
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface functionalization of a chalcogenide IR photonic sensor by means of a polymer membrane for water pollution remediation
Martin
Vrážel
a,
Raïssa Kadar
Ismail
bc,
Rémi
Courson
 d,
Abdelali
Hammouti
e,
Marek
Bouška
a,
Amélie
Larrodé
b,
Marion
Baillieul
a,
William
Giraud
f,
Stéphane
Le Floch
f,
Loïc
Bodiou
e,
Joël
Charrier
e,
Kada
Boukerma
d,
Karine
Michel
c,
Petr
Němec
a and
Virginie
Nazabal
*ba
d,
Abdelali
Hammouti
e,
Marek
Bouška
a,
Amélie
Larrodé
b,
Marion
Baillieul
a,
William
Giraud
f,
Stéphane
Le Floch
f,
Loïc
Bodiou
e,
Joël
Charrier
e,
Kada
Boukerma
d,
Karine
Michel
c,
Petr
Němec
a and
Virginie
Nazabal
*ba
aDepartment of Graphic Arts and Photophysics, Faculty of Chemical Technology, University of Pardubice, Studentska 573, 53210 Pardubice, Czech Republic
bUniv Rennes, CNRS, ISCR - UMR6226, F-35000 Rennes, France. E-mail: virginie.nazabal@univ-rennes.fr
cBRGM, Direction Eau, Environnement et Ecotechnologies, 45100 Orleans, France
dIFREMER, Laboratoire Détection, Capteurs et Mesures, 29280 Plouzané, France
eUniv Rennes, CNRS, Institut Foton - UMR 6082, F-22305 Lannion, France
fCEDRE, Research, 29218 Brest, France
First published on 24th July 2024
Abstract
Rapid, simultaneous detection of organic chemical pollutants in water is an important issue to solve for protecting human health. This study investigated the possibility of developing an in situ reusable optical sensor capable of selective measurements utilizing a chalcogenide transducer supplemented by a hydrophobic polymer membrane with detection based on evanescent waves in the mid-infrared spectrum. In order to optimise a polyisobutylene hydrophobic film deposited on a chalcogenide waveguide, a zinc selenide prism was utilized as a testbed for performing attenuated total reflection with Fourier-transform infrared spectroscopy. To comply with the levels mentioned in health guidelines, the target detection range in this study was kept rather low, with the concentration range extended from 50 ppb to 100 ppm to cover accidental pollution problems, while targeted hydrocarbons (benzene, toluene, and xylene) were still detected at a concentration of 100 ppb. Infrared measurements in the selected range showed a linear behaviour, with the exception of two constantly reproducible plateau phases around 25 and 80 ppm, which were observable for two polymer film thicknesses of 5 and 10 μm. The polymer was also found to be reusable by regenerating it with water between individual measurements by increasing the water temperature and flow to facilitate reverse exchange kinetics. Given the good conformability of the hydrophobic polymer when coated on chalcogenide photonic circuits and its demonstrated ability to detect organic pollutants in water and to be regenerated afterwards, a microfluidic channel utilising water flow over an evanescent wave optical transducer based on a chalcogenide waveguide and a polyisobutylene (PIB) hydrophobic layer deposited on its surface was successfully fabricated from polydimethylsiloxane by filling a mold prepared via CAD and 3D printing techniques.
1. Introduction
Owing to the high population density of urbanized and industrialized societies, the world nowadays faces problems related to wastewater management, degradation of the water quality as well as microbial and aquatic flora.1,2 Consequently, there has been a push to closely monitor the type and amount of a variety of organic compounds in water as it is an important aspect for ensuring the safety of the local population. While a number of techniques capable of detecting these compounds exist,3–6 there are concerns regarding the accuracy and speed with which crucial data can be obtained. One major issue is related to where the measurement takes place. The development of in situ analysis devices has been receiving much attention recently, and the analysis methods can be classified into four major categories: monitoring stations, portable instruments, electrochemical devices, and optical devices. These can be further categorized into devices in direct contact with the sample (in-line) and devices located at a fixed location near periodically checked environments (on-line). A third variation is a device not located near the site, which requires sampling (off-line). Important aspects of any sensor are calibration procedures, which ensure data quality and reliability, and in-line sensors are rated among the most difficult to calibrate, making their development a challenging process.7 While laboratory analyses are generally more accurate (e.g. HS-GC, GC-MS, HP-LC, and HS-SPME), they lose to in situ detectors in terms of time consumption and the need for water sampling. All water samples, especially those of surface water and wastewater, are susceptible to changes as a result of physical, chemical or biological reactions occurring between the time of sampling and the start of analysis, which may result in obtaining inaccurate data.8,9 Alternatively, existing in situ chemical sensors suitable for applications of contaminants monitoring (e.g. portable gas chromatographs, surface acoustic wave sensors, optical instruments)3–6 are often lacking in other areas. In situ water sensors are characterized by their sensitivity and selectivity towards chemical pollutants, speed of detection, and reliability for timely measurements; however, increasingly important factors today include the need for portability, versatility, ease-of-use, and cost reduction.5,8,10 Furthermore, despite achieving sensitivities comparable to laboratory methods (ng to μg L−1 scale), the ability of portable chemical sensors to identify a specific molecule in a mixture remains a significant challenge when analyzing real environmental samples.3,4 Mahmud et al. noted that despite the recent development of methods for the detection of nutrients in aquatic environments based on potentiometric, amperometry, and optical sensors, key problems still persist; notably, sensitivity and selectivity towards the measured molecules in water, and fouling prevention, showing the enduring need for reusable or renewable sensors.11 To meet all the challenges mentioned above, we developed a portable microfluidic optical sensor, based on a chalcogenide mid-infrared transducer and functionalized by a hydrophobic polymer, which was capable of analyte extraction and regeneration. The final aim was to achieve the real-time monitoring of multiple organic contaminants in various water body forms, especially in regard to cases of accidental pollution. Benzene, toluene, and xylene (BTX) were chosen as the target molecules for analysis, as they pose a serious threat to human health and the determination of their presence in water is crucial in deciding, whether the environment is safe for other lifeforms. Their accidental presence in the environment can be either from natural causes (crude oil, emissions from forest fires) or as a result of man-made pollution (engine emissions, oil processing, environmental disasters).12,13While there exist multiple in situ mid-infrared (MIR) techniques (IR spectroscopic ellipsometry, reflection absorption IR spectroscopy, attenuated total reflectance (ATR) operating with Fourier-transform IR spectroscopy) with multiple geometries (such as simple reflection, transmission, and fiber waveguide), MIR evanescent spectroscopy was selected as a suitable method for detecting various organic priority substances, thanks mainly to its high sensitivity, good selectivity, and non-destructive method of measurement.5,14 The objective was to develop a microsensor with both improved selectivity and sensitivity to organic chemical pollutants using high MIR transparency chalcogenide glass-based transducers. The high transparency window of such a system includes the IR signatures of the fundamental vibrations of organic species and biomolecules (λ ≥ 6.7 μm or 1500 cm−1), providing a simple, reliable, fast, and non-destructive method for detecting and determining the composition of complex samples.2
Generally, chalcogenide glasses have very specific characteristics, such as a broad transmission range, high linear and non-linear refractive indices, photosensitivity, shaping ability (being able to produce fibres or thin films), and phase changes from amorphous to crystalline states. They can be doped by rare earth elements, making them suitable for a variety of applications, such as reconfigurable lenses or photonic integrated circuits, optical memory, optical signal processing, (bio)chemical sensors, or for on-chip mid-IR emission.15,16 Chalcogenide glasses used for their optical properties are usually based on germanium and/or arsenic and/or antimony in varying ratios to tune their optical properties, especially their refractive index. In recent years, there has been an emergence of MIR spectroscopy platforms based on materials suitable for the MIR spectral range, such as Si, SiNx, Ge, and GaAs.17 Considering the above-mentioned challenges, chalcogenide glass materials and their amorphous films have key advantages: (i) different compositions available, from sulfides to tellurides, for tailoring the chemical and physical properties of amorphous waveguides; (ii) broad transparent window up to λ = 20 μm depending on the composition; (iii) tuneable refractive index in the range of ∼2.2–3.2 at 1.55 μm allowing for fine opto-geometric design for IR propagation, and (iv) easy deposition of various chalcogenide glass films of various thicknesses on Si or other flexible substrates by physical vapour deposition (PVD).18,19 A previously conducted theoretical study aimed at detecting toluene in aqueous media using a ridged, chalcogenide waveguide enhanced by polymer functionalization showed that the detection threshold could theoretically reach a value equal to 26 μg L−1 at λ = 6.66 μm (ref. 20) by means of an IR microsensor, achieving a lower LOD than unoptimized IR-ATR or IR-fibre spectroscopy (60–339 μg L−1).21,22
