
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced anticancer effect of lysozyme-functionalized metformin-loaded shellac nanoparticles on a 3D cell model: role of the nanoparticle and payload concentrations†
Anheng
Wang
 ab,
Leigh A.
Madden
c and
Vesselin N.
Paunov
*d
ab,
Leigh A.
Madden
c and
Vesselin N.
Paunov
*d
aInstitute of Chinese Medical Sciences & State Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Macau SAR, China
bZhuhai UM Science and Technology Research Institute, University of Macau, Hengqin, Guangdong, China
cCentre for Biomedicine, Hull York Medical School, University of Hull, HU67RX, UK
dDepartment of Chemistry, Nazarbayev University, 53 Kabanbay Batyr Avenue, Astana, 010000, Kazakhstan. E-mail: vesselin.paunov@nu.edu.kz
First published on 31st July 2024
Abstract
Here we used a 3D human hepatic tumour cell culture model to assess the in vitro efficacy of “active” metformin-loaded nanoparticles (NPs) as anticancer therapeutics. The metformin nanocarrier design was repurposed from previous studies targeting bacterial and fungal biofilms with antimicrobials loaded in protease-coated nanoparticles. These active nanocarriers were constructed with shellac cores loaded with metformin as the anticancer agent and featured a surface coating of the cationic protease lysozyme. The lysozyme's role as a nanocarrier surface coating is to partially digest the extracellular matrix (ECM) of the 3D tumour cell culture which increases its porosity and the nanocarrier penetration. Hep-G2 hepatic 3D clusteroids were formed using a water-in-water (w/w) Pickering emulsion based on an aqueous two-phase system (ATPS). Our specific metformin nano-formulation, comprising 0.25 wt% lysozyme-coated, 0.4 wt% metformin-loaded, 0.2 wt% shellac NPs sterically stabilized with 0.25 wt% Poloxamer 407, demonstrated significantly enhanced anticancer efficiency on 3D hepatic tumour cell clusteroids. We examined the role of the lysozyme surface functionality of the metformin nanocarriers in their ability to kill both 2D and 3D hepatic tumour cell cultures. The anticancer efficiency at high metformin payloads was compared with that at a high concentration of nanocarriers with a lower metformin payload. It was discovered that the high metformin payload NPs were more efficient than the lower metformin payload NPs with a higher nanocarrier concentration. This study introduces a reliable in vitro model for potential targeting of solid tumours with smart nano-therapeutics, presenting a viable alternative to animal testing for evaluating anticancer nanotechnologies.
1. Introduction
Targeting solid tumours remains very difficult because of their closely packed tissue layers and chaotic vasculature, which leads to extremely high rates of incidence.1–3 Although low molecular weight anti-tumour agents have enabled advances in cancer chemotherapy over the decades, poor water solubility, unfavourable pharmacokinetics and undesirable side effects of many molecules with potential anti-tumour properties have hampered their clinical application.4–6 The major obstacles to the development of new therapies are multifactorial, including the difficulty of constructing realistic in vitro tumour models and the formulation of suitable drug delivery systems.Traditional two-dimensional (2D) models have played a crucial role in in vitro cancer drug testing.7 However, their limitations lie in their inability to replicate the three-dimensional (3D) tumour growth observed in vivo, complete with the specific architecture and diverse signals governing cellular processes, especially in solid tumours.8,9 Among 3D culture models, cell spheroids have recently gained widespread usage.10 Various 3D culture techniques have been developed to facilitate their formation11–14 which often necessitate expensive equipment, and achieving high throughput with both homogeneity and substantial size in cell spheroid generation remains a challenge. Recently, our group pioneered a low-cost, high-throughput method for preparation of large amounts of cell spheroids based on Pickering emulsions formed from aqueous two-phase systems (ATPS) stabilized by solid particles.15–17 This method was demonstrated to be well suited for testing various anticancer as well as antibiofilm therapies.9 High-throughput testing on tumour models in nanomedicine, which involves passive or active drug delivery to tumours,20–22 has led to the development of a growing pipeline of anticancer nano-therapies already progressing to more advanced clinical investigations.23,24 The dense extracellular matrix (ECM) primarily comprises loosely organized and interconnected collagen lattices, along with polysaccharides like glycosaminoglycans (Scheme 1).25–27
 | ||
| Scheme 1 In our approach, nanoparticles with a high payload of metformin can penetrate deeply into the interior of the clusteroids, thanks to the protease activity of their lysozyme surface functionality on the extracellular matrix (ECM). This allows for the delivery of a high payload of metformin in depth of the 3D cell culture. In contrast, NPs with a low payload of metformin NPs can also penetrate the ECM and are mostly trapped within it, and even if some enter the interior, their lower payload limits their functional efficacy. | ||
Cancer-targeting nanotherapy encompasses a wide array of nanocarrier platforms, including polymeric nanoparticles, dendrimers, metallic nanoparticles, liposomes, quantum dots, and carbon nanotubes, among others. These systems offer unique advantages for the precise delivery of therapeutics to cancer cells while minimizing toxic effects on healthy tissues. Polymeric nanoparticles with tuneable size, shape, and surface chemistry can enhance their tumour-targeting capabilities and drug loading capacity.28–33 Dendrimers, with their highly branched and multivalent structure, enable the conjugation of multiple functional moieties for targeted drug delivery and tumour imaging.34–36 Solid nanoparticles, such as gold and zinc nanoparticles, exhibit unique optical and photothermal properties that can be harnessed for cancer therapy and diagnostics.37–44 Liposomes, being biocompatible and biodegradable, have been extensively explored as versatile drug delivery platforms, allowing for the encapsulation of both hydrophilic and hydrophobic payloads.39,45–51 Quantum dots, with their size-dependent photoluminescence properties, have shown great potential in cancer imaging and theranostics.52–55 Lastly, carbon nanotubes, with their high aspect ratio and exceptional mechanical and electrical properties, have been investigated as promising platforms for targeted drug delivery and photothermal therapy.56
This study follows the recent trend in pharmaceutical science of repurposing old drugs for new therapies. Metformin is the first-line recommended treatment for Type II diabetes mellitus, an oral biguanide drug that was licensed by the US FDA in 1994.57–59 This antidiabetic medication is getting a lot of attention as a possible anticancer treatment due to retrospective findings that diabetics taking metformin had better survival rates across a variety of cancers.60,61 The anticancer effect of metformin against multiple cancer types was validated using data from in vitro and preclinical investigations; metformin has been proved to be efficient in inducing Hep-G2 cell apoptosis through the regulation of ER stress and the AMPK/p53/miR-23a/FOXA1 pathway.62,63 Here we took this further with the development of active metformin nanocarriers to enhance the metformin anticancer effect (Scheme 1).
An additional novelty of this study also resides in repurposing of the nano-carrier system which was originally developed for targeting fungal and bacterial biofilms based on a protease coating that facilitates the penetration of the nanoparticles into the extracellular polymeric substance (EPS) of the biofilm.64–71 Shellac NPs loaded with antimicrobials have been recently proven to perform with high efficacy to disrupt both bacterial and fungal biofilms and kill the residing microbial cells.72–76 A rationale to target cancer with the same nanocarrier system is that the cancer cells tend to have increased expression of phosphatidyl serine (a negatively charged phospholipid) and tumours possess an ECM, like a biofilm. Here this active nanocarrier approach was repurposed to facilitate the nanoparticle penetration into the ECM of solid tumours, which allows enhanced delivery of metformin into the tumour interior. The solid tumour environment was modelled by using 3D hepatic cell culture. Cancer cells have a negative surface charge which helps the metformin nanocarrier functionalized with a cationic protease (lysozyme) to accumulate on the cells and to partially digest their ECM.
In this work, we develop lysozyme-functionalized metformin-loaded shellac nanoparticles (Ly-NPs) and test their in vitro anticancer efficiency on 3D hepatic cell culture. These smart nanoparticles were designed to deliver a very high payload of metformin to the interior of the clusteroids, producing a highly potent anticancer effect. In this study, we repurpose these ideas for anticancer research to potentially target solid tumours which are effectively modelled with large 3D cell clusteroids. We examine the influence of the lysozyme surface functionality of the nanocarriers on their capacity to effectively disrupt and eliminate 3D cancer cell cultures. Fig. S1 (ESI†) illustrates the two steps of the metformin-loaded shellac nanocarrier preparation and their subsequent coating by electrostatically driven adhesion of the cationic protease (lysozyme).
The advantage of the shellac nanocarrier system is that it is USFDA approved as a biocompatible material and consists of a mixture of natural resin esters, primarily polyhydroxy triterpene acids. These compounds contain abundant hydroxyl (–OH) and carboxyl (–COOH) groups in their molecular structure. These carboxyl functional groups of the shelloic acids partially dissociate in aqueous solutions and facilitate a high degree of encapsulation of the positively charged metformin, thus achieving high payloads. We also address a very important question about what makes the metformin nano-formulation more effective as anticancer treatment: (i) the concentration of metformin in the nanocarriers (the payload) or (ii) the nanocarrier concentration at a fixed overall metformin concentration? The drug concentration is the most easily controllable parameter which determines the therapeutic effect of the treatment. Therefore, in this nanocarrier-based metformin study we focused on using the same preparation with a fixed shellac-to-metformin ratio but increasing metformin concentration. We explore the efficacy of the protease-coated metformin-loaded shellac nanoparticles at (i) high and low metformin payloads as well as (ii) as a function of the nanoparticle concentration. We conduct a comprehensive comparative analysis of the anticancer efficacy between nanocarriers with high metformin payloads and high concentrations of nanocarriers containing lower quantities of metformin. This comparison aims to elucidate the optimal balance between drug loading and nanocarrier concentration to maximize the therapeutic potential of metformin in cancer treatment.
2 Experimental
2.1 Materials
CellTrace Far Red, CFSE green fluorescence dye, EasYFlasks and NUNC cell culture 24-well plates were purchased from Thermo Fisher Scientific (China). Dextran (DEX) (M.W. 500 kDa) and polyethylene glycol (PEG) (MW. 500 kDa) were purchased from Alfa Aesar Chemicals (China). Sodium chloride (99.8%) and calcium chloride (99.8%), Eagle's Modified Eagle Medium (EMEM), and Trypsin-EDTA were sourced from Gibco (China). Foetal bovine serum (FBS) was sourced from Gibco (Australia). Trypsin-EDTA was purchased from Thermo Fisher Scientific (China). The HepG2 cell line was sourced from Promocell, Ltd (UK). CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS) and CellTiter-Glo 3D Cell Viability Assay were purchased from Promega (UK). StemPro™ Accutase™ Cell Dissociation Reagent was sourced from Thermo Fisher Scientific (China). The 2 wt% gelatin suspension was produced from porcine gelatin sourced from Sigma Aldrich (UK). Matrigel was purchased from Corning (China). Whey protein was sourced from No. 1. Supplements (Suffolk, UK). All the other chemicals were of analytical grade. Deionized water was purified using a deionized water system (Millipore) with a surface tension of 71.9 mN m−1 at 25 °C, and a resistivity higher than 18 MΩ cm−1.2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well). This can be calculated based on the initial amount of cells added in generating the cell clusteroids as the cells proliferate very slowly in the emulsion template. The treatment was added to each well with medium. After incubation for 2 h at 37 °C, the clusteroids were tested using 10 μL 3D cell proliferation assay. For the Promega CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) the cells were collected by either a plate centrifuge or centrifugation in PCR tubes. The clusteroids were disintegrated by organoid dissociation solution (15 min). 10 μL of the MTS reagent were incubated with the disintegrated clusteroids in PBS.
:1, 1:2, 1:3) for 2 h each. The spheroids were embedded in paraffin wax, sectioned to a thickness of 5 μm using a microtome, and mounted on glass slides. The sections were deparaffinized in xylene, rehydrated in a series of ethanol solutions, and stained with Hematoxylin and Eosin (H&E) for general histology and Masson's Trichrome for collagen visualization. Stained sections were mounted with a mounting medium and covered with a cover slip for microscopy analysis.
000 cells per well). This can be calculated based on the initial amount of cells added in generating the cell clusteroids as the cells proliferate very slowly in the emulsion template. The treatment was added to each well with medium. After incubation for 2 h at 37 °C, the clusteroids were tested using 10 μL 3D cell proliferation assay. For the Promega CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) the cells were collected by either a plate centrifuge or centrifugation in PCR tubes. The clusteroids were disintegrated by organoid dissociation solution (15 min). 10 μL of the MTS reagent were incubated with the disintegrated clusteroids in PBS.
:1, 1:2, 1:3) for 2 h each. The spheroids were embedded in paraffin wax, sectioned to a thickness of 5 μm using a microtome, and mounted on glass slides. The sections were deparaffinized in xylene, rehydrated in a series of ethanol solutions, and stained with Hematoxylin and Eosin (H&E) for general histology and Masson's Trichrome for collagen visualization. Stained sections were mounted with a mounting medium and covered with a cover slip for microscopy analysis.
3 Results and discussion
3.1 Cell clusteroid formation in DEX-in-PEG w/w emulsions
3D cell culture has attracted a lot of researcher's attention in the past few decades, especially in various in vitro drug testing. An emerging potential area of 3D cell models is employing cell spheroids as building blocks to form complex tissues. Spheroids serve as a good ex vivo model for mini-organ simulations, representing high gene and cytokine levels.2 The creation of clusteroids involves encapsulating cells within a DEX/PEG w/w emulsion, which is stabilized using biocompatible whey protein particles. Previous studies have shown that cells remain contained within the dextran phase. The concentration of cells and the volume fraction of the two phases play crucial roles in clusteroid formation. Illustratively, Fig. 1 demonstrates how we utilize the w/w emulsion template. Initially, the cell concentration was set at 106 cells per mL, but larger clusteroids can be generated by increasing this number. Once the w/w emulsion encapsulating cells is formed, it is promptly transferred to a more concentrated PEG solution to induce cell shrinkage into clusteroids. Notably, the cell density is intermediate between the 5.5 wt% dextran solution and higher concentrations of the PEG solution phases. Gravity aids in driving the DEX phases containing cells through the denser PEG solution phase. Subsequently, the precipitation process compels the cells to aggregate into clusteroids, as shown in Fig. 1E and F. To demonstrate the efficiency of the cell encapsulation, we performed preliminary observations using a fluorescence microscope. For the observation purposes, the Hep-G2 cells were stained with DAPI (blue fluorescence) and CFSE (green fluorescence), respectively. As shown in Fig. 2, the cells are packed in multiple droplets and, after the shrinking process at higher PEG concentrations, successfully compressed into the clusteroids without influencing the cell ratio. | ||
| Fig. 1 Optical brightfield microscopy images (A, C) and fluorescence microscopy images (B, D) of the individual Hep-G2 cell clusteroids encapsulated in the w/w Pickering emulsions (5.5 wt% DEX, 5.5 wt% PEG). These clusteroids were stained with DAPI. SEM images of clusteroids (E, F). The scale bar is 50 μm for (A, B) and 100 μm for (C–F). | ||
 | ||
| Fig. 2 (A) The mean particle diameter and zeta-potential of Ly-NPs at different metformin payloads. (B) The encapsulation efficiency of Ly-NPs at different pH levels. (C) SEM image of 0.2 wt% shellac-0.25 wt% lysozyme–0.1 wt% metformin–0.25 wt% P407 NPs. (D) SEM image of 0.2 wt% shellac–0.25 wt% lysozyme–0.2 wt% metformin–0.25 wt% P407 NPs. (E) SEM image of 0.2 wt% shellac–0.25 wt% lysozyme–0.4 wt% metformin–0.25 wt% P407 NPs. The scale bar is 100 nm. | ||
The compressed clusteroids had structural integrity and were polydisperse in size (Fig. 1C and D). The size of the cell clusteroids collected from the Pickering emulsion template was commonly not uniform. This feature makes the clusteroids more suitable for the tissue engineering applications instead of precise drug susceptibility testing. The more concentrated PEG phase depletes the DEX aqueous droplets encapsulating the cells with water as it moves to restore the osmotic equilibrium, and this causes them to shrink. Hence the interfacial tension of the shrinking droplets promotes the cell–cell interactions and allows them to form more contacts. Notably, the possibility of direct cell co-culture in the ATPS-based w/w emulsion was not mentioned in any previous reports. The results indicate the advantages of the ATPS Pickering emulsion template in the high-throughput fabrication of co-cultured cell clusteroids.
3.2 Characterization of the nanocarriers
To address the efficiency of our clusteroid layer as an alternative to the real human liver, we designed a colloidal delivery system based on shellac nanoparticles for inducing cancer cell death. Several reports have shown that these nanoparticles exhibited good efficiency in killing the cancer cells with low toxicity. The Ly-NPs were designed based on the electrostatic deposition of the enzyme on the particle surface at a specific pH value. The shellac nanoparticles held a negative charge due to the partial deprotonation of the carboxylic groups in the shelloic acids, nearly −30 mV at pH 6. As the lysozyme is positively charged at pH 6 (IEP > 11), this facilitated its immobilization on the shellac nanoparticles at this pH due to electrostatic adhesion. The zeta-potential characterization shown in Fig. 2A demonstrated that at a low lysozyme concentration (below 0.1 wt%), the shellac NPs were highly negative charged (−12 mV). Fig. S3 (ESI†) shows the shellac nanocarrier zeta-potential at different lysozyme concentrations. In our experiments, we have found that excessively high concentrations of lysozyme do not further enhance the electrical properties of the nanoparticles, with the zeta potential reaching saturation at around +18 mV (in DI water) – see also Fig. S2B and C (ESI†). We also conducted SEM imaging of the Ly-NPs which is shown in Fig. S4 (ESI†), revealing an oval-shaped morphology and particle diameters around 60 nm, in line with the dynamic light scattering measurements. The encapsulation efficiency reached 90%, which could be attributed to the electrical binding of metformin and shellac (Fig. 2B). The bare shellac nanoparticles had a size of around 50 nm as shown in SEM figures (Fig. 2C and S4 (ESI)†). Ly-NPs showed strong aggregation on the sample support with no obvious size change (Fig. 2D and E). An estimate of the number of metformin molecules per Ly-NP is given in the ESI.†We examined the shellac-to-metformin ratio as shown in Fig. 3D for 1×, 2×, 3× and 4× payloads of metformin at the same overall shellac concentration. Since we have already repurposed a successfully designed active nanocarrier whose architecture we tested in biofilm studies,66,71,76 we did not aim to reassess all possible parameters of the nanocarrier system, such as the enzyme surface coating density, as this coating worked well to digest through the biofilm EPS and deliver its payload. We checked that the metformin encapsulation efficiency is over 90% (see Fig. 2B) which is in line with previous studies with antibiotic drugs encapsulated in the same nanocarrier.66,76 The results in Fig. S3 (ESI†) show that above 0.05 wt% lysozyme concentration, the particle zeta potential levels off, i.e. further addition of the enzyme leads to saturation of the nanocarrier surface with a dense layer of protease.
 | ||
| Fig. 3 Anti-cancer effect of the lysozyme-coated metformin-loaded P407-stabilised shellac nanoparticles and their individual components (A, C) and at different concentrations of the treatment corresponding to multiples of the original stock solution (1×) (B, D) on 2D monolayer cells (A, B) or the 3D cell Hep-G2 clusteroids and (C, D) for 2D Hep-G2 cell culture tested by the MTS assay. The 2D mono-layer cells and 3D clusteroids were treated for 48 h. Control at 0 h corresponds to 100%. The initial cell number was 10000 per well. | ||
As we repurpose an already developed nanocarrier system for targeting biofilms with protease-coated antibiotic-loaded nanoparticles and explore their application in the case of targeting cancers (modelled here as cell clusteroids) with metformin payload, we adhered to the same concentrations of shellac, poloxamer and protease used previously. This allowed us to maintain the shellac nanoparticle system stability while studying the metformin anticancer effect when encapsulated in this nanocarrier. Regarding the rationale for the selection of working concentrations of metformin, we used the same range of concentrations as the ones of antibiotics and antimicrobials previously reported, i.e. typically 0.1 wt%.66,71,76 However, we varied the metformin concentrations from 0.1 wt% up to 0.4 wt% to study their effects on 3D hepatic cancer cell cultures.
3.3. Anti-cancer action of Ly-NPs on hepatic cell clusteroids
These particles would preferably hetero-coagulate on the clusteroids’ outer cell layer, which might be attributed to the attraction between the protease-functionalized nanogel particles and the anionic surface of the clusteroid cells. As a result, the local release of metformin from the nanocarrier directly onto the clusteroid layers appears to increase its anti-cancer action. Fig. 3B shows the concentration dependence of the Ly-NPs on the Hep-G2 clusteroid viability. The co-cultured Ly-NPs/clusteroid layers were incubated in a series of suspensions obtained by multiple dilutions of a stock suspension with EMEM complete medium using 2D MTS assay. Note that the shellac nanocarrier formulation has a clearly stronger anticancer effect with an increasing concentration of metformin.A 90% decrease in the clusteroid viability was observed after 24 h incubation, demonstrating the high anti-cancer efficiency of these NPs. An increased concentration of metformin significantly enhances the anticancer capabilities of the particles.
Compared to the action of its individual components, the particles formed by this electrostatically driven adhesion exhibit remarkably potent anticancer abilities (Fig. 3A). The obtained results from the conventional MTS assays may not accurately reflect the true cell proliferation activity of the complex 3D clusteroids. Therefore, we utilized 3D proliferation assays to assess the activity of these clusteroids post-treatment. Consistent with the results obtained with MTS assays in 2D cell cultures, the 3D proliferation tests also demonstrated that the NPs significantly outperform conventional components in anticancer efficacy (Fig. 3C). The increased metformin payload also showed an enhanced anticancer efficacy, yet overall, the NPs exhibited a diminishing effect on cancer cell eradication within clusteroids, reaching a maximum of 80%.
This outcome could potentially be attributed to the complexity of 3D tissue structures. The SEM observation of the 3D cell culture after treatment with Ly-NPs clearly demonstrates the degradation of the clusteroids and how their morphology was changed after the treatment (Fig. 4A–C). At the outset of processing, the Ly-NPs adhere to the surface of the clusteroids, likely due to electrostatic adhesion. After 24 h of treatment, the clusteroids exhibited noticeable collapse and the emergence of a porous structure (Fig. 4B). Subsequently, by 48 h, the cellular structure was largely compromised and showed clear signs of degradation and enhanced porosity (Fig. 4C). We further examined the structure of the clusteroids after 48 h of treatment with metformin at varying payloads.
 | ||
| Fig. 4 SEM images of the Hep-G2 cell clusteroids after treatment with 0.2 wt% shellac–0.25 wt% lysozyme–0.1 wt% metformin–0.25 wt% P407 (1× payload of metformin): (A) 0 h, (B) 24 h, (C) 48 h treatment; and Hep-G2 cell clusteroids treated with (D) 1× payload of metformin, (E) 2× payload of metformin, (F) 3× payload of metformin, and (G) 4× payload of metformin. The scale bar is 50 μm. | ||
Upon increase of the nanocarriers’ metformin payload, it was evident that the size of the clusteroids notably decreased in addition to exhibiting more porous structure (Fig. 4D–G).
To elucidate the challenges faced by nano-delivery systems operating within high particle concentration yet low payload environments, SEM imaging was employed to investigate the impact of high-concentration low-payload particles on Hep-G2 clusteroids. Our observations revealed a gradual accumulation of particles in the vicinity of the clusteroids. Unlike their high-payload counterparts, low-metformin payload Ly-NPs did not induce the formation of a porous structure within the clusteroids (Fig. 5A and B) nor did they exert a pronounced inhibitory effect on cellular proliferation (see Fig. 3B). Rather, these particles exhibited a tendency to accumulate around the periphery of the clusteroids, a phenomenon that may serve as a critical determinant influencing their functional efficacy (Fig. 5C). According to these data, we can infer that the combination of metformin payload and lysozyme-surface functionality can kill the 3D Hep-G2 cell culture. However, Ly-NPs with the same amount of individual components exhibited an exponentially stronger effect in inducing cell death in 3D cell clusteroids, which could be explained by the deeper penetration through the ECM by local degradation of the glycoproteins of the matrix by the lysozyme coating on the nanocarrier.
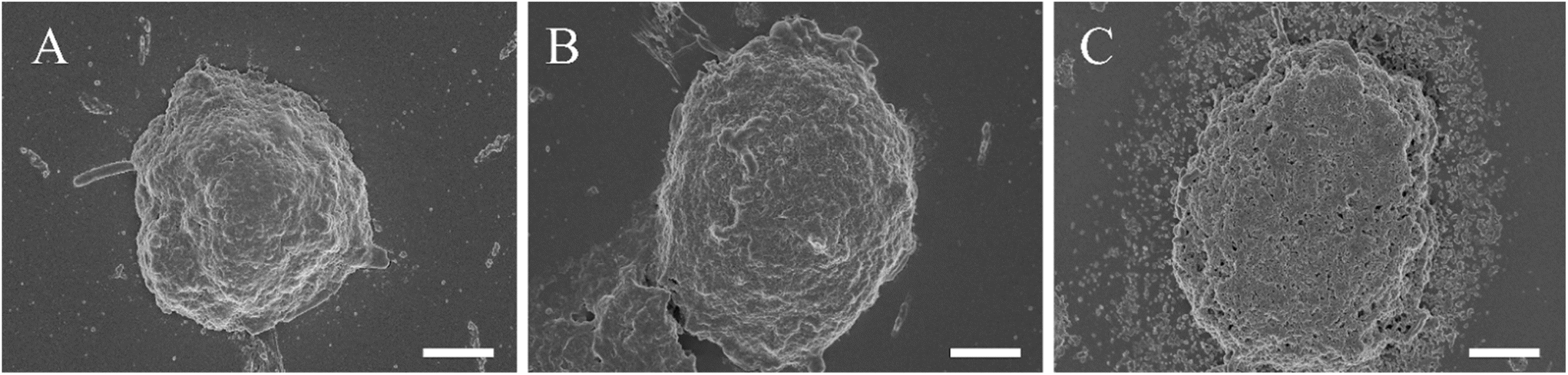 | ||
| Fig. 5 SEM images of the Hep-G2 cell clusteroids after treatment with 1 wt% shellac–1 wt% lysozyme–0.1 wt% metformin–1 wt% P407 (high NP amount): for A: 0 h B: 24 h C: 48 h. The scale bar is 50 μm. | ||
The electrostatic attraction between the cationic protease-coated Ly-NPs and the anionic ECM may further allow the released metformin to disrupt locally the cells causing their death. It was evident that the clusteroid layer gradually disintegrated over time (Fig. 6). This is also consistent with the paraffin sectioned images of 3D Hep-G2 clusteroids followed by Masson and H&E staining (Fig. 7), where one can see the increased porosity of the clusteroids after 24 h of treatment with Ly-NPs compared with after 12 h of treatment.
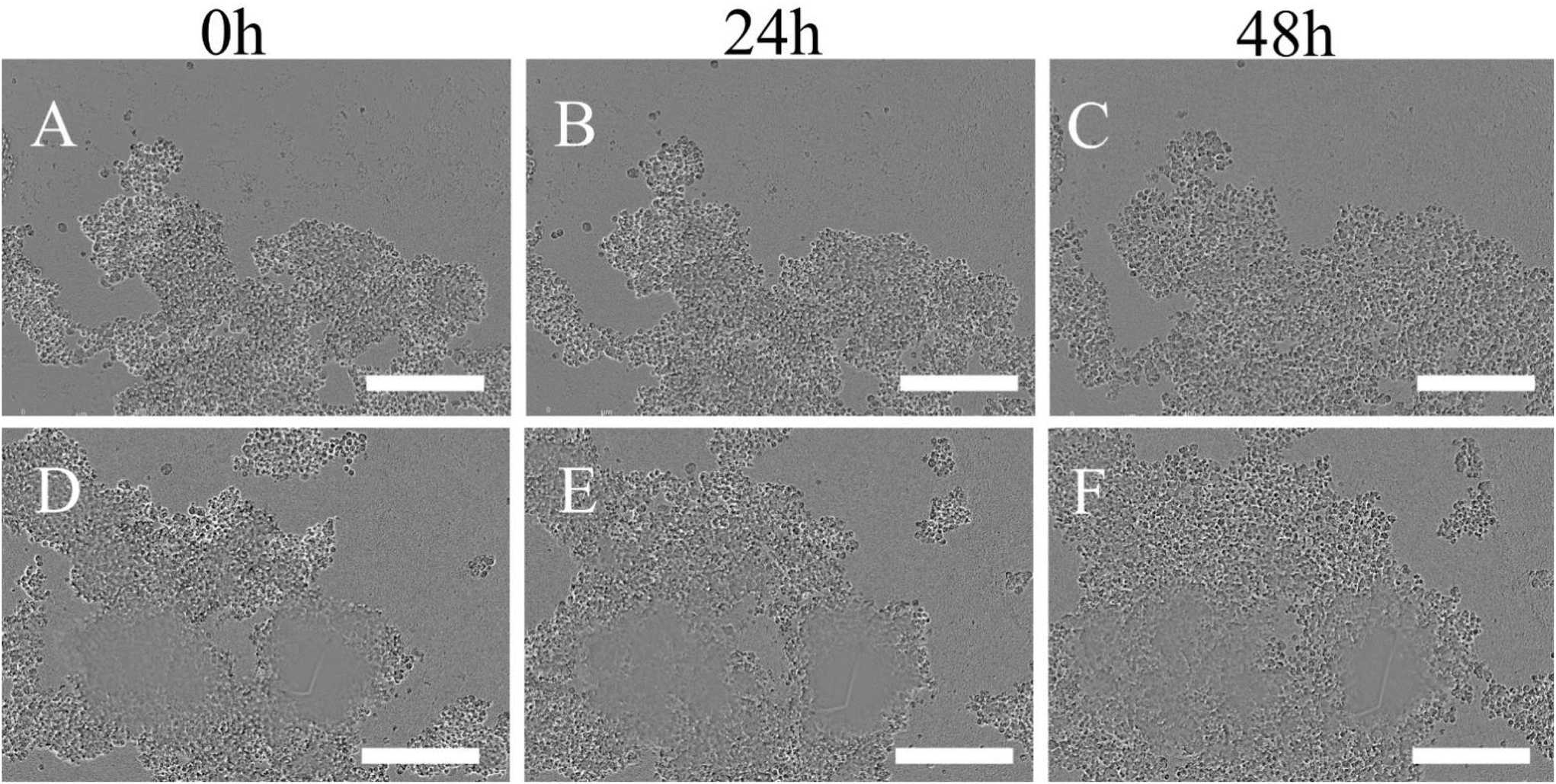 | ||
| Fig. 6 Incucyte live tracking of Hep-G2 clusteroids disassociation using 2× payload Ly-NPs solution at different time points (A–C) and 4× payload Ly-NPs (D–F). The scale bar is 100 μm. | ||
 | ||
| Fig. 7 Paraffin section images of the Hep-G2 cell clusteroids after treatment with 0.2 wt% shellac–0.25 wt% lysozyme–0.4 wt% metformin–0.25 wt% P407 (4× payload of metformin) for (A) 12 h, (B) 12 h, (C) 24 h, (D) 24 h with different dyes. The scale bar is 50 μm. | ||
Confocal observations further substantiated these findings, demonstrating disruption of internal structures such as F-actin and albumin within the clusteroids, accompanied by noticeable misalignment of nuclear distribution (Fig. 8A–D). These research findings validate the occurrence of intracellular vascular structures in cells following exposure to high-payload particles. Interestingly, the inhibitory impact crucial for restraining cancer growth was found to be compromised in the case of low-payload high-concentration particles due to mutual interference within the particle system.
 | ||
| Fig. 8 Confocal laser scanning microscopy images of paraffin-sectioned clusteroids after treatment with 0.2 wt% shellac–0.25 wt% lysozyme–0.4 wt% metformin–0.25 wt% P407 (4× payload of metformin). The scale bar is 50 μm. | ||
4 Conclusions
We have developed a novel 3D cell clusteroid platform for testing the NPs’ anticancer effect. We used the previously reported lysozyme-functionalized shellac nanocarrier as the testing unit. This nanocarrier showed a strong enhancement in anticancer efficiency compared to the non-coated metformin or lysozyme. We utilized these Ly-NPs to assess their anti-cancer properties, and the results were remarkably promising. The Ly-NPs exhibited strong anti-cancer capabilities due to their electrostatic properties. We demonstrated that increasing the payload was more effective than increasing the concentration of nanoparticles. Excessive accumulation of NPs may not necessarily lead to successful delivery of metformin to the core of the clusteroids. This is likely because upon reaching a critical concentration, the Ly-NPs have nowhere else to adsorb to, or because nanoparticles with high payloads can penetrate the interior of the clusteroids by partially digesting the ECM through the lysozyme coating delivering a higher local concentration of metformin. Low-metformin payload NPs may also penetrate the ECM, and even if some enter the clusteroids’ interior, their lower payload limits their anticancer efficacy.This study breaks new ground by delving into two pivotal aspects of metformin nanocarriers in the context of cancer treatment. The novelty of this research lies in its comprehensive approach to investigating the impact of protease surface functionality on the nanocarriers’ ability to effectively dismantle 3D cell cultures, which more closely mimic in vivo tumour conditions compared to conventional 2D monolayer cultures. Furthermore, this study pioneers a thorough comparative evaluation of the anticancer efficacy of nanocarriers with high metformin payload versus high concentrations of nanocarriers with lower metformin content. This innovative comparison seeks to determine the optimal equilibrium between drug loading and nanocarrier concentration, aiming to unleash the full therapeutic potential of metformin in the fight against cancer. By shedding light on these crucial aspects, this study paves the way for the rational design of effective metformin nanocarrier systems that can target and eliminate cancer cells in solid tumours, ultimately contributing to the advancement of cancer nanomedicine. Such active metformin nanocarriers could potentially be administered locally by direct injection into solid tumours. The authors will seek to examine the use of these NPs in vivo in a future study.
Author contributions
Conceptualization, L. A. M. and V. N. P.; methodology, A. W., L. A. M. and V. N. P.; software, A. W.; validation, A. W., L. A. M. and V. N. P.; formal analysis, A. W., L. A. M. and V. N. P.; investigation, A. W.; resources, L. A. M. and V. N. P.; data curation, A. W., L. A. M. and V. N. P.; writing – original draft preparation, A. W.; writing – review and editing, A. W., L. A. M. and V. N. P.; visualization, A. W., L. A. M. and V. N. P.; supervision, L. A. M. and V. N. P.; project administration, L. A. M. and V. N. P.; funding acquisition, L. A. M. and V. N. P. All authors have read and agreed to the published version of the manuscript.Data availability
All data regarding this manuscript are already presented in the graphs of the main paper and the ESI.†Conflicts of interest
No conflicts of interest to declare.Acknowledgements
This research was funded by the Committee of Science of the Ministry of Science and Higher Education of the Republic of Kazakhstan (Grant No. AP19677474). A. W. thanks the Chinese Scholarship Council for the financial support of his Ph.D. studies (CSC No. 201908210332) and HUAFA groups for the funding.References
- J. De Las Rivas, A. Brozovic, S. Izraely, A. Casas-Pais, I. P. Witz and A. Figueroa, Arch. Toxicol., 2021, 95, 2279–2297 CrossRef CAS PubMed; E. Hanna, J. Quick and S. Libutti, Oral Dis., 2009, 15, 8–17 CrossRef PubMed.
- E. Hanna, J. Quick and S. Libutti, Oral Dis., 2009, 15, 8–17 CrossRef CAS PubMed.
- A. Thomas-Schoemann, B. Blanchet, C. Bardin, G. Noé, P. Boudou-Rouquette, M. Vidal and F. Goldwasser, Crit. Rev. Oncol. Hematol., 2014, 89, 179–196 CrossRef PubMed.
- P. L. Bedard, D. M. Hyman, M. S. Davids and L. L. Siu, Lancet, 2020, 395, 1078–1088 CrossRef CAS PubMed.
- G. Awada, H. R. Kourie and A. Awada, Discovery Med., 2015, 20, 33–41 Search PubMed.
- R. K. Jain, Adv. Drug Delivery Rev., 2012, 64, 353–365 CrossRef PubMed.
- C. Wang, Z. Tang, Y. Zhao, R. Yao, L. Li and W. Sun, Biofabrication, 2014, 6, 022001 CrossRef PubMed.
- A. Nyga, U. Cheema and M. Loizidou, J. Cell Commun. Signaling, 2011, 5, 239–248 CrossRef PubMed.
- L. C. Kimlin, G. Casagrande and V. M. Virador, Mol. Carcinog., 2013, 52, 167–182 CrossRef PubMed.
- A. Wang, L. A. Madden and V. N. Paunov, J. Mater. Chem. B, 2020, 8, 10487–10501 RSC.
- A. Albiol, A. Albiol and C. Sánchez de Merás, Sensors, 2021, 21, 2232 CrossRef PubMed.
- J. H. Park, J.-R. Lee, S. Park, Y.-J. Kim, J.-K. Yoon, H. S. Park, J. Hyun, Y. K. Joung, T. I. Lee and S. H. Bhang, Biomater. Res., 2023, 27, 51 CrossRef CAS PubMed.
- H. Zhao, Y. Chen, L. Shao, M. Xie, J. Nie, J. Qiu, P. Zhao, H. Ramezani, J. Fu and H. Ouyang, Small, 2018, 14, 1802630 CrossRef PubMed.
- J.-Z. Wang, Y.-X. Zhu, H.-C. Ma, S.-N. Chen, J.-Y. Chao, W.-D. Ruan, D. Wang, F.-g. Du and Y.-Z. Meng, Mater. Sci. Eng., C, 2016, 62, 215–225 CrossRef CAS PubMed.
- A. Wang, Advanced biomedical applications of cell clusteroids based on aqueous twophase Pickering emulsion systems, Thesis, University of Hull, 2022, https://hull-repository.worktribe.com/output/4249064.
- A. Wang, L. A. Madden and V. N. Paunov, Bioengineering, 2022, 9, 126 CrossRef CAS PubMed.
- A. Wang, L. A. Madden and V. N. Paunov, ACS Appl. Bio Mater., 2022, 5, 1804–1816 CrossRef CAS PubMed.
- A. A. K. Das, B. W. Filby, D. A. Geddes, D. Legrande and V. N. Paunov, Mater. Horiz., 2017, 4, 1196–1200 RSC.
- S. B. Celik, S. R. Dominici, B. W. Filby, A. A. Das, L. A. Madden and V. N. Paunov, Biomimetics, 2019, 4, 50 CrossRef CAS PubMed.
- S. Li, W. Su, H. Wu, T. Yuan, C. Yuan, J. Liu, G. Deng, X. Gao, Z. Chen and Y. Bao, Nat. Biomed. Eng., 2020, 4, 704–716 CrossRef CAS PubMed.
- M. Dobbelstein and U. Moll, Nat. Rev. Drug Discovery, 2014, 13, 179–196 CrossRef CAS PubMed.
- N. Bery, A. Miller and T. Rabbitts, Nat. Commun., 2020, 11, 3233 CrossRef CAS PubMed.
- C. L. Flugel, R. G. Majzner, G. Krenciute, G. Dotti, S. R. Riddell, D. L. Wagner and M. Abou-el-Enein, Nat. Rev. Clin. Oncol., 2023, 20, 49–62 CrossRef CAS PubMed.
- I. Melero, E. Castanon, M. Alvarez, S. Champiat and A. Marabelle, Nat. Rev. Clin. Oncol., 2021, 18, 558–576 CrossRef CAS PubMed.
- M. Kanapathipillai, A. Brock and D. E. Ingber, Adv. Drug Delivery Rev., 2014, 79, 107–118 CrossRef PubMed.
- N. Essa, F. O'Connell, A. Prina-Mello, J. O'Sullivan and S. Marcone, Cancer Lett., 2022, 525, 1–8 CrossRef CAS PubMed.
- M. Li, Y. Zhang, Q. Zhang and J. Li, Mater. Today Bio, 2022, 16, 100364 CrossRef CAS PubMed.
- F. Masood, Mater. Sci. Eng., C, 2016, 60, 569–578 CrossRef CAS PubMed.
- L. Palanikumar, S. Al-Hosani, M. Kalmouni, V. P. Nguyen, L. Ali, R. Pasricha, F. N. Barrera and M. Magzoub, Commun. Biol., 2020, 3, 95 CrossRef CAS PubMed.
- N. Amreddy, A. Babu, R. Muralidharan, J. Panneerselvam, A. Srivastava, R. Ahmed, M. Mehta, A. Munshi and R. Ramesh, Adv. Cancer Res., 2018, 137, 115–170 CrossRef CAS PubMed.
- M. Chatterjee and N. Chanda, Mater. Adv., 2022, 3, 837–858 RSC.
- F. da Silva Feltrin, T. Agner, C. Sayer and L. M. F. Lona, Adv. Colloid Interface Sci., 2022, 300, 102582 CrossRef PubMed.
- B. Yadav, M. Chauhan, S. Shekhar, A. Kumar, A. K. Mehata, A. K. Nayak, R. Dutt, V. Garg, V. Kailashiya and M. S. Muthu, Int. J. Pharm., 2023, 633, 122587 CrossRef CAS PubMed.
- V. Singh and P. Kesharwani, J. Biomater. Sci., Polym. Ed., 2021, 32, 1882–1909 CrossRef CAS PubMed.
- S. K. Dubey, M. Kali, S. Hejmady, R. N. Saha, A. Alexander and P. Kesharwani, Eur. J. Pharm. Sci., 2021, 164, 105890 CrossRef CAS PubMed.
- T. Wei, C. Chen, J. Liu, C. Liu, P. Posocco, X. Liu, Q. Cheng, S. Huo, Z. Liang and M. Fermeglia, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2978–2983 CrossRef CAS PubMed.
- H. Samadian, S. Hosseini-Nami, S. K. Kamrava, H. Ghaznavi and A. Shakeri-Zadeh, J. Cancer Res. Clin. Oncol., 2016, 142, 2217–2229 CrossRef CAS PubMed.
- S. Rajeshkumar, J. Genet. Eng. & Biotechnol., 2016, 14, 195–202 Search PubMed.
- X. Ding, C. Yin, W. Zhang, Y. Sun, Z. Zhang, E. Yang, D. Sun and W. Wang, Nanoscale Res. Lett., 2020, 15, 1–17 CrossRef PubMed.
- J. Wang, J. S. Lee, D. Kim and L. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 39971–39984 CrossRef CAS PubMed.
- R. I. Priyadharshini, G. Prasannaraj, N. Geetha and P. Venkatachalam, Appl. Biochem. Biotechnol., 2014, 174, 2777–2790 CrossRef CAS PubMed.
- G. Bisht and S. Rayamajhi, Nanobiomedicine, 2016, 3, 9 CrossRef PubMed.
- S. E. González, E. Bolaina-Lorenzo, J. Pérez-Trujillo, B. Puente-Urbina, O. Rodríguez-Fernández, A. Fonseca-García and R. Betancourt-Galindo, 3 Biotech, 2021, 11, 1–12 CrossRef PubMed.
- L. Bayón-Cordero, I. Alkorta and L. Arana, Nanomaterials, 2019, 9, 474 CrossRef PubMed.
- A. Jain, A. Agarwal, S. Majumder, N. Lariya, A. Khaya, H. Agrawal, S. Majumdar and G. P. Agrawal, J. Controlled Release, 2010, 148, 359–367 CrossRef CAS PubMed.
- S. Satapathy and C. S. Patro, Adv. Pharm. Bull., 2022, 12, 298 CAS . Solid lipid.
- A. K. Othman, R. El Kurdi, A. Badran, J. Mesmar, E. Baydoun and D. Patra, RSC Adv., 2022, 12, 11282–11292 RSC.
- V. P. Chavda, D. Vihol, B. Mehta, D. Shah, M. Patel, L. K. Vora, M. Pereira-Silva and A. C. Paiva-Santos, Nanomedicine, 2022, 17, 547–568 CrossRef CAS PubMed; H. A. Hussein and M. A. Abdullah, Appl. Nanosci., 2022, 12, 3071–3096 CrossRef.
- H. A. Hussein and M. A. Abdullah, Appl. Nanosci., 2022, 12, 3071–3096 CrossRef CAS.
- F. Rommasi and N. Esfandiari, Nanoscale Res. Lett., 2021, 16, 95 CrossRef CAS PubMed.
- D. Iannazzo, I. Ziccarelli and A. Pistone, J. Mater. Chem. B, 2017, 5, 6471–6489 RSC.
- M.-X. Zhao and B.-J. Zhu, Nanoscale Res. Lett., 2016, 11, 1–9 CrossRef PubMed.
- M. Alavi, T. J. Webster and L. Li, Micro Nano Bio Asp, 2022, 1, 1–11 Search PubMed.
- C. Wang, C. Wu, X. Zhou, T. Han, X. Xin, J. Wu, J. Zhang and S. Guo, Sci. Rep., 2013, 3, 2852 CrossRef PubMed.
- W. Wu, R. Li, X. Bian, Z. Zhu, D. Ding, X. Li, Z. Jia, X. Jiang and Y. Hu, ACS Nano, 2009, 3, 2740–2750 CrossRef CAS PubMed.
- Y. Wang, A. Santos, G. Kaur, A. Evdokiou and D. Losic, Biomaterials, 2014, 35, 5517–5526 CrossRef CAS PubMed.
- M. Aljofan and D. Riethmacher, Future Sci. OA, 2019, 5, FSO410 CrossRef CAS PubMed.
- M. Kheirandish, H. Mahboobi, M. Yazdanparast, W. Kamal and M. A. Kamal, Curr. Drug Metab., 2018, 19, 793–797 CrossRef CAS PubMed.
- A. Vancura, P. Bu, M. Bhagwat, J. Zeng and I. Vancurova, Trends Pharmacol. Sci., 2018, 39, 867–878 CrossRef CAS PubMed.
- I. Pernicova and M. Korbonits, Nat. Rev. Endocrinol., 2014, 10, 143–156 CrossRef CAS PubMed.
- S. Thakur, B. Daley and J. Klubo-Gwiezdzinska, J. Mol. Endocrinol., 2019, 63, R17–R35 CAS.
- D.-S. Kim, S.-K. Jeong, H.-R. Kim, D.-S. Kim, S.-W. Chae and H.-J. Chae, Immunopharmacol. Immunotoxicol., 2010, 32, 251–257 CrossRef CAS PubMed.
- Y. Sun, C. Tao, X. Huang, H. He, H. Shi, Q. Zhang and H. Wu, OncoTargets Ther., 2016, 8, 2845–2853 Search PubMed.
- E. O. Asare, E. A. Mun, E. Marsili and V. N. Paunov, J. Mater. Chem. B, 2022, 10, 5129–5153 RSC.
- P. J. Weldrick, A. Wang, A. F. Halbus and V. N. Paunov, Nanoscale, 2022, 14, 4018–4041 RSC.
- P. J. Weldrick, S. San and V. N. Paunov, ACS Appl. Nano Mater., 2021, 4, 1187–1201 CrossRef CAS; E. O. Asare, A. Seidakhanova, D. Amangeldinova, E. Marsili and V. N. Paunov, ACS Appl. Nano Mater., 2023, 6, 22792–22806 CrossRef.
- E. O. Asare, A. Seidakhanova, D. Amangeldinova, E. Marsili and V. N. Paunov, ACS Appl. Nano Mater., 2023, 6, 22792–22806 CrossRef CAS.
- M. J. Al-Awady, A. Fauchet, G. M. Greenway and V. N. Paunov, J. Mater. Chem. B, 2017, 5, 7885–7897 RSC.
- M. J. Al-Awady, P. J. Weldrick, M. J. Hardman, G. M. Greenway and V. N. Paunov, Mater. Chem. Front., 2018, 2, 2032–2044 RSC.
- P. J. Weldrick, S. Iveson, M. J. Hardman and V. N. Paunov, Nanoscale, 2019, 11, 10472–10485 RSC.
- P. J. Weldrick, M. J. Hardman and V. N. Paunov, Mater. Chem. Front., 2021, 5, 961–972 RSC.
- S. S. M. Al-Obaidy, G. M. Greenway and V. N. Paunov, Nanoscale Adv., 2019, 1, 858–872 RSC.
- S. S. M. Al-Obaidy, A. F. Halbus, G. M. Greenway and V. N. Paunov, J. Mater. Chem. B, 2019, 7, 3119–3133 RSC.
- S. S. M Al-Obaidy, G. M. Greenway and V. N. Paunov, Pharmaceutics, 2021, 13, 1389 CrossRef CAS PubMed.
- B. W. Filby, P. J. Weldrick and V. N. Paunov, ACS Appl. Bio Mater., 2022, 5, 3826–3840 CrossRef CAS PubMed.
- P. J. Weldrick, M. J. Hardman and V. N. Paunov, Adv. NanoBiomed Res., 2021, 1, 2000027 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (i) Fig. S1: Schematic for the preparation of lysozyme coated metformin-loaded shellac NPs; (ii) Fig. S2: Zeta-potential distribution of 0.2 wt% shellac–0.1 wt% metformin–0.25 wt% P407 coated at different concentrations of lysozyme; (iii) Fig. S3: Zeta-potential of 0.2 wt% shellac–0.1 wt% metformin–0.25 wt% P407 coated at different concentrations of lysozyme; (iv) Fig. S4: SEM images of 0.2 wt% shellac–0.25 wt% lysozyme–0.2 wt% metformin–0.25 wt% P407 NPs at different magnifications; (v) Estimate of the metformin loading per shellac nanoparticle. See DOI: https://doi.org/10.1039/d4bm00692e |
| This journal is © The Royal Society of Chemistry 2024 |