poly(MDO-co-NIPAAm) hydrogels increased with increasing number of cycles compared with poly(NIPAAm-co-MDO)-g-PEG hydrogels. Moreover, the adsorption of FITC-FBN tended to be similar to that of BSA. When PEG was present on the gel surface, the amount of adsorbed proteins remained constant regardless of the number of removals, suggesting that the protein removal ability was maintained due to the hydration effect of the PEG chains and the excluded volume effect. After 5 cycles of degradation, the swelling ratio of the poly(NIPAAm-co-MDO)-g-PEG hydrogels was increased to 4.9 at 37 °C. However, the swelling ratio of the poly(MDO-co-NIPAAm) hydrogels without PEG chains was 0.23 at 37 °C. It is considered that hydrogel degradation was induced by PEG chains owing to the increased swelling ratio. These results suggested that the PEG chains also influenced effective protein removal under these preparation conditions. Therefore, the prepared hydrogel is expected to be applicable as a thermoresponsive degradable hydrogel that exhibits protein removal behavior upon degradation and maintains its surface properties even after degradation.
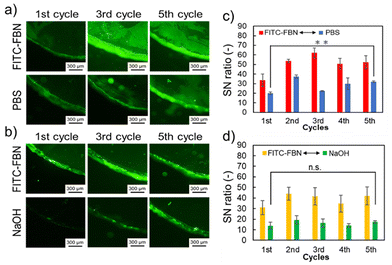 |
| | Fig. 6 Evaluation of protein removal ability (sample 1 in Table 1). (a) Fluorescence images of the hydrogel surface adsorbed with FITC-FBN after each cycle and after soaking with PBS after FITC-FBN adsorption (bottom). (b) Fluorescence images of the hydrogel surface adsorbed with FITC-FBN after each cycle (top) followed by hydrolysis in NaOH solution (bottom). All scale bars: 300 μm. (c) SN ratio of adsorbed FITC-FBN on the hydrogel surface. Red bars: SN ratio of the hydrogel immersed in PBS solution containing FITC-FBN in each cycle. Blue bars: SN ratio of the hydrogel immersed in PBS in each cycle after 1 h of incubation in PBS solution containing FITC-FBN. Data are expressed as the mean ± SD (n = 3, *: p < 0.05). (d) SN ratio of adsorbed FITC-FBN on the hydrogel surface. Yellow bars: SN ratio of the hydrogel immersed in PBS solution containing FITC-FBN in each cycle. Green bars: SN ratio of the hydrogel immersed in NaOH solution in each cycle after 1 h of soaking in PBS solution containing FITC-FBN. Data are expressed as the mean ± SD (n = 3), n.s.: not significant. | |
4. Conclusions
Thermoresponsive and degradable poly(NIPAAm-co-MDO)-g-PEG hydrogels were synthesized via radical polymerization. The prepared hydrogels exhibited thermoresponsive properties derived from PNIPAAm. The hydrogels exhibited degradable behavior via the degradation of ester groups in the polymer backbone. The degradable behavior was controlled by the thermoresponsive behavior, swelling, and shrinking. These results suggested that surface degradation was observed in the shrunken state of the hydrogels. The regenerative protein removal properties were observed by surface degradation. The synthesized hydrogels are expected to serve as new biomaterials with regenerative protein removal properties.
Author contributions
S. K. and A. K. designed this research and developed the hypothesis, and wrote and edited the manuscript. N. K. and S. K. performed the preparation and sample characterization. All authors discussed the obtained results. All authors have read and approved the submitted manuscript.
Data availability
All the data supporting the findings will be made public and can be obtained by contacting the corresponding author A. Kikuchi.
Conflicts of interest
There are no conflicts of interests to declare.
Acknowledgements
Part of this work was supported by the KAKENHI Grant-in-Aid for Young Scientists (23K13845) from the Japan Society for Promotion of Science (JSPS).
References
- B. R. Young, W. G. Pitt and S. L. Cooper, J. Colloid Interface Sci., 1998, 124, 28–43 CrossRef.
- W. R. Gombotz, G. H. Wang, T. A. Horbett and A. S. Hoffman, J. Biomed. Mater. Res., 1991, 25, 1547–1562 CrossRef PubMed.
- Y. Nagasaki, Polym. J., 2011, 43, 949–958 CrossRef.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef.
- H. D. W. Roesink, M. A. M. Beerlarge, W. Potman, Th. Van den Boomgaard, M. H. V. Mulder and C. A. Smolders, Colloids Surf., 1991, 55, 231–243 CrossRef.
- S. Robinson and P. A. Williams, Langmuir, 2002, 18, 8743–8748 CrossRef.
- K. Fujimoto, M. Minato, H. Tadokoro and Y. Ikada, J. Biomed. Mater. Res., 1993, 27, 335–343 CrossRef PubMed.
- K. Ishihara, N. P. Ziats, B. P. Tierney, N. Nakabayashi and J. M. Anderson, J. Biomed. Mater. Res., 1991, 25, 1397–1407 CrossRef PubMed.
- W. Feng, X. Gao, G. McClung, S. Zhu, K. Ishihara and J. L. Brash, Acta Biomater., 2011, 7, 3692–3699 CrossRef PubMed.
- W. N. Yu, D. H. N. Manik, C. Huang and L. Chau, Chem. Commun., 2017, 53, 9143–9146 RSC.
- D. E. Heath and S. L. Cooper, Acta Biomater., 2012, 8, 2899–2910 CrossRef CAS PubMed.
- J. Wu, Z. Xiao, C. He, J. Zhu, G. Ma, G. Wang, H. Zhang, J. Xiao and S. Chen, Acta Biomater., 2016, 40, 172–181 CrossRef CAS.
- N. B. Omali, L. N. Subbaraman, C. C. Brennan, Z. Fadli and L. W. Jones, Optom. Vis. Sci., 2015, 92, 750–757 CrossRef.
- A. Schulze, D. Breite, Y. Kim, M. Schmidt, I. Thomas, M. Went, K. Fischer and A. Prager, Polymers, 2017, 9, 97, DOI:10.3390/polym9030097.
- J. Ma, W. Lin, L. Xu, S. Lin, W. Xue and S. Chen, Langmuir, 2020, 36, 3251–3259 CrossRef CAS PubMed.
- G. Dai, Q. Xie, X. Ai, C. Ma and G. Zhang, ACS Appl. Mater. Interfaces, 2019, 13, 13735–13743 CrossRef PubMed.
- T. Kamiya, S. Komatsu and A. Kikuchi, ACS Appl. Bio Mater., 2021, 4, 8498–8502 CrossRef PubMed.
- S. Komatsu, T. Asoh, R. Ishihara and A. Kikuchi, Polymer, 2017, 130, 68–73 CrossRef.
- S. Komatsu, T. Asoh, R. Ishihara and A. Kikuchi, Polymer, 2019, 179, 121633, DOI:10.1016/j.polymer.2019.121633.
- S. Komatsu, T. Sato and A. Kikuchi, Polym. J., 2021, 53, 731–739 CrossRef.
- S. Komatsu, S. Ishida and A. Kikuchi, React. Funct. Polym., 2022, 177, 105321, DOI:10.1016/j.reactfunctpolym.2022.105321.
- M. L. T. Zweers, G. H. M. Engbers, D. W. Grijpma and J. Feijen, J. Controlled Release, 2004, 10, 347–356 CrossRef PubMed.
- M. Heskins and J. E. Guillet, J. Macromol. Sci: Part A- Chem., 1968, 2, 1441–1455 CrossRef.
- H. Cui, Y. Zhang, Y. Shen, S. Zhu, J. Tian, Q. Li, Y. Shen, S. Liu, Y. Cao and H. C. Shum, Adv. Mater., 2022, 2205649, DOI:10.1002/adma.202205649.
- T. Maeda, M. Takenouchi, K. Yamamoto and T. Aoyagi, Biomacromolecules, 2006, 7, 2230–2236 CrossRef PubMed.
- J. Lian, H. Xu, S. Duan, X. Ding, Y. Hu, N. Zhao, X. Ding and F. Xu, Biomacromolecules, 2020, 21, 732–742 CrossRef.
- J. Wang, Y. Chen, J. An, K. Xu, T. Chen, P. M. Buschbaum and Q. Zhong, ACS Appl. Mater. Interfaces, 2017, 9, 13647–13656 CrossRef PubMed.
- W. J. Bailey, Z. Ni and S. R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef.
- S. Komatsu, S. Yamada and A. Kikuchi, ACS Biomater. Sci. Eng., 2024, 10, 897–904 CrossRef.
- B. R. Kordes, L. Ascherl, C. Rüdinger, T. Melchin and S. Agarwal, Macromolecules, 2023, 56, 1033–1044 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Naoki
Kamei
a and
Akihiko
Kikuchi
*a
ab,
Naoki
Kamei
a and
Akihiko
Kikuchi
*a