Polyisobutylene (PIB) was chosen as a hydrophobic polymer because of its high transmittance in the investigated wavelength range and its ease of preparation. The functionalized waveguide can utilize its permeability to extract the analytes and thus bring them closer to the evanescent field, while excluding water from the analyzed solution. PIB was previously successfully evaluated for detecting hydrocarbons in natural and complex water samples.19,21,23–27
The, currently under development, optical microdevice equipped with a PIB membrane was created with a view to its on-field use, and with improved sensitivity, reusability, and reduced costs compared to other field devices. The aim of this study was to participate in such a development by addressing the aspects needed for creating a robust, portable, capable of in situ measurements, and reusable sensor using renewable hybrid materials for on-site environmental monitoring with a short acquisition time (<30 min) and low LOD range (75–200 ppb depending on the organic chemical pollutants targeted). As part of this development, there is a requirement to validate the sensor use in cases of accidental pollution in a large concentration range, as well as to validate its analysis time and limit of detection (previously reported as 250 ppb for BTX by ATR-FTIR spectroscopy).27 The possible regeneration of the polymer must also be proven for such a high concentration usage in order to reduce the negative environmental impact from the manufacturing process. It is also essential to optimize its regeneration by ensuring cycle repeatability, to transfer the polymer to the selenide transducer, and to develop a microfluidic cell compatible with the selenide transducer functionalized by a layer of hydrophobic polymer as a basis for a future microfluidic MIR sensor based on chalcogenides.
2. Experimental
The polyisobutylene for deposition was sourced from Sigma-Aldrich and was used as provided (average Mw = 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). For dissolving the PIB, a mixture of xylene isomers in ethylbenzene was utilized (≥98.5% xylenes in ethylbenzene basis, Sigma-Aldrich).
000 g mol−1). For dissolving the PIB, a mixture of xylene isomers in ethylbenzene was utilized (≥98.5% xylenes in ethylbenzene basis, Sigma-Aldrich).
A stock solution of BTX was prepared by mixing benzene (99%), toluene (>99.5%), O-xylene (97%), M-xylene (>99%), and P-xylene (>99%), all obtained from Sigma-Aldrich and used as provided, at a concentration of 50000 mg L−1 each (weight ppm) in methanol (anhydrous, >99.8%, Sigma-Aldrich). To obtain aqueous solutions of the chosen concentrations ranging from hundreds of weight ppb to a hundred of weight ppm, the appropriate volume of stock solution was diluted with distilled water.
ATR crystals (80 mm × 10 mm × 4 mm) made from ZnSe (Innovation Photonics), with an angle of incidence of 45° were used as the testing ground for the ATR experiments with the polymer. A 10% w/w PIB solution for coating was prepared by dissolving the polymer into a mixture of xylenes and ethylbenzene, which was the maximum concentration at which the viscosity of prepared solution was still low enough to be reproducibly deposited by spin-coating. Spin-coating (using a WS-400B-6NPP-Lite Spin Processor by Laurell Technologies) was performed by spreading 0.5 ml of solution across the substrate and spinning for 30 s at 590 rpm for obtaining a thinner film (5 μm) and at 185 rpm for a thicker film (10 μm) and then left overnight to allow evaporation of the solvent under a fume hood at room temperature. Solutions of 9%, 9.5%, and 10% w/w were prepared for the viscosity measurements (using a RV1_HAAKE rotational viscometer in a cylinder-in-cylinder configuration), and then tested after a period of one month to determine the potential change in viscosity caused by solution ageing; however, no degradation of the solutions occurred over this period.
Contact angle goniometry (using an OCA 50EC system, DataPhysics Instruments) using a static sessile drop method was used to determine the water contact angle of the polymer film deposited on a Si substrate.
By spin-coating, PIB films with thicknesses of 5.1 ± 0.3 μm and 10.2 ± 0.3 μm were deposited on the ZnSe crystal. The thickness of the film was measured using a gravimetric technique and determined according to eqn (1):
 | (1) |
The two thicknesses of polymer were selected considering the depth of penetration of an evanescent wave, which directly depends on the wavelength of the radiation and the refractive index of the prism and the polymer film. This penetration depth can be used to calculate appropriate thickness of the film using eqn (2):28
 | (2) |
ATR measurements were performed on a Nicolet Fourier-transform infrared spectrometer (iS50 Spectrometer by Thermo Fisher), equipped with an ATR accessory (HATR produced by PIKE technologies). The parameters of the measurement were the same throughout all the experiments: 32 scans performed over 47 s in the 650–4500 cm−1 spectral range and the aperture opened to 8 mm. A flow cell, containing ZnSe crystal coated by PIB, was installed onto the accessory and the tested solution was pumped across the surface by a peristaltic pump.
The infrared spectra of the molecules of benzene, toluene, and the three isomers of xylene included numerous characteristic absorption bands between 650–3100 cm−1. The strongest IR signals could be found in the range between 650–900 cm−1,29 where we could easily discern between individual molecules, and since the sensitivity of ATR-FTIR spectrometry is lower than that of a microsensor, this range was used to characterize and optimize the PIB films by the ATR method. The second (950–1200 cm−1) and third (1450–1650 cm−1) regions29 could also be exploited for detecting BTX molecules, albeit with over 70% loss in signal strength. However, these spectral regions are favourable for the development of selenide microsensors involving a quantum cascade laser source and infrared detector. Therefore, it would be interesting to compare them with the more conventional range of detection operations when employing ATR-FTIR spectrometry. Moreover, depending on the polymer used, those regions might become important if there is a strong IR signal disturbance in the first area. As mentioned above, most discerning bands occurred between 650–900 cm−1. Thus, the characteristic absorption bands chosen for the detection of BTX using ATR-FTIR spectrometry for optimization of the PIB layer were located at 674 cm−1 for benzene, 692 and 727 cm−1 for toluene, 741 cm−1 for O-xylene, 690 and 767 cm−1 for M-xylene, and 794 cm−1 for P-xylene.26,29Fig. 1 shows the differentiation of each band associated with a BTX molecule as they were separately analyzed in individual 5 ppm solutions. It is important to mention that the absorption band at 690 cm−1 for M-xylene is interfering with the band for toluene at 692 cm−1, making it more intensive. When comparing the peaks for the toluene solution with those for the BTX solution, the M-xylene increased the size of the toluene peak at 692 cm−1 by 55%. To clarify the quantification of the polluting molecules while maintaining a nomenclature close to previous papers, this band peaking at 692 cm−1 was named toluene M-X (toluene with M-xylene).26,27 In the second region (950–1200 cm−1), the absorption bands were more numerous and often interfered with each other. Despite that, the bands for benzene (1038 cm−1), toluene (1035 and 1085 cm−1), O-xylene (985, 1020, 1052, and 1120 cm−1), M-xylene (1048, 1100, and 1170 cm−1), and P-xylene (1022, 1040, 1100, and 1120 cm−1)29 can be used for calculating the concentration of hydrocarbons present in the water solution, particularly within the framework of a multivariate analysis approach. Absorption bands in the third region could be found for benzene at 1480 and 1525 cm−1, for toluene at 1465, 1500, and 1615 cm−1, for O-xylene at 1470, 1495, and 1605 cm−1, for M-xylene at 1500 and 1620 cm−1, and for P-xylene at 1450 and 1520 cm−1.29 Indeed, this range (1450–1650 cm−1) can be used for the detection of individual molecules. However, this region is not optimal for measuring a solution containing several hydrocarbons in addition to xylenes, as this is a less selective spectral region, where a chemometric approach is absolutely necessary. This would make it fairly easy to identify a mixture of BTX and track their degradation, while multivariate analysis should make it easier to exploit the data in this spectral range.
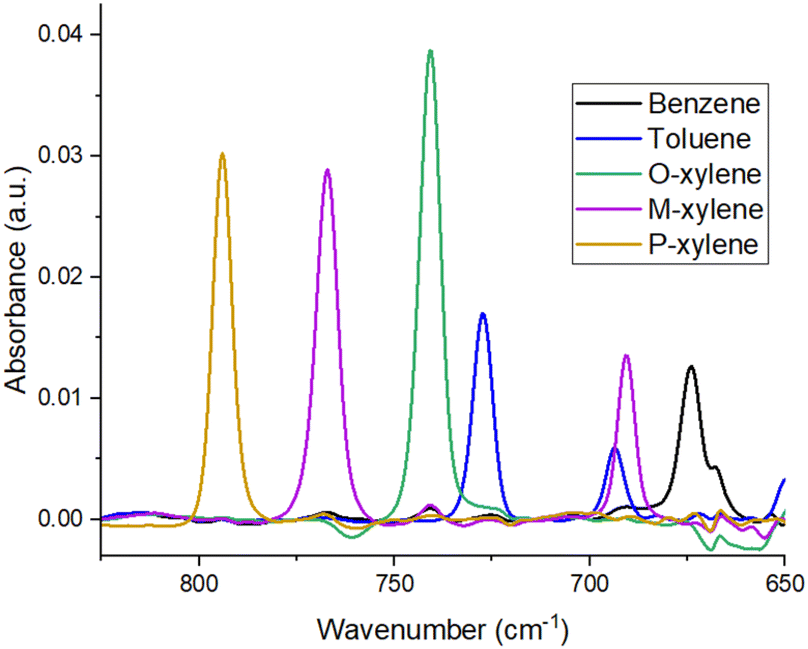 | ||
| Fig. 1 Absorbance spectra of solutions of benzene, toluene, and xylene molecules, each individually diluted in water to a concentration of 5 ppm. The measurement was taken after 90 min using 5 μm-thick polyisobutylene films. | ||
For all the spectra, polymer-coated ZnSe crystal was used as a background. The absorption bands intensities were analyzed using OriginLab (version 2022, OriginLab, Northampton, MA, USA) and GRAMS/AI© software (version 9.3, Thermo Fisher Scientific Inc., Waltham, MA, USA). OriginLab was used for the initial quantitative analysis, and the results were then validated with GRAMS. The baseline was set by the User Defined method, while the individual anchor points were detected by the 2nd derivative (zeroes) method. This let us utilize the fact that the baseline area had a smaller curvature than the absorption band area. After Adjacent-Averaging smoothing, the second derivative at each point was calculated. All the data points whose second derivative approached 0 (under the tolerance) were used to perform a second-order polynomial fit. The points lying closest to the fitted line as anchor points were adopted and they were connected by interpolation. This process creates a baseline that enables peaks to be manually selected and the areas beneath them to be integrated for quantitative calculation. However, as some molecules shared a band foot overlap, a more precise method was needed to calculate the exact areas associated with their absorption bands. This could be achieved by refining the spectrum envelope by considering a Gaussian sum to best describe the area under each spectral band associated with a molecule, which thus allowed us to separate the contribution of each (software GRAMS, Fig. 2), for instance. The Application Baseline Correction feature was used to remove the background noise using a polynomial function of the third-degree. The Peak Fitting application was then used to de-summarize the peaks using Gaussian functions.
 | ||
| Fig. 2 Fitting of the measured peaks using Gaussian bands, depicting the measurement of 5 ppm benzene, toluene, xylene solution after 90 min using a 5 μm-thick polyisobutylene film. | ||
While measurements were being made on the ATR-FTIR spectrometer, another series of measurements was carried out in parallel on a gas chromatograph combined with mass spectrometry (GC-MS 7890A system from Agilent Technologies). The aim was to determine the actual concentration of the solutions by GC-MS, in order to establish a validated calibration curve for the ATR-FTIR spectrometry. It was also necessary to determine the period of time during which the BTX solutions prepared could be used without any change in concentration, given the high volatility of the molecules to be detected. The measured periods were one and two weeks for a stock solution of 50000 ppm, 2 h for the daughter solution of 2500 ppm, and 90 min for a solution of 1 ppm, corresponding to the ATR-FTIR analysis time in the latter case. The results showed that the 1 ppm and 2500 ppm solutions did maintain the required concentration during the time frames tested. Surprisingly, the stock solution, which was kept at 4 °C after being prepared, also showed no significant decrease in concentration over the period of two weeks. Despite that, as a precautionary measure, the stock solution was renewed at the beginning of each week, to ensure repeatability of the results.
To control the consistency of the measurements regarding ATR-FTIR, a series of measurements were performed in order to determine any errors with which the BTX solutions were prepared. A series of ten 5 ppm solutions were prepared and measured on newly prepared PIB films. Out of these 10 solutions, we obtained very similar values of the average value of the absorption band areas: benzene: 0.055 ± 0.001 (deviation 2.4%), toluene M-X: 0.086 ± 0.001 (deviation 1.7%), toluene: 0.071 ± 0.002 (deviation 4.1%), O-xylene: 0.232 ± 0.007 (deviation 3%), M-xylene: 0.185 ± 0.004 (deviation 2.1%), and P-xylene: 0.190 ± 0.004 (deviation 2.2%). As the deviations were below 5%, this proved that we had prepared samples with consistent concentrations.
Next, the topography of the deposited films was measured by amplitude modulated atomic-force microscopy (AFM, Solver Next, NT-MDT). Tapping mode imaging was used on an area of 5 μm × 5 μm and 10 μm × 10 μm at a scanning rate of 1 line per 2 s, with the whole image consisting of 512 lines. We used root mean squared (RMS) values based on the median for the surface analysis.
To ascertain the adhesion between the polymer and the Ge–Sb–Se selenide photonic circuit and to observe the microstructure of the polymer film, scanning electron microscopy (SEM) was utilized to analyze the cross-sections of the deposited films. The used equipment (JEOL JSM 7100 F) was operated at a 8.5 mm working distance, with an accelerating voltage of 10 kV. Since PIB is a sticky material, the wafer with the deposited polymer could not be simply cut, as it would shear the polymer from the target. Therefore, the polymer had to be frozen under liquid nitrogen and then the wafer could be carefully broken at the place of interest.
3. Results and discussion
3.1. Polyisobutylene membrane deposition on the optical transducers
It has been pointed out in the literature that polymer membranes with interesting organic compounds extraction and diffusion rates are generally amorphous and rubbery, making polyisobutylene a good choice for the sensor.30–32 Since the polyisobutylene was used to form the surface of the optical transducer, it was characterized not only for its sensitivity towards aromatic hydrocarbons but also for its reproducibility, and reusability. As mentioned before, the PIB was successfully deposited on a chalcogenide photonic circuit.26 In this study, the reproducibility of the PIB film deposition on a selenide photonic circuit based on a Ge–Sb–Se glass system was verified as having a different chemical composition than previously reported, which was richer in antimony. The microsensor consisted of a Se4 (Ge19Sb17Se64) guiding layer and was deposited by radiofrequency magnetron sputtering on a Se2 (Ge28Sb6Se66) confinement layer. The latter was at least 5 μm thick to ensure the confinement of the mid-infrared wave in the Se4 waveguide with a lower refractive index than in the case of the preceding antimony-rich guiding layer. Several PIB films were deposited on the Se4 chalcogenide layer via spin-coating with differences in thickness below 5%, and using PIB solution less than one month old. As shown in Fig. 3, the polymer fully adhered to the chalcogenide Se4 waveguide previously etched by ICP-RIE, and the spin-coating deposition was fully compliant with the integrated photonic circuit. No air bubbles or defects in the polymer film could be observed, particularly at the interfaces with the Se4 chalcogenide–etched waveguide. The polymer appeared to have a homogeneous and dense microstructure, as could be observed by the amorphous layers. This suggested that there should be no negative influence associated with polymer film deposition on the optical measurements using this optical transducer, except for its own intrinsic absorption bands in the mid-IR spectral range. The optical transducer currently has losses in the order of 2–3 dB cm−1 at around 7 μm, which makes it possible to consider detection measurements. However, measurements of the optical losses in the MIR are still in progress in order to estimate the impact of the overall losses of the transducer with the PIB polymer. | ||
| Fig. 3 SEM image of a polyisobutylene film about 5 μm thickness, deposited on a Ge19Sb17Se64 (Se4/thickness of about 2 μm) chalcogenide waveguide on a Ge28Sb6Se66 (Se2/thickness of about 5 μm) planar film covering a silicon wafer. | ||
To check that the PIB layer met our expectations in terms of hydrophobicity, water contact angle measurements were carried out with the thickest layer. A series of 10 measurements was made with a 10 μm film, spin-coated onto a silicon wafer, using a static sessile drop method. These measurements gave a contact angle of 110° ± 3°, demonstrating the hydrophobicity of PIB under the conditions selected in this study. This value is also in line with other studies, which have reported contact angle values between 100°–110°.33,34
For analysis of the surface topography of the deposited polymer film, several measurements were conducted, using a profilometer and AFM. For this measurement, the polymer was deposited on a silicon wafer via spin-coating, using the same conditions as for the film deposition on the ZnSe prism. The thickness was also kept the same (5 μm). With the profilometer, the measured areas were 10 μm × 10 μm and 30 μm × 30 μm, since the profilometer is less sensitive, and was used to primarily gain the overall shape of the surface. The surface appeared generally flat with a few waves, which were caused mainly by the fluid viscosity, the surface tension, the parameters used in the spin-coating, and the solvent drying process. They may also have been caused by the rubber nature of the polymer, making the probe slightly jump at certain points, creating a possibly slightly distorted image. This might have also contributed to the higher value of RMS of 40 ± 10 nm, as we were expecting lower values given the method of deposition. AFM scans (10 μm × 10 μm and 5 μm × 5 μm) of the polyisobutylene films showed rather smooth surfaces with the exception of a few “cones”, which were about 9 nm in height and were randomly scattered across the surface (Fig. 4). The nature of these cones remains, as of now, unknown. The experiments carried out so far with PIB films on optical transducers had not been able to highlight this type of morphology, since SEM hardly allows such an observation at this size scale on a polymer layer. Consequently, several experiments were conducted in order to determine their nature or at least to reduce their presence in order to evaluate their possible impact on BTX detection. Here, the PIB film was cast using PIB solutions of different concentrations (9, 9.5, and 10% w/w) and different temperatures (20 °C, 25 °C, and 50 °C) in order to determine if the cones would change or disappear based on the viscosity and temperature of the PIB solution. The reason was to determine if those cones were inherent to the polymer or if there was an issue with dissolving the polymer wholly at a concentration of 10% w/w, which was the limit of solubility. However, despite those changes in the PIB solubilisation parameters, the cones still appeared and with the same size (in the range of 9–16 nm) and similar frequency. Another possibility for the nature of these cones were impurities present in the starting polymer. To eliminate this possibility, the 10% w/w solution was heated up to 60 °C and then filtered through 20 nm filters made from nylon or polyethylene terephthalate. However, the polymers prepared by this filtration process still featured the aforementioned cones; albeit, they appeared with less frequency and were smaller (8–9 nm for nylon and 4–7 nm for polyethylene terephthalate). Nevertheless, the original AFM measurements (with 10% w/w solution deposited at ambient temperature), despite the presence of the cones, showed a low RMS surface roughness of 0.4 ± 0.1 nm, which was not significantly modified by filtration and was within the expected range for spin-coated thin polymer films, based on literature.35,36 It should be noted that this roughness is of the order of that reported for sputtered chalcogenide films deposited on a silicon substrate to form integrated photonic circuits required for infrared microsensor development.37
 | ||
| Fig. 4 AFM scan of a 5 μm × 5 μm area of the 10% polyisobutylene film. | ||
3.2. Characterization of the PIB polymer membrane detection efficiency to transpose it onto a Ge–Sb–Se selenide microsensor
As mentioned, the ultimate goal of this project was to be able to use the PIB on a selenide microsensor in a wavelength range suitable for a QCL laser and an infrared detector in a relatively comfortable way, i.e. in the range around 650–1650 cm−1 where BTX have defined absorption bands, which would make it possible to identify them and to possibly monitor their degradation. To qualify the PIB polymer suitable for Ge–Sb–Se photonic integrated circuit transducers and determine its effectiveness, durability, and optimize its sensitivity via the choice of a suitable thickness, the most relevant optical spectroscopy method to achieve a transfer to an evanescent wave technology remains ATR-FTIR spectroscopy using a selenide-based prism, which can allow relatively good simulation of the selenide optical transducers for infrared microsensors. In order to optimize the analysis conditions for quantitative validation of the PIB polymer membrane, we carried out a study that would allow us to get closer to the optical sensor in use and to continue to optimize the PIB polymer by estimating the potential range of detection, the sensitivity limit, the resolution, and its capacity for recycling and the eventual incorporation of a microfluidic cell. The main objective of the ATR-FTIR measurements was to determine the dependence of the absorption band areas of the hydrocarbons on their concentration in an aqueous solution in order to establish a calibration curve and its range of validity at the concentrations studied. In order to determine the suitability of the ATR-FTIR sensor for real-time measurement and analysis, a number of measurements were performed at room temperature by measuring a variety of water solutions containing different concentrations of BTX as well as individual compounds.The first objective was to confirm that the temporal response of the BTX-related absorption bands followed a logical relationship with their concentration as a function of the hydrocarbon–polymer partition coefficient, and that the dependence of the absorption band areas was linear for the selected PIB (molecular weight, concentration, RMS roughness, nano-morphology, contact angle, thickness). For this, a large number of solutions were tested, ranging in concentrations from 50 ppb to 100 ppm of BTX. A new polymer film was prepared for every concentration and the solution flow was set to 3 ml min−1, to ensure the same conditions as in previous works and thus to simulate the same diffusion rate.25,27,30 Regarding other conditions of the solutions (e.g. interference from other pollutants in water or influence of pH), they were not investigated in this study, because the combination of a hydrophobic polymer and nonpolar molecules makes the influence of pH negligible, and the impact of complex water has already been tested, using seawater, wastewater, and groundwater matrices, proving that PIB can be used with good reliability for hydrocarbon detection.25,27 The amount of analyte dissolved in the polymer film depends on its distribution between water and polymer, with a partition coefficient (K) defined as the ratio of the equilibrium concentration of the analyte in the film to that in solution. Thus, the experiments were run until the polymer reached maximum saturation, with individual measurements taken every 10 minutes (Fig. 5). Two thicknesses, namely 5 and 10 μm, of PIB films were tested to evaluate the influence of the film's thickness on the sensitivity and the diffusion kinetics of the pollutant molecules. While the main focus was on the first region of the infrared spectra (650–900 cm−1) in order to ensure the best possible precision and to obtain the maximum amount of transposable information, the remaining two spectral regions of interest were checked as well, with the same overall trends as presented below. In the case of a thinner PIB film (5 μm) and for a concentration of a BTX mixture of 20 ppm for the first spectral range (Fig. 5a), benzene reached the saturation level of the IR signal (named T100) after only 20 min, toluene after 50 minutes, and xylenes after 80 minutes, according to the logically different diffusion coefficients associated with these molecules. For comparison, a thicker PIB film (10 μm) was also evaluated, for which benzene reached saturation after 70 min, toluene after 120 min, and xylenes after 160 min (Fig. 5b). The difference in response times between molecules was mainly caused by the different molecules sizes, whereby larger molecules have a slower diffusion rate, in line with the literature. The diffusion time of the molecules was necessarily longer given the increasing thickness of the film; however, it should be noted that this increase in time necessary to reach the saturation level was non-linear and depended on the molecule considered. However, it is important to note that the thicker film provided equally strong IR signals (absorption band areas) at 90 min as the thinner film, with the difference reduced to only around 5%. On the other hand, the time required to reach saturation was greater, as previously mentioned, but with a stronger final IR signal (>10%–15%). The amount of molecules inserted into the polymer film therefore seemed greater before reaching equilibrium with the water above. Strictly speaking, the fact that a higher IR signal at saturation could be observed in the case of the thicker film is debatable. This may seem surprising if we consider that the partition coefficient K is governed solely by the thermodynamics of a given water/polymer system and that, all other things being equal, it should be independent of the film thickness at first sight.38 The slight variation of K assumed with the thickness of the film was not dependent on the fact that the equilibrium had not been reached, as this has been verified. This variation was therefore essentially linked to the polymer film, as the flow and the fluid cell had not yet evolved and therefore had no impact on the fluid mechanics parameters of the water. This is one of the reasons why the morphology of PIB films, the contact angles, the roughness, the evolution of the viscosity over time of the solution used for the spin-coating deposition were examined and did not indicate any notable variation from the first examination. The influence of the polymer surface, i.e. its interface with the water, compared with the polymer volume, could have had an impact on the partition coefficient and diffusion coefficient, particularly in the case of inhomogeneous swelling. A zone of water/polymer equilibrium could be observed at the surface of a volume that could be described as a buffer zone, but the deeper volume presented a slightly different BTX concentration near the polymer/selenide interface. A non-symmetrical system, in terms of the chemical gradient, the mechanical, electrical, and chemical properties between the polymer surface in contact with the water and the polymer surface in contact with the selenide transducer could have an impact on the partition coefficient and diffusion coefficient as a function of film thickness. This increase in thickness could have an interesting influence for improving the detection limit, as the IR signal (absorption band area) at equilibrium increases. However, this limit of detection obtained during saturation, i.e. an established equilibrium between water and polymer, must be greatly weighted by the detection time in the case of a thick film. Indeed, in terms of the detection speed, if we consider the first 30 min of measurement for the development of a detection methodology, the area under the absorption band decreased by at least half (depending on the molecule) for the thickest film. This was mainly due to the distribution of the infrared evanescent field in the volume following the exponential decay and the diffusion coefficient of the molecules covering it over time. The polymer film (5 μm) just covered the tail of the evanescent field in the first spectral region (650–900 cm−1) and the molecules then diffused efficiently towards the polymer/selenide interface to quickly cover the entire evanescent field. The thinner film seemed more promising as it allowed a good compromise between the strength of the acquired IR absorption signal, the time it took to reach full saturation of the polymer, and the required penetration depth.
 | ||
| Fig. 5 Dependence of the absorbance bands areas on time for 20 ppm BTX solution using: (a) 5 μm-thick and (b) 10 μm-thick polyisobutylene films. | ||
The kinetics associated with the detection were evaluated by considering the temporal evolution of the IR signal as a function of the hydrocarbon concentration and by considering the thickness of the polymer film. Five concentrations of BTX solution (5, 20, 30, 40, and 50 ppm) were selected and the periods of time necessary to reach 30% (T30) and 90% (T90) of the maximum absorbance, using 5 μm and 10 μm polyisobutylene films were investigated (the concentration of 5 ppm was evaluated with the thinner film only). Fig. 6 shows the plots for toluene as a representative, as we know that benzene behaves in the same way, and M-xylene, which is representative of the behaviour of the isomers of xylene. The times required to reach the T30 and T90 signal values varied not only depending on the molecule chosen, but also significantly depending on the concentration itself. As shown in Fig. 6, for toluene (as well as for benzene), the T30 and T90 times started at higher values for both thicknesses, then dropped significantly until reaching 30 ppm and then continue almost unchanged, showing a horizontal plot that no longer evolved for higher concentrations. The xylenes showed the same behaviour for the thickest film; however, the thinner film showed a rather opposite behaviour, as it started at a lower time value and then increased and finally stabilized once the concentration reached 30 ppm. For benzene and toluene, the T30 and T90 values were reached much faster than for the xylenes at the highest concentrations, regardless of the film thickness. However, this was not necessarily for concentrations below 30 ppm, where such a difference between the molecules was not shown, and the times for one molecule or another were relatively close when the concentrations were lower than 20–30 ppm. There did not seem to be a big difference in detection regime ultimately between the BTX molecules for concentrations lower than 20–30 ppm by any size effect, although detection could potentially be differently impacted by the flow of water in circulation and the coefficient of partition for lower concentrations. For the xylenes, the difference in detection time when increasing the concentration was not as significant in the case of a thicker film, whereas for a thinner film, this dependence was exactly the opposite, and the T30 and T90 values were reached more quickly for the lowest concentrations, probably without any competition effect. This shows an interesting diffusion dynamics of molecules that occurs inside the film that depends strongly on the concentration with a threshold effect, but is also impacted by the thickness of the film and of course the nature of the molecules.
 | ||
| Fig. 6 Time dependence of the IR signal (at 30% and 90% of the saturated signal, denoted as T30 and T90) as a function of the concentration for: (a) toluene and (b) M-xylene using 5 and 10 μm-thick polyisobutylene films. | ||
Fig. 7a, using a 5 μm-thick film, shows that the dependence of the absorption bands areas at saturation on the concentration was not perfectly linear. There are two regions in the figure – namely between 20–30 ppm and 70–80 ppm – where the slope becomes flat and then continues to rise afterwards. These two plateaus persisted during multiple measurements. The cause of this phenomenon remains unknown at present, although an effort has been made in this study to address the problem and understand its origin. Specifically, a thicker (10 μm) PIB film was also tested in the concentration range of 15–45 ppm, i.e. covering the first plateau. Although the areas of the absorption bands increased, the plateau at 20–30 ppm remained present, and thus it could not be linked to a simple saturation problem. This was partly because higher concentrations were easily measurable once the plateau had been exceeded, but also because increasing the thickness of the polymer did not eliminate this plateau effect by increasing the volume of polymer available for storing pollutant molecules. The observation of the plateau could be related to previous observations, where a threshold effect was observed at 20–30 ppm when detecting the IR signal at T30 and T90 (Fig. 6).
 | ||
| Fig. 7 Dependence of absorption band areas on the concentration of BTX solution after 90 min for a 5 μm-thick polyisobutylene film: (a) in the range of 0–100 ppm, (b) 0–5 ppm, and (c) 0–1 ppm and (d) after 60 min of measurements using only the chosen values of concentration. | ||
At low concentrations (Fig. 7b and c), BTX could be reliably detected down to 150 ppb, which could be considered a limit of quantification for this detection method. At lower concentrations, we could reliably determine isomers of xylenes down to 100 ppb, while benzene and toluene were hard to quantify. At 50 ppb, the IR signal became very weak, and while xylenes could still be observed, the peaks representing toluene and benzene disappeared. All this means that for the whole BTX mixture, 150 ppb was set as the limit of quantification, while for xylenes only, this limit could be decreased to 100 ppb, comparable but lower than the literature.39 Meanwhile, the limit of detection for benzene and toluene was set at 100 ppb and for xylenes at 50 ppb.
The aforementioned plateaus in the measured data represent a potential problem during future calibration of the sensor in these specific concentration areas. However, when the results were compared to the literature,23,25,40 it could be noted that linear slopes were instead reported, but with a lower concentration sampling than that used. After confirming the results by several experiments to verify their reproducibility, the absorption band area of the molecules was plotted as a function of concentration using data found in the literature, and using more spaced samplings in concentration in accordance with the specifications reported in the literature in terms of the concentration range, which were taken after 60 min of measurement.35 With these comparable, reported conditions, a linear dependence could be obtained (with R2 values for each line reaching over 0.99) for the absorption band areas as a function of concentration as seen in Fig. 7d. Since we carried out a series of more precise measurements in terms of concentration steps, it was possible to observe where the plateaus were positioned, which is valuable information and provides a better understanding of the prediction limits of the optical selenide transducer in these specific concentration ranges. The PIB film can still be used to detect aromatic hydrocarbons in water; however, the plateau phases must be considered for future calibrations.
3.3. Regeneration of the PIB film to extend its life cycle
An important characteristic of a sensor is its reusability. A key way to minimize the environmental impact is by promoting the longest possible life cycle, and to avoid single use scenarios. In order to test whether the thinner PIB film could be used multiple times, a series of experiments was performed. The first type of experiments consisted of continuous regeneration comprising multiple cycles. Each cycle consisted of a 60 min flow of BTX solution and 90 min flow of distilled water. The flow for the BTX cycle was set to 3 ml min−1, while the water cycle ran at 6 ml min−1, the highest speed achievable with the used set-up. The concentration of BTX was kept at 20 ppm.During the initial measurement, when the water used was at room temperature, the first three BTX cycles showed comparable results; nevertheless, the fourth and fifth cycles showed different results each, showing unstable conditions in these cycles. It should also be noted that with each measurement, the regeneration during the water cycle became less effective as more molecules remained inside the polymer. Further tests were performed with an aim to improve the regeneration process. Since further increasing the water flow was not possible due to the limitation of the used water pump, two other ways were tried out: to increase the time in which the polymer was being regenerated and to utilize water at a higher temperature, which was maintained throughout the whole regeneration process. For this experiment, three temperatures were tested, namely 30 °C, 40 °C, and 50 °C, as these temperatures could be easily obtained and controlled during the process.
Fig. 8 shows that the regeneration with water at 40 °C provided good results in the fourth cycle, as the absorption bands areas for all four cycles showed the same values. We could thus conclude that utilizing warm water for regeneration significantly enhanced the regeneration capabilities of the PIB film, and within a reasonable time window. While the results with the 30 °C water did not differ much from those using water at room temperature, the 40 °C experiment showed promising results, in agreement with the literature.41 The 50 °C experiment also proved effective; however, during the fourth cycle we noticed an interference in the area of interest. Since this might have been caused by the degradation of the polymer due to the water temperature, 40 °C was chosen for the experiments going forward.
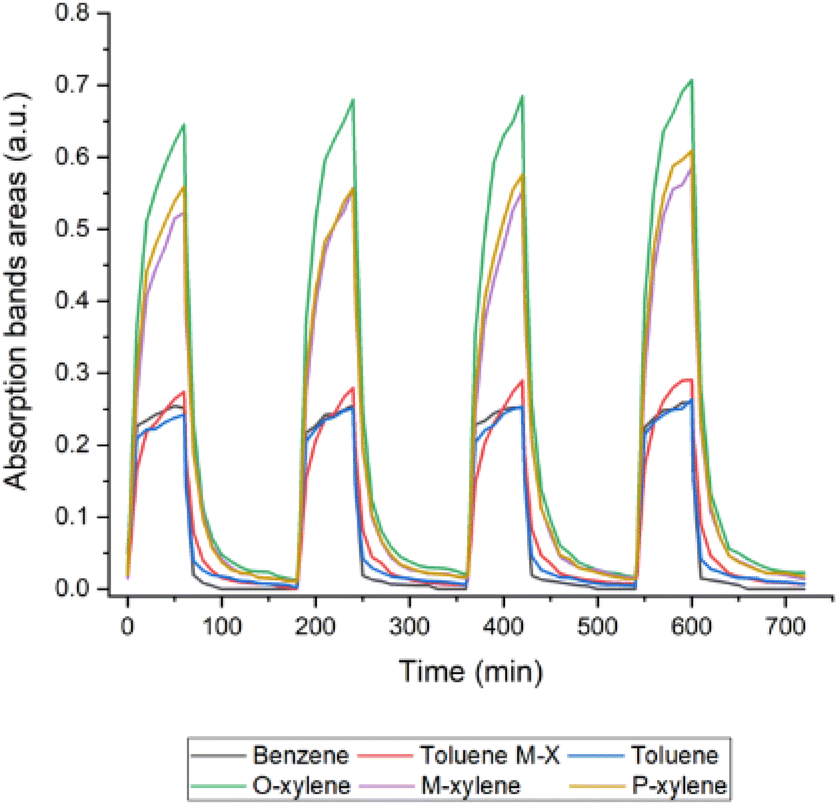 | ||
| Fig. 8 Regeneration of 20 ppm BTX solution using a 5 μm polyisobutylene film and 40 °C distilled water for the regeneration cycle. | ||
Another point was to try discontinuous regeneration, to assess how the regeneration could be managed in field. The conditions of the measurement were kept the same, and the only change was a 48 h delay between individual cycles. Two kinds of experiments, regarding the state in which the prism was kept in the meantime, were performed. For the first one, droplets of solution were removed from the polymer and the prism was kept in a dry cell. For the second one, the prism with the polymer was kept under distilled water during the period. The first experiment proved successful, as the consequent measurements kept the same characteristics as with the continuous regeneration mentioned previously. However, the second method showed significant changes and a fluctuation of the results, especially for the xylenes, where the obtained absorption band areas were not the same as in the previous cycle. The cause of this change most likely lays with the swelling of the deposited polymer, which changed its properties. Since the first method showed satisfactory results, and promises better handling of the sensor, this “dry” version of the regeneration seems to be a better option for future use.
3.4. Fabrication of a microfluidic cell on a Ge–Sb–Se selenide microsensor
A microfluidic channel designed to contain the flow of a water on top of the chalcogenide microsensor was created from polydimethylsiloxane (PDMS) by filling a mold specially prepared by CAD and 3D printing techniques. The PDMS device, after forming the mold, was directly placed onto the microstructured chalcogenide glass waveguide deposited onto a 2-inch silicon substrate. To minimize the influence of water in the MIR region, the PIB was already deposited on top of the chalcogenide PIC substrate (Fig. 9) and the channel was therefore grafted on top of the PIB layer. Moreover, an adhesive was applied to prevent leakage between PIB and PDMS. This was done by a double tape adhesive structured by xurography. The first tests proved the practicality of the prepared microfluidic channel, as it was able to pump the solution without any problems or leakage, proving that the concept of the device was feasible and will make it possible to carry out future BTX detection tests in the field using PIB polymer on a chalcogenide microsensor. In addition to the use of a different spectral domain for the Ge–Sb–Se microsensor allowing us to consider thinner polymer films, and exacerbated evanescent field of the selenide photonic integrated circuit compared to that of the selenide-based prism for the infrared transducer, it will also be necessary to take into account a different water fluidic condition using the microfluidic cell, which would likely impact the detection limits and detection speeds. | ||
| Fig. 9 PDMS microfluidic device grafted onto polyisobutylene-functionalised chalcogenide photonic integrated circuits on top of a 2-inch silicon substrate: (a) after deposition of the PDMS mold and (b) after connection to the water pump. | ||
4. Conclusions
In this study, it was demonstrated that mid-infrared evanescent wave spectroscopy can be successfully used for the in situ detection of environmentally harmful organic pollutants in water. This method was used in combination with a PIB film-functionalized ATR prism, which reduces the high absorbance of water in the IR spectrum and allows extracting hydrophobic pollutant compounds from the solution. The polymer was deposited by spin-coating and despite the appearance of small cones on the surface of the film, the polymer created a homogeneous, dense, and smooth hydrophobic membrane. The thinner film of PIB (5 μm) showed satisfactory sensitivity towards the measured hydrocarbons, while the time required to discern their presence in water was relatively short (only a few minutes) compared to with a thicker film (10 μm). The detection time strongly depended on the thickness of the film allowing the molecules to diffuse more quickly through the polymer towards the surface of the infrared waveguide for a thickness close to the penetration depth of the evanescent wave. The time periods to reach 30% and 90% of the maximum IR absorbance signal were analyzed, where the threshold value between 20 and 30 ppm seemed to be a concentration zone at which the behaviour changes. Beyond 30 ppm, these two periods of time thus reach a constant value, which no longer changed as a function of the concentration following a significant increase or decrease in its value for lower concentrations, depending on the nature of the molecule and the thickness of the film. It is important to note the observation of plateau phases – around concentrations of 25 and 80 ppm BTX – which have not been previously reported and should be considered in future calibrations. The limit of quantification was lowered, compared to previous reports, to 150 ppb, while the limit of detection was set at 100 ppb for all the molecules in a BTX mixture. When measuring only the isomers of xylenes, the limit of quantification was considered to be 100 ppb, while the limit of detection reached 50 ppb. These results demonstrate that this optical method has the potential to measure relatively high down to low values of concentrations of organic pollutants, which makes it perfectly suited to monitoring the degradation of polluting molecules during an accidental pollution event. The spectral region (1450–1650 cm−1) could be used for the detection of BTX, as we observed the thinner PIB film displaying the same trends as the first spectral region (650–900 cm−1) with a necessarily higher concentration range, of the order of a few tens of ppm. However, this spectral region has limitations for measuring a solution containing several hydrocarbons in addition to xylenes. On the other hand, it would be interesting for technological transfer to use microsensors. This more accessible spectral domain would make it relatively easy to identify a mixture of BTX and to monitor their degradation, while multivariate analysis should facilitate the exploitation of data in this spectral range. It was also proven that the PIB film could be regenerated using unpolluted water and that if water at 40 °C were used, the film could be used over several cycles without difficulty, providing IR signals that are easy to analyze without interference. This was also evaluated for measurements with an interval of 48 h between individual cycles, which promises good handling in the field. For future use, this fact also demonstrated that the possible degradation of PIB film is negligible at temperatures between 20 °C and at least 40 °C. Finally, a PDMS microfluidic channel grafted onto a chalcogenide transducer in the form of an integrated photonic circuit, previously functionalized with polyisobutylene, was successfully manufactured and its viability was proven, opening prospects for the development of this type of infrared microsensor for the detection of BTX in water.Author contributions
Martin Vrážel: experiment, methodology, data analysis, writing – original draft, review & editing; Raïssa Kadar Ismail: PIB experiment, GRAMS data analysis; Amelie Larrode: regeneration experiment; Remi Courson & Kada Boukerma: creation and application of PDMS microfluidic device; Abdelali Hammouti, Loïc Bodiou, Joel Charrier: fabrication of chalcogenide waveguide; Karine Michel, Marek Bouška: experiment supervision, writing – review & editing; Marion Baillieul: experiment supervision, Stéphane Le Floch, William Giraud: measurements on gas chromatograph and advice for end users; Petr Němec & Virginie Nazabal: conceptualization, funding acquisition, supervision, formal analysis, writing – review & editing.Data availability
All the data in this article are available on the secure cloud (Directory: Article Analyst – Martin Vrazel – 2024) of the Information Systems Department (DSI) of the University of Rennes.The data can be recovered without difficulty following an official request to the DSI.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge IBAIA (101092723) Horizon Europe project, ANR AQUAE (ANR-21-CE04-0011-04) project of French National Research Agency (ANR), and project No. 22-05179S of Czech Science Foundation (GA ČR) for financial support.References
- J. C. G. Sousa, A. R. Ribeiro, M. O. Barbosa, M. F. R. Pereira and A. M. T. Silva, A review on environmental monitoring of water organic pollutants identified by EU guidelines, J. Hazard. Mater., 2018, 344, 146–162 CrossRef CAS PubMed.
- O. Ogbeide, I. Tongo and L. Ezemonye, Risk assessment of agricultural pesticides in water, sediment, and fish from Owan River, Edo State, Nigeria, Environ. Monit. Assess., 2015, 187(10), 654 CrossRef PubMed.
- M. Alahi and S. C. Mukhopadhyay, Detection methods of Nitrate in water: A Review, Sens. Actuators, A, 2018, 280, 210–221 CrossRef CAS.
- M. E. E. Alahi and S. C. Mukhopadhyay, Smart nitrate sensor: Internet of Tings enabled real-time water quality monitoring, Smart Sensors, Measurement and Instrumentation, Springer, Springer Nature, Cham, 2019, Available from: DOI:10.1007/978-3-030-20095-4_1.
- C. Kratz, A. Furchner, G. Sun, J. Rappich and K. Hinrichs, Sensing and structure analysis by in situ IR spectroscopy: from mL flow cells to microfluidic applications, J. Phys.: Condens.Matter, 2020, 32(39), 393002 CrossRef CAS PubMed.
- Opportunities for photonic integrated circuits in optical gas sensors - IOPscience. Available from: https://iopscience.iop.org/article/10.1088/2515-7647/ab6742.
- N. Jornet-Martínez, et al., Trends for the Development of In Situ Analysis Devices. in Encyclopedia of Analytical Chemistry. John Wiley & Sons, Ltd, 2017, pp. 1–23 Search PubMed.
- A. M. Nightingale, S. Hassan, B. M. Warren, K. Makris, G. W. H. Evans and E. Papadopoulou, et al. A Droplet Microfluidic-Based Sensor for Simultaneous in Situ Monitoring of Nitrate and Nitrite in Natural Waters, Environ. Sci. Technol., 2019, 53(16), 9677–9685 CrossRef CAS PubMed.
- M. Gavrilescu, K. Demnerová, J. Aamand, S. Agathos and F. Fava, Emerging pollutants in the environment: present and future challenges in biomonitoring, ecological risks and bioremediation, New Biotechnol., 2015, 32(1), 147–156 CrossRef CAS PubMed.
- B. Mizaikoff, Waveguide-enhanced mid-infrared chem/bio sensors, Chem. Soc. Rev., 2013, 42(22), 8683–8699 RSC.
- M. A. Mahmud, F. Ejeian, S. Azadi, M. Myers, B. Pejcic and R. Abbassi, et al. Recent progress in sensing nitrate, nitrite, phosphate, and ammonium in aquatic environment, Chemosphere, 2020, 259, 127492 CrossRef CAS.
- U.S. Department of Health and Human Services, Interaction profile for: Benzene, Toluene, Ethylbenzene, and Xylenes (BTEX), GPO, 2004 Search PubMed.
- A. Masih, A. S. Lall, A. Taneja and R. Singhvi, Exposure levels and health risk assessment of ambient BTX at urban and rural environments of a terai region of northern India, Environ. Pollut., 2018, 242, 1678–1683 CrossRef CAS PubMed.
- T. Schädle, B. Pejcic and B. Mizaikoff, Monitoring dissolved carbon dioxide and methane in brine environments at high pressure using IR-ATR spectroscopy, Anal. Methods, 2016, 8(4), 756–762 RSC.
- J. Hu, et al. Feature issue introduction: mid-infrared optical materials and their device applications, Opt. Mater. Express, 2018, 8(7), 2026–2034 CrossRef CAS.
- Chalcogenide glasses: preparation, properties and applications, ed. J. L. Adam and X. Zhang, Woodhead publishing, 2014 Search PubMed.
- M. Sieger, et al. Mid-Infrared Spectroscopy Platform Based on GaAs/AlGaAs Thin-Film Waveguides and Quantum Cascade Lasers, Anal. Chem., 2016, 88(5), 2558–2562 CrossRef CAS PubMed.
- A. Gutierrez-Arroyo, et al. Theoretical study of an evanescent optical integrated sensor for multipurpose detection of gases and liquids in the Mid-Infrared, Sens. Actuators, B, 2017, 242, 842–848 CrossRef CAS.
- E. Baudet, Development of an evanescent optical integrated sensor in the mid-infrared for detection of pollution in groundwater or seawater, Adv. Device Mater., 2017, 3(2), 23–29 CrossRef CAS.
- J. Charrier, M. L. Brandily, H. Lhermite, K. Michel, B. Bureau and F. Verger, et al. Evanescent wave optical micro-sensor based on chalcogenide glass, Sens. Actuators, B, 2012, 173, 468–476 CrossRef CAS.
- C. Dettenrieder, et al. Determination of Volatile Organic Compounds in Water by Attenuated Total Reflection Infrared Spectroscopy and Diamond-Like Carbon Coated Silicon Wafers, Chemosensors, 2020, 8(3), 75 CrossRef CAS.
- M. Sieger and B. Mizaikoff, Toward On-Chip Mid-Infrared Sensors, Anal. Chem., 2016, 88(11), 5562–5573 CrossRef CAS PubMed.
- R. Stach, B. Pejcic, E. Crooke, M. Myers and B. Mizaikoff, Mid-Infrared Spectroscopic Method for the Identification and Quantification of Dissolved Oil Components in Marine Environments, Anal. Chem., 2015, 87(24), 12306–12312 CrossRef CAS PubMed.
- R. Stach, B. Pejcic, C. Heath, M. Myers and B. Mizaikoff, Mid-infrared sensor for hydrocarbon monitoring: the influence of salinity, matrix and aging on hydrocarbon–polymer partitioning, Anal. Methods, 2018, 10(13), 1516–1522 RSC.
- B. Pejcic, L. Boyd, M. Myers, A. Ross, Y. Raichlin and A. Katzir, et al. Direct quantification of aromatic hydrocarbons in geochemical fluids with a mid-infrared attenuated total reflection sensor, Org. Geochem., 2013, 55, 63–71 CrossRef CAS.
- M. Baillieul, E. Baudet, K. Michel, J. Moreau, P. Němec, K. Boukerma, F. Colas, J. Charrier, B. Bureau and E. Rinnert, et al. Toward Chalcogenide Platform Infrared Sensor Dedicated to the In Situ Detection of Aromatic Hydrocarbons in Natural Waters via an Attenuated Total Reflection Spectroscopy Study, Sensors, 2021, 21, 2449 CrossRef CAS PubMed.
- M. Baillieul, Capteurs infrarouges de polluants aquatiques: synthèse, optimisation et qualification, Doctroral Thesis, University of Rennes, 2018 Search PubMed.
- C. Vigano, J. Ruysschaert and E. Goormaghtigh, Sensor applications of attenuated total reflection infrared spectroscopy, Talanta, 2005, 65(5), 1132–1142 CrossRef CAS PubMed.
- P. J. Linstrom and W. G. Mallard, Evaluated Infrared Reference Spectra, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, Coblentz Society, Inc., National Institute of Standards and Technology, Gaithersburg MD, 20899 (accessed 8 Feb 2024).
- K. Flavin, H. Hughes, V. Dobbyn, P. Kirwan, K. Murphy, H. Steiner, B. Mizaikoff and P. McLoughlin, A comparison of polymeric materials as pre-concentrating media for use with ATR/FTIR sensing, Int. J. Environ. Anal. Chem., 2006, 86, 401–415 CrossRef CAS.
- P. Heinrich, et al. Determination of organic compounds by IR/ATR spectroscopy with polymer-coated internal reflection elements, Appl. Spectrosc., 1990, 44, 1641–1646 CrossRef CAS.
- R. Göbel, R. Krska, K. Kellner, R. W. Seitz and S. A. Tomellini, Investigation of different polymers as coating materials for IR/ATR spectroscopic trace analysis of chlorinated hydrocarbons in water, Appl. Spectrosc., 1994, 48, 678–683 CrossRef.
- Y. Du, C. Li, J. Jin, C. Li and W. Jiang, Surface modification of polyisobutylene via grafting amino acid-based poly (acryloyl-6-aminocaproic acid) as multifunctional material, Colloids Surf., B, 2018, 161, 73–82 CrossRef CAS PubMed.
- Y. Shao, S. Yan, J. Li, Z. Silva-Pedraza, T. Zhou and M. Hsieh, et al. Stretchable encapsulation materials with high dynamic water resistivity and tissue-matching elasticity, ACS Appl. Mater. Interfaces, 2022, 14(16), 18935–18943 CrossRef CAS PubMed.
- M. Hildebrandt, E. Y. Shin, S. Yang, W. Ali, S. Altinpinar and J. S. Gutmann, Investigation of roughness correlation in polymer brushes via X-ray scattering, Polymers, 2020, 12(9), 2101 CrossRef CAS PubMed.
- S. Hosseini, F. Ibrahim, L. Koole and I. Djordjevic, Polyacrylate Spin-coated Surfaces with Controllable Functionality: Potential Biochips for Diagnostic Devices, in 2014 IEEE Conference on Biomedical Engineering and Sciences, Miri, Sarawak, Malaysia, 2014, December 8–10.
- T. Halenkovič, M. Baillieul, J. Gutwirth, P. Němec and V. Nazabal, Amorphous Ge-Sb-Se-Te chalcogenide films fabrication for potential environmental sensing and nonlinear photonics, J Materiomics., 2022, 8(5), 1009–1019 CrossRef.
- B. Pejcic, M. Myers and A. Ross, “Mid-infrared sensing of organic pollutants in aqueous environments, Sensors, 2009, 9(8), 6232–6253 CrossRef CAS PubMed.
- J. Yang and S. S. Tsai, Cooled internal reflection element for infrared chemical sensing of volatile to semi-volatile organic compounds in the headspace of aqueous solutions, Anal. Chim. Acta, 2002, 462, 235–244 CrossRef CAS.
- C. Heath, B. Pejcic and M. Myers, Block Copolymer-Coated ATR-FTIR Spectroscopic Sensors for Monitoring Hydrocarbons in Aquatic Environments at High Temperature and Pressure, ACS Appl. Polym. Mater., 2019, 1(8), 2149–2156 CrossRef CAS.
- C. Heath, M. Myers and B. Pejcic, The effect of pressure and temperature on mid-infrared sensing of dissolved hydrocarbons in water, Anal. Chem., 2017, 89(24), 13391–13397 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |