
Stromal and tumor immune microenvironment reprogramming through multifunctional cisplatin-based liposomes boosts the efficacy of anti-PD-1 immunotherapy in pancreatic cancer†
Hang
Yu‡
,
Wenting
Zhu‡
,
Caiyan
Lin
,
Menglei
Jia
,
Xiaoxiao
Tan
,
Zhongwen
Yuan
,
Senglin
Feng
and
Pengke
Yan
 *
*
Department of Pharmacy, Biomedicine Research Center, Guangdong Provincial Key Laboratory of Major Obstetric Diseases, Guangdong Provincial Clinical Research Center for Obstetrics and Gynecology, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangzhou 510150, China. E-mail: gysyypk@126.com
First published on 3rd November 2023
Abstract
The dense stromal barrier in pancreatic cancer tissues blocks intratumoral delivery and distribution of chemotherapeutics and therapeutic antibodies, causing poor chemoimmunotherapy responses. We designed a multi-targeted pH-sensitive liposome which encapsulates cisplatin (Pt) in its water core (denoted as ATF@Pt Lps) and shows high affinity for uPAR receptors in pancreatic cancer cells, tumor-associated macrophages, and cancer-associated fibroblasts. Systemic administration of ATF@Pt Lps enabled overcoming the central stromal cellular barrier and effective drug delivery into tumor cells, resulting in a strong therapeutic response in a Panc02 cell derived transplanted tumor mouse model. More importantly, ATF@Pt Lps degradation of collagen contributes to the infiltration of CD8+ T cells into tumors as well as an enhanced accumulation of anti PD-1 monoclonal antibodies. Furthermore, the killing of tumor cells by Pt also leads to the release of tumor antigens, which promote the proliferation of immune cells, especially CD83+ cells, Th1 CD4+ cells, and CD8+ cytotoxic T cells, that converted an immunoscore “cold” pancreatic cancer into a pro-immune “hot” tumor. A further combination with an immune checkpoint agent, anti PD-1 antibodies that inhibit PD-1, can enhance tumor specific cytotoxic T cell response. Accordingly, ATF@Pt Lps displays multi-targeting, controlled drug release, stromal disruption, enhanced penetration, killing of cancer cells, modification of the immunosuppressive microenvironment, and enhancement of immunity. This study provides important mechanistic information for the further development of a combination of ATF@Pt Lps and anti PD-1 antibodies for the effective treatment of pancreatic cancer.
Introduction
Pancreatic cancer accounts for almost as many deaths (466![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) as cases (496000) because of its poor prognosis.1,2 Due to the fact that approximately 85% of pancreatic cancer patients receive their diagnosis in the late stages of the disease, surgically removing the tumor is extremely difficult.3 According to the guidelines of the American Cancer Society, the chemotherapeutic agent gemcitabine, alone or in combination with 5-fluorouracil (5-FU), oxaliplatin, irinotecan (CPT-11) and folic acid (FOLFIRINOX), is the standard clinical treatment strategy for pancreatic cancer.4,5 The immune checkpoint therapy has shown promising therapeutic responses in many types of human cancers.6 Nevertheless, pancreatic cancer patients had a poor response to immunotherapy.7–9 Increasing evidence shows that the dense stromal barrier in pancreatic cancer tissues blocks intratumoral delivery and distribution of therapeutic drugs.10 Stromal physical and immune suppressive biological barriers in the tumor microenvironment further limit the number and function of cytotoxic CD8+ T cells and prevent infiltration of cytotoxic T cells in the tumor.11,12 Several other factors contribute to the failure of immunotherapy in pancreatic cancer, such as the absence of immune checkpoint receptors, low immunogenicity of tumors (“cold tumors”), and the accumulation of regulatory T-cells (Tregs).5,13
000) as cases (496000) because of its poor prognosis.1,2 Due to the fact that approximately 85% of pancreatic cancer patients receive their diagnosis in the late stages of the disease, surgically removing the tumor is extremely difficult.3 According to the guidelines of the American Cancer Society, the chemotherapeutic agent gemcitabine, alone or in combination with 5-fluorouracil (5-FU), oxaliplatin, irinotecan (CPT-11) and folic acid (FOLFIRINOX), is the standard clinical treatment strategy for pancreatic cancer.4,5 The immune checkpoint therapy has shown promising therapeutic responses in many types of human cancers.6 Nevertheless, pancreatic cancer patients had a poor response to immunotherapy.7–9 Increasing evidence shows that the dense stromal barrier in pancreatic cancer tissues blocks intratumoral delivery and distribution of therapeutic drugs.10 Stromal physical and immune suppressive biological barriers in the tumor microenvironment further limit the number and function of cytotoxic CD8+ T cells and prevent infiltration of cytotoxic T cells in the tumor.11,12 Several other factors contribute to the failure of immunotherapy in pancreatic cancer, such as the absence of immune checkpoint receptors, low immunogenicity of tumors (“cold tumors”), and the accumulation of regulatory T-cells (Tregs).5,13
The combination therapy approach to treat cancer may be an effective way to remodel the tumor immunosuppressive microenvironment and improve response to immune checkpoint inhibitors (ICIs).14 For example, cisplatin (Pt) plus anti-programmed cell death 1 (anti PD-1) antibodies enhanced the treatment efficacy in advanced esophageal squamous cell carcinoma15 and head and neck squamous cell carcinoma.16 More interestingly, Pt was found to be more toxic to tumor cells than oxaliplatin; however, its use has been limited due to neurotoxicity, ototoxicity, and nephrotoxicity.17,18 Therefore, novel strategies are needed to minimize the systemic toxicity of Pt and effectively boost the effect.
Recently, nanomedicine delivery systems have played an important role in promoting the aqueous solubility of anticancer drugs, reducing side effects, and increasing tumor uptake of chemotherapeutic drugs.19,20 Liposomes are composed of phospholipid bilayers, which can encapsulate both hydrophobic and hydrophilic drugs with good safety.21 In addition, recent studies have demonstrated the efficacy of liposomal irinotecan (Onivyde) in patients with metastatic pancreatic cancer who have progressed on gemcitabine.22 The study provided the first clinical evidence supporting liposomal formulations in pancreatic cancer.23 However, liposomal drugs can only be passively targeted through the high permeability and retention effect of solid tumors, and cannot deliver drugs and kill tumor cells across the tumor stromal barrier inside the tumor.
A promising approach is to improve drug delivery efficiency by preparing a nanosystem that targets multiple cell types.24 uPAR is overexpressed in malignant tumor cells, angiogenic tumor endothelial cells, tumor-associated fibroblasts and macrophages, and is very low in normal tissues and organs, making it an ideal target molecule.25 The uPAR amino-terminal receptor binding peptide (ATF) has been shown to be effective in targeting uPAR.26 Lily Yang further demonstrated that ATF peptide-modified magnetic iron oxide nanoparticles carrying gemcitabine and doxorubicin can target tumor mesenchymal and tumor cells in pancreatic cancer and kill tumor cells more efficiently by destroying tumor mesenchymal cells and entering deeper into the tumor tissue.26,27 Noteworthily, ATF peptide-decorated liposomes co-loaded with cisplatin and rapamycin provide a new approach for treating metastatic pancreatic cancer.28 In another study, Zhai et al. found that ATF-modified liposomal delivery of β-elemene in combination with Pt was effective in improving the treatment of bladder cancer.29
Here, we propose the development of a multi-targeted pH-responsive liposome nanodrug (ATF@Pt Lps) that reduces immunosuppressive fibroblasts, overcomes the tumor mesenchymal barrier and promotes immune cell infiltration into mouse pancreatic tumors, specifically CD83+ cells, Th1 CD4+ cells and CD8+ cytotoxic T cells. In addition, tumor targeting increases cellular uptake of cisplatin, which efficiently delivers the drug to kill tumor cells and leads to the release of tumor antigens and activation of cytotoxic T cells, that converts an immunoscore “cold” pancreatic cancer into a pro-immune “hot” tumor. A further combination with an immune checkpoint blocker, anti PD-1 antibodies, can enhance tumor specific cytotoxic T cell response, providing a novel combination therapy for the effective treatment of pancreatic cancer (Scheme 1).
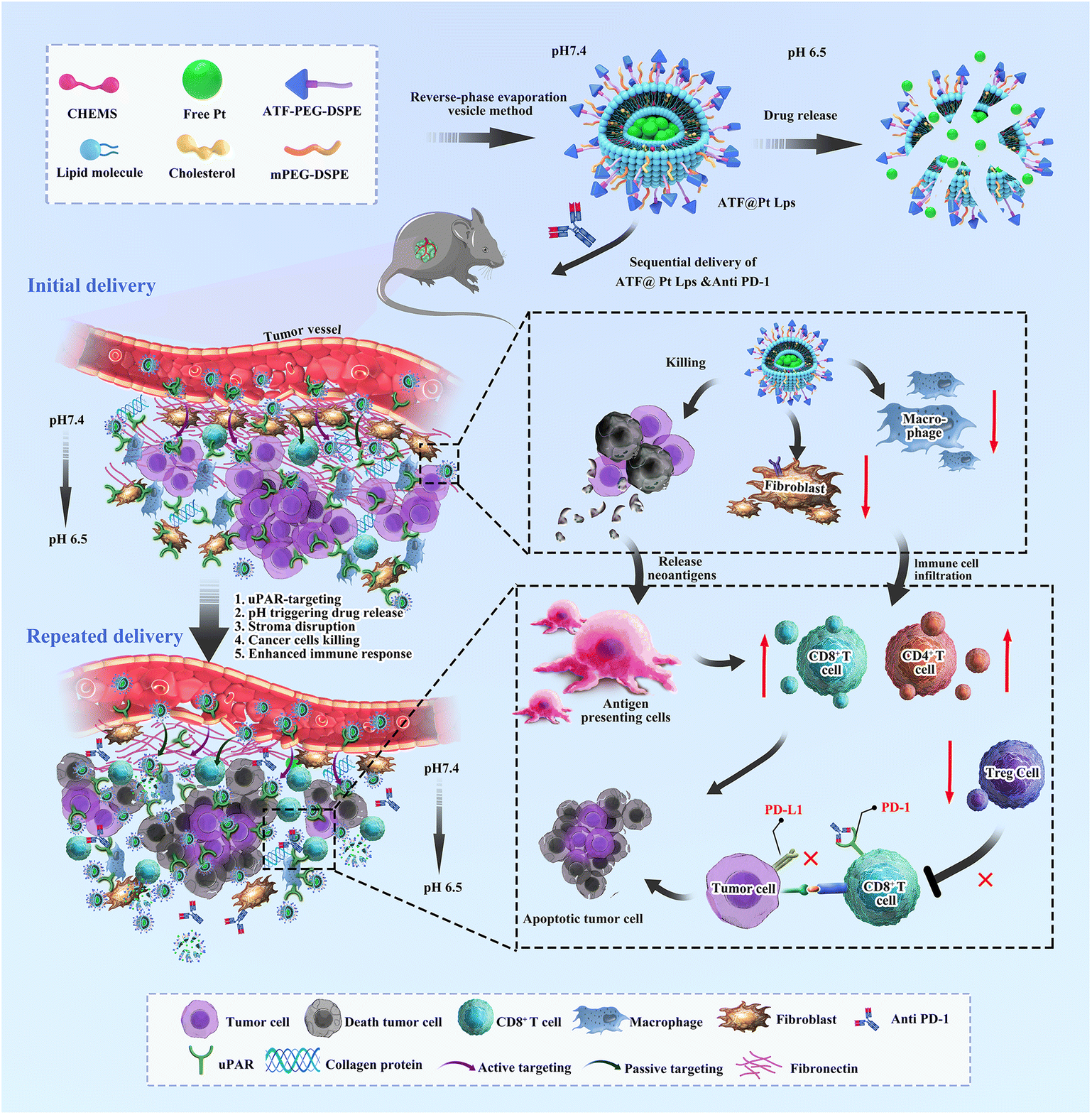 | ||
| Scheme 1 A new approach to potent anti PD-1 therapies based on multifunctional liposomes containing cisplatin. Using sequential delivery of the ATF peptide decorated cisplatin liposomes and anti PD-1 to treat pancreatic cancer in mice showed improved efficacy by enhancing drug penetration and remodelling the immunosuppressive microenvironment. | ||
Results
Preparation and characterization of ATF@Pt Lps
A schematic diagram of ATF@Pt Lps preparation and in vitro release is shown in Fig. 1A. DSPE–PEG2K–ATF was added to the liposomes to provide an active uPAR targeting effect. The TEM images of the two liposomes generally display a spheroid shape with a size of 50 and 70 nm, respectively (Fig. 1B and C), which was smaller than the diameters of 96.32 ± 0.4957 nm and 100.5 ± 0.2517 nm detected by dynamic light scattering (Fig. 1D), because of the drying process of particles during TEM sample preparation eliminating the expanded micelle of the particle core. The PDI values were 0.1488 ± 0.008 (Table S1†), indicating a homogeneous population of phospholipid vesicles. The zeta potential was −12.77 ± 0.416 mV, indicating a negatively charged surface on the liposomes (Fig. 1E). The encapsulation rate of the two liposomes can reach more than 90%, and the drug loading capacity of the two liposomes was about 5% (Table S1†). The Pt content of Pt Lps was 229.95 ± 22.53 μg mL−1. The average content of Pt in ATF@Pt Lps was 203.52 ± 17.95 μg mL−1. When the pH value of the suspension system was adjusted to about 6.5, the regular spherical shape of ATF@Pt Lps liposomes was destroyed (Fig. S2†), indicating that the pH value in the tumor environment affected the stability of the liposomes.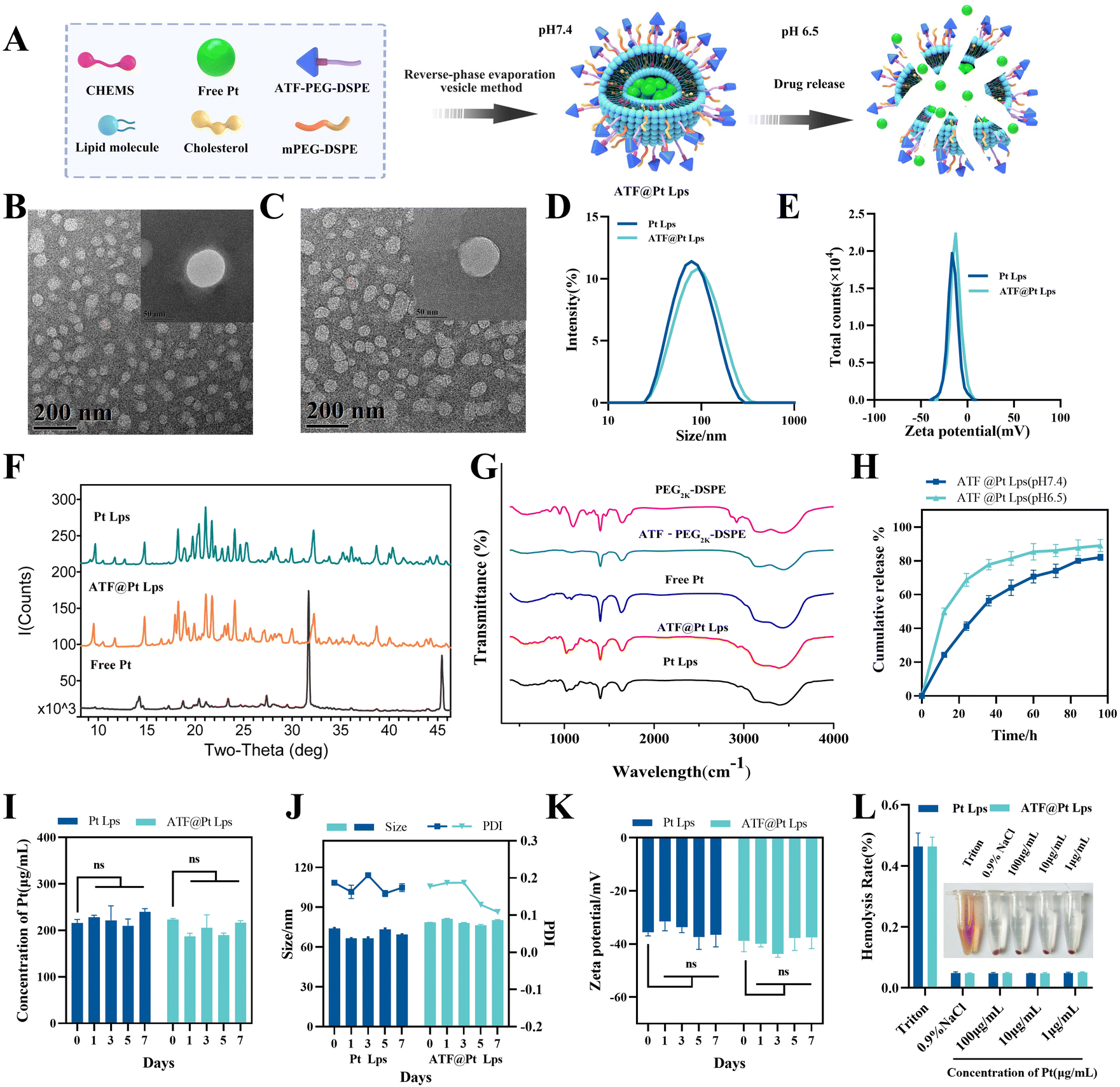 | ||
| Fig. 1 (A) Schematic illustration of the development and structure of cisplatin-loaded multifunctional liposomes (ATF@Pt Lps). (B) Transmission electron microscopy images of Pt Lps. (C) Transmission electron microscopy images of ATF@Pt Lps. (D) Size distribution of Pt Lps and ATF@Pt Lps. (E) Zeta potential diagram of Pt Lps and ATF@Pt Lps. (F) XRD spectrum of free Pt, Pt Lps and ATF@Pt Lps. (G) FTIR spectrum of mPEG2k–DSPE, DSPE–PEG2k–ATF, free Pt, Pt Lps and ATF@Pt Lps. (H) In vitro release kinetics of cisplatin in PBS at 37 °C under different pH from liposomes. (I) The content stability of liposomes stored at 4 °C detected by HPLC. (J) PDI, particle size and (K) zeta potential of liposomes obtained by dynamic light scattering. (L) Hemolysis test of Pt Lps and ATF@Pt Lps. | ||
The X-ray diffraction diagram of free Pt, Pt Lps and ATF@Pt Lps is shown in Fig. 1F. It shows that free Pt had a series of characteristic peaks at θ values of 14.28, 31.73, 45.49 and 56.50°, respectively. The XRD spectra of Pt Lps and ATF@Pt Lps exhibited no peak at a θ value of 31.73° because the drug was encapsulated in the liposomes, which significantly differed from the peaks of the free Pt. In particular, ATF@Pt Lps showed an extra peak at a θ value of 17.95° compared to Pt Lps.
The FTIR spectra obtained for cisplatin for injection, PEG2K–DSPE, ATF@PEG2K–DSPE, Pt Lps and ATF@Pt Lps are shown in Fig. 1G. In the spectra, the bands at 1750, 1450 cm−1 and 3280 cm−1 indicated the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO, and NH2 in PEG2K–DSPE and ATF@PEG2K–DSPE. The symmetric/asymmetric stretching vibration of the PO group was identified at 1190 cm−1 and 1250 cm−1 in PEG2K–DSPE. Upon the addition of Pt, the asymmetric stretching vibration spectrum of the P = 0 group of Pt Lps and ATF@Pt Lps shifted to a low frequency from 1250 cm−1, and the peak shape changed. In addition, the stretching vibration bands of P–O–C at 840 cm−1 appearing in the spectrum of PEG2K–DSPE also vanished due to the same reason. Moreover, the spectra of ATF@Pt Lps were similar to those of Pt Lps and free Pt, indicating that the presence of the ATF peptide did not affect the formation of liposomes.
C, CO, and NH2 in PEG2K–DSPE and ATF@PEG2K–DSPE. The symmetric/asymmetric stretching vibration of the PO group was identified at 1190 cm−1 and 1250 cm−1 in PEG2K–DSPE. Upon the addition of Pt, the asymmetric stretching vibration spectrum of the P = 0 group of Pt Lps and ATF@Pt Lps shifted to a low frequency from 1250 cm−1, and the peak shape changed. In addition, the stretching vibration bands of P–O–C at 840 cm−1 appearing in the spectrum of PEG2K–DSPE also vanished due to the same reason. Moreover, the spectra of ATF@Pt Lps were similar to those of Pt Lps and free Pt, indicating that the presence of the ATF peptide did not affect the formation of liposomes.
As shown in Fig. 1H, ATF@Pt Lps showed a slower release under physiological conditions (pH 7.4), with a release amount of 50% at 36 h. The liposomes at pH 6.5 showed a burst Pt release, with 40% of Pt released at 10 h, which indicated the pH-responsiveness of ATF@Pt Lps.
Physical stability studies of the liposomal formulations were performed by storage at 4 °C for a week. The particle size, zeta potential and drug content of Pt in the liposomes were determined at the end of the experiment. Changes in liposome size, zeta potential and drug content of Pt are presented in Fig. 1I–K. The liposomes stored at 4 °C had no risk of aggregation, fusion and drug leaking. Therefore, liposomes could provide an effective formulation to maintain drug leakage from the carrier at refrigerator temperature and a higher percentage of the drug was maintained in liposomal formulations for convenient drug delivery. In addition, in vitro hemolysis experiments showed that cisplatin concentrations of 1–100 μg mL−1 showed favorable hemocompatibility.
pH sensitivity-mediated ATF-modified liposomes enhance in vitro tumor sphere penetration and tumor and fibroblast cellular uptake
To investigate whether ATF peptide modified liposomes could increase the penetration of the drug, we constructed tumor spheres mixed with Panc02 and NIH/3T3 cells in vitro. As shown in Fig. 2A, in the normal physiological environment (pH 7.4), the green fluorescence of the coumarin 6 liposome (Cou-6 Lps) group was mostly enriched in the superficial layer of the tumor spheres, and the fluorescence of the ATF decorated coumarin 6 liposome (ATF@Cou-6 Lps) group penetrated deep into the tumor. In the tumor microenvironment at pH 6.5, due to the addition of CHEMS during the preparation of ATF@Cou-6 Lps, it was able to accelerate the release of the drug under the protonation of a low-pH situation, which made the fluorescence in the deep part of the tumor spheroid enhanced. Alternatively, this effect was diminished in the free ATF peptide pre-incubation group due to competitive binding to the uPAR receptor, diminishing its targeting capacity. The effect of ATF peptide modification and pH sensitive protonation on cellular uptake efficiency was then assessed by laser scanning confocal microscopy.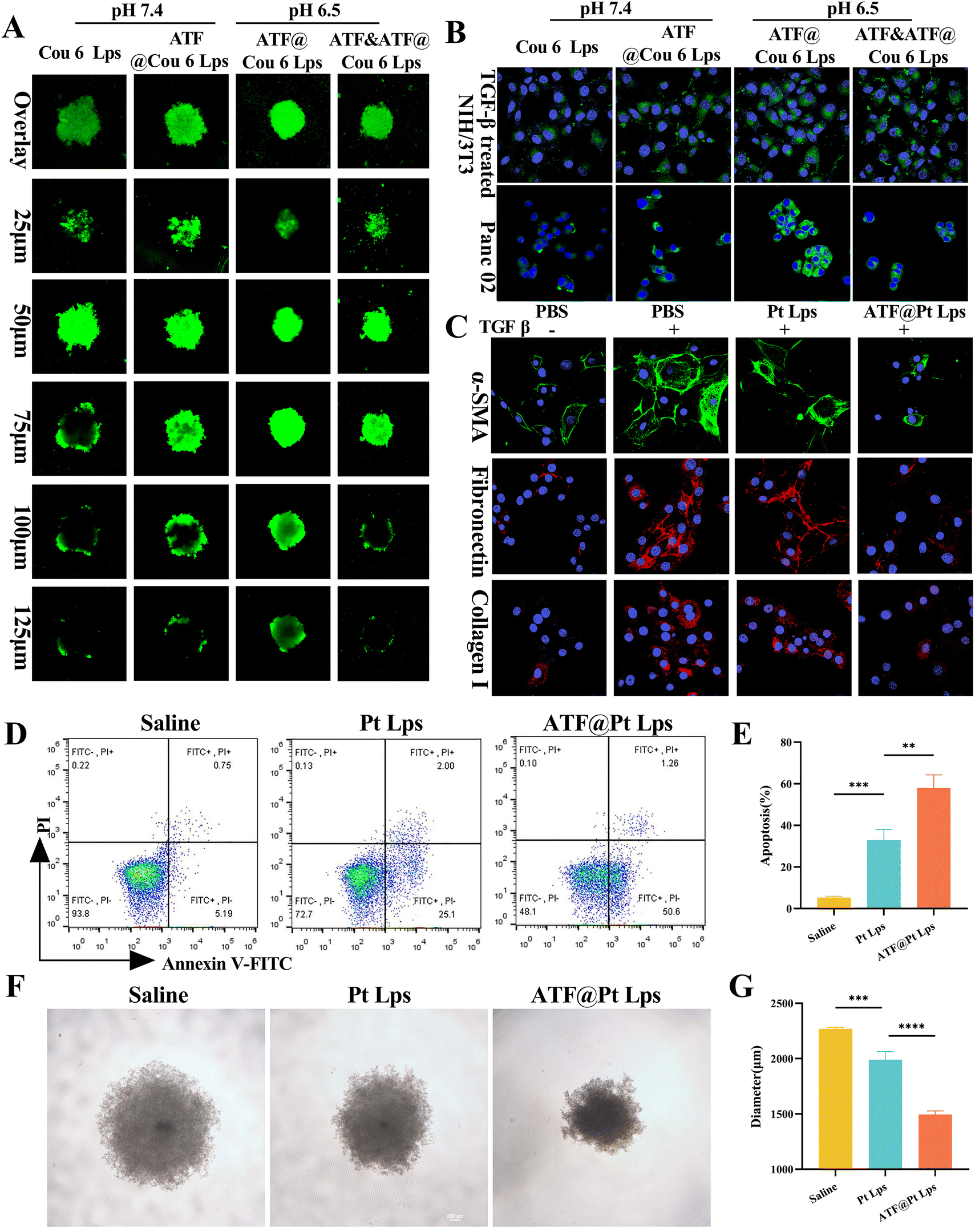 | ||
| Fig. 2 Antitumor efficacy in vitro of ATF@Pt Lps. (A) Z-axis continuous top-down scanning confocal images of the 3D tumor spheres treated with different liposomes loaded with coumarin 6 (Cou-6) under 10× magnification. The scanning interval is 25 μm. (B) Confocal images of TGF-β treated NIH/3T3 cells or Panc02 incubated with different preparations of Cou-6 under 60× magnification. (C) Fluorescence microscopy images of α-SMA, collagen I and fibronectin in TGF-β induced NIH/3T3 cells treated with different preparations of Cou-6 under 60× magnification. (D) Flow cytometry analysis of the killing ability of Pt Lps or ATF@Pt Lps to Panc02 cell and (E) the data analysis of the apoptosis rate. (F) Light microscopy images (magnification = 20×) of 3D tumor spheres of Panc02 cell treated with various formulations and (G) the diameter analysis of 3D tumor spheres (*p < 0.05, **p < 0.01, and ***p < 0.001). | ||
Fig. S3† illustrates that Panc02 cells express uPAR on their membrane. So, the uptake of ATF@Cou-6 Lps into Panc02 cells was slightly higher than that of Cou-6 Lps at pH 7.4 (Fig. 2B). However, pH 6.5 treatment significantly increased the cellular uptake of ATF@Cou-6 Lps compared to pH 7.4. Consistent with the Panc02 uptake results, pH sensitive protonation at pH 6.5 also increased the endocytosis efficiency of ATF@Cou-6 Lps on TGF-β induced NIH/3T3 activated fibroblasts (mimicking CAFs). Furthermore, pre-incubation with the free ATF peptide at pH 6.5 significantly reduced the intracellular fluorescence intensity in the ATF@Cou-6 Lps treated group in both cell lines. Overall, multi-targeted ATF peptide modification and CHEMS-mediated pH-sensitive protonation were shown to be the main factors promoting the intracellular accumulation of drugs in tumor cells and CAFs.
In vitro potency of ATF@Pt Lps against CAFs and tumor cells
Since the ATF peptide increased the uptake of liposomes by tumor cells and CAF cells, we examined the cytotoxic effects of ATF@Lps, Pt Lps and ATF@Pt Lps on Panc02 cells using the MTT assay, and the results are shown in Fig. S4.† ATF@Lps was essentially non-toxic to Panc02 cells as expected. Conversely, Pt Lps and ATF@Pt Lps showed toxicity that was concentration-dependent, and the IC50 of ATF@Pt Lps (0.27 μg mL−1) was lower than that of Pt Lps (0.33 μg mL−1) in Panc02 cells. We also further verified that ATF@Pt Lps inhibited tumor cell proliferation more strongly than Pt Lps by the cell cloning assay (Fig. S5†). In addition, the apoptosis results of FITC and PI double-stained cells (Fig. 2D and E) showed that the apoptosis rate of ATF@Pt Lps (57.95 ± 6.33%) was higher than that of the Pt Lps group (32.88 ± 5.12%).In vitro activation of NIH/3T3 cells was induced with TGF-β. We found that TGF-β promotes cell growth of NIH/3T3, while Pt Lps and ATF@Pt Lps had cytotoxic effects on TGF-β-induced NIH/3T3 cells in a concentration-dependent manner, and ATF@Pt Lps was stronger than Pt Lps in cytotoxicity (Fig. S6†). In addition, we also examined the effects of Pt Lps and ATF@Pt Lps on markers of activated CAFs by ICC, and the results are shown in Fig. 2C. ATF@Pt Lps significantly decreased the expression of α-SMA, collagen I, and fibronectin, which indicated that ATF@Pt Lps had a destructive effect on the ECM matrix. The growth inhibition of tumor spheres is shown in Fig. 2F and G; compared with the untreated group, the tumor diameters of Pt Lps and ATF@Pt Lps groups were 2269.12 ± 12.73 μm, 1618.75 ± 18.89 μm, and 1495.03 ± 33.28 μm, respectively, and the ATF@Pt Lps group inhibited the growth of 3D tumor spheres more effectively than Pt Lps.
In vivo distribution of ATF@Dir Lps and drug penetration in tumor tissues
To study the biodistribution of multi-targeted ATF peptide modified liposomes, a matrix-rich xenograft nude mouse model was developed in this study. Mice were injected intravenously with Dir Lps or ATF@Dir Lps, and then analyzed using an in vivo fluorescence imaging system after 2, 4, 8, and 24 hours. As shown in Fig. 3A and B, both Dir Lps and ATF@Dir Lps could reach the tumor quickly, but the accumulation of ATF@Dir Lps in the tumor was significantly higher than that of Dir Lps, which was probably due to the decoration of uPAR and the pH sensitive characteristic. Meanwhile, the fluorescence intensity of tumors in ATF@Dir Lps treated mice at each time point was all higher than that of the Dir Lps group (Fig. 3B), which confirmed the uPAR targeting feature again. Notably, both liposomes showed a relatively longer sustained release confirmed by the higher fluorescence intensity after 24 h of treatment with Dir compared with the initial values in concord with the feature of in vitro release of the liposomes. In the isolated organ samples, the fluorescence signal was distributed in the liver and spleen, which was attributed to the innate capture of the reticuloendothelial system associated with liposomes (Fig. 3C). Interestingly, the tumor treated with ATF@Dir Lps showed an overwhelming fluorescence signal than that with Dir Lps, but other organs such as the liver showed no differences (Fig. 3C and D), which manifested the effective tumor targeting endowed by the successful decoration of uPAR.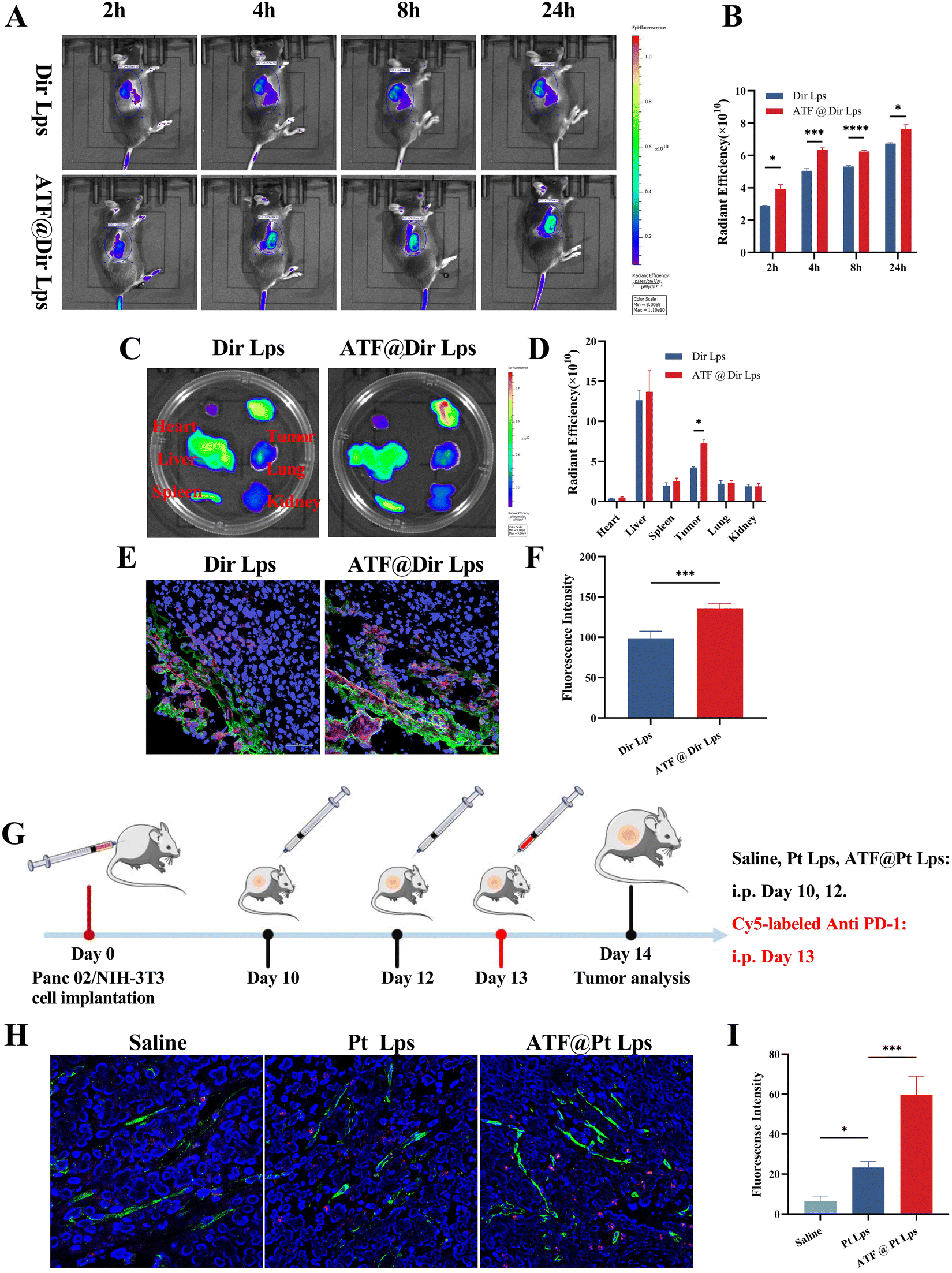 | ||
| Fig. 3 Enhanced tumor accumulation and penetration of model drug delivered by ATF decoration of liposomes. (A) In vivo radiant imaging of ectopic tumor bearing mice treated with Dir Lps or ATF@Dir Lps at different time points and data analysis (B). (C) Radiant images of the organs of xenograft mice treated with different Dir preparation at the end point and the relative radiant efficiency analyze (D). (E) Confocal images (magnification = 40×) of the distribution of the Dir in the tumor slice and the data analysis (F). (G) Schematic schedule of treatment for tumor accumulation and penetration analysis of anti PD-1 in Panc02 tumor-bearing mice. (H) Colocalization fluorescence images (magnification = 40×) of Cy5-labeled anti PD-1 (red) with endothelial cells labeled with FITC-conjugated CD31 antibodies (green) in tumor sections and (I) relative anti PD-1 antibodies in tumors of Panc02 tumor-bearing mice (*p < 0.05, **p < 0.01, and ***p < 0.001). | ||
Next, CAFs were labeled with green α-SMA antibodies and the intra-tumor penetration of various agents labeled with red was further investigated by CLSM. As shown in Fig. 3E and F, Dir Lps accumulated mainly in the CAFs, the outer layer of the tumor. In contrast, ATF peptide modification not only helps liposomes to accumulate in tumors, but the signals of ATF@Dir Lps within tumor tissues overlap significantly with those of CAFs, and these indicate that ATF@Dir Lps can be distributed around CAFs.
ATF@Pt Lps efficiently enhance tumor penetration of anti PD-1 antibodies
The abundant ECM in the pancreatic cancer tumor tissue severely affects tumor accumulation and penetration of anti PD-1 antibodies. Based on the ECM-damaging ability of ATF@Pt Lps, we evaluated the effect of ATF@Pt Lps on tumor penetration of anti PD-1 antibodies in Panc02 tumor-bearing mice. The administration schematic is shown in Fig. 3G, where Panc02 tumor-bearing mice were injected intravenously with saline, Pt Lps, or ATF@Pt Lps every other day, followed by intraperitoneal injection of Cy5-modified anti PD-1 antibodies. 24 hours after injection, the tumor tissues were stained with FITC-coupled CD31 antibodies and then analyzed by confocal microscopy (Fig. 3H and I). Due to the dense tumor ECM, the anti PD-1 antibodies co-localized with CD31-labeled tumor vessels in the saline group, indicating less anti PD-1 penetration toward the extravasation in the untreated group. In the Pt Lps treated group, extravasation of Cy5-PD-1 was also not significantly increased. Surprisingly, treatment with ATF@Pt Lps resulted in a wide distribution of anti PD-1 antibodies in the tumor tissue, indicating that anti PD-1 antibodies exuded from tumor vessels and then infiltrated into the tumor tissue. Additionally, Fig. S7† indicated that there was no difference between groups in terms of the number of new blood vessels (FITC-green fluorescence optical density) and that antibody penetration gradually decreased in deeper tissues. In the ATF group, the total number of antibodies that penetrated was clearly greater than in the other two groups. These results reveal that tumor penetration of anti PD-1 antibodies is through the disruption of the ECM matrix in the tumor tissue by ATF@Pt Lps.ATF@Pt Lps combined with anti PD-1 monoclonal antibodies inhibits pancreatic cancer tumor growth via regulations both on tumor cells and CAFs
Pt exerts a better anti-tumor effect when administered continuously at high doses, but can also produce serious side effects. Our results shown in Fig. S8† confirmed this argument. Administration of 2.5 mg kg−1, 5 mg kg−1 and 7.5 mg kg−1 Pt significantly reduced the body weight of mice while slowing down the tumor growth; especially in the 7.5 mg kg−1 group, the mice lost about 35% of their body weight (Fig. S9†). The results of HE staining of kidney tissues (Fig. S10†) showed that both 5 mg kg−1 and 7.5 mg kg−1 Pt administration caused acute kidney injury in mice. As a result, we employed nanocarriers to deliver Pt, at a dosage of 2.5 mg kg−1.The regimen for the combination of ATF@Pt Lps and anti PD-1 monoclonal antibodies is shown in Fig. 4A. In Fig. 4B, anti PD-1 monoclonal antibodies (1709.389 ± 127.445 mm3) and Pt Lps (1061.336 ± 143.090 mm3) exhibited slight inhibition of tumor growth, probably due to poor target penetration and low dose. In contrast, the tumor volume of ATF@Pt Lps (601.156 ± 90.762 mm3) was reduced, but the combined administration of ATF@Pt Lps and anti PD-1 monoclonal antibodies significantly inhibited tumor growth in the Panc02 mouse pancreatic cancer model, which had the smallest tumor volume (356.270 ± 58.776 mm3) compared to the other groups. The tumor inhibition rate of 80% was calculated for the ATF@Pt Lps + anti PD-1 monoclonal antibody group. Similarly, the tumor bulk map was observed (Fig. 4C) and the removed tumors were weighed, which showed that the tumor weight in the ATF@Pt Lps group (0.85 ± 0.22) was smaller than that in the Pt Lps group (1.33 ± 0.12), while the mean tumor weight of mice in the ATF@Pt Lps + anti PD-1 monoclonal antibody treatment group (0.48 ± 0.05) was significantly lower than that in the single treatment group (Fig. 4D), indicating that ATF modification significantly increased the concentration of the drug at the tumor site as well as the effectiveness of the combination treatment.
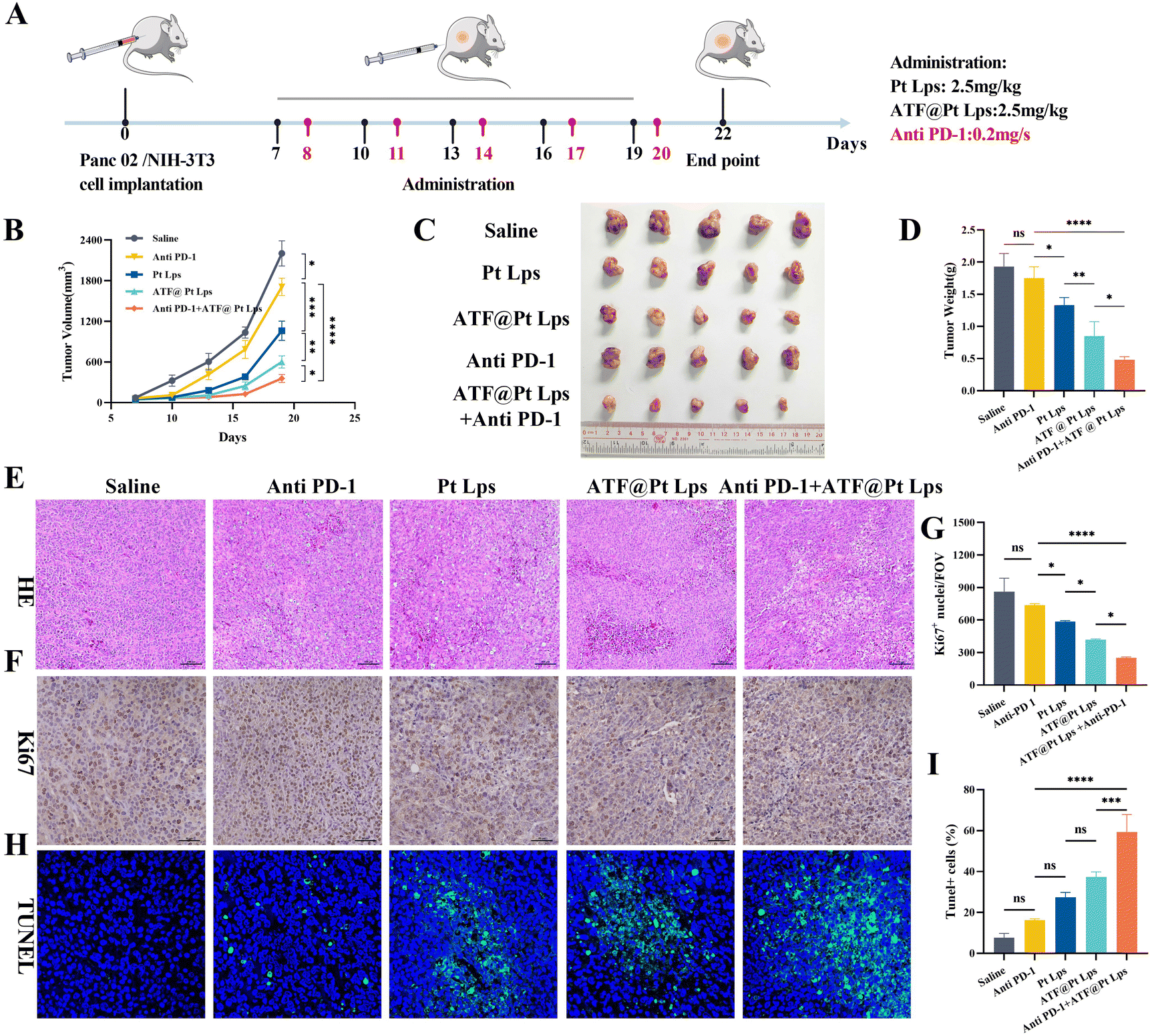 | ||
| Fig. 4 In vivo therapeutic efficacy of ATF@Pt Lps. (A) Schematic representation of the treatment regimen. (B) Tumor volume curve diagram measured throughout the study. (C) Photograph of the ex vivo tumor and (D) tumor weights measured at the endpoint of the efficacy studies. (E) Photograph of the tumor sections stained with HE under 20× magnification. (F) Ki67 expression in the tumor tissue was detected by immunohistochemical staining images (magnification = 40×) and data analysis (G). (H) TUNEL staining images (magnification = 40×) and data analysis (I) showed the apoptotic cells. All quantitative data are represented as mean ± SD, and quantified by 3 randomly selected observation fields using ImageJ software (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). | ||
As shown in Fig. 4E, H&E staining images show significant tumor necrosis in the ATF@Pt Lps + anti PD-1 monoclonal antibody group. In addition, we further verified the effects on tumor cell proliferation and apoptosis after drug treatment by Ki67 staining and TUNEL staining. The Ki67-positive area of tumor sections after ATF@Pt Lps + anti PD-1 monoclonal antibody treatment was significantly lower than that of the other groups (Fig. 4F and G). The TUNEL assay confirmed that the combination treatment induced a significant amount of tumor cell apoptosis (Fig. 4H and I). Overall, the therapeutic effect of ATF@Pt Lps was superior to that of the Pt Lps group, while the combination of ATF@Pt Lps and anti PD-1 monoclonal antibodies achieved overwhelming anti-cancer performance in all treatments.
As the largest stromal cell population in the TME, CAFs secrete collagen and integrate through fibronectin to constitute the dense extracellular matrix. As shown in Fig. 5A, the targeting of CAFs by ATF@Pt Lps significantly disrupted the stroma in the tumor microenvironment. Firstly, Masson staining results (Fig. 5B and C) showed that ATF@Pt Lps significantly reduced collagen fibers in tumor tissues compared with Pt Lps. Following this, the changes in ECM proteins were further examined by IHC, as illustrated in Fig. 5D–I. Compared with the saline group, α-SMA, collagen I, and fibronectin in tumors of ATF@Pt Lps treated mice were significantly attenuated compared with the saline group. In summary, the tumor cell and CAF targeting ability of ATF@Pt Lps overcame the tumor mesenchymal barrier and efficiently delivered drugs to kill tumor cells.
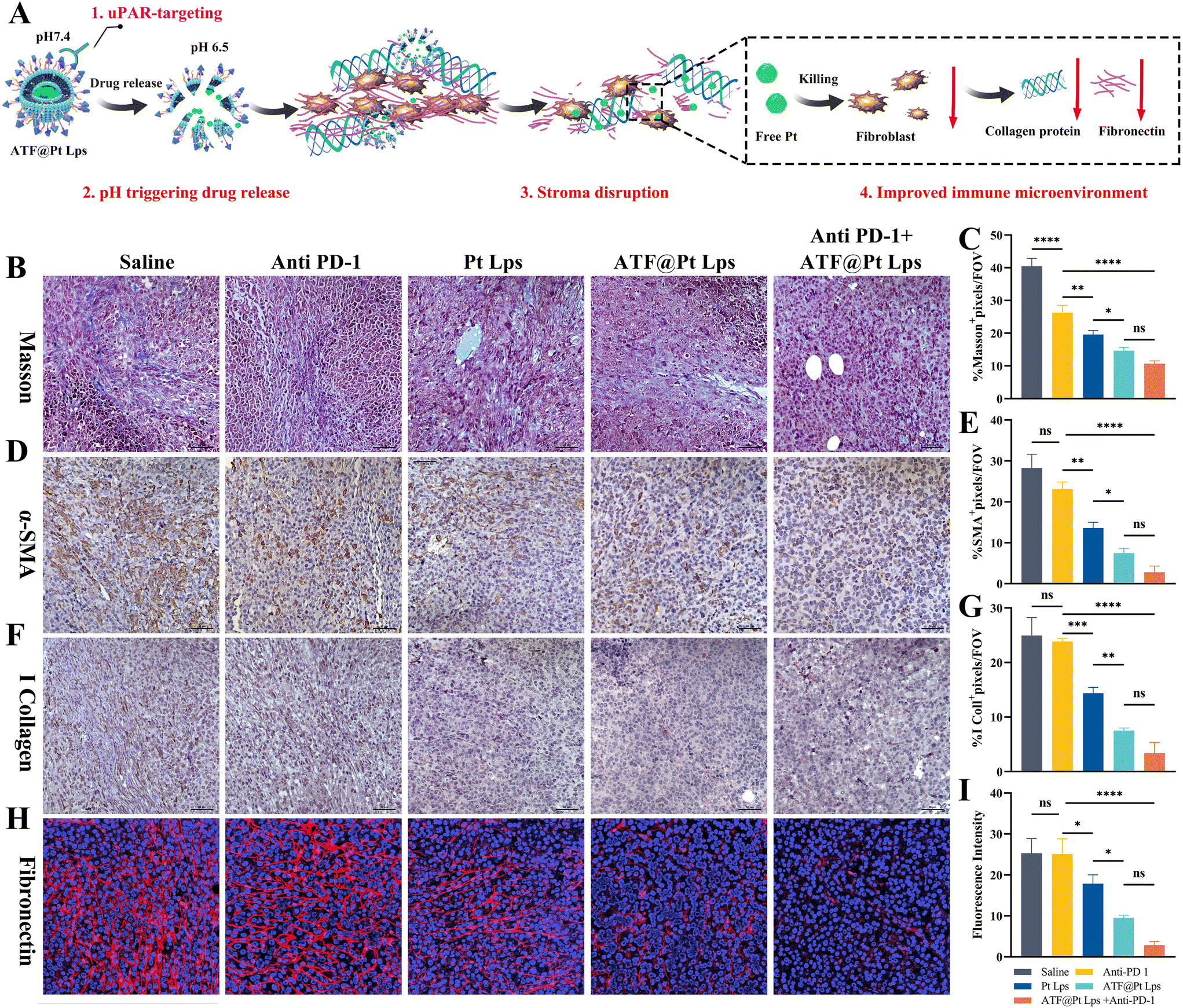 | ||
| Fig. 5 (A) Schematic illustration of the ATF@Pt Lps-remodelled tumor microenvironment by inhibiting the expression of collagenous fiber, α-SMA and fibronectin to decrease the secretion of collagen I. (B) Tumor sections were stained with Masson Trichrome and quantitative analysis was carried out (C). (D) Immunohistochemical staining of tumor slices stained with the α-SMA antibody and the relative content analysis (E). (F) Images of collagen I staining of the tumor slices and content analysis (G). (H) Confocal images of fibronectin immunofluorescence staining of tumor tissues and quantitative analysis of fluorescence intensity in tumor tissues (I) (magnification = 40×, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). | ||
ATF@Pt Lps remodel the immune microenvironment to potentiate anti PD-1 therapy in pancreatic cancer
To determine the effect of ATF@Pt Lps treatment on immune cells and immune responses in the tumor microenvironment, tumor tissue sections from mice with pancreatic cancer after receiving five ATF@Pt Lps treatments were examined for CD83+, CD4+ and CD8+ T cell numbers by IHC or IF. The results showed that ATF@Pt Lps treatment significantly increased the infiltration of CD83+, CD4+ and CD8+ T cells in the tumors (Fig. 6A–F). Furthermore, many immunosuppressive cells were present at high levels in the tumor tissue (Fig. 6G–J), such as Foxp3+ regulatory T cells and CD163+ M2 type macrophages. However, after ATF@Pt Lps treatment, the number of these immunosuppressive cells was significantly reduced in the tumor tissue. | ||
| Fig. 6 Immunofluorescence of tumor sections demonstrating CD83 cell infiltration (A) and corresponding data analysis (B). Immunohistochemical staining and semi-quantification analysis of CD4 (C and D), CD8 (E and F), CD163 (G and H) and Treg (I and J) cells in tumor tissues of mice treated with various formulations. The tumor infiltrated with CD4 (K and L) and CD8 (M and N) cells detected by flow cytometry and corresponded quantification analysis. All images were observed under 40× magnification. (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001). (O) Enhanced chemoimmunotherapy of ATF Pt Lps combined with anti PD-1 by regulating the immune cell populations. | ||
The functional status of various T cells was further determined by using flow analysis, as shown in Fig. 6K–N. Compared to saline (0.247 ± 0.046%), anti-PD-1 (0.512 ± 0.096%), Pt Lps (0.369 ± 0.085%) and ATF@Pt Lps (1.334 ± 0.896%)-treated mice, ATF@Pt Lps + anti PD-1-treated mice (2.159 ± 0.778%) showed that CD8+ TIL infiltration in their tumors (CD8+, CD3+) was significantly increased, indicating reactivation of depleted T cells, and CD4+ T results were consistent with them, suggesting that uPAR-targeted ATF@Pt Lps activates T cell activity and that combination therapy with anti-PD-1 monoclonal antibodies resulted in a significant increase in CD4+ and CD8+ T cell levels in tumor tissues. Moreover, the levels of IFN-γ and TNF-α in tumor tissues were also measured after treatment, as shown in Fig. S11 and S12.† Compared with mice treated with saline, no significant changes in IFN-γ and TNF-α levels were observed in mice treated with anti PD-1 and Pt Lps, while ATF@Pt Lps + anti PD-1 treated mice showed a significant increase, suggesting activation of T-cell antitumor activity.
Overall, the disruptive effect of ATF@Pt Lps on the tumor barrier increases the infiltration of cytotoxic T cells in pancreatic cancer tumor tissues. Additionally, the effective killing of tumor cells by ATF@Pt Lps could release tumor-associated antigens, increase antigen-presenting cells in the tumor microenvironment, and promote CD4+ and CD8+ cell proliferation, while anti PD-1 monoclonal antibodies in combination with ATF@Pt Lps would further boost tumor-specific cytotoxic T cell responses (Fig. 6O). These responses can lead to increased anti-tumor immunity, and eventually inhibit tumor development and progression.
The safety of ATF@Pt Lps combined with anti PD-1 monoclonal antibodies in vivo
In this experiment, we chose 2.5 mg kg−1 as the dose of Pt Lps and ATF@Pt Lps administration, and liposome-loaded Pt modified by the ATF peptide had no effect on mouse body weight compared with the saline group, and the combination with anti PD-1 monoclonal antibodies did not show significant weight loss (Fig. 7A).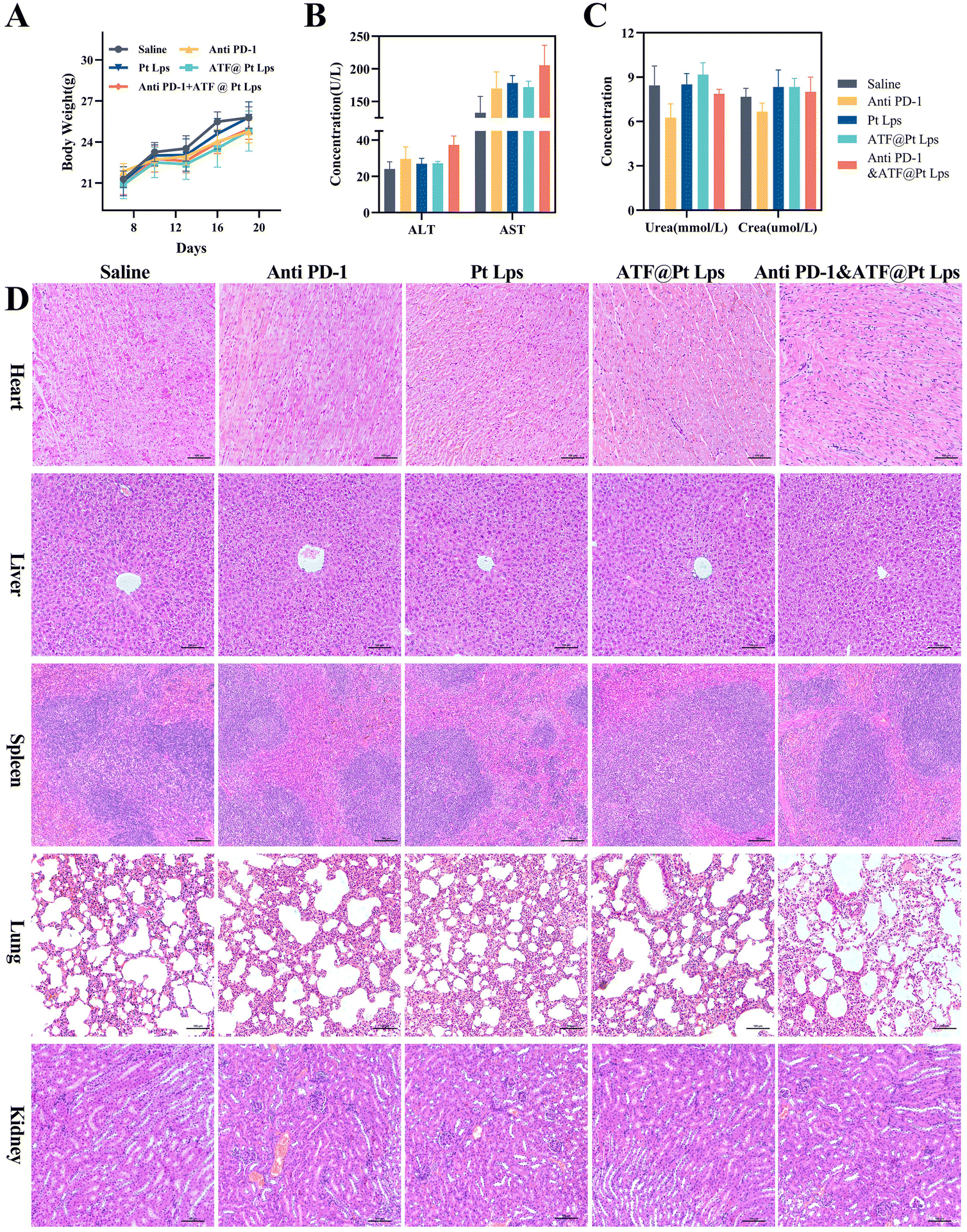 | ||
| Fig. 7 The safety of various Pt formulations by intraperitoneal injection. (A) The body weight change curve of tumor-bearing mice. The concentration analysis of makers of liver (B) and kidney (C) function. (D) Photograph of H&E staining of the heart, liver, spleen, lung, and kidneys (20×). | ||
Moreover, blood biochemical tests were performed to evaluate the safety of the combination, and the results showed no significant liver function abnormalities, including ALT and AST, and creatinine and urea were not elevated in the administration group (Fig. 7B and C). H&E staining further showed no significant abnormalities in the major organs (including the heart, liver, spleen, lungs, and kidneys) in each group (Fig. 7D).
Discussion
In this study, we developed a multifunctional cisplatin-based liposome in combination with the anti PD-1 antibodies for the treatment of pancreatic cancer. ATF@Pt Lps, which targets both tumor cells and CAFs with highly expressed receptors, overcomes the mesenchymal barrier and increases the penetration of anti PD-1 antibodies in the tumor tissue and the number and distribution of cytotoxic T cells in the tumor microenvironment. Moreover, tumor targeting is highly effective at killing tumor cells, leading to the release of tumor antigens and modulating the immune microenvironment to enhance tumor-specific immune responses, and further combination with the immune checkpoint inhibitor anti PD-1 abs enhances tumor-specific cytotoxic T cell responses. Thus, we confirmed that the combination strategy may induce potent anti-tumor immunity to inhibit pancreatic cancer growth.The dense stromal barrier in pancreatic cancer tissues is characterized by reduced normal blood vessels and a significant increase in the extracellular matrix as well as increased macrophage numbers and proliferation of activated fibroblasts.36 Creating physical and immunosuppressive biological barriers in the tumor microenvironment, not only blocks the delivery of chemotherapeutic agents and therapeutic antibodies to the tumor tissue, but they also limit the number and function of cytotoxic T cells (CD8+) within the tumor tissue and prevent direct contact between cytotoxic T cells and tumor cells.12,37 Currently, nanomedicine-based immunotherapies attempt to target tumor cells,38,39 cancer mesenchymal fibroblasts,40 or immune cells.39,41,42 The immunological activity of multi-targeted nano pharmaceuticals therapy28,43 is greater than that of single-targeted nanoparticles for tumor cells.44 ATF@Pt Lps were successfully prepared in our study, and in vitro tumor sphere infiltration and live imaging experiments demonstrated that liposomes modified with ATF peptides enriched drugs in deep tumors, suggesting that the ATF peptide targets mesenchymal tumor and cancer cells and disrupts the stromal barrier, decreasing the expression of α-SMA, collagen I, and fibronectin. Tumor penetration of anti PD-1 antibodies in vivo further confirms that the disruption of the tumor mesenchymal barrier by ATF@Pt Lps improves anti PD-1 antibody intra-tumor delivery and distribution. This is consistent with the previous studies,33,45 the collagen-degrading capacity of Met@Man-MPs contributes to the infiltration of CD8+ T cells into tumor interiors and enhances tumor accumulation and penetration of anti PD-1 antibodies. Nevertheless, the pancreatic mesenchyme is considered the “guardian” against tumor metastasis, and an intact outer stroma can prevent tumor spread effectively.46 According to Na Yu et al., nanomedicine-based strategies for matrix homeostasis remodelling appear promising for treating pancreatic cancer.47 The liposomes we used are matrix-targeted and pH-responsive liposomes, which should, most likely, result in the destruction of only the central mesenchyme, while leaving the external mesenchyme unharmed.
Furthermore, the tumor-targeting nanodrug ATF@Pt Lps can directly kill tumor cells. As determined by flow cytometry and in vitro cell cloning, ATF@Pt Lps inhibited cell proliferation and induced cell apoptosis more effectively than Pt Lps. The Ki67 staining and TUNEL staining also confirmed that the combined treatment effectively inhibited cancer cell proliferation and induced apoptosis in vivo. Novel experimental evidence suggests that the anti-tumour activity of chemotherapy is not only due to the direct cytotoxic effects on cancer cells but also potentially due to the stimulation of the host immune response.48 Tumor cells that are apoptosizing or necrotic release tumor antigens, which are taken up by more antigen-presenting cells and processed and presented to CD4+ and CD8 T+ cells, leading to their activation and proliferation.49,50 Interestingly, our results show that tumor cells are killed by ATF@Pt Lps and that increased expression of CD83+ in the tumor microenvironment. The CD83 molecule is a key marker molecule that appears on the surface of dendritic cells (DCs) and activates the function of cytotoxic T cells by stimulating their growth. Moreover, several previous studies have also demonstrated the mechanism by which Pt enhances tumor immunogenicity. For example, Pt nanoparticles significantly increased the release of the chemokine (C–X–C motif) ligand 10 (CXCL10), which correlated with further tumor infiltration by CD8+ T cells.51 Besides, Fournel et al. found that Pt treatment could synergize with PD-1/PD-L1 blockade to increase the clinical response, in particular through early and sustainable enhancement of PD-L1 expression.52
In cancer immunotherapy, nanomedicines suppress immunosuppressive cells (tumor-associated macrophages) and reduce immunosuppressive molecule expression, transforming the immunosuppressive TME into immune-supportive cells.44,53,54 uPAR is expressed in macrophages, our study showed that ATF@Pt Lps significantly decreased the number of M2-type macrophages (CD163+) and suppressive regulatory T cells (Treg, FOXp3+), while increasing the number of CD4+T and cytotoxic T cells (CD8+T) in the tumor microenvironment. As a result of the reduction of immunosuppressive cells, the function of these cells in suppressing CD4+ T and CD8+ T cell activation and proliferation is diminished, ultimately remodelling the immunosuppressive tumor microenvironment. The combination of PD-1 inhibitors further enhances the activation of tumor antigen-specific T cells and their ability to kill tumor cells directly.
In general, immunochemotherapy using PD-1/PD-L1 antibodies in combination with chemotherapeutic agents has become a mainstream treatment for cancer patients.14,55–57 For example, a combination of gemcitabine and anti PD-1 antibodies enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis.58 However, Glorieux showed that Pt-based chemotherapy is an excellent choice for combination with immune checkpoint antibody to achieve favorable clinical outcome than the combination of gemcitabine with immunotherapy.59 More importantly, we confirmed that ATF@Pt Lps can effectively reduce the toxicity of cisplatin, which is consistent with the report of Lily Yang et al.27 Taken together, the ATF@Pt Lps prepared in this study solved the dilemma of conventional liposomes by increasing active targeting and bioavailability. In addition, the combination of ATF@Pt Lps with anti PD-1 antibodies is a very promising treatment for pancreatic cancer.
Conclusion
In this study, we developed ATF peptide-decorated liposomes carrying cisplatin, denoted as ATF@Pt Lps, and combined them with anti PD-1 antibodies for the therapy of a murine model of pancreatic cancer. The ATF@Pt Lps decorated with the ATF peptide could target malignant tumor cells, tumor-associated macrophages, and cancer-associated fibroblasts. Meanwhile, upon the incorporation of pH-responsive materials into the shell of ATF@Pt Lps, the shell collapsed under the acidic tumor microenvironment to release the chemical drug, but remained stable under normal physiological conditions. Collectively, the sequential administration of chemical nano-drugs and anti PD-1 antibodies enabled the therapy strategy to possess multifunctionality, including uPAR targeting, pH triggered drug release, disruption of the stroma, killing of cancer cells, enhancement of the immune response, and remodelling of the immunosuppressive microenvironment. Furthermore, the liposome is able to effectively decrease the toxicity of cisplatin without compromising its antitumor efficacy. In conclusion, ATF@Pt Lps provided an effective approach for targeted delivery of cisplatin, which is conjugated with immune checkpoint blockers to effectively combat pancreatic cancer.Experimental
Materials
The following reagents were obtained: ATF24 peptides Cys-Leu-Asn-Gly-Gly-Thr-Cys-Val-Ser-Asn-Lys-Tyr-Phe-Ser-Asn-Ile-His-Trp-Cys-Asn-Cys-Pro-Lys-Lys (purity >97.16%, ChinaPeptides, Shanghai, China), soybean lecithin (A.V. T, Shanghai, China), cholesterol (A.V. T, Shanghai, China), cholesterol hemic succinate (A.V. T, Shanghai, China), PEG2K-DSPE (A.V. T, Shanghai, China), CD4 antibody (Cat: 14-0041-82, Lot: 2212874, Invitrogen, UA), CD8 antibody (Cat: 14-0081-82, Lot: 2297466, Invitrogen, UA), Foxp3 antibody (Cat: ab215206, Lot: GR3292034-9, Abcam, UK), CD83 antibody (Cat: AF5233, RRID: AB_2837719, Affinity Biosciences, China), FITC anti-mouse CD3ε (Cat: 100203, Lot: B371019, Biolegend, UA), APC anti-mouse CD8a (Cat: 100712, Lot: B374670, Biolegend, UA), APC-anti-mouse CD4 antibody (Cat: 100412, Lot: B372226, Biolegend, UA), CD163 antibody (Cat: ab182422, Lot: GR3339055-16, Abcam, UK), α-SMA antibody (Cat: ab7817, GR3356520-4 Abcam, UK), collagen I antibody (Cat: ab260043, Lot: GR3440276-6, Abcam, UK), Ki67 antibody (Cat: GB121141, Lot: AC220621048, Servicebio, China), and fibronectin (Cat: 15613-1-AP, Lot: 00102536, Proteintech, China). ATF–PEG2k–DSPE was synthesized by Xi'an Rui Xi Biotechnology (Fig. S1†). Pt was purchased from QILU PHARMACEUTICAL (Shandong, China).Cell cultures
Mouse Panc02 pancreatic cancer cells were purchased from FengHuiShengWu (Changsha), mouse embryonic fibroblast NIH/3T3 cells were purchased from Procell Life Science & Technology Co., Ltd (Wuhan, China). Both cells were cultured in the RPMI 1640 medium (Gibico, UK) supplemented with 10% fetal bovine serum (FBS, Procell, Wuhan) in a humidified incubator at 37 °C containing 5% CO2.Animals
Male c57/BL mice (6–8 weeks) were purchased from BesTest Bio-Tech (Zhuhai, China) and kept in the SPF environment. All animal procedures were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee of the Third Affiliated Hospital of Guangzhou Medical University (approval number: 2023-002).Preparation of ATF decorated liposomes loaded with cisplatin (ATF@Pt Lps)
Fig. 1A shows the synthesis scheme and estimated structure of ATF@Pt Lps. Cisplatin was loaded into liposomes using the reverse phase evaporation method.30 Briefly, 5 mL CHCl3 with soya bean lecithin (100 mg), CHEMS (cholesterol hemic succinate, 10 mg), sodium cholesteryl sulfate (10 mg), ATF- PEG2K–DSPE (5 mg) and PEG2K–DSPE (15 mg) was added to a rotating flask and slowly evaporated under reduced pressure using a rotary evaporator at 50 °C to form a thin dry film on the inner wall. The film was redissolved in 15 mL ether and a solution containing 0.4 mg cisplatin in 5 mL 0.9% NaCl was added. The mixture was sonicated for 5 min, swirled by hand, and sonicated again for another 5 min in an ice water bath (Ningbo Scientz Biotechnology Co., Ltd, Ningbo, China). The resultant opalescent dispersion was rotary evaporated to disrupt the formed gel immediately and ensure the removal of residual organic solvent and the raw liposomes were acquired. Contiguously, the coarse liposome suspensions were sonicated using a Vibra cell probe sonicator to obtain the homogenized liposomes. The encapsulation rate and drug loading capacity of liposomes were determined by the ultrafiltration centrifugation method. 3 mL liposome solution was added to an ultrafiltration centrifuge tube (10000 Da), centrifuged at 3000 rpm for 20 min, and the filtrate and supernatant were precisely absorbed and unpackaged with 0.1% Triton. After dilution at an appropriate ratio of 1:5, the solution was filtered using a 0.45 μm filter membrane, the cisplatin content was determined by HPLC, and the encapsulation rate (EE) and drug load (DL) of the liposome were calculated.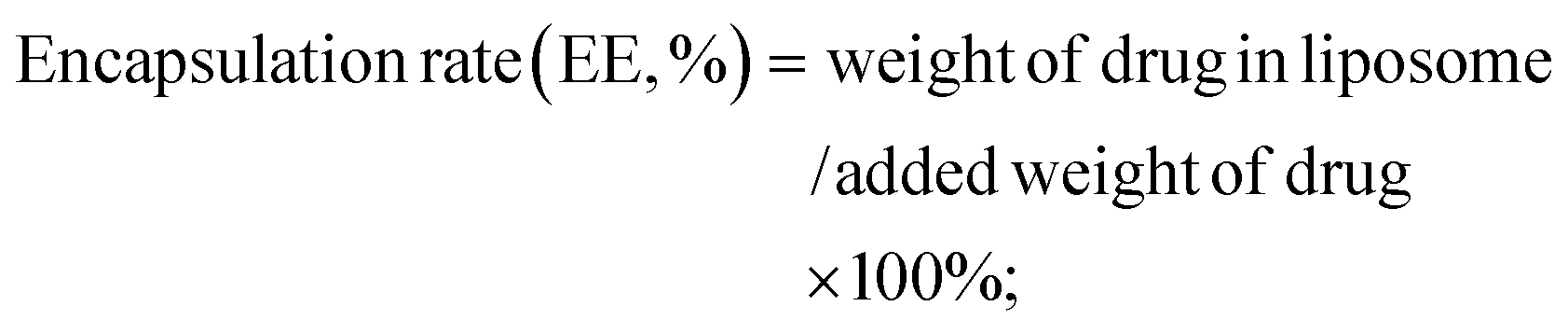
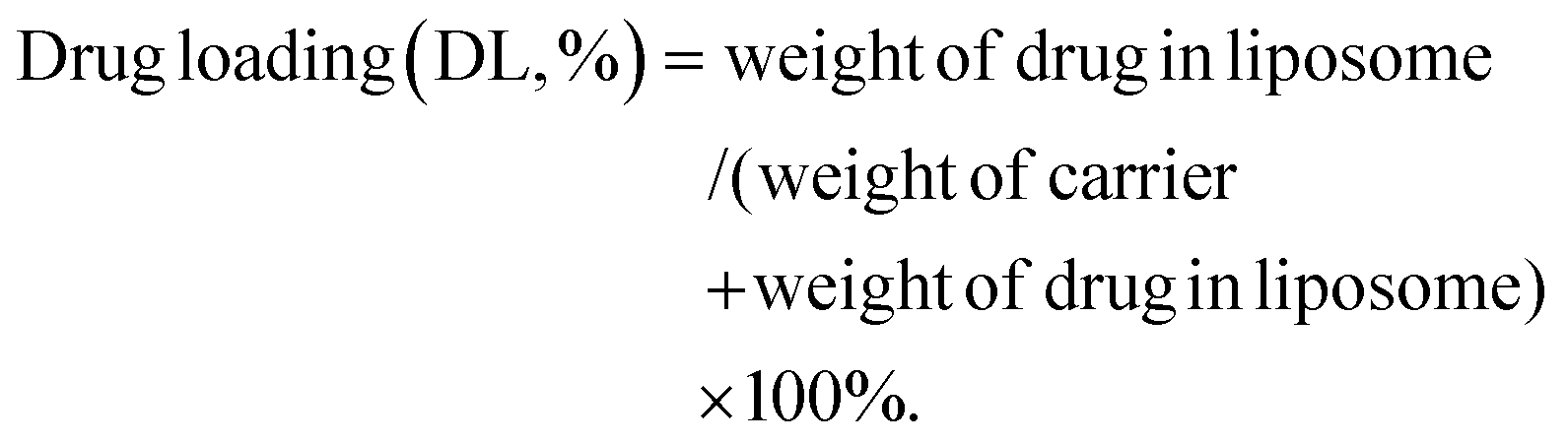
Liposomes loaded with Dir/Cou-6 were prepared by using the same method except that 2 mg of Dir or Cou-6 was added to the oil phase. The liposomal dispersions were mixed with 5% glucose and 5% mannitol and lyophilized to obtain the powder for storage. The powder of liposomes was redispersed with 2 mL 0.9% NaCl and passed through a 0.22 μm membrane filter before use.
Characterization of ATF decorated liposomes loaded with cisplatin (ATF@Pt Lps)
ATF@Pt Lps was dissolved in 0.9%NaCl at 150 μg mL−1 and filtered through a 0.45 μm filter. The particle size, polydispersity index, and ζ-potential of the liposomes were analyzed using a Zeta Sizer nano-ZS90 (Malvern, UK). The morphology of the liposomes was investigated using transmission electron microscopy (TEM) after the samples were deposited on a copper grid and stained with 1% acetic acid glaze. The FTIR spectra of the free Pt, Pt Lps, and ATF@Pt Lps were obtained using an FTIR spectrophotometer. The powder X-ray diffraction (PXRD) patterns were recorded on a diffractometer.In vitro stability and drug release assay of liposomes
Pt concentration was determined by HPLC, using a CLC-ODS-18 column (5 cm, 4.6 × 150 mm; Waters Corp., Milford, MA) maintained at 25 °C, with an ultraviolet detector at 277 nm. The mixture of 60% acetonitrile and 40% water (v/v) was used as a mobile phase and delivered at a flow rate of 0.5 mL min−1. The injection volume was 20 μL, and the retention time was about 5 min. A total volume of 1 mL ATF@Pt Lps suspension with a cisplatin concentration of 0.2 mg mL−1 was loaded in a dialysis bag with a molecular weight cutoff of 3000 Da and dialyzed against 100 mL of PBS (pH 7.4) in a temperature-controlled thermo shaker with a stirring speed of 200 rpm at 37 °C under different pH values of 7.4 and 6.5. At predetermined time intervals, 5 mL samples of the release medium were withdrawn and concentrated for the detection. The samples taken for the measurement were replaced with fresh medium, and the cumulative amount of drug released into the media at each time point was calculated as the percentage of total drug released to the initial amount of the drug. All experiments were performed in duplicate and the data reported as the mean of three individual experiments.Physicochemical stability and hemolysis analysis of liposomes
To investigate the stability of liposomes, the particle size, zeta potential and drug content of the prepared liposomes were analyzed eventually at 4 °C over one week.The hemolysis assay was conducted as follows: 1.5 mL of liposome solutions (Pt concentration: 100, 10, and 1 μg mL−1) were respectively mixed with 50 μL of 2% erythrocyte pellets solution in an EP tube, and the mixture was incubated for 1 h at 37 °C. Eventually, the tubes were centrifuged at 1500 rpm for 10 min to obtain the supernatant and the OD value was detected at 545 nm.
Penetration in multicellular 3D tumor spheroids
Panc02 and NIH/3T3 cells were mixed in a 3:1 ratio and inoculated with 8 × 103 cells spliced into U-shaped plates (FULA962-1pc, Beyotime Biotechnology), which grew into large uniform tumor spheres in 5–7 days according to the previous report.31 To simulate the tumor pH microenvironment, the cell culture medium was supplemented with 25 mmol L−1 PIPES (pH adjusted to 7.4) and 25 mmol L−1 of HEPES (pH adjusted to 6.5). The tumor spheres were treated with Cou-6 Lps, ATF@Cou-6 Lps and ATF peptide pretreated ATF@Cou-6 Lps (the concentration of Cou-6 was 2.5 μg mL−1) at different pH values for 6 h, and then transferred to confocal dishes for stereo scanning with a confocal microscopy (Nikon, A1R + N-STORM, Tokyo, Japan) at 25 μm per layer.
Cell uptake assay
The Panc02 pancreatic cancer cells or NIH/3T3 cells were inoculated separately in confocal dishes, and NIH/3T3 cells were induced into tumor-associated fibroblasts with TGF-β1 (10 ng mL−1). The uptake of Cou-6 Lps, ATF@Cou-6 Lps and ATF pretreated ATF@Cou-6 Lps by both cells at pH 7.4 and 6.5 was examined (the concentration of Cou-6 was 2.5 μg mL−1 for both), and after 4 h of action, they were fixed with 4% paraformaldehyde, followed by DAPI staining and confocal microscopy (Nikon, A1R + N-STORM, Tokyo, Japan) observation.Immunocytochemistry (ICC) staining
NIH/3T3 cells were inoculated in confocal dishes, and after the cells adhered to the wall, TGF-β (10 ng mL−1) was added for 12 h. The culture was continued with Pt Lps and ATF Pt Lps at pH 6.5 for 24 h. The collected cells were fixed with 4% PFA or methanol for 15 min, permeabilized with 0.1% Triton x – 100 for 10 min and blocked with 1% BSA at room temperature for 30 min. After washing with PBS, cells were incubated with α-SMA, collagen I, fibronectin, and primary antibodies (1:100) overnight at 4 °C. After washing with PBS, the cells were incubated with Alexa Fluor 488 or Alexa Fluor 649 labeled secondary antibodies (1:200) for 1 h at room temperature. Before observing by confocal microscopy (Nikon, A1R + N-STORM, Tokyo, Japan), the cells were stained with DAPI for 5 min and washed with PBS three times.
Cytotoxicity, cell cloning assay and anti-proliferation against multicellular 3D tumor spheroids
The 3 × 103 Panc02 or TGF-β treated NIH-3T3 cells were inoculated in 96-well plates and treated with free Pt, Pt Lps, and ATF@Pt Lps for 48 h with the same Pt concentrations (10, 5, 2.5, 1.25, 0.625, 0.312, 0.156, 0.078, 0.039, and 0 μg mL−1), followed by cell viability detection by MTT assay. The absorbance of each well was recorded at 490 nm using a microplate reader (BioTek, Vermont, UK).The 1 × 103 Panc02 cells were inoculated in 6-well plates and treated with free Pt, Pt@Lps, and ATF@Pt Lps for 7–10 days with the same Pt concentration. Then fixed with paraformaldehyde, stained with crystal violet and photographed.
3D tumor spheroids were treated with free Pt, Pt Lps, and ATF@Pt Lps for 96 h, respectively. The concentration of Pt was set as 0.125 μg mL−1. Subsequently, the shape of the tumor was observed with an inverted microscope (Nikon, ECLIPSE Ti, Tokyo, Japan) and the diameters of the tumor were calculated.
Cell apoptosis assay
1 × 104 Panc02 cells were seeded in a 6-well plate and treated with free Pt, Pt@Lps, and ATF@Pt Lps for 48 h with the same concentration of Pt (1.5 μg mL−1), followed by staining with the Annexin-FITC/PI double-staining apoptosis kit (Beyotime Biotechnology, Shanghai), and apoptosis was detected by flow cytometry (Attune NxT, Life).Biodistribution analysis
The distribution of Dir liposomes in vivo after ATF modification was detected by in vivo imaging according to a previous study.32 When tumor volume of Panc02 tumor-bearing mice reached around ∼400 mm3, the mice were intravenously injected with free Dir, Dir Lps, and ATF@Dir Lps at a Dir dosage of 0.8 mg kg−1. The distribution of liposomes in vivo was obtained using a Caliper IVIS Lumina II in vivo imaging system (PerkinElmer, Waltham, MA, USA) at 2 h, 4 h, 8 h and 24 h. The mice were sacrificed by pentobarbital overdose and cervical dislocation. And then, tumor, heart, liver, spleen, lung, and kidney tissues were collected to detect the distribution of liposomes.After the preparation of frozen sections, the anti-α-SMA antibody was employed to observe the cancer associated fibroblasts (CAFs) by CLSM.
Tumor penetration of anti PD-1 antibodies in vivo
The tumor penetration of anti PD-1 antibodies in vivo experimental procedure refered to the literature method and made minor modifications.33 When tumor volume of Panc02 tumor-bearing mice reached around 250 mm3, the mice were intravenously injected with saline, Pt Lps, and ATF@Pt Lps at a Pt dosage of 2.5 mg kg−1 every other day two times. Then the mice were intraperitoneally injected with Cy5-labeled anti-PD-1 antibodies (100 μg). At 24 h after injection, the mice were sacrificed by pentobarbital overdose and cervical dislocation and tumor tissues were collected, washed with PBS and frozen sectioned. The tumor sections were incubated with FITC-conjugated CD31 antibodies (1:200, Biolegend) at 37 °C for 30 min, washed with PBS and then detected using a confocal microscope (Nikon, A1R + N-STORM, Tokyo, Japan). The penetration capacity of anti PD-1 antibodies was expressed as the ratio of Cy5 in the tumor parenchyma to that within the blood vessels.
Anticancer efficacy in stromal-rich xenograft tumor models
To establish the stromal-rich xenograft models,34 Panc02 and NIH/3T3 (3:1) 1 × 106 cells were injected in the right flank of C57/BL mice and the tumors were expected to grow to 50 mm3. Mice bearing Panc02 tumor were treated with saline, Pt, Pt@Lps, ATF@Pt Lps, anti PD-1 mb and ATF@Pt Lps + anti PD-1 mb (n = 6 per group). Pt was administered at the same dose (2.5 mg kg−1) and anti PD-1 mb was administered at 200 μg per mice every 3 days, and both Pt and anti PD-1 mb were administered by intraperitoneal injection for a total of 21 days. We measured the tumor volume (V = length × width2/2), body weight, and tumor volume of mice during their treatment. Afterward, the mice were sacrificed by pentobarbital overdose and cervical dislocation and the tumors, liver, and kidney tissues were collected.
H&E and Masson trichrome staining
Paraffin-embedded tumor sections were deparaffinized and rehydrated. The slides were then stained using a Masson trichrome kit (Solarbio, Beijing) and H&E staining (Servicebio, Wuhan) according to the manufacturer's instructions. Then, the staining results were visualized with an upright microscope (Nikon, Ni-u, Tokyo, Japan).Immunohistochemistry (IHC) and immunofluorescence (IF) staining
Tumor sections for IHC were de-paraffinized, rehydrated, and subjected to antigen retrieval under high pressure. After antigen retrieval, slides were blocked in Tris-buffered saline (pH 7.4) with 10% goat serum and incubated with primary antibodies (CD3, CD4, CD8, Foxp3, α-SMA, collagen I, Ki67) overnight at 4 °C. The 3,3′-diaminebenzidine (DAB) system was used to visualize staining according to the manufacturer's instructions. Hematoxylin was used to counterstain the slide and a bright-field microscope was used to observe the slide at 40× magnification.CD83 and fibronectin were subjected to IF staining, CD83 was stained with Alexa Fluor® 488-goat-anti-mouse, fibronectin was stained with Alexa Fluor® 649-goat-anti-rabbit at room temperature for 1 hour and counterstained with DAPI. A confocal microscope (Nikon, A1R + N-STORM, Tokyo, Japan) was used to observe the slide.
TUNEL assay
The TdT-dependent dUTPbiotin nick end labelling (TUNEL) assay was performed to detect apoptosis of the tumor using the FITC-labeled TUNEL detection kit (KeyGEN BioTECH, China) according to the manufacturer's instructions. Slides were then rinsed with PBS and incubated with DAPI (Solarbio, Beijing). All staining was evaluated and digital images were acquired using a confocal laser scanning microscope (Nikon, Ni-u, Tokyo, Japan) under 40× magnification.Flow cytometry and cytokine analysis
The tumors were cut into small pieces and digested with 0.8 mg mL−1 collagenase and 5 μg mL−1 DNase I for 1 h at 37 °C. After incubation, homogenates were washed with PBS and passed through a 70 μm cell filter to obtain single cell suspensions. For the analysis of CD8+ T cells, the cells were double-labeled with fluorescently labeled FITC-anti-CD3ε (1:100) and APC-anti-CD8 (1:100), and for the analysis of CD4+ T cells, the cells were double-labeled with fluorescently labeled FITC-anti-CD3 (1:100) and APC-anti-CD4 (1:100). Unstained controls and FMO were used as gating controls to accurately distinguish between positive and negative signals. All antibodies were diluted according to the instructions and incubated with the cells for 30 min at room temperature. Finally, the cells were analyzed by flow cytometry.35
The concentrations of the cytokines of IFN-γ and TNF-α of tumors were detected using ELISA kits (Elabscience, Wuhan) according to the manufacturer's instructions.
Hematological analysis
Blood samples were collected from the orbital cavity on day 20 after drug treatment. Alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (Cr), blood urea nitrogen (BUN) and uric acid (UA) were measured using an Exigo automatic blood analyzer (Boule, Swedish).Statistical analysis
All animal studies were performed after randomization. One-way ANOVA of variance with GraphPad Prism 8 was used for the analysis of variance, with significance set at p < 0.05. All values were reported as mean ± SD, n ≥ 3.Abbreviation
| Pt | Cisplatin |
| ATF | The uPAR amino-terminal receptor binding peptide |
| Pt Lps | Cisplatin-loaded liposomes |
| ATF@Pt Lps | ATF-modified cisplatin-loaded liposomes |
| Anti PD-1 | Anti-programmed cell death 1 |
| Dir Lps | Dir-loaded liposomes |
| ATF@Dir Lps | ATF-modified DIR-loaded liposomes |
| Cou-6 | Coumarin 6 |
| Cou-6 Lps | Cou-6-loaded liposomes |
| ATF@Cou-6 Lps | ATF-modified Cou-6-loaded liposomes |
Author contributions
Hang Yu: Conceptualization, data curation, formal analysis, investigation, resources, software, validation, visualization, writing – original draft, and writing – review & editing. Wenting Zhu: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing – original draft, and writing – review & editing. Cai Yan Lin: methodology and software. Menglei Jia: methodology and data curation. Xiaoxiao Tan: visualization and software. Zhongwen Yuan: validation and software. Senglin Feng: formal analysis and methodology. Pengke Yan: conceptualization, project administration, supervision, visualization, writing – review & editing, and funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81971742), the Natural Science Foundation of Guangdong Province (No. 2022A1515010199), and Lin He's New Medicine and Clinical Translation Academician Workstation Research Fund (2021HLKY07).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- C. J. Halbrook, C. A. Lyssiotis, D. M. M. Pasca and A. Maitra, Cell, 2023, 186, 1729–1754 CrossRef CAS PubMed.
- L. D. Wood, M. I. Canto, E. M. Jaffee and D. M. Simeone, Gastroenterology, 2022, 163, 386–402 CrossRef PubMed.
- T. Conroy, F. Desseigne, M. Ychou, O. Bouche, R. Guimbaud, Y. Becouarn, A. Adenis, J. L. Raoul, S. Gourgou-Bourgade, C. de la Fouchardiere, J. Bennouna, J. B. Bachet, F. Khemissa-Akouz, D. Pere-Verge, C. Delbaldo, E. Assenat, B. Chauffert, P. Michel, C. Montoto-Grillot and M. Ducreux, N. Engl. J. Med., 2011, 364, 1817–1825 CrossRef CAS PubMed.
- J. P. Neoptolemos, J. Kleeff, P. Michl, E. Costello, W. Greenhalf and D. H. Palmer, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 333–348 CrossRef PubMed.
- N. Kirchhammer, M. P. Trefny, D. M. P. Auf, H. Laubli and A. Zippelius, Sci. Transl. Med., 2022, 14, o3605 CrossRef PubMed.
- H. M. Kolbeinsson, S. Chandana, G. P. Wright and M. Chung, J. Invest. Surg., 2023, 36, 2129884 CrossRef PubMed.
- X. Li, M. Gulati, A. C. Larson, J. C. Solheim, M. Jain, S. Kumar and S. K. Batra, Semin. Cancer Biol., 2022, 86, 14–27 CrossRef CAS PubMed.
- R. Balsano, V. Zanuso, A. Pirozzi, L. Rimassa and S. Bozzarelli, Curr. Oncol., 2023, 30, 3871–3885 CrossRef PubMed.
- R. Rebelo, C. Xavier, E. Giovannetti and M. H. Vasconcelos, Trends Mol. Med., 2023, 29, 439–453 CrossRef CAS PubMed.
- D. Thomas and P. Radhakrishnan, Mol. Cancer, 2019, 18, 14 CrossRef PubMed.
- M. R. Goulart, K. Stasinos, R. Fincham, F. R. Delvecchio and H. M. Kocher, World J. Gastroenterol., 2021, 27, 7956–7968 CrossRef CAS PubMed.
- N. Martinez-Bosch, J. Vinaixa and P. Navarro, Cancers, 2018, 10, 6 CrossRef PubMed.
- I. T. Chyuan, C. L. Chu and P. N. Hsu, Cancers, 2021, 13, 1188 CrossRef CAS PubMed.
- W. Lin, J. Qian, H. Wang, L. Ren, Y. Yang, C. Chen, X. Chen, Y. Huang, J. Liu, N. Xu and L. Teng, Am. J. Cancer Res., 2022, 12, 451–468 CAS.
- L. Tran, C. T. Allen, R. Xiao, E. Moore, R. Davis, S. J. Park, K. Spielbauer, C. Van Waes and N. C. Schmitt, Cancer Immunol. Res., 2017, 5, 1141–1151 CrossRef CAS PubMed.
- S. Manohar and N. Leung, J. Nephrol., 2018, 31, 15–25 CrossRef CAS PubMed.
- A. Fernandes, G. A. de Brito, A. L. Baptista, L. Andrade, M. H. Imanishe and B. J. Pereira, Health Sci. Rep., 2022, 5, e479 CrossRef PubMed.
- Y. N. Diep, T. J. Kim, H. Cho and L. P. Lee, J. Controlled Release, 2022, 351, 1017–1037 CrossRef CAS PubMed.
- D. Iyengar, K. Tatiparti, N. S. Gavande, S. Sau and A. K. Iyer, Drug Discovery Today, 2022, 27, 1554–1559 CrossRef CAS PubMed.
- F. Raza, L. Evans, M. Motallebi, H. Zafar, M. Pereira-Silva, K. Saleem, D. Peixoto, A. Rahdar, E. Sharifi, F. Veiga, C. Hoskins and A. C. Paiva-Santos, Acta Biomater., 2023, 157, 1–23 CrossRef CAS PubMed.
- A. Wang-Gillam, R. A. Hubner, J. T. Siveke, D. D. Von Hoff, B. Belanger, F. A. de Jong, B. Mirakhur and L. T. Chen, Eur. J. Cancer, 2019, 108, 78–87 CrossRef CAS PubMed.
- J. E. Frampton, Drugs, 2020, 80, 1007–1018 CrossRef CAS PubMed.
- Y. Han, P. Wen, J. Li and K. Kataoka, J. Controlled Release, 2022, 345, 709–720 CrossRef CAS PubMed.
- F. Blasi and P. Carmeliet, Nat. Rev. Mol. Cell Biol., 2002, 3, 932–943 CrossRef CAS PubMed.
- L. Yang, H. Mao, Z. Cao, Y. A. Wang, X. Peng, X. Wang, H. K. Sajja, L. Wang, H. Duan, C. Ni, C. A. Staley, W. C. Wood, X. Gao and S. Nie, Gastroenterology, 2009, 136, 1514–1525 CrossRef CAS PubMed.
- G. Y. Lee, W. P. Qian, L. Wang, Y. A. Wang, C. A. Staley, M. Satpathy, S. Nie, H. Mao and L. Yang, ACS Nano, 2013, 7, 2078–2089 CrossRef CAS PubMed.
- W. Zhu, H. Yu, M. Jia, C. Lin, Z. Yuan, X. Tan and P. Yan, Int. J. Pharm., 2023, 644, 123316 CrossRef CAS PubMed.
- B. Zhai, P. Chen, W. Wang, S. Liu, J. Feng, T. Duan, Y. Xiang, R. Zhang, M. Zhang, X. Han, X. Chen, Q. Li, G. Li, Y. Liu, X. Huang, W. Zhang, T. Pan, L. Yan, T. Jin, T. Xie and X. Sui, Cancer Biol. Med., 2020, 17, 676–692 CAS.
- J. Lee, G. Jang, S. Ma, C. U. Lee, J. Son, W. Jeong and J. Moon, Small, 2022, 18, e2202159 CrossRef PubMed.
- Y. Li, Z. Zhao, C. Y. Lin, Y. Liu, K. F. Staveley-Ocarroll, G. Li and K. Cheng, Theranostics, 2021, 11(5), 2182–2200 CrossRef CAS PubMed.
- Y. Q. Chen, W. T. Zhu, C. Y. Lin, Z. W. Yuan, Z. H. Li and P. K. Yan, Int. J. Nanomed., 2021, 16, 269–281 CrossRef PubMed.
- Z. Wei, X. Zhang, T. Yong, N. Bie, G. Zhan, X. Li, Q. Liang, J. Li, J. Yu, G. Huang, Y. Yan, Z. Zhang, B. Zhang, L. Gan, B. Huang and X. Yang, Nat. Commun., 2021, 12, 440 CrossRef CAS PubMed.
- X. Geng, L. Li, Y. Luo, W. Yang, J. Hu, Z. Zhao, C. Cheng, T. Zhang, Y. Zhang, L. Liu, Y. Xie, G. Li, D. Liu, R. Bai, X. Bai, G. Wang, H. Chen, Y. Wang, H. Chen and B. Sun, Adv. Sci., 2023, 10(10), e2203324 CrossRef PubMed.
- G. Rao, K. Latha, M. Ott, A. Sabbagh, A. Marisetty, X. Ling, D. Zamler, T. A. Doucette, Y. Yang, L. Y. Kong, J. Wei, G. N. Fuller, F. Benavides, A. M. Sonabend, J. Long, S. Li, M. Curran and A. B. Heimberger, Clin. Cancer Res., 2020, 26, 4699–4712 CrossRef CAS PubMed.
- X. Liu, J. Iovanna and P. Santofimia-Castano, J. Physiol. Biochem., 2023, 79, 213–222 CrossRef PubMed.
- M. L. Stone and G. L. Beatty, Pharmacol. Ther., 2019, 201, 202–213 CrossRef CAS PubMed.
- Y. J. Kang, C. K. Holley, M. R. Abidian, A. B. Madhankumar, J. Connor and S. Majd, Adv. Healthcare Mater., 2021, 10, e2001261 CrossRef PubMed.
- X. Liu, F. Wu, K. Cai, Z. Zhao, Z. Zhang, Y. Chen, Y. Liu, J. Cheng and L. Yin, Biomater. Sci., 2021, 9, 1301–1312 RSC.
- Z. Xiao, Y. Tan, Y. Cai, J. Huang, X. Wang, B. Li, L. Lin, Y. Wang, X. Shuai and K. Zhu, J. Controlled Release, 2023, 356, 360–372 CrossRef CAS PubMed.
- X. Han, Y. Xu, M. Geranpayehvaghei, G. J. Anderson, Y. Li and G. Nie, Biomaterials, 2020, 232, 119745 CrossRef CAS PubMed.
- R. Manchanda, A. Fernandez-Fernandez, S. Paluri and B. R. Smith, Adv. Pharmacol., 2021, 91, 293–335 CAS.
- X. H. Ren, D. Han, X. Y. He, T. Guo, X. S. Chen, X. Pang and S. X. Cheng, Adv. Healthcare Mater., 2023, 12, e2202155 CrossRef PubMed.
- C. H. Hsieh, H. C. Hsieh, F. S. Shih, P. W. Wang, L. X. Yang, D. B. Shieh and Y. C. Wang, Theranostics, 2021, 11, 7072–7091 CrossRef CAS PubMed.
- S. Guo, C. M. Lin, Z. Xu, L. Miao, Y. Wang and L. Huang, ACS Nano, 2014, 8, 4996–5009 CrossRef CAS PubMed.
- X. Chen, W. Zhou, C. Liang, S. Shi, X. Yu, Q. Chen, T. Sun, Y. Lu, Y. Zhang, Q. Guo, C. Li, Y. Zhang and C. Jiang, Nano Lett., 2019, 19, 3527–3534 CrossRef CAS PubMed.
- N. Yu, X. Zhang, H. Zhong, J. Mu, X. Li, T. Liu, X. Shi, X. J. Liang and S. Guo, Nano Lett., 2022, 22, 8744–8754 CrossRef CAS PubMed.
- L. Galluzzi, A. Buque, O. Kepp, L. Zitvogel and G. Kroemer, Cancer Cell, 2015, 28, 690–714 CrossRef CAS PubMed.
- L. Zitvogel, O. Kepp, L. Senovilla, L. Menger, N. Chaput and G. Kroemer, Clin. Cancer Res., 2010, 16, 3100–3104 CrossRef CAS PubMed.
- E. Catanzaro, O. Feron, A. G. Skirtach and D. V. Krysko, Front. Immunol., 2022, 13, 925290 CrossRef CAS PubMed.
- Y. Wang, N. Shen, Y. Wang, M. Li, W. Zhang, L. Fan, L. Liu, Z. Tang and X. Chen, Biomater. Sci., 2021, 9, 3019–3027 RSC.
- L. Fournel, Z. Wu, N. Stadler, D. Damotte, F. Lococo, G. Boulle, E. Segal-Bendirdjian, A. Bobbio, P. Icard, J. Tredaniel, M. Alifano and P. Forgez, Cancer Lett., 2019, 464, 5–14 CrossRef CAS PubMed.
- Y. Cheng, S. Song, P. Wu, B. Lyu, M. Qin, Y. Sun, A. Sun, L. Mu, F. Xu, L. Zhang, J. Wang and Q. Zhang, Adv. Healthcare Mater., 2021, 10, e2100590 CrossRef PubMed.
- S. S. Kim, J. B. Harford, M. Moghe, C. Doherty and E. H. Chang, Cells, 2022, 11, 3434 CrossRef CAS PubMed.
- Y. Duan, X. Zhang, H. Ying, J. Xu, H. Yang, K. Sun, L. He, M. Li, Y. Ji, T. Liang and X. Bai, Oncogene, 2023, 42, 2061–2073 CrossRef CAS PubMed.
- M. Yi, X. Zheng, M. Niu, S. Zhu, H. Ge and K. Wu, Mol. Cancer, 2022, 21, 28 CrossRef CAS PubMed.
- M. Wu, Q. Huang, Y. Xie, X. Wu, H. Ma, Y. Zhang and Y. Xia, J. Hematol. Oncol., 2022, 15, 24 CrossRef PubMed.
- T. Ho, A. Nasti, A. Seki, T. Komura, H. Inui, T. Kozaka, Y. Kitamura, K. Shiba, T. Yamashita, T. Yamashita, E. Mizukoshi, K. Kawaguchi, T. Wada, M. Honda, S. Kaneko and Y. Sakai, J. Immunother. Cancer, 2020, 8, e001367 CrossRef PubMed.
- C. Glorieux, X. Xia, X. You, Z. Wang, Y. Han, J. Yang, G. Noppe, C. Meester, J. Ling, A. Robert, H. Zhang, S. P. Li, H. Wang, P. J. Chiao, L. Zhang, X. Li and P. Huang, J. Adv. Res., 2022, 40, 109–124 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01118f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |