
Pristine gelatin incorporation as a strategy to enhance the biofunctionality of poly(vinyl alcohol)-based hydrogels for tissue engineering applications†
Alessia
Longoni
*a,
Gretel S.
Major
a,
Shaoyuan
Jiang
b,
Brooke L.
Farrugia
 c,
David C.
Kieser
a,
Tim B. F.
Woodfield
a,
Jelena
Rnjak-Kovacina
b and
Khoon S.
Lim
*ad
c,
David C.
Kieser
a,
Tim B. F.
Woodfield
a,
Jelena
Rnjak-Kovacina
b and
Khoon S.
Lim
*ad
aDepartment of Orthopaedic Surgery and Musculoskeletal Medicine, University of Otago Christchurch, New Zealand. E-mail: A Longoni alessia.longoni@otago.ac.nz; KS Lim khoon.lim@sydney.edu.au
bGraduate School of Biomedical Engineering, UNSW Sydney, Sydney 2052, Australia
cSchool of Biomedical Engineering, University of Melbourne, Australia
dLight-Activated Biomaterials Group, School of Medical Sciences, University of Sydney, Australia
First published on 31st October 2023
Abstract
Synthetic polymers, such as poly(vinyl alcohol) (PVA), are popular biomaterials for the fabrication of hydrogels for tissue engineering and regenerative medicine (TERM) applications, as they provide excellent control over the physico-chemical properties of the hydrogel. However, their bioinert nature is known to limit cell-biomaterial interactions by hindering cell infiltration, blood vessel recruitment and potentially limiting their integration with the host tissue. Efforts in the field have therefore focused on increasing the biofunctionality of synthetic hydrogels, without limiting the advantages associated with their tailorability and controlled release capacity. The aim of this study was to investigate the suitability of pristine gelatin to enhance the biofunctionality of tyraminated PVA (PVA-Tyr) hydrogels, by promoting cell infiltration and host blood vessel recruitment for TERM applications. Pure PVA-Tyr hydrogels and PVA-Tyr hydrogels incorporated with vascular endothelial growth factor (VEGF), a well-known pro-angiogenic stimulus, were used for comparison. Incorporating increasing concentrations of VEGF (0.01–10 μg mL−1) or gelatin (0.01–5 wt%) did not influence the physical properties of PVA-Tyr hydrogels. However, their presence within the polymer network (>0.1 μg mL−1 VEGF and >0.1 wt% gelatin) promoted endothelial cell interactions with the hydrogels. The covalent binding of unmodified gelatin or VEGF to the PVA-Tyr network did not hamper their inherent bioactivity, as they both promoted angiogenesis in a chick chorioallantoic membrane (CAM) assay, performing comparably with the unbound VEGF control. When the PVA-Tyr hydrogels were implanted subcutaneously in mice, it was observed that cell infiltration into the hydrogels was possible in the absence of gelatin or VEGF at 1- or 3-weeks post-implantation, highlighting a clear difference between in vitro an in vivo cell-biomaterial interaction. Nevertheless, the presence of gelatin or VEGF was necessary to enhance blood vessel recruitment and infiltration, although no significant difference was observed between these two biological molecules. Overall, this study highlights the potential of gelatin as a standalone pro-angiogenic cue to enhance biofunctionality of synthetic hydrogels and provides promise for their use in a variety of TERM applications.
1. Introduction
Hydrogels are highly-hydrated, three-dimensional (3D) networks of cross-linked polymers, which are currently used in a wide range of tissue engineering and regenerative medicine (TERM) applications, including as cell-instructive scaffolds, bioinks, drug delivery platforms and for functional grafting purposes.1 While different polymers can be used as the base biomaterial for hydrogel fabrication, synthetic polymers, like poly(vinyl alcohol) (PVA) or poly(ethylene glycol) (PEG) offer several advantages.1,2 Compared to natural polymers, synthetic polymers allow for greater control over the physical and chemical properties of the hydrogel, including stiffness, permeability, availability of functional groups and degradation rate, which can all be tailored for a specific application.2 Given their synthetic origin, material synthesis can be tightly controlled resulting in enhanced batch-to-batch consistency, higher reproducibility and greater uniformity in the 3D structure compared to natural polymers-derived hydrogels.2 Despite the enhanced control and reproducibility offered by synthetic polymers, their applications are limited by their bioinert nature,2,3 given that the polymer backbone lacks moieties for the interaction with host cells. As a consequence, cellular infiltration and integration of synthetic hydrogels with the surrounding tissue is reduced, potentially hindering their therapeutic effect.2,4,5 This is particularly challenging when aiming at TERM applications, where timely blood vessel ingrowth within engineered hydrogels is essential for the survival of the colonizing cells and for the regeneration of the target tissue.4–8Several strategies have been used to promote cellular and vascular ingrowth within synthetic hydrogels. For instance, the introduction of microporous structures within synthetic dextran vinyl sulfone hydrogels enhanced the formation of lumenized sprouts and cell infiltration in vivo.6 Another effective approach to enhance host cell infiltration and interactions is to increase the biofunctionality of synthetic polymers by incorporating native extracellular matrix (ECM) components (e.g., collagen, fibrinogen, fibronectin and laminins) or their specific bioactive peptide sequences (e.g., arginine-glycine-aspartic acid – RGD – motif) within the network.7,9–12 A particularly promising bioactive macromolecule is gelatin, which is a bioactive natural polymer derived from the hydrolysis of collagen. Gelatin retains cell adhesion motifs (RGD), which are typical of collagen, promoting the attachment of different cell types and cell migration.13–18 An advantage of gelatin over its collagen precursor is the low immunogenicity of both the natural polymer and its degradation products, which reduces the risk of an immune reaction upon implantation.13 Additionally, because of the harsh acidic or basic denaturation processes involved in its preparation, the risks of pathogen transmittance, which are a concern for natural polymers, are also reduced.14 Finally, gelatin is also attractive from a translational and economic perspective given that it is already considered safe by the Food and Drug administration, possesses a wide track record for use in the food/pharmaceutical industry and, compared to other biomaterials, it is relatively inexpensive.14
To ensure the survival of infiltrating cells and promote tissue maturation, recruitment of host vasculature within engineered constructs of clinically relevant-size is crucial.4–8 To guide blood vessel infiltration, a popular method has been to functionalize synthetic hydrogels with pro-angiogenic growth factors (GFs).8,19,20 Vascular endothelial growth factor (VEGF) is a widely exploited pro-angiogenic GF for tissue engineering (TE) applications due to its role in driving the early stages of angiogenesis, regulating the loosening of endothelial cell junctions and promoting endothelial cell survival.21,22 Nevertheless, the main drawback of VEGF is its short half-life (±50 minutes).23,24 Thus, to achieve the desired biological effect, supra-physiological doses of VEGF need to be administered. This has shown to lead to rapid and dysregulated angiogenesis, which negatively impacts new vessel quality and function.25–28 High doses of VEGF can also induce severe side effects including undesired vascularization at non-target sites (i.e. in tumours), angioma-like vascular tumours in muscle, heart and other tissues, hypotension and oedema.25,29,30 To remain within a safe therapeutic window, strategies have been implemented to control the release of VEGF from hydrogels through covalent binding or using affinity-based methods.19–21,31 Nevertheless, the covalent incorporation of GFs like VEGF requires their chemical modification, potentially leading to a reduction of their biological activity.21,32,33 On the other hand, methods that rely on bioaffinity require the presence of additional specific molecules, such as heparin or fibrin/fibrinogen, to form binding complexes with the GFs of interest and control their release.34 However, different GFs isoforms (e.g. VEGF 120, 165 or 188) can present different binding affinities.35 Furthermore, ECM molecules binding with a specific molecules can occur.35,36 While the optimal strategy for the delivery of therapeutic levels of VEGF is still under investigation, efforts have focussed on the identification of safer alternatives, which include the incorporation of other pro-angiogenic cues for TERM applications. It is known that gelatin promotes endothelial cell attachment in 2D cell culture,37 however only a limited number of studies have investigated the potential role of pristine gelatin as a stand-alone pro-angiogenic cue.38–40 These studies suggest a positive influence on endothelial cell survival, proliferation, and migration in vitro and angiogenesis in vivo.38–40 Nevertheless, the biological implications of gelatin incorporation on vascularization in synthetic hydrogels remain underexplored.
Therefore, the aim of this study was to evaluate the suitability of gelatin to enhance biofunctionality of synthetic hydrogels in terms of both cellular infiltration and host vessel recruitment for TE applications. To achieve this goal, we fabricated a synthetic hydrogel network by grafting tyramines onto the PVA backbone, as previously described by our group.41 Hydrogel crosslinking occurred in the presence of ruthenium (Ru) and sodium persulfate (SPS) photoinitiators and photopolymerization was triggered by exposure to visible light.17,41,42 More specifically, pristine gelatin was covalently incorporated in the polymer network through bi-phenol bond formation between native tyrosine moieties and the tyramine groups grafted onto the PVA (PVA-Tyr GT). In order to investigate the pro-angiogenic potential of gelatin, pristine VEGF (also containing native tyrosine moieties) was covalently crosslinked into PVA-Tyr hydrogels in an identical fashion and included in the study as comparison (PVA-Tyr VEGF). In order to evaluate the tailorability of the hydrogel system, we systematically investigated the impact of gelatin and VEGF concentrations on the physical properties of the hydrogel. The effect of hydrogel biofunctionalization on the interaction with endothelial cells was then investigated in vitro. Finally, angiogenesis induced by the delivered bioactive molecules was compared using two in vivo models: the chorioallantoic membrane (CAM) angiogenesis model in fertilized chicken eggs and a subcutaneous implantation model in mice. A summary of the experimental groups and set up in included in Fig. 1.
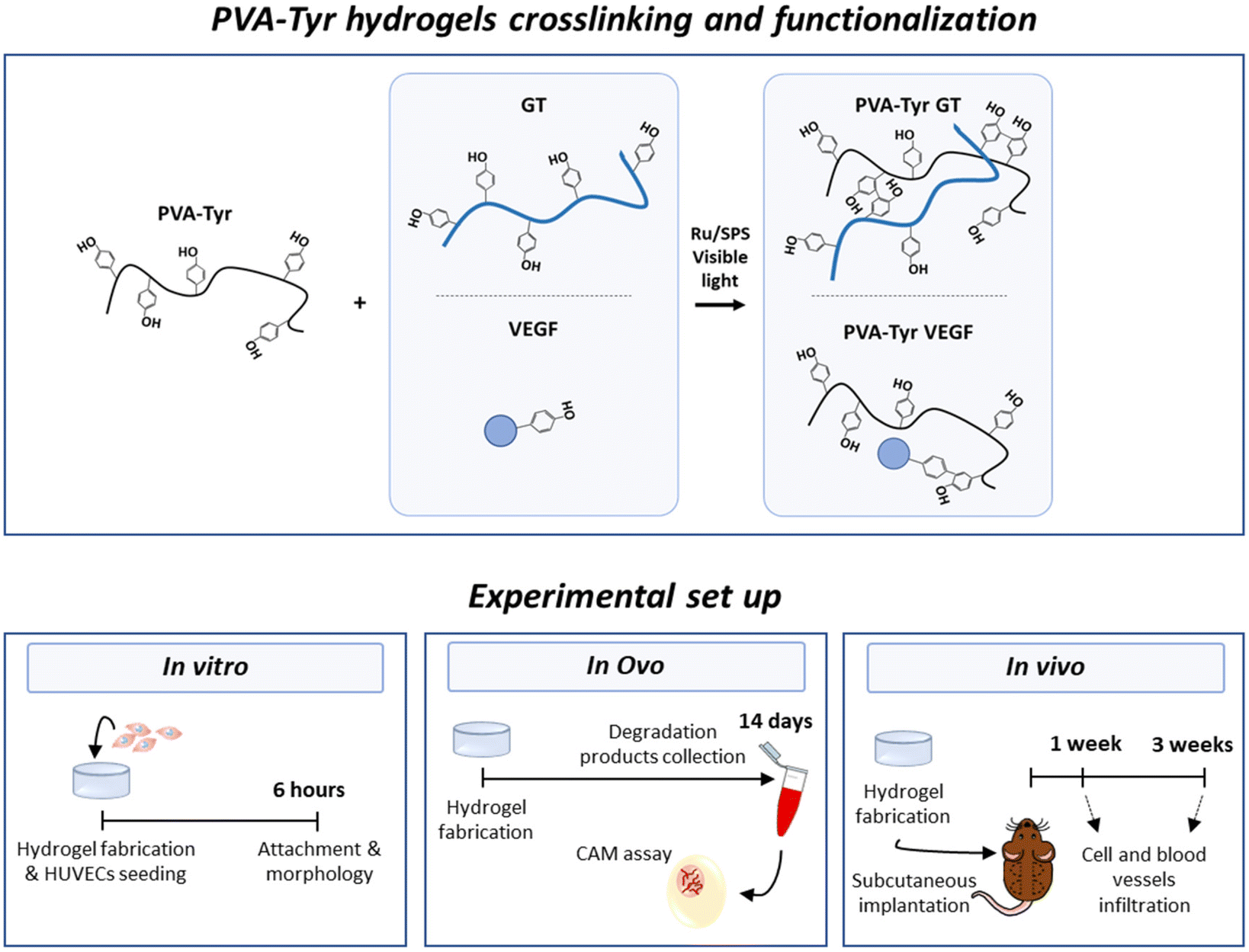 | ||
| Fig. 1 Schematic outline of the crosslinking chemistry and the experimental set up. | ||
2. Materials and methods
2.1 Materials
Type-B gelatin from porcine skin (bloom strength 225 g), PVA (13–23 kDa, 98% hydrolyzed), dimethyl sulfoxide, succinic anhydride (SA), triethylamine (TEA), 1,3-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), tyramine (Tyr), sodium persulfate (SPS), 1,1-carbonyldiimidazole, tris(2,2-bipyridyl)dichlororuthenium(II) hexahydrate (Ru), phosphate buffered saline (PBS), dialysis (10 kDa molecular weight cutoff), phalloidin–tetramethylrhodamine B isothiocyanate, were purchased from Sigma-Aldrich and used as received. Rh-VEGF-165 (VEGF) was purchased from Gibco (PHC9394). Human umbilical vein endothelial cells (HUVECs), vascular Basal Media, endothelial Cell Growth Kit-VEGF, trypsin for primary cells, and trypsin neutralizing solution were purchased from ATCC. Trichrome Stain Kit, CD31 antibody and Goat Anti-Rabbit IgG H&L (HRP) were purchased from Abcam. Vimentin antibody was purchased from Cell Signalling Technology. Pierce BCA protein assay kit was purchase from ThermoFisher Scientific and VEGF ELISA and the ancillary kit were bought from R&D system.2.2 Synthesis of PVA-Tyr
PVA-Tyr was synthesised following a two-step reaction as previously described.17,41 Briefly, to achieve a carboxylation of 2% (7 carboxyl groups per PVA chain), 1 g of PVA was dissolved in 5.5 ml of dry dimethyl sulfoxide (DMSO) under a nitrogen atmosphere at 60 °C. SA (45 mg) and TEA (61 μl) were added to the reaction mixture, and stirred at 60 °C for 24 h. The resultant PVA-COOH was purified by dialysis against water. Following freeze-drying, Tyr moieties were conjugated to the carboxylated PVA (PVA-COOH) using a carbodiimide-coupling reaction, aiming to achieve a tyramination of 2% (7 tyramine groups per PVA chain). Afterwards, 1 g of PVA-COOH was dissolved in 6 ml of DMSO, 0.271 g of DCC, 0.151 g of NHS and 0.121 g of tyramine were added to the reaction. The resulting PVA-Tyr solution was further purified by dialysis (10 kDa molecular weight cut-off) against water and freeze-dried. The resulting amounts of carboxyl groups in PVA-COOH and tyramine groups in PVA-Tyr were quantified using 1H nuclear magnetic resonance (NMR).2.3 Hydrogel fabrication
Dried PVA-Tyr was dissolved in PBS at 56 °C. Upon complete dissolution, mixed with Ru and SPS stock solutions to achieve a final concentration of 5 wt% PVA-Tyr and 0.5/5 mM Ru/SPS. Gelatin (0.01–5 wt%) or growth factor stock solutions (0.01–10 μg mL−1) were mixed with the PVA-Tyr solution prior to the addition of Ru and SPS, after allowing the PVA-Tyr solution to reach a maximum of 37 °C. All samples were photo-crosslinked using visible light (OmniCure® S1500, Excelitas Technologies with a Rosco IR/UV filter 400–450 nm, 3 min, 30 mW cm−2) in an open environment. For in vitro and in ovo studies, cylindrical moulds (h = 1 mm, ∅ = 6 mm) were used, whereas for subcutaneous implantation studies a rectangular mould was used (w = 7.5 mm, l = 7.5 mm, h = 1 mm).2.4 Swelling and mass loss analysis
After crosslinking, each hydrogel (30 μL) was weighed (minitial,t0). For each hydrogel composition, three samples were lyophilised immediately after their preparation to evaluate their initial dry weights (mdry,t0) and the actual macromer weight fraction. The actual macromer weight fraction was defined as the ratio between the initial dry weight and the initial weight (eqn (1)). | (1) |
The initial dry weight of the remaining samples was determined using the actual macromer fraction of eqn (1). and the individual initial weight of each hydrogel. To determine the mass loss, samples were allowed to swell in PBS at 37 °C. Swollen hydrogels were collected and the wet weight was recorded (mswollen). Following lyophilization, the freeze-dried weight (mdry) was used to calculate the mass loss and mass swelling ratio (q) according to eqn (2)–(4):
| mdry,t0 = minitial × actual macromer fraction | (2) |
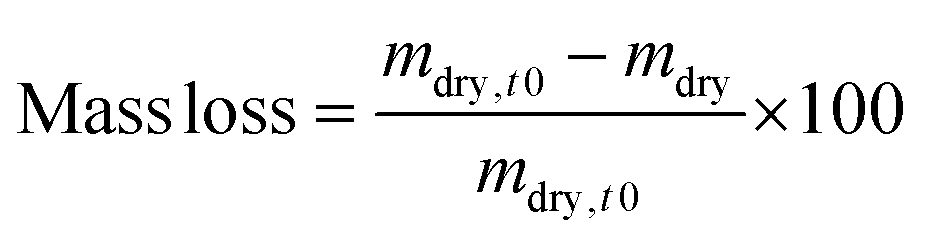 | (3) |
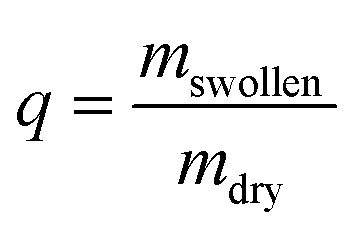 | (4) |
The sol fraction was defined as the percent of macromers that were not cross-linked into the hydrogel network, and it was determined as the mass loss after equilibrium swelling (after 24 h).
2.5 Cell attachment and morphology analysis
Hydrogels with increasing concentration of gelatin and VEGF were prepared as described in 2.3 and transferred to a 96 well-plate. HUVECs were cultured in vascular Basal Media supplemented with endothelial Cell Growth Kit-VEGF (ATCC). Upon reaching passage 3 to 4, cells were trypsinized and seeded on the hydrogels at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per hydrogel. After 6 h, hydrogels were washed two times with PBS and fixed using 4% w/v formaldehyde for 1 h at room temperature. Samples were permeabilized with 0.25% Triton-X for 30 min. To visualize actin filaments, samples were stained with phalloidin–tetramethylrhodamine B isothiocyanate (TRITC 1:250) for 1 h at room temperature. After washing twice with PBS, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI 1:1000) for 15 min at room temperature. Constructs were visualized with Zeiss Axioimager Z1 fluorescence microscope. Four images per samples (four samples per condition) were used to quantify cell attachment. Cell circularity was defined with ImageJ using the equation below:
000 cells per hydrogel. After 6 h, hydrogels were washed two times with PBS and fixed using 4% w/v formaldehyde for 1 h at room temperature. Samples were permeabilized with 0.25% Triton-X for 30 min. To visualize actin filaments, samples were stained with phalloidin–tetramethylrhodamine B isothiocyanate (TRITC 1:250) for 1 h at room temperature. After washing twice with PBS, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI 1:1000) for 15 min at room temperature. Constructs were visualized with Zeiss Axioimager Z1 fluorescence microscope. Four images per samples (four samples per condition) were used to quantify cell attachment. Cell circularity was defined with ImageJ using the equation below: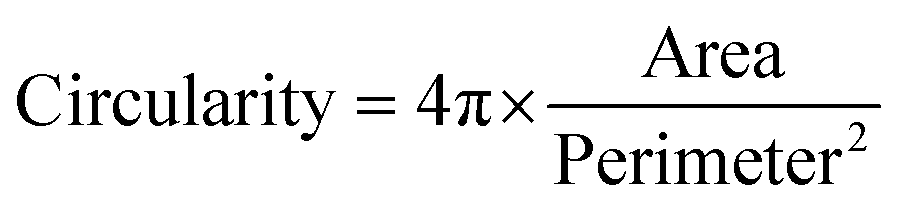 | (5) |
2.6 Release profile of bioactive molecules
Samples incorporating 1 wt% gelatin or 100 ng mL−1 VEGF were prepared as described in 2.3. Retention of the bioactive molecules was calculated as the percentage of the gelatin or VEGF retained in the hydrogel 24 h after preparation, following incubation in PBS at 37 °C. PBS (0.5 mL) was collected and refreshed at each timepoint until all samples had fully degraded. The cumulative release was defined as the sum of the released macromolecules at each time point, relative to the release achieved after complete degradation of the samples (100% release). Gelatin release was assessed using the Pierce BCA protein assay kit (ThermoFisher Scientific) according to manufacturer's instructions. Known concentrations of gelatin were used to create a standard curve. Absorbance was measured at 562 nm. VEGF release was assessed by ELISA (R&D Systems), according to manufacturer's instructions. Known concentrations of VEGF were used to create a standard curve. Absorbance was measured at 450 nm (correction at 570 nm). Supernatant collected from plain PVA-Tyr hydrogels was used as a control for both assays.2.7 Chorioallantoic membrane (CAM) assay
CAM assay experimental protocols were approved by the University of New South Wales Animal Care and Ethics Committee (ACEC 18/16A). All surgical procedures were performed under aseptic conditions as previously described.43 Fertilized eggs were cleaned on embryonic day 1 (E1) with 80% ethanol and incubated in a poultry incubator (Multiquip, Australia) at 37.5 °C with 40–50% humidity. The eggs were rotated every 5 h for the first 3 days. On E4, 3–4 mL of albumen was aspirated with a 19G needle, to detach the CAM from the eggshell and create an air sac directly over the CAM. A 1 cm2 circular window was opened on the eggshell above the air sac using scissors and sealed with Aquafilm transparent dressing (Livingstone Laboratory Supplies, Australia) to prevent dehydration and possible infections. At E8, the window was reopened and a single 9 mm inner diameter silicone ring was placed on top of the CAM in each egg. 100 μL of each sample (n = 10 per condition) was pipetted into the silicone ring and the window was resealed prior to eggs being returned to the incubator. Samples tested included PBS (negative control), PVA-Tyr, PVA-Tyr supplemented with 1 wt% gelatin, PVA-Tyr supplemented with VEGF (final concentration 100 ng mL−1) and the same concentration of unbound VEGF as positive control. Hydrogels were degraded in a total of 1 mL sterile PBS at 37 °C. The degradation products were collected at multiple timepoints as describes in section 2.6, pooled together and the total degradation product was used for the assay. At E12, the CAM membrane was excised and the area inside the silicone ring was imaged using a dissecting microscope at 2.5× magnification (Leica M80, Germany). Blood vessel density was quantified from each captured image using an image processing method implemented in the Matlab language (R2017b, Mathworks, USA). Briefly, raw images were decomposed into the red, green, and blue channels. The red channel was zeroed, and contrast stretching was performed on the green and blue channels individually such that the bottom 1% and the top 1% of all pixel values were mapped to between 0 and 1, respectively. The resulting image was converted to grayscale by eliminating the hue and saturation information while retaining the luminance. Finally, a median filter (3 × 3 pixel kernel) was applied to the resulting image, followed by contrast stretching, resulting in the pre-processed image. A minimum (Smin) and maximum (Smax) feature size in pixels were input by the user. For each feature size between Smin and Smax, in steps of two pixels, morphological bottom-hat filtering was applied to the pre-processed image with a disk-shaped structuring element and a radius equal to the feature size. A threshold, determined using the Otsu method,44 was applied to the resulting image, producing a logical image where the pixel value at each location is true if that pixel belonged to a feature that is the same size as the current feature size, or false otherwise. Finally, a logical OR operation was applied to the logical images for each feature size to generate the final logical image in which a pixel with a value of true represents the positive identification of a blood vessel. The number of true pixels in the final logical image represents the extent of vascularization and is presented as fold-change relative to the negative control (PBS). The number of blood vessel branch points was manually counted using ImageJ.2.8 Subcutaneous implantation
Animal experiments were performed with the approval of the University of Otago Animal Ethics Committee (AUP 21–24 and AUP 21–152) and in accordance with the Animal Welfare Act and the ARRIVE guidelines.45 10–12 weeks old female Balb/c mice were housed in groups (3–4 mice per cage) in individually ventilated cages at the Christchurch Animal Research Area (CARA) of the University of Otago. Animals received standard food pellets and water ad libitum, under climate-controlled conditions (≈22 °C; 12 hours light/12 hours darkness). Prior to surgery, mice received a dose of Temgesic subcutaneously (0.1 mg per kg body weight) and a local injection of lidocaine (4 mg per kg body weight). Subcutaneous pockets were created aseptically under general anaesthesia (2–3.5% isoflurane in oxygen) from 8 mm dorsal incisions and blunt dissection as previously described.46 In each pocket, one casted hydrogel (w = 7.5 mm, l = 7.5 mm, h = 1 mm) composed of 5 wt% PVA-Tyr with or without 1 wt% gelatin or 10 μg mL−1 VEGF and 0.5/5 mM Ru/SPS was implanted. A maximum of 4 pockets were created per mouse. The skin was closed with Mono Q resorbable 5–0 sutures. As pain relief treatment, mice received one additional Temgesic injection subcutaneously (0.1 mg per kg bodyweight) 4–6 h post-surgery and a single subcutaneous injection of Carprofen (5 mg per kg of body weight) for the following 2 days. Mice were euthanized with an intraperitoneal overdose of barbiturates (phenobarbital; 100 mg per kg body weight) 1 or 3 weeks post-surgery (n = 7 for each timepoint). Implants were retrieved and fixed in 4% w/v formaldehyde.To visualize perfusable blood vessels in the euthanized mice a lateral incision was made through the integument and abdominal wall, and the diaphragm was cut along the entire length of the rib cage to expose the pleural cavity. The rib cage was cut, the sternum was lifted away to expose the heart, and a cardiac puncture was performed as previously described.47 Specifically, a 23G catheter was inserted into the left ventricle, while an incision was made in the right atrium to create an outlet for blood (exsanguination) and the solutions were perfused in the circulatory system. Heparinised PBS (20 U mL−1), followed by 10% neutral buffered formalin, was perfused using a peristaltic pump at 5 mL min−1 for 2 min. After fixation, 5 mL of radiopaque contrast agent (Microfill MV-122, Flow Tech) was prepared and perfused as per manufacturer instructions, and the compound was polymerised O/N at 4 °C. Following Microfill polymerisation, grafts were explanted and post-fixed in 10% neutral buffered formalin for 24 h. To measure the volume of perfused vessels within each graft, the surrounding tissue was removed and the implants were scanned individually using Skyscan 1172 micro-CT (Bruker, Kontich, Belgium) (n = 2 per group). Hydrogel volumes were manually drawn and segmented with a global threshold. The vessel volume was measured using image processing software 3D slicer and expressed as a percentage of the total hydrogel volume. Three-dimensional reconstructions of the grafts with blood vessels were based on the micro-CT data and created using Meshlab.
2.9 Histological immunohistochemical analysis
Tissue structure, integration and remodelling was investigated via histological analysis. Samples were sequentially rinsed in PBS with 0.3 M glycine, PBS and embedded in a tissue freeze medium (Leica Biosystems, Germany). Tissue was cryosectioned to 10 μm using a Leica Biosystems cryostat microtome (CM1860). Sections were stained with H&E (Richard-Allen Scientific) and Masson's Trichrome following manufacturer's instructions (Abcam, Connective Tissue Stain, ab150686). For CD31 (ab281583) and vimentin (5741S) staining, endogenous peroxidase activity was blocked by incubating the samples in 0.3% H2O2 for 10 min. Non-specific blocking was performed incubating the samples with 5% BSA-PBS for 30 min at room temperature. Primary antibodies were incubated O/N at 4 °C, with a final dilution of 1:2000 and 1:200 for the CD31 and vimentin antibody, respectively. Samples were then incubated with an HRP-conjugated secondary antibody (ab205718) diluted in 5% BSA for 1 h at room temperature (1:5000). Isotypes were used as negative controls at concentrations matching with those of the primary antibodies (Fig. S1†). Slides were visualised by DAB oxidation and nuclei were counterstained with haematoxylin. Slides were washed, dehydrated in graded ethanol (70–100% EtOH) and mounted in DPX. Slides were imaged at 20× using an Aperio CS2 Slide scanner.
Cell infiltration was evaluated in the entire graft at two different depths, approximately 250 μm apart. Two sections of the whole constructs were used per depth for each sample, thus cell infiltration was evaluated on a total of 28 sections per group (total cell infiltration). Additionally, cell infiltration in the inner part of the construct was evaluated by excluding the cells present in the first 150 μm of the hydrogel closer to the border (cell infiltration in the center). Cell numbers were calculate using QuPath48 and normalized to the construct surface area (mm2) to compare cell densities.
CD31 positive staining was quantified using Adobe Photoshop C6. A range of pixels defining the positive DAB staining was selected and applied to all the samples. The number of pixels for each area was quantified via the function “recording measurement” and expressed as a percentage of the ROI. For the samples analysed 1 week post-implantation, the ROI was represented by an area of 50 μm surrounding the implanted constructs (i.e. recruited vessels). For the samples analysed 3 weeks post-implantation, the ROI was the entire hydrogel construct. Vessels size was defined by manually tracing out the perimeter of all the blood vessels (determined by the presence of an obvious lumen, red blood cells and CD31 positive staining) present in each image using ImageJ.
2.10 Statistical analysis
All in vitro experiments were performed in triplicate and repeated three times to account for experiment variability. Data are expressed as mean ± standard error of the mean. Using GraphPad Prism7 (GraphPad Prism 7, San Diego, CA, USA), groups of data were compared by one-way ANOVA with Tukey post-hoc test, when normality assumption was met, or by Kruskal–Wallis test followed by a Dunn's post hoc test, when normality assumption was not met. For the frequency of vessels infiltrating in the hydrogels, the area under the curve of samples between groups was compared. The presence of outliers was checked (ROUT method, Q = 1%). Differences were considered to be statistically significant at p < 0.05.3. Results and discussion
3.1 Characterization of gelatin and VEGF influence on PVA-Tyr hydrogels properties
While biofunctionalizing synthetic hydrogels may provide enhanced cellular interactions, the presence of biological molecules can introduce a discrepancy in macromer molecular weight distribution, structure and charge, which can negatively impact the crosslinking reaction. To assess whether covalent incorporation of gelatin and VEGF into PVA-Tyr hydrogels altered the resulting physico-chemical properties, the effect of incorporating a range of gelatin (0.01–5 wt%) and VEGF (0.01–10 μg mL−1) concentrations into PVA-Tyr hydrogels was systematically investigated. No statistical differences were observed in swelling ratio and sol fraction with ≤1 wt% gelatin (Fig. 2A and B). When 5 wt% gelatin was incorporated, significant changes in hydrogel physical properties were observed, with swelling ratio increasing from 18.1 ± 0.49 (unmodified PVA-Tyr) to 29.94 ± 1.1 and the sol fraction increasing from 22% ± 2.22 to 40.46% ± 2.1, respectively. Furthermore, the degradation profile was greatly affected, as the hydrogel containing 5 wt% of gelatin was fully degraded within 3 days, a significantly shorter timeframe compared to the 14 ± 2 days required for all the other groups (Fig. 2C). These results are in-line with those previously obtained by Lim et al., where the incorporation of <1 wt% gelatin did not affect the physico-chemical properties of 20 wt% PVA-Tyr hydrogels crosslinked with the same Ru/SPS photoinitiator system.17 While for the current study a complete degradation of the hydrogel is not desirable, as it will result in the burst release of the bioactive molecule, these results highlight the potential of exploiting gelatin incorporation as a tool to tailor PVA-Tyr network properties. Specifically, combining different PVA macromer, photoinitiators and gelatin concentrations allowed fine tuning of the hydrogel physical characteristics, which could be adapted according to the required application.17,41,42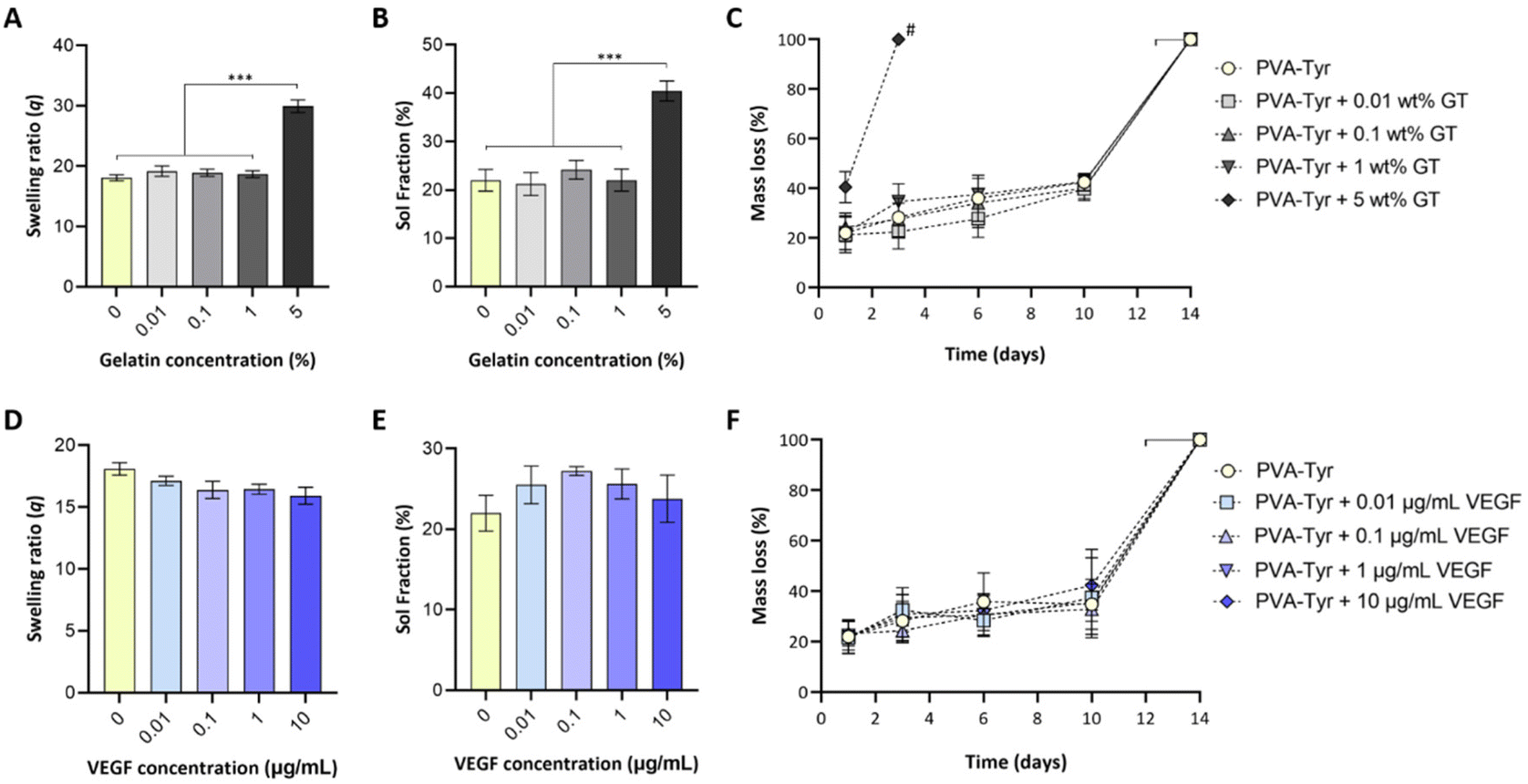 | ||
| Fig. 2 Effect of bioactive molecule incorporation on hydrogel physical properties. Overview of the changes in swelling ratio (A and D), sol faction (B and E) and degradation rate (C and F) of 5 wt% PVA-Tyr hydrogels crosslinked with 0.5/5mM Ru/SPS when increasing concentrations of gelatin or VEGF were incorporated into the polymer network. *** p < 0.001. # significantly different from all other groups. | ||
Incorporation of VEGF ≤10 μg ml−1 did not affect PVA-Tyr hydrogels properties (Fig. 2D–F). There were no significant differences observed in sol fraction, swelling ratio and degradation profile in all PVA-Tyr-VEGF hydrogels evaluated (Fig. 2D–F). Atienza-Roca et al. have previously shown similar results when different unmodified GFs (VEGF, basic fibroblast growth factor -bFGF and brain-derived neurotrophic factor-BDNF) were incorporated into a comparable PVA-Tyr delivery platform, although the concentration range of GFs used were much narrower (25–100 ng mL−1).41 Here, the feasibility of biofunctionalizing the hydrogels with ≤10 μg mL−1 of VEGF was demonstrated (100× more than previous studies) without impacting the physico-chemical properties of the PVA-Tyr hydrogels, confirming the high tunability and versatility of this system.
3.2 In vitro evaluation of cell interactions with the biofunctionalized hydrogels
There is an overwhelming amount of evidence demonstrating that gelatin (from 0.0002 to 20%) enhances the attachment of different cell types including endothelial cells, fibroblasts, smooth muscle cells and mesenchymal stem cells to 3D scaffolds.12,15–17,49–52 However, there are conflicting results regarding the influence of VEGF on endothelial cell adhesion.53,54 For instance, Monchaux et al. demonstrated that the immobilization of VEGF alone was not sufficient to induce attachment of HUVECs on a low-fouling carboxy-methyl-dextran layer, but it enhanced cell adhesion when co-immobilized with RGD sequences.53 In contrast, a more recent study by Saotome et al. showed that the fabrication of hydrogels with transgenic silk-VEGF fusion proteins promoted HUVEC attachment and proliferation compared to the unmodified silk.54 The use of hydrogel systems with different physico-chemical properties, immobilization strategies and VEGF isoforms may explain these discrepancies. Thus, to tailor the concentration of bioactive molecules required to enhance biofunctionality of PVA-Tyr hydrogels, here we evaluated the influence of increasing concentrations of gelatin (0.01–5 wt%) and VEGF (0.01–10 μg mL−1) on cell adhesion and morphology. Endothelial cells were selected as a model, as recruitment of the host vasculature is crucial when developing platforms for TERM applications.In-line with previous reports, the bioinert nature of PVA due to the lack of cell adhesion motifs limited endothelial cell attachment (6 ± 0.8 cells per FOV) and prevented cell spreading on the hydrogel surface.12,17,51,52 The incorporation of increasing gelatin concentrations led to an increase in HUVEC attachment, where significant differences in cell adhesion were already evident at 0.1 wt% gelatin (34.7 ± 4.8 cells per FOV). Interestingly, increasing gelatin concentration from 1 to 5 wt% did not result in any further improvement in cell attachment (63.8 ± 9.4 cells per FOV and 67.9 ± 8 cells per FOV, respectively) (Fig. 3A and C). Nevertheless, significant differences were evident in cell morphology, as endothelial cells were elongating and forming cell–cell interactions at a faster rate when 1 and 5 wt% gelatin were present (Fig. 3D). This is consistent with reports by Lim et al., where gelatin incorporation in PVA-Tyr polymer networks crosslinked with the Ru/SPS photoinitiating system enhanced L929 fibroblast attachment and spreading.17 In that study, the incorporation of as little as 0.01% of gelatin markedly increased the bioactivity of the hydrogels, already reaching 60% of the maximum efficiency achieved in the study.17 Similarly, Zhang et al. also demonstrated that 0.0128% of gelatin incorporation in physically crosslinked PVA hydrogels significantly promoted NIH3T3 fibroblasts attachment and spreading, reducing cell circularity.15 Differences in the concentration of gelatin required to induce a significant biological effect might be due to the intrinsic differences in adhesion molecule affinities for ECM binding sites between cell types.53,55–57 Consistent with our results, other studies indicated that higher percentages of gelatin are required in order to promote HUVEC spreading.51,58 For instance, 1.5–4 wt% of gelatin was included in a chitosan-based scaffold in order to promote HUVECs spreading over time.58 Thus, the optimal concentration of incorporated bioactive molecules should be customized according to the targeted cell type.
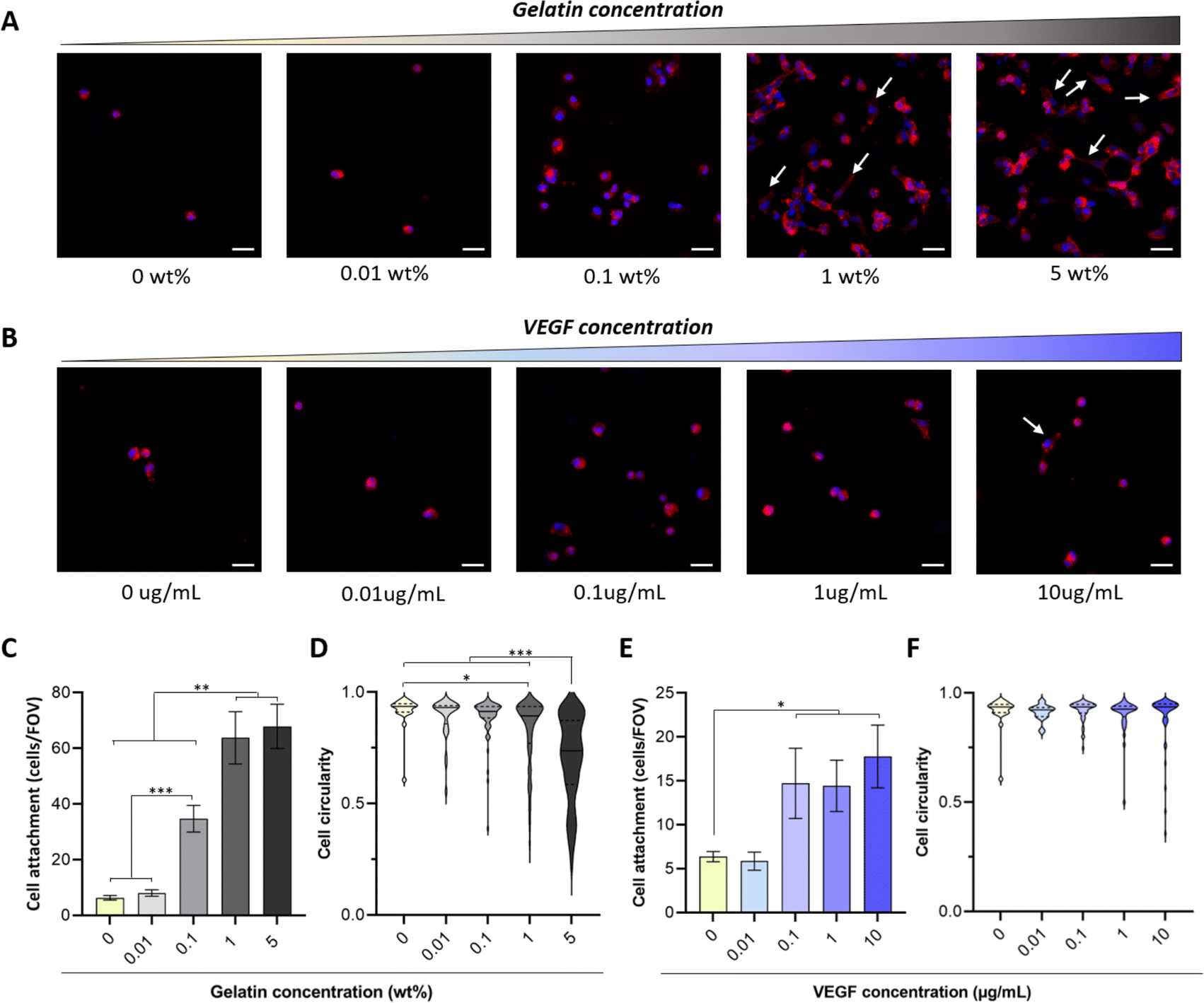 | ||
| Fig. 3 Bioactive molecule incorporation affects cell attachment and shape. Representative images of HUVECS attached to the PVA-Tyr hydrogels with or without increasing concentrations of gelatin (A) and VEGF (B) 6 hours after seeding. Cell spreading on the hydrogels is highlighted with white arrows. Quantification of the attached cells (C and E) and overview of average cell shape, with values equal to 1 indicating perfect circularity and thus less cell spreading (D and F). Scalebar: 10 μm. * p < 0.05, **p < 0.01, ***p < 0.001. FOV-field of view. | ||
The presence of VEGF concentrations between 0.1 and 10 μg mL−1 also enhanced cell adhesion, although to a lesser extent (5× less) compared to 1–5 wt% gelatin (Fig. 3B and E). The lower impact of VEGF on HUVEC attachment and morphological changes can be explained by the difference in adhesive motifs present on the two bioactive molecules. Gelatin contains RGD motifs, which are recognized by αvβ3 and α5β1 integrins present on the endothelial cells and promote their attachment.53,59 VEGF, on the other hand, does not possess such motifs. Nevertheless, integrins including αvβ3 and α9β1 can also directly bind to the 165 isoform of VEGF-A, which has been used in this study.60,61 Additionally, an extensive crosstalk exists between VEGF receptor-2 (VEGFR-2) pathway, which is activated via the interaction with VEGF-A, and integrin αvβ3 pathway, potentially enhancing VEGF-mediated endothelial cell attachment.62,63 Interestingly, no change in cell circularity was observed at any of the considered VEGF concentrations (Fig. 3F). Similar results were achieved by Traub et al. when comparing HUVEC attachment on 2D surfaces either coated with a cell adhesive domain from fibronectin, VEGF or a combination of the two. Higher attachment was observed in the presence of the fibronectin compared to VEGF. Furthermore, HUVEC spreading was observed only when fibronectin's adhesion motif was present alone or in combination with the pro-angiogenic GF.62
Considering the capability of retaining the physico-chemical properties of the hydrogel while enhancing the interactions with endothelial cells in vitro, gelatin was incorporated at 1 wt% into PVA-Tyr hydrogels (PVA-Tyr GT) for subsequent experiments. For VEGF-functionalized hydrogels, the concentration incorporated into the PVA-Tyr hydrogels was selected based on the optimal range required for the subsequent experiments (PVA-Tyr VEGF), at either 100 ng mL−1 (CAM assay) or 10 μg mL−1 (subcutaneous mouse model), respectively.
3.3 Bioactive molecules incorporation, release and angiogenic potential in ovo
To understand the incorporation efficiency and release profiles of both gelatin- and VEGF-functionalised hydrogels, longitudinal studies were performed (Fig. 4A–C). In the presence of the Ru and SPS photoinitiating system, covalent-incorporation of both gelatin and VEGF into PVA-Tyr hydrogels was successfully achieved, with an initial retention of ∼70%, and both molecules showing comparable release profiles (Fig. 4A–C). This is particularly interesting, considering that VEGF and gelatin contain a different percentage of tyrosines (∼2.5% and ∼0.5%, respectively).64,65 Nevertheless, the 3D protein structural conformation also affects the availability of such moieties. Thus, it is possible that only a fraction of the tyrosine groups was available for crosslinking. The molecular weight (38.6 kDa for VEGF and up to 400 kDa for gelatin) and hydrodynamic radius of the macromolecules may have also influenced the initial retention within the polymer network and release.66,67 Finally, as the incorporation of these bioactive components did not influence the physico-chemical properties of the hydrogels, it is likely that the release of both bioactive molecules, is mainly dependent on the degradation of the PVA-Tyr network. This offers the possibility for tailoring gelatin and GF release profiles by regulating the degradation rate of the PVA-Tyr backbone, for instance by changing the degree of PVA tyramination, or the macromer and photoinitiator concentrations.41,42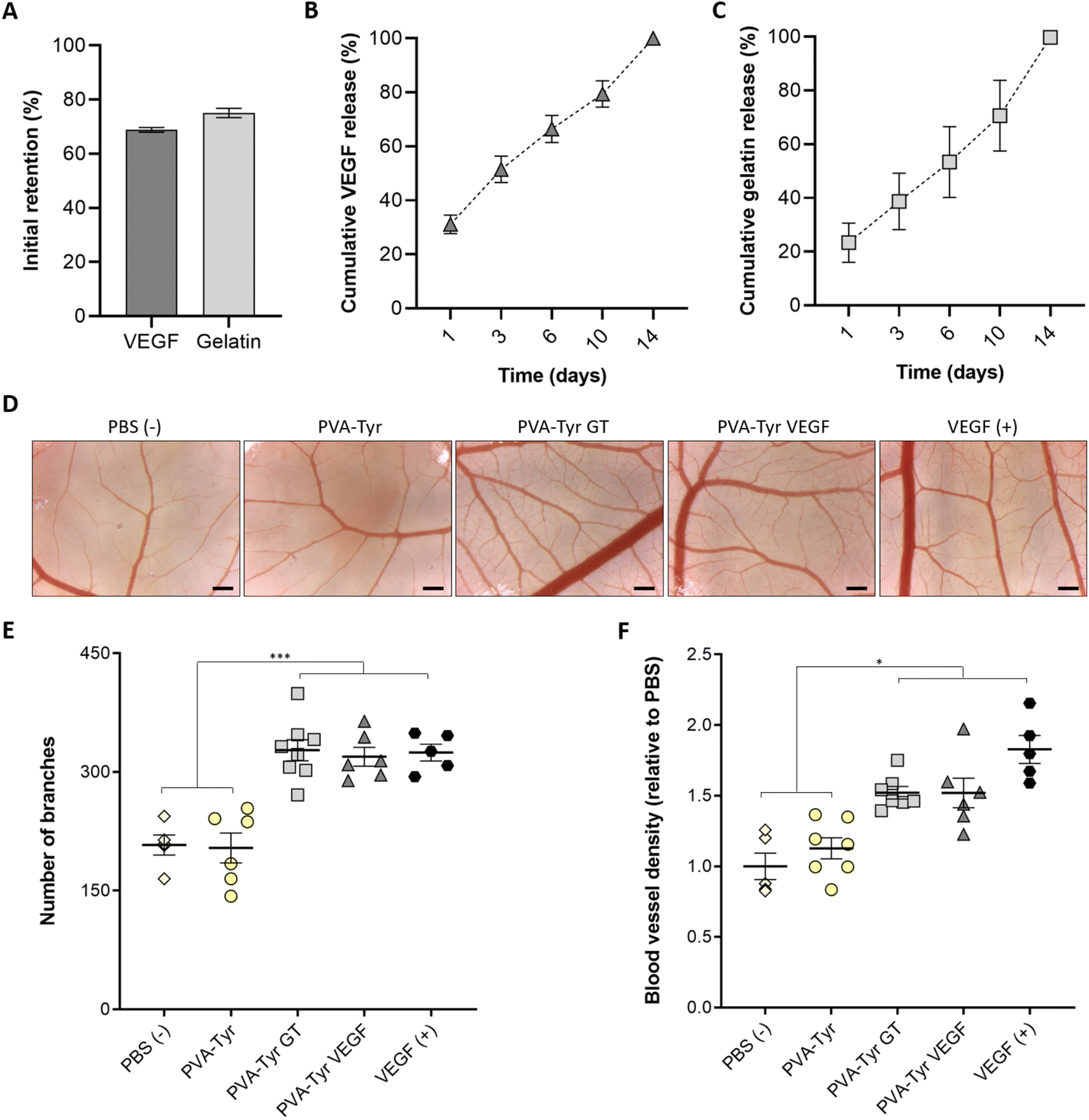 | ||
| Fig. 4 Overview of bioactive molecules retention, release pattern and retention of bioactivity. Initial retention of gelatin and VEGF (A) was expressed as a function of the total released molecules after the complete hydrogel degradation. Cumulative release profile of the bioactive molecules (B and C) and evaluation of the retention of their bioactivity via CAM assay (D–F). Representative images of the angiogenesis induced by each experimental group (D) and quantification of the number of branches observed (E) and vessel density relative to the PBS group (F). PBS and unbound VEGF represent the negative and positive control of the experiment, respectively. Scalebar: 500 μm. * p < 0.05, **p < 0.01, ***p < 0.001. | ||
Covalent binding of GFs often requires them to be put through chemical modification processes, which may lead to reduction of their biological activity.21,32,33 Furthermore, since it is not possible to selectively control the reaction sites on the GFs, the immobilization process may alter the 3D protein conformation, potentially decreasing their bioactivity.32 Hu et al. compared the retention of VEGF biological activity on endothelial cells after covalent immobilization on a hyaluronic acid–catechol coating via a reaction involving activated carboxylic acid groups or by affinity binding to a heparin-catechol coating.21 Although the same amount of VEGF was initially retained and the two systems displayed similar release profiles, higher endothelial cell metabolic activity, CD31 and Von Willebrand factor expression, and capillary formation were observed in the group where affinity binding was employed. This suggests that VEGF immobilization reduced its bioactivity.21 The reaction conditions for the covalent conjugation of VEGF, such as the reaction buffer, pH and step or bulk immobilization process also greatly affected GF pro-angiogenic effects of growth factors on endothelial cells.31
In our work, we evaluated the capability of the released bioactive molecules to induce angiogenesis in a more complex in ovo model, comparing them with the same concentration of unbound VEGF (positive control). The CAM assay is a well-established model, widely used to examine the angiogenic response to constructs for vascular TE because, being minimally-invasive to the chick embryo, it represents an intermediate step in between in vitro and in vivo. Hence it is considered a refinement model for animal research.68,69 While no additional angiogenesis was induced by the pure PVA-Tyr compared to the PBS control, a significant increase in vessel branches and an overall higher blood vessel density was observed in the PVA-Tyr GT and PVA-Tyr VEGF groups, with no significant differences observed between them (Fig. 4D–F). This is particularly interesting because, while the effects of VEGF on vessel formation in the CAM assay are well-known, this is one of the first reports indicating that 1 wt% pristine gelatin released from PVA-Tyr hydrogels triggers an identical response, highlighting the potential value of gelatin as a stand-alone pro-angiogenic cue. Additionally, the branches and the vessel density induced by the PVA-Tyr GT and PVA-Tyr VEGF groups were comparable to the samples where the same concentration of unbound VEGF was used as positive control, confirming that the incorporation process did not alter their functionality. The conjugation of the GF to a biomaterial might have protected it from degradation, extending the GF half-life and guaranteeing a more prolonged therapeutic effect.32,70,71 This is in-line with our previous results, where different growth factors (i.e. VEGF, FGF and BDNF) were able to withstand the oxidative stress and co-polymerisation to PVA-Tyr without losing their ability to trigger cell proliferation, migration and vasculogenesis.41
Finally, it is important to underline that no negative effects on angiogenesis were induced by the degradation products of the PVA-Tyr delivery platform, as no differences we observed compared to the PBS control. This further validates previous observations where no cell cytotoxicity was observed for the PVA-Tyr macromer and the concentration of Ru/SPS used for this study.46,72,73
3.4 Hydrogel–host cells interactions in vivo
After evaluating the angiogenic potential in ovo, PVA-Tyr hydrogels with or without bioactive VEGF and gelatin molecules were implanted subcutaneously in an immunocompetent mouse model to assess cell-hydrogel interactions. Cell infiltration, biomaterial degradation and matrix synthesis were evaluated after 1 and 3 weeks (Fig. 5 and 6). Based on our in vitro results, higher number of infiltrating cells were expected in the PVA-Tyr GT hydrogels, followed by the PVA-Tyr VEGF group and limited cell interaction was expected within the pure PVA-Tyr. Nevertheless, similar amounts of infiltrating cells were observed in the three groups, with 676.9 ± 168 cells per mm2 for the PVA-Tyr group and 527.3 ± 179 and 528.139 ± 139.8 cells per mm2 for the PVA-Tyr GT and PVA-Tyr VEGF groups, respectively. To assess the speed and depth of cell penetration within the hydrogel, only the cells that were at a minimum distance of 150 μm from the hydrogel edge were included in the count. Interestingly, the presence of molecules that promoted cell adhesion did not influence travel depth, as comparable numbers of cells were observed, with 383.6 ± 89.3, 202.5 ± 88.9 and 154.5 ± 65.12 cells per mm2 for the PVA-Tyr, PVA-Tyr GT and PVA-Tyr VEGF hydrogels, respectively (Fig. 5B). For all the groups, an increase in cellular infiltration was observed after 3 weeks, with an average number of 5805 ± 823, 6888 ± 422 and 7288 ± 478 cells per mm2 detected within the PVA-Tyr, PVA-Tyr GT and PVA-Tyr VEGF hydrogels, respectively (Fig. 6B). However, more variation was observed for the pure PVA-Tyr group, with a standard error of the mean that was almost double compared to the other two groups (823 vs. 422 and 478). Additionally, the pure PVA-Tyr group demonstrated a slower increase in cell infiltration between the two timepoints, with an average increase of 8.5× compared to the 13× and 13.5× for the PVA-Tyr GT and PVA-Tyr VEGF groups. Similar to the 1-week timepoint, no significant differences were observed in the number of cells infiltrating into the central areas of the constructs after 3 weeks. The presence of vimentin positive, elongated cells at both timepoints (Fig. 5A and 6A) suggests that at least a proportion of the cells infiltrating inside the constructs were fibroblasts. Their presence, together with the initial collagen deposition observed in the Masson Trichrome staining in all the groups, is a positive indication of the host cells remodelling the hydrogels.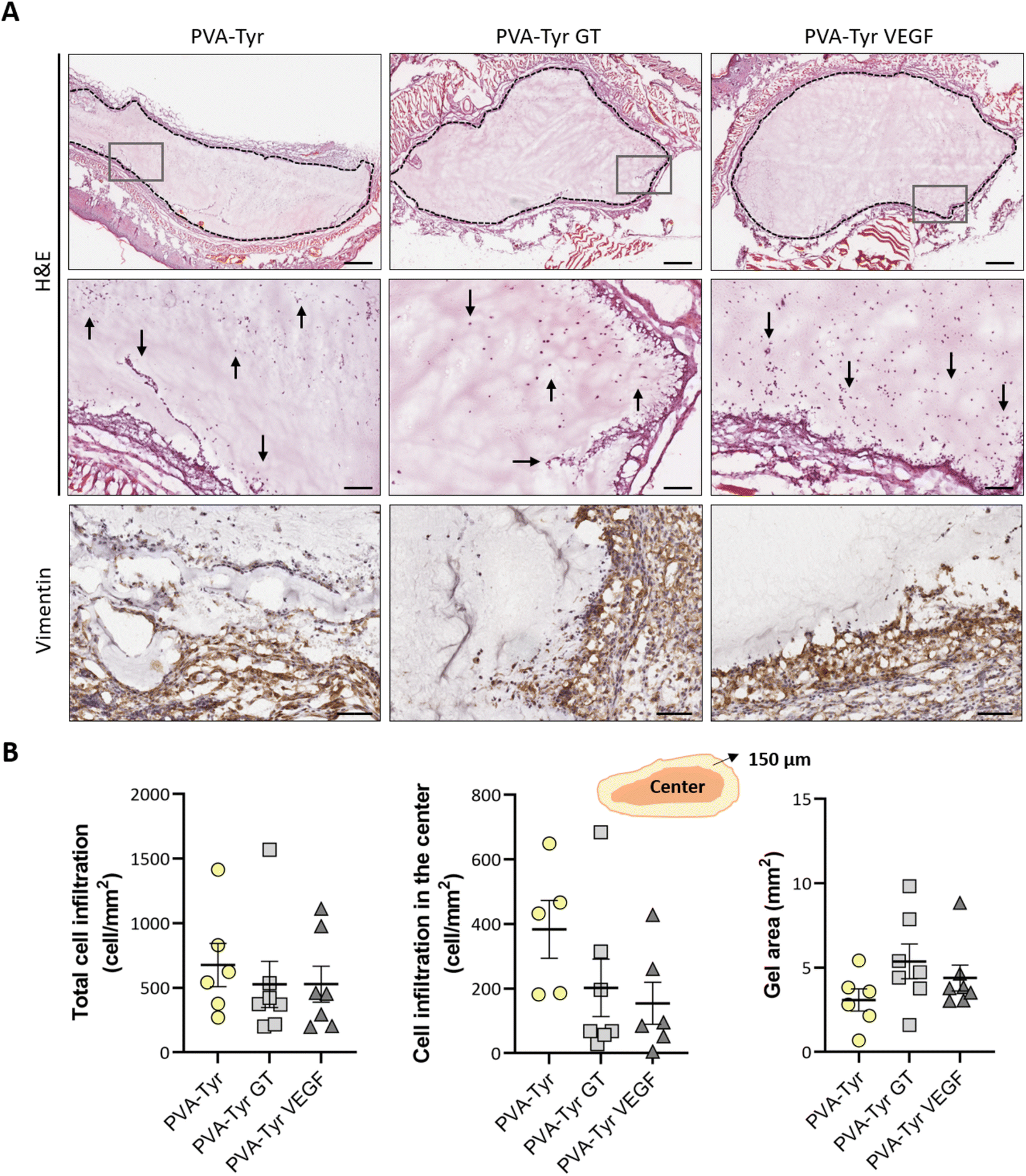 | ||
| Fig. 5 Cell infiltration within the PVA-Tyr hydrogels with and without functionalization 1-week post-implantation. Representative images of the hydrogels and infiltrating cells (A). The black dotted lines indicate the edges of the constructs whereas the grey boxes highlight the area depicted in the higher magnification pictures in the second row (H&E). Black arrows indicate areas of infiltrating cells. Vimentin staining indicates the presence of fibroblast-like cells. Quantitative evaluation of cell infiltration within the entire construct or excluding the border (150 μm) and hydrogel size (B). Scale bar in the first H&E row: 500 μm. Scale bar in the second H&E row and vimentin: 100 μm.* p < 0.05. | ||
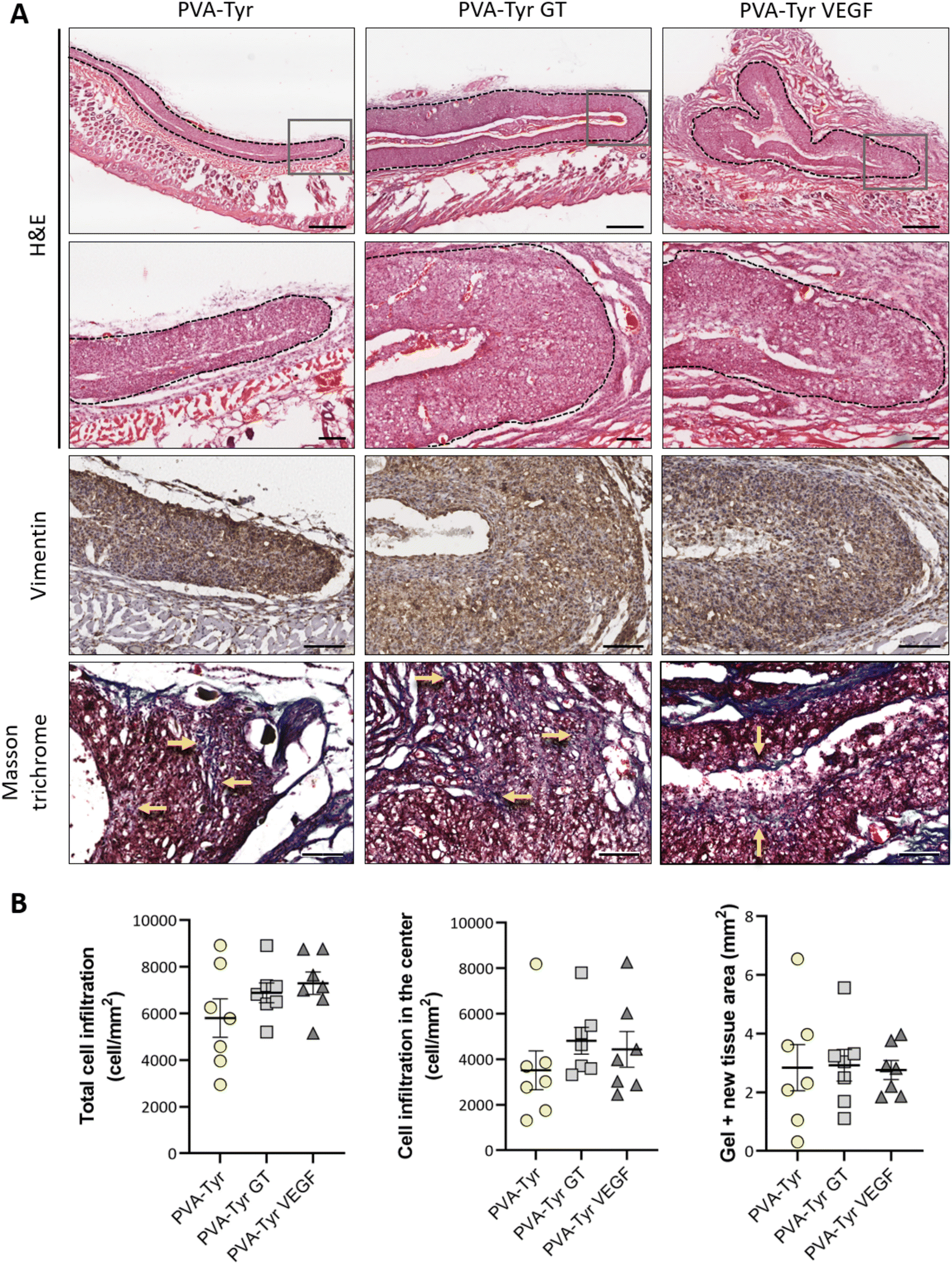 | ||
| Fig. 6 Cell infiltration within the PVA-Tyr hydrogels with and without functionalization 3-week post-implantation. Representative images of the hydrogels, infiltrating cells and matrix deposition (A). The black dotted lines indicate the edges of the area of interest, with the hydrogels and the newly formed tissue. The grey boxes highlight the area depicted in the higher magnification pictures in the second row (H&E). Vimentin staining indicates the presence of fibroblast-like cells infiltrating in the constructs. Masson trichrome staining highlights the beginning of collagen deposition within the construct (blue staining, highlighted by the yellow arrows). Quantitative evaluation of cell infiltration within the entire construct or excluding the border (150 μm) and hydrogel size (B). Scale bar in the first H&E row: 500 μm. Scale bar in the second H&E row, vimentin and Masson trichrome: 100 μm.* p < 0.05. | ||
Taken together, these results suggest that, while adhesive sequences are required to achieve PVA-Tyr and endothelial cell interaction in vitro, this is not the case in vivo. One of the reasons for this difference is that, upon implantation, proteins such as fibrinogen and vitronectin are adsorbed on the surface of the hydrogel, providing a provisional matrix with which host cells can interact with.74,75 Additional cell infiltration within the center of the PVA-Tyr hydrogels might have occurred as a consequence of hydrogel degradation over time. In addition to hydrolysis, in vivo degradation might have also been mediated by macrophages and other immune cells recruited at the implantation site. Specifically, the production of reactive oxygen species from immune cells might have led to the oxidation and degradation of the PVA polymer and of the crosslinked network.76 Consistent with the in vitro results, similar hydrogel sizes were observed between groups in vivo after 1 and 3 weeks, explaining the similar cell infiltration and new tissue deposition observed. This is interesting, as the incorporation of ECM macromolecules or bioactive motifs commonly results in altered degradation profiles of synthetic hydrogels, due to the introduction of motifs susceptible to enzymatic degradation.2,4,70,77 For instance, Lan et al. showed that the presence of 2 mg mL−1 chondroitin sulfate and 5 wt% of collagen type II in 5 wt% PVA hydrogels prepared with the freeze–thaw method promoted faster degradation compared to the non-functionalized construct in a rabbit osteochondral defect.78 The absence of differences in our work suggests the ability of PVA-Tyr hydrogels to protect incorporated proteins from proteases and confirms that the hydrogel properties are mainly dependant on the PVA-Tyr polymer network.
3.5 Angiogenesis induced by bioactive molecules in vivo
Slow blood vessel infiltration within TE constructs and the lack a functional microvasculature connected to the host blood supply is considered to be one of the main reasons behind TE implant failure.8 Several strategies have been developed to overcome this challenge, including pre-vascularization of the engineered tissues in vitro prior to implantation to facilitate anastomosis with the host vasculature, manipulation of scaffold architecture or the incorporation of pro-angiogenic cues (e.g. VEGF) recruitment.6,8,34,79 Another interesting option to promote vascularization is to develop smart biomaterials with cell-instructive properties, which will guide the neo-vascularization process.8 Here, we evaluated the capability of gelatin to promote host blood vessel infiltration within an otherwise bioinert synthetic hydrogel.The angiogenic potential of gelatin observed in the CAM assay was confirmed in a subcutaneous mouse model, to an extent that was comparable to the VEGF concentration selected for this study. After 1 week, almost 50% more CD31 positive cells were recruited in the area surrounding the PVA-Tyr VEGF and PVA-Tyr GT hydrogels compared to the pure PVA-Tyr control (6.9 ± 0.44% for the PVA-Tyr VEGF and 7.02 ± 0.71% for the PVA-Tyr GT vs. 4.92 ± 0.44% for the PVA-Tyr) (Fig. 7). Similar differences were observed after 3 weeks, with an average of 1.103 ± 0.1% of CD31 positive cells infiltrating within the pure PVA-Tyr hydrogel compared to 2.52 ± 0.26% and 2.64 ± 0.32% of the PVA-Tyr GT and PVA-Tyr VEGF groups, respectively (Fig. 8A and B). In addition to promoting blood vessel recruitment, it is also crucial that the vasculature network attracted by the bioactive factors is healthy and functional. For instance, the uncontrolled release of VEGF at supraphysiological doses causes the growth of large, aberrant, leaky vascular structures.26,27 To evaluate the capability of gelatin to trigger healthy angiogenesis, blood vessel perimeter (an indicator for aberrant vessels) was compared between groups. A larger perimeter was observed in the PVA-Tyr VEGF group (53.13 ± 4.1 μm) when compared to the PVA-Tyr GT and PVA-Tyr groups (41.41 ± 3.37 μm and 44.39 ± 3.01 μm, respectively), although this difference was not significant (Fig. 8C). This is in-line with previous studies, where the selected VEGF concentration led to healthy angiogenesis.70,80 The average vessel perimeter values observed in our study are also within the same range reported in a similar experimental set up, where blood vessel infiltration was evaluated in a porous silk-based hydrogel after subcutaneous implantation in female mice of comparable age.81 Furthermore, in all groups a similar distribution in blood vessel size was observed (Fig. 8B). Finally, Karvinen et al. highlighted that aberrant blood vessels could not be properly perfused, as in their histological analysis red blood cells were not present in the vessel lumen.28 In our study we confirmed the functionality of the infiltrating vessels histologically, by identifying red blood cells within the lumen. Blood vessel functionality was also qualitatively confirmed by perfusing mice vasculature with a radiopaque resin (n = 2). The 3D reconstructions of the hydrogels indicate that perfusable vessel are present within the explanted hydrogel area, presenting a trend that is consistent with the CD31 staining results (Fig. 8C).
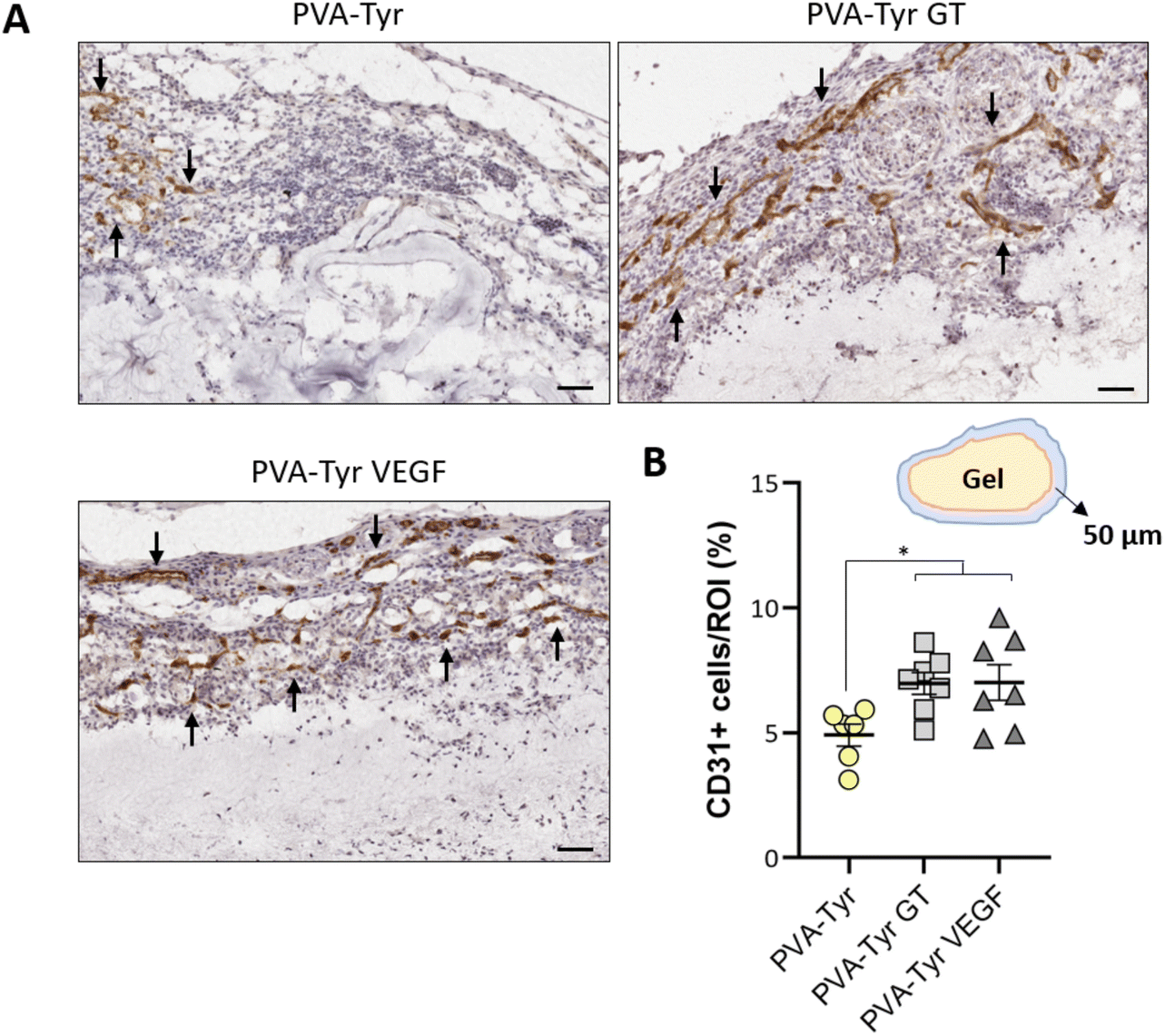 | ||
| Fig. 7 Blood vessel recruitment around the PVA-Tyr hydrogels with and without functionalization 1 week post-implantation. Representative images of the hydrogels and the tissue surrounding (A). Black arrows indicated the CD31 positive vessels. Quantitative evaluation CD31 positive cells present within the 50 μm of tissue surrounding the hydrogel platform (B). Scale bar: 100 μm. * p < 0.05. | ||
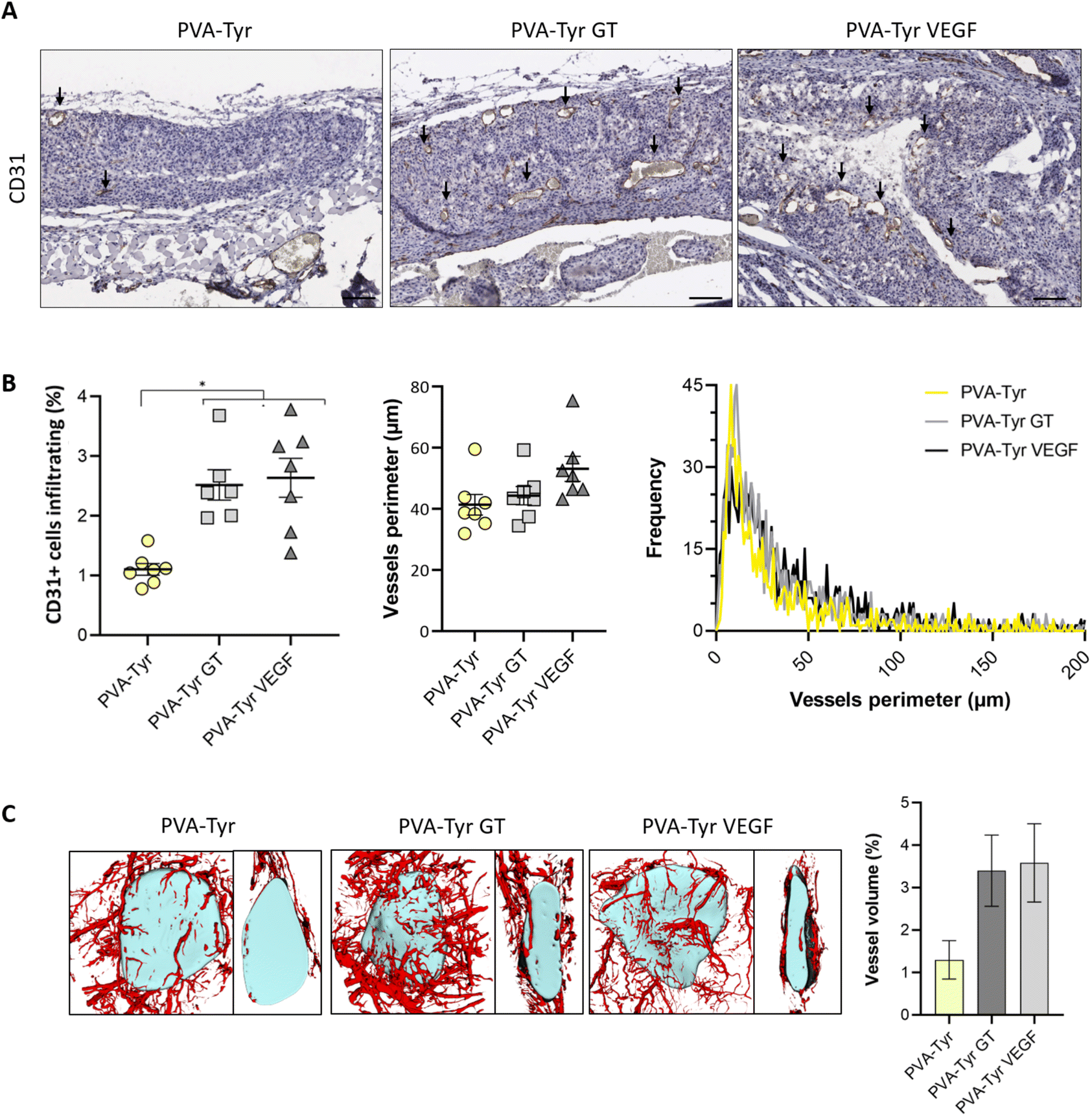 | ||
| Fig. 8 Blood vessel recruitment within the hydrogels 3-week post-implantation. Representative images of the hydrogels and infiltrating blood vessels (black arrows) (A). Quantitative evaluation CD31 positive cells and blood vessels present within the hydrogel platform, vessel perimeter and relative frequency (B). 3D reconstruction of the perfusable blood vessels around and infiltrating the hydrogels and the relative quantification of the volume of vessels infiltrating in the hydrogels (n = 2) (C). Scale bar: 100 μm. * p < 0.05. | ||
Few studies have reported on the potential of gelatin as a stand-alone bioactive cue for TE applications.38,40 Dreesmann et al. described comparable pro-angiogenic effects when evaluating the vessel recruitment triggered by a 12 wt% gelatin sponge crosslinked with a methanal-based method in a CAM assay and a subcutaneous model.38 However, the gelatin sponges were only compared to collagen control samples and, therefore, it was not possible to establish whether the pro-angiogenic effects were the result of an active function of the gelatin sponges (i.e. cell instructive), or they were simply more permissive compared to the collagen constructs.38 Similarly, Mony et al. reported enhanced vascularization in a CAM assay and in a subcutaneous diabetic mouse model when a cholecyst-derived scaffold was functionalized by covalently incorporating gelatin.40 In our study, a direct comparison of VEGF and gelatin incorporated in a PVA-Tyr network in an identical fashion was performed, validating the pro-angiogenic potential of this molecule. We hypothesize that this effect is associated with the extensive crosstalk existing between VEGF-R2 and integrin αvβ3, the integrin responsible for cell interactions with gelatin.82 Furthermore, Ma et al. recently demonstrated that activating the focal adhesion kinase (FAK)/P38 signalling pathway via integrin αVβ3 promotes the expression of the cellular (c)-Myc. As a consequence, c-Myc increases histone acetylation levels of the VEGF promoter, ultimately promoting VEGF expression.83 Thus, the interaction with gelatin might have ultimately led to the activation of the same pathways as VEGF, explaining the pro-angiogenic effects shown in the PVA-Tyr GT group.
Exploring the potential of gelatin and ECM proteins to mediate angiogenesis is a particularly promising approach for promoting blood vessel recruitment for TE applications, as the utilization of these macromolecules may reduce safety concerns associated with the use of VEGF. Future studies will focus on evaluating whether gelatin functionalization will induce sufficient recruitment of a microvascular network in more challenging environments (i.e. ischemia or necrosis). Interestingly, different GFs or bioactive cues could be incorporated in the gelatin-functionalized PVA-Tyr hydrogels, to trigger tissue specific effects, offering a broad range of applications for this platform. For instance, incorporating a combination of bFGF and gelatin could be used to promote wound closure for soft tissue regeneration. Additionally, the inclusion of specific topological cues (e.g. microchannels or micro-grooves) within the PVA-Tyr GT hydrogels by utilizing biofabrication techniques such as stereolithography, digital light processing or microfluidic systems can further enhance blood vessels recruitment and provide guidance for endothelial cells migration and microcapillary formation in specific areas of the engineered constructs.84,85 Nevertheless, the use of different fabrication methods and introduction of topological cues into these hydrogels might also affect their physico-chemical properties (swelling, degradation etc.). For this reason, these parameters should be evaluated and optimized depending on the intended application.
4. Conclusions
In the present study, we investigated the potential of using gelatin to enhance the biofunctionality of PVA-Tyr synthetic hydrogels, by promoting cell infiltration and host blood vessel recruitment for TE applications. With our focus on vascularization, we compared the pro-angiogenic potential of gelatin with VEGF. Firstly, we demonstrated that a wide range of unmodified gelatin and VEGF concentrations can be covalently incorporated without affecting the properties of the PVA-Tyr hydrogel network or their bioactivity. Interestingly our findings indicated that, although biofunctionalization is required to promote endothelial cell interaction with the synthetic biomaterial in vitro, this is not the case in vivo, as cells were able to infiltrate the pure PVA-Tyr hydrogels to a comparable extent of the functionalized ones after subcutaneous implantation. This suggests that, in vivo, the presence of additional cell recognition motifs is not as crucial for cell interactions as it is in vitro. Nevertheless, the presence of gelatin or VEGF is essential to recruit the targeted cell type, endothelial cells, with no differences in angiogenesis triggered by the two bioactive cues. These results highlight the potential of using pro-angiogenic ECM-based cues such as gelatin as alternatives to GFs for TE applications.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
K. L. would like to acknowledge funding support from the Health Research Council of New Zealand (Sir Charles Hercus Health Research Fellowship 19/135, Project Grant 20/508), and Royal Society Te Apārangi (Marsden Fast Start Grant MFP-UOO1826). K. L. and J. R-K. would like to acknowledge funding from NSW Health (H22/98586). J. R-K is supported by the Australian Research Council Future Fellowship (FT210100668) and the UNSW Scientia Program.References
- F. Xu, C. Dawson, M. Lamb, E. Mueller, E. Stefanek, M. Akbari and T. Hoare, Front. Bioeng. Biotechnol., 2022, 10, 849831 CrossRef PubMed.
- E. J. Bolívar-Monsalve, M. M. Alvarez, S. Hosseini, M. A. Espinosa-Hernandez, C. F. Ceballos-González, M. Sanchez-Dominguez, S. R. Shin, B. Cecen, S. Hassan, E. Di Maio and G. Trujillo-de Santiago, Mater. Adv., 2021, 2, 4447–4478 RSC.
- A. Z. Unal and J. L. West, Bioconjugate Chem., 2020, 31, 2253–2271 CrossRef CAS PubMed.
- C. Chokoza, C. A. Gustafsson, K. P. Goetsch, P. Zilla, N. Thierfelder, F. Pisano, M. Mura, M. Gnecchi, D. Bezuidenhout and N. H. Davies, ACS Biomater. Sci. Eng., 2019, 5, 5430–5438 CrossRef CAS.
- M. Mori da Cunha, B. Arts, L. Hympanova, R. Rynkevic, K. Mackova, A. W. Bosman, P. Y. W. Dankers and J. Deprest, Acta Biomater., 2020, 106, 82–91 CrossRef CAS PubMed.
- W. Y. Wang, R. N. Kent, 3rd, S. A. Huang, E. H. Jarman, E. H. Shikanov, C. D. Davidson, H. L. Hiraki, D. Lin, M. A. Wall, D. L. Matera, J. W. Shin, W. J. Polacheck, A. Shikanov and B. M. Baker, Acta Biomater., 2021, 135, 260–273 CrossRef CAS PubMed.
- S. Trujillo, C. Gonzalez-Garcia, P. Rico, A. Reid, J. Windmill, M. J. Dalby and M. Salmeron-Sanchez, Biomaterials, 2020, 252, 120104 CrossRef CAS.
- R. Chapla and J. L. West, Prog. Biomed. Eng., 2021, 3, 012002 CrossRef.
- D. Barros, P. Parreira, J. Furtado, F. Ferreira-da-Silva, E. Conde-Sousa, A. J. García, M. C. L. Martins, I. F. Amaral and A. P. Pêgo, Biomaterials, 2019, 192, 601–611 CrossRef CAS.
- J. Jia, R. C. Coyle, D. J. Richards, C. L. Berry, R. W. Barrs, J. Biggs, C. James Chou, T. C. Trusk and Y. Mei, Acta Biomater., 2016, 45, 110–120 CrossRef CAS PubMed.
- N. Huettner, T. R. Dargaville and A. Forget, Trends Biotechnol., 2018, 36, 372–383 CrossRef CAS PubMed.
- E. H. Nafea, L. A. Poole-Warren and P. J. Martens, J. Biomed. Mater. Res., Part A, 2015, 103, 3727–3735 CrossRef CAS.
- K. Su and C. Wang, Biotechnol. Lett., 2015, 37, 2139–2145 CrossRef CAS PubMed.
- J. Van Hoorick, L. Tytgat, A. Dobos, H. Ottevaere, J. Van Erps, H. Thienpont, A. Ovsianikov, P. Dubruel and S. Van Vlierberghe, Acta Biomater., 2019, 97, 46–73 CrossRef CAS.
- R. Zhang, D. Zhang, X. Sun, X. Song, K. C. Yan and H. Liang, Int. J. Biol. Macromol., 2022, 219, 672–684 CrossRef CAS PubMed.
- K. Kopeć, M. Wojasiński, M. Eichler, H. Genç, R. P. Friedrich, R. Stein, R. Singh, C. Alexiou, H. Hlawaty, T. Ciach and I. Cicha, Biomater. Adv., 2022, 134, 112544 CrossRef.
- K. S. Lim, M. H. Alves, L. A. Poole-Warren and P. J. Martens, Biomaterials, 2013, 34, 7097–7105 CrossRef CAS PubMed.
- M. C. Echave, C. Pimenta-Lopes, J. L. Pedraz, M. Mehrali, A. Dolatshahi-Pirouz, F. Ventura and G. Orive, Int. J. Pharm., 2019, 562, 151–161 CrossRef CAS.
- G. Marchioli, A. D. Luca, E. de Koning, M. Engelse, C. A. Van Blitterswijk, M. Karperien, A. A. Van Apeldoorn and L. Moroni, Adv. Healthcare Mater., 2016, 5, 1606–1616 CrossRef CAS.
- V. Mastrullo, W. Cathery, E. Velliou, P. Madeddu and P. Campagnolo, Front. Bioeng. Biotechnol., 2020, 8, 188 CrossRef PubMed.
- X. Hu, K. G. Neoh, J. Zhang, E.-T. Kang and W. Wang, Biomaterials, 2012, 33, 8082–8093 CrossRef CAS PubMed.
- S. M. Anderson, S. N. Siegman and T. Segura, Biomaterials, 2011, 32, 7432–7443 CrossRef CAS PubMed.
- Y. Mizuno and T. Taguchi, J. Tissue Eng. Regener. Med., 2019, 13, 2291–2299 CrossRef CAS.
- D. F. Lazarous, M. Shou, M. Scheinowitz, E. Hodge, V. Thirumurti, A. N. Kitsiou, J. A. Stiber, A. D. Lobo, S. Hunsberger, E. Guetta, S. E. Epstein and E. F. Unger, Circulation, 1996, 94, 1074–1082 CrossRef CAS PubMed.
- R. Gianni-Barrera, N. Di Maggio, L. Melly, M. G. Burger, E. Mujagic, L. Gürke, D. J. Schaefer and A. Banfi, Stem Cells Transl. Med., 2020, 9, 433–444 CrossRef.
- V. Sacchi, R. Mittermayr, J. Hartinger, M. M. Martino, K. M. Lorentz, S. Wolbank, A. Hofmann, R. A. Largo, J. S. Marschall, E. Groppa, R. Gianni-Barrera, M. Ehrbar, J. A. Hubbell, H. Redl and A. Banfi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6952–6957 CrossRef CAS PubMed.
- C. R. Ozawa, A. Banfi, N. L. Glazer, G. Thurston, M. L. Springer, P. E. Kraft, D. M. McDonald and H. M. Blau, J. Clin. Invest., 2004, 113, 516–527 CrossRef CAS.
- H. Karvinen, E. Pasanen, T. T. Rissanen, P. Korpisalo, E. Vähäkangas, A. Jazwa, M. Giacca and S. Ylä-Herttuala, Gene Ther., 2011, 18, 1166–1172 CrossRef CAS PubMed.
- Q. Tan, H. Tang, J. Hu, Y. Hu, X. Zhou, Y. Tao and Z. Wu, Int. J. Nanomed., 2011, 6, 929–942 CrossRef CAS PubMed.
- T. Qaum, Q. Xu, A. M. Joussen, M. W. Clemens, W. Qin, K. Miyamoto, H. Hassessian, S. J. Wiegand, J. Rudge, G. D. Yancopoulos and A. P. Adamis, Invest. Ophthalmol. Visual Sci., 2001, 42, 2408–2413 CAS.
- L. L. Chiu, R. D. Weisel, R. K. Li and M. Radisic, J. Tissue Eng. Regener. Med., 2011, 5, 69–84 CrossRef CAS PubMed.
- K. S. Masters, Macromol. Biosci., 2011, 11, 1149–1163 CrossRef CAS PubMed.
- B. K. Mann, R. H. Schmedlen and J. L. West, Biomaterials, 2001, 22, 439–444 CrossRef CAS PubMed.
- J. Fu, C. Wiraja, H. B. Muhammad, C. Xu and D.-A. Wang, Acta Biomater., 2017, 58, 225–237 CrossRef CAS PubMed.
- M. M. Martino, S. Brkic, E. Bovo, M. Burger, D. J. Schaefer, T. Wolff, L. Gürke, P. S. Briquez, H. M. Larsson, R. Gianni-Barrera, J. A. Hubbell and A. Banfi, Front. Bioeng. Biotechnol., 2015, 1, 3–45 Search PubMed.
- Z. Wang, Z. Wang, W. W. Lu, W. Zhen, D. Yang and S. Peng, NPG Asia Mater., 2017, 9, e435–e435 CrossRef CAS.
- A. B. Bello, D. Kim, D. Kim, H. Park and S.-H. Lee, Tissue Eng., Part B, 2020, 26, 164–180 CrossRef CAS.
- L. Dreesmann, M. Ahlers and B. Schlosshauer, Biomaterials, 2007, 28, 5536–5543 CrossRef CAS PubMed.
- P. Wang, X. Meng, R. Wang, W. Yang, L. Yang, J. Wang, D.-A. Wang and C. Fan, Adv. Healthcare Mater., 2022, 11, 2102818 CrossRef CAS.
- M. P. Mony, S. J. Shenoy, R. Raj, C. S. Geetha, K. V. Pratheesh, R. S. Nair, C. Purnima and T. V. Anilkumar, ACS Appl. Bio Mater., 2021, 4, 3320–3331 CrossRef CAS PubMed.
- P. Atienza-Roca, D. C. Kieser, X. Cui, B. Bathish, Y. Ramaswamy, G. J. Hooper, A. N. Clarkson, J. Rnjak-Kovacina, P. J. Martens, L. M. Wise, T. B. F. Woodfield and K. S. Lim, Biomater. Sci., 2020, 8, 5005–5019 RSC.
- K. S. Lim, Y. Ramaswamy, M. H. Alves, R. A. Green, L. A. Poole-Warren and P. J. Martens, Cham, 2015, 15(10), 1423–1432 CAS.
- X. Lin, F. Tang, S. Jiang, H. Khamis, A. Bongers, J. M. Whitelock, M. S. Lord and J. Rnjak-Kovacina, Adv. Sci., 2020, 7, 2000900 CrossRef CAS.
- N. Otsu, IEEE Trans. Syst., Man, Cyber., 1979, 9, 62–66 Search PubMed.
- N. Percie du Sert, V. Hurst, A. Ahluwalia, S. Alam, M. T. Avey, M. Baker, W. J. Browne, A. Clark, I. C. Cuthill, U. Dirnagl, M. Emerson, P. Garner, S. T. Holgate, D. W. Howells, N. A. Karp, S. E. Lazic, K. Lidster, C. J. MacCallum, M. Macleod, E. J. Pearl, O. H. Petersen, F. Rawle, P. Reynolds, K. Rooney, E. S. Sena, S. D. Silberberg, T. Steckler and H. Würbel, PLos Biol., 2020, 18, e3000410 CrossRef CAS PubMed.
- B. G. Soliman, A. Longoni, M. Wang, W. Li, P. N. Bernal, A. Cianciosi, G. C. J. Lindberg, J. Malda, J. Groll, T. Jungst, R. Levato, J. Rnjak-Kovacina, T. B. F. Woodfield, Y. S. Zhang and K. S. Lim, Adv. Funct. Mater., 2023, 33, 2210521 CrossRef CAS.
- M. Koolen, A. Longoni, J. van der Stok, O. Van der Jagt, D. Gawlitta and H. Weinans, Eur. Cells Mater., 2019, 38, 94–105 CrossRef CAS PubMed.
- P. Bankhead, M. B. Loughrey, J. A. Fernández, Y. Dombrowski, D. G. McArt, P. D. Dunne, S. McQuaid, R. T. Gray, L. J. Murray, H. G. Coleman, J. A. James, M. Salto-Tellez and P. W. Hamilton, Sci. Rep., 2017, 7, 16878 CrossRef PubMed.
- K. T. Shalumon, S. Deepthi, M. S. Anupama, S. V. Nair, R. Jayakumar and K. P. Chennazhi, Int. J. Biol. Macromol., 2015, 72, 1048–1055 CrossRef CAS PubMed.
- W. Ramadhan, G. Kagawa, Y. Hamada, K. Moriyama, R. Wakabayashi, K. Minamihata, M. Goto and N. Kamiya, ACS Appl. Bio Mater., 2019, 2, 2600–2609 CrossRef CAS PubMed.
- V. M. Merkle, P. L. Tran, M. Hutchinson, K. R. Ammann, K. DeCook, X. Wu and M. J. Slepian, Acta Biomater., 2015, 27, 77–87 CrossRef CAS PubMed.
- H. Jeong, D. Y. Lee, D. H. Yang and Y.-S. Song, Macromol. Res., 2022, 30, 223–229 CrossRef CAS.
- E. Monchaux and P. Vermette, Biomacromolecules, 2007, 8, 3668–3673 CrossRef CAS PubMed.
- T. Saotome, H. Hayashi, R. Tanaka, A. Kinugasa, S. Uesugi, K.-i. Tatematsu, H. Sezutsu, N. Kuwabara and T. Asakura, J. Mater. Chem. B, 2015, 3, 7109–7116 RSC.
- A. J. Campillo-Fernández, R. E. Unger, K. Peters, S. Halstenberg, M. Santos, M. S. Sánchez, J. M. M. Dueñas, M. M. Pradas, J. L. G. Ribelles and C. J. Kirkpatrick, Tissue Eng., Part A, 2008, 15, 1331–1341 CrossRef PubMed.
- N. P. Ziats and J. M. Anderson, J. Vasc. Surg., 1993, 17, 710–718 CrossRef CAS.
- D. Hao, Y. Fan, W. Xiao, R. Liu, C. Pivetti, T. Walimbe, F. Guo, X. Zhang, D. L. Farmer, F. Wang, A. Panitch, K. S. Lam and A. Wang, Acta Biomater., 2020, 108, 178–193 CrossRef CAS PubMed.
- Y. Huang, S. Onyeri, M. Siewe, A. Moshfeghian and S. V. Madihally, Biomaterials, 2005, 26, 7616–7627 CrossRef CAS PubMed.
- A. Post, E. Wang and E. Cosgriff-Hernandez, Ann. Biomed. Eng., 2019, 47, 366–380 CrossRef PubMed.
- N. E. Vlahakis, B. A. Young, A. Atakilit, A. E. Hawkridge, R. B. Issaka, N. Boudreau and D. Sheppard, J. Biol. Chem., 2007, 282, 15187–15196 CrossRef CAS PubMed.
- H. Hutchings, N. Ortega and J. Plouët, FASEB J., 2003, 17, 1520–1522 CrossRef CAS.
- S. Traub, J. Morgner, M. M. Martino, S. Höning, M. A. Swartz, S. A. Wickström, J. A. Hubbell and S. A. Eming, Biomaterials, 2013, 34, 5958–5968 CrossRef CAS PubMed.
- T. V. Byzova, C. K. Goldman, N. Pampori, K. A. Thomas, A. Bett, S. J. Shattil and E. F. Plow, Mol. Cell, 2000, 6, 851–860 CAS.
- P. Guerrero, I. Zugasti, A. Etxabide, H. N. Bao, T. Trang Si, M. Peñalba and K. de la Caba, Polymers, 2020, 12, 570 CrossRef CAS PubMed.
- W. G. Cobbett, A. W. Kenchington and A. G. Ward, Biochem. J., 1962, 84, 468–474 CrossRef CAS PubMed.
- H. Babavalian, A. M. Latifi, M. M. Sepantafar, H. Mohammadi, F. Shakeri and S. Khodi, J. Appl. Biotechnol. Rep., 2014, 1, 83–96 Search PubMed.
- Y. Lyu and H. S. Azevedo, Molecules, 2021, 26(4), 873 CrossRef CAS PubMed.
- D. Ribatti, T. Annese and R. Tamma, Microvasc. Res., 2020, 131, 104026 CrossRef CAS PubMed.
- D. Ribatti, R. Tamma and T. Annese, Exp. Cell Res., 2021, 405, 112716 CrossRef CAS PubMed.
- Z. Li, T. Qu, C. Ding, C. Ma, H. Sun, S. Li and X. Liu, Acta Biomater., 2015, 13, 88–100 CrossRef CAS PubMed.
- O. Oliviero, M. Ventre and P. A. Netti, Acta Biomater., 2012, 8, 3294–3301 CrossRef CAS.
- J. Tang, X. Cui, Z. Zhang, Y. Xu, J. Guo, B. G. Soliman, Y. Lu, Z. Qin, Q. Wang, H. Zhang, K. S. Lim, T. B. F. Woodfield and J. Zhang, Adv. Healthcare Mater., 2022, 11, 2100312 CrossRef CAS.
- H. Adelnia, R. Ensandoost, S. Shebbrin Moonshi, J. N. Gavgani, E. I. Vasafi and H. T. Ta, Eur. Polym. J., 2022, 164, 110974 CrossRef CAS.
- M.-N. Abdallah, S. D. Tran, G. Abughanam, M. Laurenti, D. Zuanazzi, M. A. Mezour, Y. Xiao, M. Cerruti, W. L. Siqueira and F. Tamimi, Acta Biomater., 2017, 54, 150–163 CrossRef CAS.
- J. Kim, Colloids Surf., B, 2020, 188, 110756 CrossRef CAS.
- B. Cerroni, R. Cicconi, L. Oddo, M. Scimeca, R. Bonfiglio, R. Bernardini, G. Palmieri, F. Domenici, E. Bonanno, M. Mattei and G. Paradossi, Heliyon, 2018, 4, e00770 CrossRef PubMed.
- M. V. Turturro, M. C. Christenson, J. C. Larson, D. A. Young, E. M. Brey and G. Papavasiliou, PLoS One, 2013, 8, e58897 CrossRef CAS PubMed.
- W. Lan, M. Xu, M. Qin, Y. Cheng, Y. Zhao, D. Huang, X. Wei, Y. Guo and W. Chen, Mater. Des., 2021, 204, 109652 CrossRef CAS.
- C.-H. Chuang, R.-Z. Lin, H.-W. Tien, Y.-C. Chu, Y.-C. Li, J. M. Melero-Martin and Y.-C. Chen, Acta Biomater., 2015, 19, 85–99 CrossRef CAS PubMed.
- E. A. Phelps, K. L. Templeman, P. M. Thulé and A. J. García, Drug Delivery Transl. Res., 2015, 5, 125–136 CrossRef CAS.
- F. Tang, X. D. Manz, A. Bongers, R. A. Odell, H. Joukhdar, J. M. Whitelock, M. S. Lord and J. Rnjak-Kovacina, ACS Biomater. Sci. Eng., 2020, 6, 1476–1486 CrossRef CAS PubMed.
- N. Davidenko, C. F. Schuster, D. V. Bax, R. W. Farndale, S. Hamaia, S. M. Best and R. E. Cameron, J. Mater. Sci. Mater. Med., 2016, 27, 148 CrossRef.
- B. Ma, T. Wang, J. Li and Q. Wang, Stem Cell Res. Ther., 2022, 13, 327 CrossRef CAS.
- H. Lai, B. Gong, J. Yin and J. Qian, Mater. Des., 2022, 218, 110663 CrossRef CAS.
- K. O. Rojek, M. Ćwiklińska, J. Kuczak and J. Guzowski, Chem. Rev., 2022, 122, 16839–16909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01172k |
| This journal is © The Royal Society of Chemistry 2024 |