
Holistic food safety evaluation of herbs: methods for the determination of organic and inorganic trace contaminants in Moringa stenopetala as a case study
Ignacio
Machado
 ab,
Natalia
Gérez
b,
Analía
Bertón
b,
Horacio
Heinzen
b and
María Verónica
Cesio
*b
ab,
Natalia
Gérez
b,
Analía
Bertón
b,
Horacio
Heinzen
b and
María Verónica
Cesio
*b
aGrupo de Bioanalítica y Especiación (BioEsp), Analytical Chemistry, DEC, Facultad de Química, Universidad de la República, Montevideo, Uruguay
bGrupo de Análisis de Compuestos Traza (GACT), Departamento de Química Orgánica, Facultad de Química, Universidad de la República, Montevideo, Uruguay. E-mail: cs@fq.edu.uy
First published on 26th April 2024
Abstract
Moringa stenopetala is considered a superfood due to the many bioactive compounds that it provides to the diet. However, like all edible plants, it is mandatory to guarantee food safety. Thus it is necessary to develop analytical methods that can rapidly and accurately determine hazardous pollutants, to evaluate compliance with food regulations. In this regard, two multi-component procedures were developed trying to cover some of the main organic and inorganic potential contaminants. A microwave-assisted digestion followed by electrothermal atomic absorption spectrometry was used for arsenic, cadmium, and lead determination, while a modification of the QuEChERS protocol followed by gas chromatography-tandem mass spectrometry was employed for the determination of 55 pesticides from different families. Both analytical methods were thoroughly validated according to international guidelines. The analyzed samples obtained from the Uruguayan market showed compliance with both, national and international, food regulations. The holistic approach employed in this research is not commonly presented in the literature, thus constituting a novel way to face food safety.
Introduction
Moringa stenopetala is one of the 14 species of the Moringaceae family, indigenous to Eastern Africa and cultivated for its multipurpose uses. Its leaves are rich in protein and contain substantial amounts of essential amino acids.1 This species is widely used in Ethiopian and Kenyan traditional medicine for treating several illnesses such as diabetes, hypertension, stomach pain, malaria, leishmaniasis, leprosy, epilepsy, diarrhea, and asthma. Also, other pharmacological activities have been associated with it, such as antioxidant, anticarcinogenic, and anti-HIV. Both fresh leaves and dried powdered leaves are sold as nutritional supplements and for medicinal uses. It has been used as a “superfood” since it contains minerals, carbohydrates, essential amino acids, and vitamins.2,3 However, like all edible plants, it is necessary to guarantee its food safety. For this reason, the residues of organic and inorganic contaminants that it may contain must be analyzed, and compared with the maximum limits allowed by different regulatory entities.4 It is of utmost importance to develop analytical methods that can rapidly and accurately determine the levels of organic and inorganic contaminants in these crops and to determine compliance with national and international regulations. In this regard, new analytical methods are continuously exploring more effective sample treatments, especially when working with complex matrices, to prevent interferences and improve sensitivity.4In general terms, plants may accumulate potentially toxic elements such as arsenic (As), cadmium (Cd), and lead (Pb), among others. The elemental composition of a certain plant can be affected by genetic factors, soil characteristics, and environmental conditions.5 The geographical origin, the use of agrochemicals, the harvesting time, and the type of soil represent the main sources of inorganic contaminants in plants.6 In this regard, the MERCOSUR regulation establishes maximum limits for edible vegetables of 0.30 mg kg−1 for As and Pb, and 0.20 mg kg−1 for Cd, respectively. It also establishes maximum limits for infusion vegetables of 0.6 mg kg−1 for As and Pb, and 0.4 mg kg−1 for Cd, respectively.7
In the field of metal/metalloid analysis in food, the trend in analytical chemistry is to avoid drastic treatments and to look for efficient quantitative multi-extraction procedures under soft conditions, more aligned with the principles of Green Analytical Chemistry.8,9 In particular, microwave-assisted extraction is a very efficient strategy for sample preparation. This method has the advantage of working with closed vessels, thus reducing the risk of contamination. A microwave-assisted extraction using diluted acid, followed by electrothermal atomic absorption spectrometry (ETAAS), is herein described.6
On the other hand, the inappropriate use of pesticides can result in high levels of residues that may pose a risk on human health. Even when applying the pesticides following Good Agriculture Practices (GAPs), residues can remain on the crops.10 For this reason, several monitoring programs and legal regulations have been established to control the use of pesticides on edible crops. Pesticide residues must comply with the Maximum Residue Levels (MRL) established for each compound by international regulations.
Modern analytical methods for pesticide residue analysis in food, seek miniaturized, rapid, and cost-effective sample preparation procedures. In this regard, a versatile multi-residue method (MRM) such as the QuEChERS (Quick, Easy, Cheap, Effective, Rugged and Safe) method, can be suitable for large-scale residue analysis in a great variety of matrices.11,12 It is well known that pesticides have different chemical structures that lead to different physicochemical properties. These differences must be considered when developing an MRM. A simple, fast, and cheap method, involving a modification of the QuEChERS sample preparation protocol, followed by gas chromatography-tandem mass spectrometry (GC-MS/MS), is herein presented.12,13
This work aimed to generate proper analytical tools for the determination of potentially toxic elements and pesticide residues in Moringa stenopetala, to assess food safety using a holistic approach. Multi-contaminant procedures were developed trying to cover the main potential inorganic and organic contaminants for this crop. The two validated methods are considered highly useful tools for the surveillance of this increasingly used raw material. The holistic approach employed in this research is not commonly presented in the literature, thus constituting a novel way to face food safety.
Materials and methods
Reagents
Ultrapure water of 18.2 MΩ cm resistivity, obtained from a Millipore™ Direct Q3 UV (Merck Millipore, São Paulo, Brazil), was used throughout this work.For inorganic contaminants determinations, calibration curves were prepared by serial dilution of commercial 1000 mg per L stock solutions of As, Cd and Pb (Merck, Darmstadt, Germany) in 0.01 mol per L nitric acid (HNO3), prepared from concentrated HNO3 (Merck, Darmstadt, Germany). A commercial solution of palladium nitrate (Pd(NO3)2) (Merck, Darmstadt, Germany) containing 10.0 g L−1 and a commercial solution of magnesium nitrate (Mg(NO3)2) (Sigma-Aldrich, St. Louis, MO, USA) containing 20.0 g L−1, were used to prepare the chemical matrix modifier for As, Cd and Pb determinations. All glassware was previously soaked overnight in 1.4 mol per L HNO3 and then rinsed exhaustively with ultrapure water.
For pesticide residue determinations, LC-grade acetonitrile (MeCN) and ethyl acetate (EtOAc) were used (Pharmco, Brookfield, CT, USA). Glacial acetic acid (HAc) was purchased from Dorwil (Buenos Aires, Argentina). Magnesium sulphate (MgSO4) and sodium acetate (AcONa) were purchased from J. T. Mallinckrodt Baker, Inc. (Phillipsburg, NJ, USA). Primary secondary amine (PSA) provided by SUPELCO (Bellefonte, PA, USA). Graphitized carbon black (GCB) was provided by Supelco (Bellefonte, PA, USA). All reagents were of analytical grade.
High-purity pesticide standards were obtained from Dr Ehrenstorfer (Augsburg, Germany) and stored in the dark at −18 °C. Individual pesticide stock solutions of 2000 mg L−1 were prepared in EtOAc and stored in the dark at −18 °C. Mix solutions used for calibration and spiking procedures were prepared from the stock standards at appropriate dilutions. The working standard mix solution for spiking purposes was prepared at 10 mg L−1 in EtOAc. These solutions were then diluted with EtOAc as needed to prepare different standard solutions: 5.0, 10.0, 25.0, 50.0, 100.0, and 200.0 μg L−1 for GC-MS/MS.
Samples
Samples were purchased from local markets of Montevideo (Uruguay), during August 2022. Samples were washed with ultrapure water, dried in an oven with forced air circulation at 40 °C for 6 hours and stored at 20 °C.A certified reference material (CRM) consisting of spinach leaves (NIST 1570a) was used for trueness (EURACHEM) and precision evaluation, during the validation of inorganic contaminants determinations. Before sample preparation, dry samples were milled using a blade mill and then passed through a 425 μm sieve, to obtain a particle size as similar as possible as that of the CRM.
Sample preparation for the determination of As, Cd, and Pb
For the determination of total concentrations of As, Cd, and Pb in the samples a microwave-assisted acid digestion was carried out using a CEM Mars 6 microwave digester (CEM, Charlotte, NC, USA) provided with 12 Easy Prep Plus® vessels. For sample preparation, 0.5 g of sieved samples were accurately weighted into each vessel and 10.0 mL of 4.2 mol per L HNO3 was added. The program consisted of a 15 minute ramp time until 200 °C and then holding at that temperature for 10 min, with power varying between 400 and 1800 W. Maximum pressure achieved was 500 psi. The obtained solutions were used for analytical determinations without further dilution. Samples and reagent blanks were run in triplicate.Sample preparation for the determination of pesticide residues
A variation of the QuEChERS AOAC 2007.01 method was employed.14 A 2.0 g portion of milled and homogenised Moringa stenopetala leaves was weighed in a polypropylene conical centrifuge tube. To hydrate the sample, 10 mL of ultrapure water was added, and the suspension was vortexed for 1 minute, and then left to stand for 30 min. Next, 10 mL of 1% v/v HAc in MeCN were added to the samples and shaken by hand for 5 min. Afterwards, 4 g of MgSO4 and 1 g of AcONa were added and the samples were hand shaken again for 5 min. The extract was then centrifuged at 3700g for 5 min, and 4 mL of the supernatant was transferred to a 15 mL polypropylene centrifuge tube containing 600 mg of MgSO4 and 200 mg of PSA for clean-up. The extract was vortexed for 1 minute and centrifuged again. Two-millilitre aliquots were evaporated under a N2 stream and reconstituted to 1.0 mL with EtOAc for GC-MS/MS analysis. Before injection, samples were filtered through a 0.45 μm Millex FG PVDF filters (Millipore, Mildford, MA, USA).14Analytical determinations
Total determinations of As, Cd and Pb were performed by ETAAS using a Thermo Scientific iCE 3500 spectrometer equipped with an auto-sampler, employing Zeeman correction (Thermo Scientific, Cambridge, United Kingdom). A transversely heated graphite tube furnace was used (Thermo Scientific, Cambridge, United Kingdom). The analytical lines employed were: 193.7 nm (As), 228.8 nm (Cd), and 283.3 nm (Pb), while the signal used for quantification was integrated absorbance (peak-area). Pyrolytically coated graphite tubes from Thermo Scientific were used. The purge and protective gas used was argon 99.998% (Air Liquide, Montevideo, Uruguay). The graphite furnace heating programs and the experimental conditions used for analytical determinations are reported in Table 1.6,15 Injection volumes were 30 μL. The chemical matrix modifier used for As, Cd and Pb determinations consisted of 10 μL of a solution containing 5 μg of Pd(NO3)2 and 3 μg of Mg(NO3)2.| Stage | Temperature (°C) | Ramp rate (°C s−1) | Hold time (s) |
|---|---|---|---|
| a Ar gas flow rate was 0.2 (L min−1) in all stages (except for atomization). | |||
| Drying | 120 | 10 | 30 |
| Pyrolysis | 1200(As)/800(Cd)/1100(Pb) | 150 | 20 |
| Atomization | 2200(As)/1800(Cd)/1900(Pb) | 0 | 3 |
| Cleaning | 2600 | 0 | 3 |
Pesticide residue analyses were performed by GC-MS/MS using a Shimadzu GCMS-TQ8050 system. The instrument was equipped with a 2010 plus gas chromatograph coupled to a triple quadrupole mass spectrometer. Aliquots of 1 μL of sample extract were injected into the gas chromatograph in split-less mode. The injector temperature was 280 °C. Helium was used as the carrier gas at a constant flow rate of 1 mL min−1. The used liner was a Topaz liner, splitless: 3.5 mm × 5.0 cm × 95 mm for Shimadzu GC from Restek (Bellefonte, PA, USA). The chromatographic separation was carried out with an Rxi®-5Sil MS capillary column (5% diphenyl/95% dimethyl polysiloxane; 30 m, 0.32 mm i.d., 0.25 μm d.f.) provided by Restek (Bellefonte, PA, USA). The oven temperature was programmed as follows: 80 °C (2 min), 180 °C (20 °C min−1), 300 °C (5 °C min−1), 3 min. The total run time was 34 min. The interface temperature was 290 °C and the ion source was at 230 °C operated in electron ionization mode (70 eV). Detection was performed with a detector voltage of 1.4 kV. Argon was the collision gas at a constant pressure of 200 kPa. Tandem mass detection was performed in the Multiple Reaction Monitoring (MRM) mode using transitions and collision energies previously selected for each compound as shown in Table 2.16,17
| # | Compound | t R (min) | 1st transition | 2nd transition | ||
|---|---|---|---|---|---|---|
| m/z1 | CE (V) | m/z2 | CE (V) | |||
| 1 | 2-Phenylphenol | 8434 | 170.10 > 141.10 | 20 | 170.10 > 116.10 | 30 |
| 2 | Tecnazene | 9087 | 280.90 > 202.90 | 14 | 280.90 > 230.90 | 8 |
| 3 | Ethoprophos | 9441 | 200.00 > 158.00 | 6 | 200.00 > 114.0 | 14 |
| 4 | Trifluralin | 9654 | 306.10 > 264.10 | 10 | 306.10 > 206.10 | 16 |
| 5 | Cadusafos | 9969 | 158.90 > 130.90 | 8 | 158.90 > 97.00 | 18 |
| 6 | Gamma-BHC (lindane) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 971 971 |
218.90 > 182.90 | 8 | 218.90 > 144.90 | 20 |
| 7 | Diazinon | 11102 |
304.10 > 179.10 | 10 | 304.10 > 162.10 | 10 |
| 8 | Tefluthrin | 11507 |
177.00 > 127.10 | 18 | 177.00 > 137.10 | 16 |
| 9 | Pirimicarb | 11724 |
238.10 > 166.10 | 10 | 238.10 > 72.00 | 25 |
| 10 | Chlorpyrifos-methyl | 12353 |
285.90 > 93.00 | 20 | 285.90 > 270.90 | 15 |
| 11 | Vinclozolin | 12441 |
285.00 > 212.00 | 10 | 285.00 > 178.00 | 15 |
| 12 | Parathion-methyl | 12508 |
263.00 > 109.00 | 15 | 263.00 > 136.00 | 10 |
| 13 | Alachlor | 12548 |
188.10 > 160.10 | 10 | 188.10 > 132.10 | 18 |
| 14 | Fenchlorphos | 12816 |
284.90 > 269.90 | 18 | 284.90 > 93.00 | 24 |
| 15 | Pirimiphos-methyl | 13092 |
305.10 > 180.10 | 10 | 305.10 > 290.10 | 10 |
| 16 | Fenitrothion | 13184 |
277.00 > 260.00 | 5 | 277.00 > 109.10 | 18 |
| 17 | Malathion | 13407 |
173.10 > 99.00 | 15 | 173.10 > 127.00 | 6 |
| 18 | Chlorpyrifos | 13638 |
313.90 > 257.90 | 15 | 313.90 > 286.90 | 10 |
| 19 | Fenthion | 13816 |
278.00 > 109.00 | 20 | 278.00 > 125.00 | 20 |
| 20 | Parathion | 13856 |
291.10 > 109.00 | 15 | 291.10 > 137.00 | 5 |
| 21 | Dicofol | 14105 |
250.00 > 139.00 | 14 | 250.00 > 216.00 | 8 |
| 22 | Heptachlor-exo-epoxide | 14945 |
352.80 > 289.00 | 15 | 352.80 > 253.00 | 26 |
| 23 | Chlorfenvinphos (sum) | 14918 |
323.00 > 267.00 | 15 | 323.00 > 296.0 | 8 |
| 24 | Mecarbam | 15138 |
329.00 > 131.10 | 18 | 329.00 > 169.10 | 4 |
| 25 | Bromophos-ethyl | 15555 |
358.90 > 302.90 | 16 | 358.90 > 330.90 | 10 |
| 26 | Chlordane (cis + trans) | 15.153 | 372.80 > 263.90 | 28 | 372.80 > 336.80 | 10 |
| 15.589 | ||||||
| 27 | Kresoxim-methyl | 16.770 | 206.10 > 116.10 | 6 | 206.10 > 131.10 | 14 |
| 28 | Endrin | 16.997 | 262.90 > 191.00 | 30 | 262.90 > 193.00 | 28 |
| 29 | Fensulfothion | 17.705 | 293.00 > 153.00 | 8 | 293.00 > 125.00 | 14 |
| 30 | Ethion | 17.832 | 230.90 > 174.90 | 15 | 230.90 > 184.90 | 10 |
| 31 | p,p′-DDD | 17.719 | 235.00 > 165.00 | 24 | 235.00 > 199.00 | 14 |
| 32 | Trifloxystrobin | 18.940 | 222.10 > 190.10 | 6 | 222.10 > 162.10 | 10 |
| 33 | Endosulfan sulfate | 18.837 | 386.80 > 252.90 | 10 | 386.80 > 288.80 | 6 |
| 34 | p,p′-DDT | 18.868 | 235.00 > 165.00 | 10 | 235.00 > 199.00 | 15 |
| 35 | Bifenthrin | 20.658 | 181.10 > 166.10 | 15 | 181.10 > 163.10 | 10 |
| 36 | Bromopropylate | 20.585 | 340.90 > 182.90 | 20 | 340.90 > 184.90 | 20 |
| 37 | Chlorantraniliprole | 21.028 | 278.00 > 249.00 | 20 | 278.00 > 261.00 | 20 |
| 38 | Fenazaquin | 21.120 | 160.20 > 145.10 | 8 | 160.20 > 116.10 | 24 |
| 39 | Phosalone | 21.875 | 182.00 > 111.00 | 14 | 182.00 > 138.00 | 8 |
| 40 | Pyriproxyfen | 21.969 | 136.10 > 78.00 | 20 | 136.10 > 98.00 | 10 |
| 41 | Cyhalothrin | 22.596 | 197.00 > 141.00 | 8 | 197.00 > 161.00 | 12 |
| 42 | Mirex | 22.989 | 271.80 > 236.80 | 18 | 271.80 > 238.80 | 18 |
| 43 | Permethrin (sum) | 23.837 | 183.10 > 168.10 | 10 | 183.10 > 166.10 | 15 |
| 24.126 | ||||||
| 44 | Cyfluthrin (sum) | 25.300 | 226.10 > 206.10 | 15 | 226.10 > 199.10 | 10 |
| 45 | Boscalid | 25.788 | 342.10 > 140.10 | 14 | 342.10 > 112.10 | 28 |
| 46 | Flucythrinate (sum) | 26.100 | 199.10 > 157.10 | 10 | 199.10 > 107.10 | 22 |
| 26.434 | ||||||
| 47 | Etofenprox | 26.158 | 163.10 > 135.10 | 10 | 163.10 > 107.10 | 18 |
| 48 | Fenvalerate | 27.384 | 419.10 > 225.10 | 6 | 419.10 > 167.10 | 16 |
| 49 | Pyraclostrobin | 27.553 | 164.10 > 132.10 | 16 | 164.10 > 77.00 | 30 |
| 50 | Fluvalinate (sum) | 27.800 | 250.10 > 55.00 | 20 | 250.10 > 200 | 20 |
| 51 | Esfenvalerate | 27.787 | 419.10 > 225.10 | 6 | 419.10 > 167.10 | 12 |
| 52 | Difenoconazole | 28.333 | 323.00 > 265.00 | 15 | 323.00 > 202.00 | 30 |
| 53 | Deltamethrin (sum) | 28.784 | 252.90 > 93.00 | 20 | 252.90 > 171.90 | 8 |
| 54 | Azoxystrobin | 29751 |
344.10 > 329.10 | 15 | 344.10 > 188.10 | 25 |
| 55 | Famoxadone | 29.483 | 330.10 > 224.10 | 10 | 330.10 > 196.10 | 22 |
Results and discussion
Microwave-assisted method optimization and validation
ETAAS technique was selected for As, Cd, and Pb determinations due to the low expected levels for these contaminants in Moringa stenopetala. This technology requires the use of chemical modifiers to decrease analyte volatilization and eliminate gas-phase interferences. These modifiers allow the thermal stabilization of volatile analytes, such as the ones studied in this work, enabling the application of higher pyrolysis temperatures and thus the conversion of matrix interferences into more volatile species. Many chemical modifiers have been described for As, Cd, and Pb determinations in the literature.18The nickel modifier has been widely used for As determinations by ETAAS, however, it does not always bring optimum results. Instead, the Pd(NO3)2–Mg(NO3)2 modifier allows the use of pyrolysis temperatures up to 1200–1400 °C and an optimum atomization temperature of 2200–2500 °C, stabilizing both inorganic and organic As species. In the case of Cd, ammonium phosphate has been used as a chemical modifier in ETAAS determinations to thermally stabilize the element up to 900 °C. However, spectral interferences may appear when trying to determine low Cd concentrations in matrices with high chloride content. The Pd(NO3)2–Mg(NO3)2 modifier proved to be especially good in these cases, being the maximum pyrolysis temperature around 800 °C. In the case of Pb, the Pd(NO3)2–Mg(NO3)2 modifier has been extensively and successfully applied in a wide variety of samples. Pyrolysis temperatures of 1100–1400 °C can be employed using this modifier, enabling the separation of highly interfering concomitants. The stabilizing effect of this modifier also allows a relatively high atomization temperature of 2000 °C.6,18,19
Therefore, the Pd(NO3)2–Mg(NO3)2 modifier was evaluated for all the studied analytes, using 5 μg of Pd(NO3)2 and 3 μg of Mg(NO3)2. Pyrolysis and atomization temperatures were exhaustively optimized by constructing pyrolysis/atomization curves in the range 600–3000 °C, using 200 °C intervals. Afterwards, a fine adjustment was performed leading to the conditions described in Table 1. There were no significant differences in the optimum pyrolysis temperatures using either the standard solutions or the sample solutions. The lowest temperatures for quantitative atomization were the same in both cases, suggesting the absence of matrix effects.
The microwave-assisted method was validated for As, Cd, and Pb determinations based on Eurachem Guide recommendations.20 For trueness evaluation, a Student's t-test was performed to compare the experimental values and the certified values of the spinach leaves CRM.21 All experimental t-values were below the theoretical t (0.05, 5) = 2.57, indicating that the obtained concentrations did not differ significantly from the certified values, at the 95% confidence level. Repeatability expressed as RSD (%) for the analysis of the CRM (n = 6) was less than 10% for all the studied elements. Detection and quantification limits (LOD and LOQ) as well as linearity ranges for each element are summarised in Table 3.
QuEChERS method optimization and validation
To select the best sample preparation for pesticide residue analysis, the QuEChERS AOAC 2007.01 method adapted for matrices with low water content, was employed. Four different clean-up combinations were tested with the conventional values for the evaluated method as follows:| Option 1: PSA + MgSO4 |
| Option 2: PSA + C18 + MgSO4 |
| Option 3: PSA + GCB + MgSO4 |
| Option 4: PSA + GCB + C18 + MgSO4. |
Fortifications were assayed at 100 μg kg−1 (n = 3) for the four clean-up options described above. Trial assays were performed to check the fit-for-purpose of the four methods with 15 representative compounds, including organochlorines, organophosphates, pyrethroids and fungicides such as conazoles. The figures of merit obtained for each of the methods were compared, and the first option yielded better results.
Even though all the methods resulted in acceptable precision values (≤20%), expressed as RSD percentages, the recovery percentages were considerably better for option 1. The results of the preliminary test can be seen in Fig. 1.
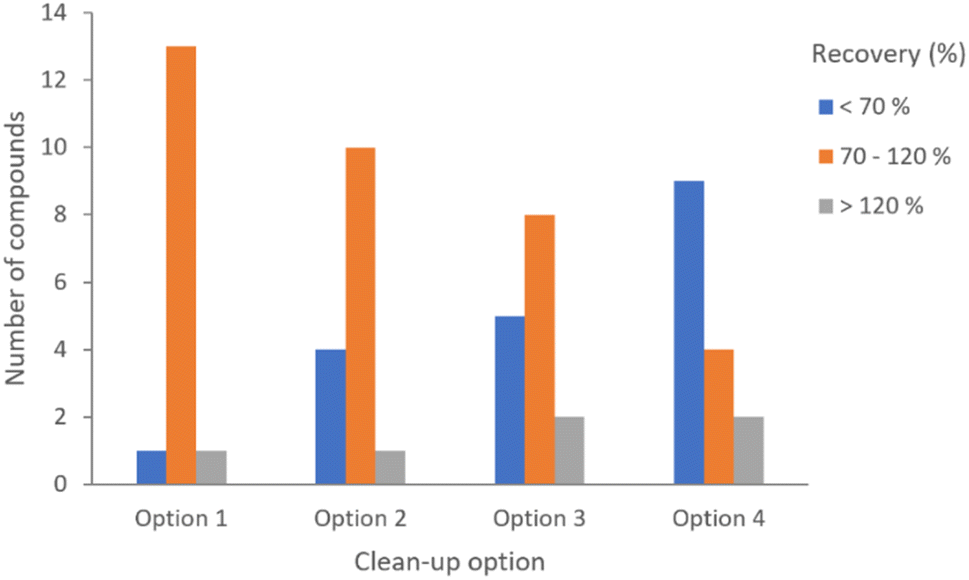 | ||
| Fig. 1 Dispersive clean-up comparison for the selection of pesticide analysis residue protocol. | ||
The method validation for option 1 was performed following DG-SANTE Guidelines for 55 analytes.22 The figures of merit evaluated were: linearity (via correlation coefficient and residual analysis), trueness (average recovery for spike levels tested), precision (RSDr: repeatability for spiked levels tested and RSDwR: within-laboratory reproducibility), limit of quantitation (LOQ) (lowest spike level meeting the method performance criteria for trueness and precision) and matrix effect (Fig. 2).
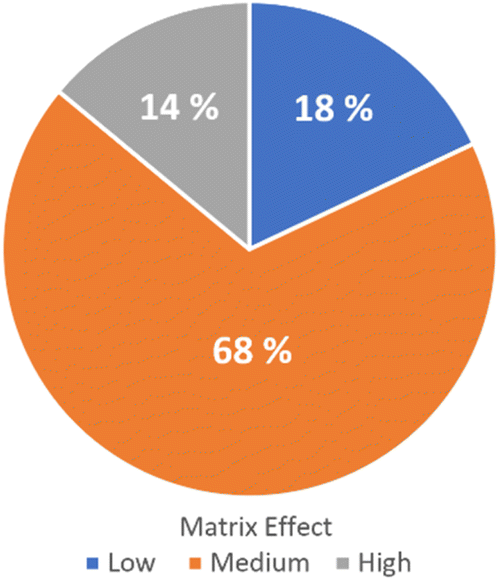 | ||
| Fig. 2 Matrix effect classification. Low effect (blue), medium effect (orange), and high effect (gray). | ||
Linearity was evaluated considering peak areas by constructing six-point calibration curves with a wide concentration range (two orders of magnitude). Good linearity was observed for all compounds at concentrations within the range tested (up to 200 μg kg−1), with determination coefficients (R2) greater than 0.99. The individual residues for each compound were studied and the deviations from the calibration curve in the corresponding region were <20% in all cases, as established by the DG-SANTE Guidelines. In the same way, the calculation of back-calculated concentration was also <20% in all cases.22
Recoveries were evaluated at four concentration levels: 10, 25, 50, and 100 μg kg−1. Five replicates were analyzed for each spiking level. Recovery values obtained at all concentration levels assayed, were in the range 70–120%. Precision expressed as RSDr and RSDwR were below 20% for all the studied pesticides. Considering the selected spiking levels, 40% of the studied analytes presented LOQ values of 10 μg kg−1, 42% presented LOQ values of 25 μg kg−1 and 18% presented LOQ values of 50 μg kg−1. Precision and trueness requirements were met in at least two of the different concentration levels evaluated, for all the analytes. The LOQ was less than or equal to the corresponding MRL value established by the European Union for Moringa species, in all cases.23
Solvent and matrix-matched calibration curves were compared, and matrix effects were quantified. Percentage matrix effect (ME%) for each compound was calculated as: ME% = [(matrix matched calibration curve slope − solvent calibration curve slope)/solvent calibration curve slope] × 100. Out of 55 pesticides tested in QuEChERS extracts, 7 exhibited a low matrix effect (ME < 20%), 34 showed a medium matrix effect (20% < ME < 50%), and 14 presented a strong matrix effect (ME > 50%). In turn, regarding the sign of the matrix effect, it was observed that it was mostly negative (51 out of the 55 analytes studied), that is, they presented suppression of the analytical signal. This relative signal suppression may be due to analytes' behavior in the GC inlet (liner and start of the column) that could be affected by the presence of other compounds from the matrix.24 Matrix components may co-elute with the target analytes, causing ionization suppression in the mass spectrometer. On the other hand, 4 analytes presented positive matrix effects, namely: dicofol, cyfluthrin, p,p′-DDT and chlorantraniliprole. For this reason, the quantification was performed using matrix-matched calibration curves prepared using a blank sample extracted with the same procedure as the recovery test samples. Blank samples were obtained from local organic producers. The selected method was fit for the intended purpose as the LOQs are below the fixed MRLs in the EU Pharmacopoeia 11 Ed., and the default value of 0.05 mg kg−1 of the EU MRLs for herbs.
As, Cd and Pb determinations in the analysed samples
Once the validation was completed, 10 Moringa stenopetala samples were analyzed. In the case of As the concentrations found were below the respective LOQ values. The average values for Cd were in the range 0.010 ± 0.001 to 0.017 ± 0.002 mg kg−1, while average values for Pb were in the range 0.21 ± 0.02 to 0.37 ± 0.03 mg kg−1. These Pb results are in line with previous reports.25 As previously stated, the mineral composition of plant-origin products depends on several factors such as genetics, soil characteristics and environmental conditions. Much information can be found in the literature about essential elements in Moringa stenopetala, however, data concerning potentially toxic elements levels is scarce.According to the obtained results, all samples complied with the requirements of MERCOSUR regulation related to these inorganic contaminants in edible vegetables, being As and Pb below 0.30 mg kg−1 and Cd below 0.20 mg kg−1.7 Furthermore, these values agree with the values of 0.02 mg kg−1 (Cd) and 2 mg kg−1 (Pb), recommended by World Health Organization (WHO) for plants in unpolluted soils (Table 3).26 Results also complied with the maximum limits established by the European Union for Moringa species.27
Vascular plants such as Moringa stenopetala take up metals and metalloids from their roots, transpiration through stomata, and deposition on the leaf surface, being deposition the main route of entry in the food chain. Therefore, proper surveillance of edible plants is of utmost importance from the toxicological point of view, mandatory when food safety is to be evaluated.6,28,29
Pesticide residues determinations in the analyzed samples
Once the validation was completed (Table 4), the analysis of the same 10 commercial samples, acquired from local markets, was carried out. No positives for any of the pesticides included in the scope of the method were detected.| # | Compound | Trueness and precision | LOQ (μg kg−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 μg kg−1 | 25 μg kg−1 | 50 μg kg−1 | 100 μg kg−1 | |||||||
| Rec (%) | RSD (%) | Rec (%) | RSD (%) | Rec (%) | RSD (%) | Rec (%) | RSD (%) | |||
| 1 | 2-Phenylphenol | 113 | 8 | 99 | 10 | 77 | 12 | 86 | 14 | 10 |
| 2 | Tecnazene | — | — | — | — | 83 | 8 | 78 | 12 | 50 |
| 3 | Ethoprophos | — | — | 94 | 14 | 88 | 6 | 85 | 9 | 25 |
| 4 | Trifluralin | 78 | 10 | 70 | 6 | 75 | 11 | 78 | 5 | 10 |
| 5 | Cadusafos | — | — | 71 | 8 | 70 | 9 | 89 | 11 | 25 |
| 6 | Gamma-BHC (lindane) | 104 | 6 | 71 | 9 | 70 | 12 | 83 | 10 | 10 |
| 7 | Diazinon | 83 | 4 | 72 | 6 | 75 | 9 | 92 | 7 | 10 |
| 8 | Tefluthrin | — | — | 75 | 10 | 74 | 12 | 81 | 8 | 25 |
| 9 | Pirimicarb | — | — | 85 | 8 | 91 | 14 | 94 | 10 | 25 |
| 10 | Chlorpyrifos-methyl | 92 | 5 | 78 | 6 | 76 | 5 | 83 | 3 | 10 |
| 11 | Vinclozolin | 72 | 8 | 87 | 9 | 70 | 7 | 100 | 10 | 10 |
| 12 | Parathion-methyl | — | — | 79 | 6 | 79 | 14 | 88 | 10 | 25 |
| 13 | Alachlor | — | — | — | — | 73 | 11 | 88 | 6 | 50 |
| 14 | Fenchlorphos | 70 | 6 | 85 | 7 | 74 | 5 | 82 | 8 | 10 |
| 15 | Pirimiphos-methyl | 77 | 9 | 110 | 8 | 76 | 11 | 85 | 4 | 10 |
| 16 | Fenitrothion | — | — | 119 | 10 | 84 | 12 | 93 | 8 | 25 |
| 17 | Malathion | — | — | 113 | 15 | 82 | 10 | 96 | 12 | 25 |
| 18 | Chlorpyrifos | 108 | 8 | 95 | 10 | 79 | 9 | 79 | 14 | 10 |
| 19 | Fenthion | 70 | 6 | 75 | 8 | 79 | 14 | 94 | 10 | 10 |
| 20 | Parathion | 82 | 10 | 87 | 9 | 87 | 12 | 96 | 7 | 10 |
| 21 | Dicofol | 94 | 14 | 72 | 16 | 102 | 9 | 103 | 14 | 10 |
| 22 | Heptachlor-exo-epoxide | — | — | 77 | 16 | 81 | 14 | 92 | 16 | 25 |
| 23 | Chlorfenvinphos (sum) | — | — | 84 | 12 | 80 | 11 | 93 | 9 | 25 |
| 24 | Mecarbam | — | — | — | — | 92 | 10 | 87 | 8 | 50 |
| 25 | Bromophos-ethyl | — | — | 70 | 11 | 85 | 14 | 79 | 8 | 25 |
| 26 | Chlordane (cis + trans) | — | — | — | — | 74 | 12 | 80 | 14 | 50 |
| 27 | Kresoxim-methyl | — | — | 72 | 8 | 92 | 10 | 91 | 8 | 25 |
| 28 | Endrin | — | — | — | — | 77 | 10 | 79 | 9 | 50 |
| 29 | Fensulfothion | — | — | 116 | 5 | 98 | 9 | 105 | 10 | 25 |
| 30 | Ethion | — | — | 81 | 8 | 85 | 9 | 89 | 5 | 25 |
| 31 | p,p′-DDD | 73 | 10 | 74 | 11 | 70 | 14 | 78 | 9 | 10 |
| 32 | Trifloxystrobin | — | — | — | — | 76 | 16 | 93 | 13 | 50 |
| 33 | Endosulfan sulfate | 76 | 13 | 100 | 8 | 78 | 14 | 95 | 17 | 10 |
| 34 | p,p′-DDT | 70 | 17 | 113 | 14 | 80 | 12 | 82 | 14 | 10 |
| 35 | Bifenthrin | — | — | 71 | 8 | 72 | 14 | 77 | 12 | 25 |
| 36 | Bromopropylate | 79 | 9 | 80 | 8 | 97 | 10 | 93 | 11 | 10 |
| 37 | Chlorantraniliprole | — | — | 98 | 13 | 87 | 13 | 92 | 8 | 25 |
| 38 | Fenazaquin | — | — | 81 | 8 | 75 | 14 | 90 | 9 | 25 |
| 39 | Phosalone | 70 | 15 | 75 | 12 | 75 | 10 | 94 | 9 | 10 |
| 40 | Pyriproxyfen | — | — | — | — | 91 | 9 | 90 | 14 | 50 |
| 41 | Cyhalothrin | — | — | 88 | 12 | 70 | 10 | 88 | 9 | 25 |
| 42 | Mirex | — | — | — | — | 72 | 9 | 70 | 15 | 50 |
| 43 | Permethrin (sum) | — | — | 70 | 11 | 83 | 9 | 88 | 14 | 25 |
| 44 | Cyfluthrin (sum) | — | — | 102 | 13 | 100 | 9 | 91 | 8 | 25 |
| 45 | Boscalid | 88 | 16 | 83 | 14 | 87 | 8 | 97 | 12 | 10 |
| 46 | Flucythrinate (sum) | — | — | 70 | 13 | 99 | 9 | 86 | 14 | 25 |
| 47 | Etofenprox | — | — | 90 | 10 | 85 | 7 | 82 | 15 | 25 |
| 48 | Fenvalerate | — | — | 70 | 9 | 82 | 9 | 89 | 11 | 25 |
| 49 | Pyraclostrobin | — | — | 117 | 12 | 78 | 11 | 98 | 9 | 25 |
| 50 | Fluvalinate (sum) | 71 | 17 | 82 | 14 | 73 | 13 | 84 | 8 | 10 |
| 51 | Esfenvalerate | — | — | — | — | 93 | 9 | 94 | 10 | 50 |
| 52 | Difenoconazole | 76 | 11 | 78 | 11 | 89 | 13 | 98 | 14 | 10 |
| 53 | Deltamethrin (sum) | — | — | — | — | 76 | 9 | 105 | 10 | 50 |
| 54 | Azoxystrobin | 72 | 12 | 79 | 9 | 110 | 9 | 84 | 14 | 10 |
| 55 | Famoxadone | 94 | — | 84 | 10 | 88 | 9 | 87 | 9 | 10 |
Although there are some reports concerning pesticide residue determination in Moringa oleifera samples,30,31 scarce information is available on this sort of analysis for Moringa stenopetala monitoring. This highlights once again the importance of the present study. Pyrethroids such as deltamethrin and permethrin have been quantified in Moringa oleifera samples with levels above the corresponding MRL, however, these were not detected in this work.32 These results reinforce the importance of the analysis of different species of natural plant products due to their ontogeny variation.
Conclusions
The optimization and validation of two multi-contaminant analytical methods for the determination of inorganic contaminants and pesticide residues in Moringa stenopetala, was successfully carried out.The proposed methods were suitable and efficient alternatives for monitoring relevant pollutants in Moringa stenopetala, being the values obtained in commercial samples analyzed to test the performance of the methods, within the limits established by national and international regulations, ensuring the food safety of these highly consumed herb. Thus, the proposed methods can be postulated as good strategies for food surveillance. The overall work highlights the importance of performing multidisciplinary studies to cover wider ranges of scope in food analysis.
Author contributions
Machado, Gérez, and Bertón were responsible for sample preparation and measurement collection. Machado and Gérez were responsible for the data processing and its interpretation, experimental design, and writing. Heinzen and Cesio were responsible for ideation, funding acquisition, data visualization, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Agencia Nacional de Investigación e Innovación (ANII) and PEDECIBA-Química.References
- A. Melesse, Y. Getye, K. Berihun and S. Banerjee, Livest. Sci., 2013, 157, 498–505 CrossRef.
- S. Habtemariam and G. K. Varghese, Beverages, 2015, 1(3), 169–182 CrossRef CAS.
- S. Habtemariam, The African and Arabian Moringa Species, Chemistry, Bioactivity and Therapeutic Applications, Elsevier, Amsterdam, 1st edn, 2017 Search PubMed.
- I. Machado and I. Dol, J. Food Compos. Anal., 2021, 103, 104090 CrossRef CAS.
- A. A. Shaltout, M. S. Abdel-Aal, B. Welz and I. N. B. Castilho, Anal. Lett., 2013, 46, 2089–2100 CrossRef CAS.
- I. Machado, I. Dol, E. Rodríguez-Arce, M. V. Cesio and M. Pistón, Microchem. J., 2016, 128, 128–133 CrossRef CAS.
- Reglamento técnico MERCOSUR (DEROGACIÓN DE LAS RES. GMC No. 102/94 y No. 35/96) MERCOSUR/GMC/RES. No. 12/11, https://www.puntofocal.gov.ar/doc/r_gmc_12-11.pdf, accessed November 2023 Search PubMed.
- F. Iaquinta, L. Lopes Fialho, J. A. Nóbrega, M. Pistón and I. Machado, J. Food Meas. Charact., 2021, 15, 4105–4111 CrossRef.
- F. Iaquinta, L. Lopes Fialho, J. A. Nóbrega, M. Pistón and I. Machado, Food Anal. Methods, 2021, 14(1), 156–164 CrossRef.
- R. Štěpán, J. Tichá, J. Hajšlová, T. Kovalczuk and V. Kocourek, Food Addit. Contam., 2005, 22(12), 1231–1242 CrossRef PubMed.
- I. Machado, N. Gerez, M. Pistón, H. Heinzen and M. V. Cesio, Food Chem., 2017, 227, 227–236 CrossRef CAS PubMed.
- M. Anastassiades, S. J. Lehotay, D. Stajnbaher and F. J. Schenck, J. AOAC Int., 2003, 86(2), 412–431 CrossRef CAS PubMed.
- S. J. Lehotay, K. Maštovská and A. R. Lightfield, J. AOAC Int., 2005, 88(2), 615–629 CrossRef CAS PubMed.
- Official Methods of Analysis of the Association of Official Analytical Chemists, Official Methods of Analysis of AOAC International, AOAC, Washington DC, 21st edn, 2019 Search PubMed.
- I. Machado, M. V. Cesio and M. Pistón, Microchem. J., 2017, 133, 663–668 CrossRef CAS.
- N. Gérez García, G. Zinola, G. Macías, M. V. Cesio and H. Heinzen, Microchem. J., 2022, 178, 107349 CrossRef.
- F. Jesús, R. Hladki, N. Gerez, N. Besil, S. Niell, G. Fernández, H. Heinzen and M. V. Cesio, Talanta, 2018, 178, 410–418 CrossRef PubMed.
- B. Welz and M. Sperling, Atomic Absorption Spectrometry, WILEY-VCH Verlag GmbH, Weinheim, 3rd edn, 1999 Search PubMed.
- F. Tissot and I. Machado, J. Food Meas. Charact., 2020, 14, 1842–1849 CrossRef.
- Eurachem Guide, The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, 2nd edn, 2014 Search PubMed.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Prentice Hall – Pearson Education Limited, Harlow, 6th edn, 2010 Search PubMed.
- European Commission, DG-SANTE, 2021, Document No. SANTE/11312/2021. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed, https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf, accessed November 2023 Search PubMed.
- European Commission, European Union Pesticides Database, 2023, https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/, accessed November 2023 Search PubMed.
- M. Soliman, M. A. Khorshid and M. M. Abo-Aly, Microchem. J., 2020, 156, 104852 CrossRef CAS.
- A. M. Yimer and M. A. Khan, Br. J. Appl. Sci. Technol., 2016, 13(5), 1–6 CrossRef CAS.
- World Health Organization (WHO), Permissible Limits of Heavy Metals in Soil and Plants, Geneva, 1996 Search PubMed.
- European Commission, Food Safety, 2023, https://food.ec.europa.eu/safety/chemical-safety/contaminants/legislation_en, accessed November 2023 Search PubMed.
- E. P. Colangelo and M. L. Guerinot, Curr. Opin. Plant Biol., 2006, 9, 322–330 CrossRef CAS PubMed.
- M. A. G. O. Azenha and M. T. S. D. Vasconcelos, Food Chem. Toxicol., 2000, 38, 899–912 CrossRef CAS PubMed.
- E. I. Adeyeye, O. A. Ibigbami, A. F. Akinsola, O. A. Akinwumi, H. O. Adubiaro and A. J. Adesina, Indian J. Public Health Res. Dev., 2020, 11(8), 261–267 Search PubMed.
- Y. Zhu, P. Du, J. Yang, Q. Yin and Y. Yang, RSC Adv., 2020, 10, 36906 RSC.
- E. Scherbaum and C. Lerch, Die Untersuchungsämter für Lebensmittelüberwachung und Tiergesundheit, Baden-Württemberg, 2017, https://www.ua-bw.de/pub/beitrag.asp?ID=2445%26subid=1, accessed November 2023 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |