
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of a graphite-paraffin carbon paste electrode and demonstration of its use in electrochemical detection strategies†
Stuart A.
Milne
 *a,
Perrine
Lasserre
b and
Damion K.
Corrigan
b
*a,
Perrine
Lasserre
b and
Damion K.
Corrigan
b
aUniversity of Strathclyde, Biomedical Engineering, Wolfson Centre, 106 Richmond St, Glasgow G1 1XQ, UK. E-mail: stuart.a.milne.2016@uni.strath.ac.uk
bUniversity of Strathclyde, Pure and Applied Chemistry, Thomas Graham Building, 295 Cathedral St, Glasgow G1 1XL, UK
First published on 2nd August 2024
Abstract
Electrochemical detection methods hold many advantages over their optical counterparts, such as operation in complex sample matrices, low-cost and high volume manufacture and possible equipment miniaturisation. Despite these advantages, the use of electrochemical detection is currently limited in the clinical setting. There is a wide range of potential electrode materials, selected for optimal signal-to-noise ratios and reproducibility when detecting target analytes. The use of carbon paste electrodes (CPEs) for electrochemical detection can be limited by their analytical performance, however they remain very attractive due to their low cost and biocompatibility. This paper presents the fabrication of an easy-to-make and use graphite powder/paraffin wax paste combined with a substrate produced via additive manufacturing and confirms its functionality for both direct and indirect electrochemical measurements. The produced CPEs enable the direct voltammetric detection of hexaammineruthenium(III) chloride and dopamine at an experimental limit of detection (ELoD) of 62.5 μM. The key inflammatory biomarker Interleukin-6 through an enzyme-linked immunosorbant assay (ELISA) was also quantified, yielding a clinically-relevant ELoD of 150 pg ml−1 in 10% human serum. The performance of low-cost and easy-to-use CPEs obtained in 0.5 hours is showcased in this study, demonstrating the platform's potential uses for point-of-need electroanalytical applications.
Introduction
A biosensor can be defined as ‘a device that uses specific biochemical reactions mediated by isolated enzymes, immunosystems, tissues, organelles or whole cells to detect chemical compounds usually by electrical, thermal or optical signals’.1Biosensors can be used in either point of care (POC), or laboratory settings. While in laboratories optical methods dominate due to their simple, highly automated nature, they can struggle in POC settings. There are many reasons for this including interference from coloured samples, long analysis times, requirements for large/power intensive equipment and large sample volumes.2 Although electrochemically active samples and especially those with changing pH can interfere with electrochemical sensors, their ability to provide fast, low-cost, specific responses3 in the field4 and general smaller size make them ideal for use in POC settings. The greatest illustration of their utility at the POC is the rise of continuous glucose monitors. Glucose biosensing testifies of evident success, being the focus of 90% of the global market for electrochemical biosensors.5 Nevertheless, electrochemical sensing has not yet been fully exploited for wider commercial and medical applications.5
The sensitivity of an electrochemical biosensor relies on the electrode material on which it is based. To this end, a substantial number of materials have been developed and tested in the literature, commonly gold6 and carbon.7 Gold materials, although more expensive,8 are often chosen due to their chemical stability9 and in the case of biosensors, their ability to be easily functionalized via strong gold-sulphur bonds.10 Carbon on the other hand is inexpensive,11 highly conductive, chemically inert and has a wide voltage range.12 Despite these advantages, carbon electrodes often require high overpotentials to reduce or oxidise molecules on their surface,11 an issue most often overcome via modification with suitable redox mediators11 or catalytic nanomaterials.13
Carbon paste electrodes (CPEs) are a popular subtype of carbon electrode with a long history, produced through the mixing of electrically conductive carbon materials with binders/solvents.14 While the use of carbon paste itself is certainly not new, being described as early as 1958,15 its simple manufacturing process of mechanical mixing imparts several benefits, permitting the in-house manufacturing of electrodes at a low cost. Traditionally, 70% of carbon pastes are made with liquid binders.16 In recent years, solid electrodes produced through the mixing of a solid binder such as paraffin wax with carbon materials have gained traction and are now commonly used, allowing the incorporation of current manufacturing methods such as additive manufacturing (3D printing). The versatility of carbon paste allows numerous modifications/enhancements to be made easily, such as the inclusion of redox mediators,17 recognition elements12 such as enzymes18,19 and binding molecules20 being common choices.
Electrochemical biosensors can function by direct detection, in which a molecule of interest is inherently electrochemically active, or by indirect detection, in which a molecule is coupled to a tag (e.g. an enzyme) which enables its electrochemical detection. A molecule often targeted by direct electrochemical detection and biosensors is dopamine, with 271 results being found from a search of ‘Dopamine’ AND ‘Biosensor’ in web of science since 2023. Dopamine is a neurotransmitter that plays a critical role in neuromodulation, being expressed, and detected in both the central and peripheral nervous systems,21 whose levels are raised in Parkinson's, Huntington's, and schizophrenia.22
Direct electrochemical detection of dopamine and other neurotransmitters of interest has been reported on many electrode materials and morphologies,23in vivo using carbon fibre electrodes24 and through clinical depth electrodes.25 Indirect electrochemical sensing of dopamine has been demonstrated extensively, for instance using a fluorescent artificial receptor26 or electrochemiluminescence using copper nanoclusters.27 However, many of these reports require complex multi-step functionalisation protocols due to well-known interferents at similar oxidation potentials.28 Given the large amount of literature showing both benchtop and in vivo detection, the ubiquity of measurement data and its challenging detection position dopamine as a good benchmark compound for electrode performance, especially because the mechanism of electron transfer and general oxidation mechanism is more complex than for the simple outer sphere ruthenium couple.
Enzyme-Linked Immunosorbent Assay (ELISA) is a commonly used method in laboratories worldwide and is reliable, sensitive, and specific, in part due to the amplification that is enabled by enzymes.29 Enzymes are used to detect and often quantify immunological interactions, either the binding of an antigen of interest to an antibody or vice versa. Some common enzyme choices include alkaline phosphatase30 and horseradish peroxidase (HRP)31 which are both conveniently able to provide an optical and electrochemical signal. A common target of both optical and electrochemical ELISA is Interleukin-6 (IL-6).32 This pro-inflammatory cytokine is a key biomarker in sepsis33 and many other diseases such as cancer, multiple sclerosis, Alzheimer's disease, rheumatoid arthritis, anaemia, inflammatory bowel disease, asthma, and inflammatory pulmonary diseases.34 Both electrochemical35,36 and optical37 detections commonly test for the presence of IL-6. The popularity of ELISA and this specific cytokine position them both as a strong benchmark measurement for any potential detection platform.
This study demonstrates the facile manufacturing of a CPE chip aided with 3D printing. Considering the strengths of electrochemical systems as affordable solutions easily transferable to the point of need, the produced CPE performance will be assessed in both direct and indirect detection strategies of dopamine as an exemplar measurement of a redox active molecule and IL-6 as an exemplar immunoassay. Alongside a basic characterisation with ruthenium hexamine chloride, these measurements showcase the potential utility of the platform in a variety of useful analytical scenarios.
Materials and methods
Chemicals
Phosphate-buffered saline tablets (P4117), dopamine hydrochloride (H8502), L-ascorbic acid (A92902), uric acid (U2625), serotonin hydrochloride (H9523), human serum (H4522) and hexaammineruthenium(III) chloride (262005) were purchased from Merck (UK). Streptavidin-tagged horseradish peroxidase, IL-6 capture and detection antibodies as well as IL-6 protein standard were taken from a Human IL-6 Duo Set enzyme linked immunosorbent assay (ELISA) kit (DY206, Bio-Techne, Abingdon, UK). Reagent diluent (DY995), wash buffer (WA126) and HRP substrate solution (DY999B) were also purchased from R&D Systems. Paraffin wax (56–60 degrees) was purchased from APC pure (Hyde, UK) and graphite powder from Inoxia Ltd (Cranleigh, UK).3D-printing of electrode base and well
An electrode base and well to sit on top were printed using a RAISE3D E2 IDEX Dual 3D printer with a nozzle diameter of 0.4 mm. Computer-aided design (CAD) files were produced in FreeCAD 0.20.1 software and exported in .stl file formats to ideaMaker 4.2.3 (RAISE3D) for slicing.Parts were printed using 1.75 mm white polyethylene terephthalate glycol (PETG) filament (ERYONE) and slicing was conducted using the slicers high quality template (0.1 mm layer height, 10% infill, 70 mm s−1 infill speed) with the 1.75 mm PETG filament option selected. Resulting .gcode and .data files were transferred to the printer via a flash drive for printing.
Ag/AgCl quasi-reference electrode
During this investigation, an Ag/AgCl quasi-reference electrode was used, produced by the submersion of Ag wire in household bleach alike other methods seen in literature.38 This electrode produces a stable voltage through its reaction with chloride ions in the surrounding solution (eqn (1)). It does however differ from a traditional reference electrode by being in direct contact with the analyte, not with a saturated chloride solution and so the voltage relies on the concentration of Cl− ions in the solution. A few things were done to attempt to keep this constant:• The only source of chloride ions was the phosphate buffered saline used.
• Measurements were performed quickly to reduce the effect of evaporation.
Half reaction of Ag/AgCl reference system:
| AgCl + e− ↔ Ag + Cl−. | (1) |
Carbon paste manufacturing
Graphite powder and paraffin wax binder were combined via heating and manual mixing in a mantle (electrothermal). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix by weight was easy to work with while still providing suitable conductivity. This paste was used for the working and counter electrodes. SEM and FTIR were performed to investigate the chemical and physical structure of the produced paste, further details are provided in the ESI (S3 and S4†).
:1 mix by weight was easy to work with while still providing suitable conductivity. This paste was used for the working and counter electrodes. SEM and FTIR were performed to investigate the chemical and physical structure of the produced paste, further details are provided in the ESI (S3 and S4†).
Chip assembly
Electrode chips were assembled in a few simple steps. Firstly, the carbon paste outlined was heated until soft before being transferred to the printed tracks via a spatula. It was then pushed into the tracks before excess was removed. While still warm, an Ag/AgCl quasi-reference electrode was added to the relevant track, producing a chip with 8 carbon paste working electrodes that share a single carbon paste counter electrode and Ag/AgCl quasi – reference electrode. A more detailed ‘recipe’ for their manufacturing is included in the ESI.†Use of phosphate buffered saline (1× PBS)
PBS used during this investigation was composed of 0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride, at a pH of 7.4. This was obtained by dissolving tablets in the volume of deionized water recommended by the supplier (200 ml per tablet).Electrochemical methods
Electrochemical procedures were performed using a PalmSens PS4 potentiostat driven by PSTrace 5.9 software (PalmSens, Houten, Netherlands). Data analysis was undertaken using Origin 2022 (OriginLab, Massachusetts, USA).Surface cleaning
Both physical and chemical cleaning methods were carried out on electrodes before use. Physical smoothing of the electrode surface was first performed by rubbing chips in a figure of eight motion on white printer paper until a glassy surface was obtained. Chemical cleaning was then performed by cyclic voltammetry (CV) cycling in PBS. 10 cycles were performed between potentials of −0.6 and 1.3 V at a speed of 100 mV s−1 and step of 0.02 V. Chips were then rinsed with deionised water, dried using compressed air and used.Hexaammineruthenium(III) chloride characterisation
CV, square wave voltammetry (SWV) and electrochemical impedance spectroscopy (EIS) in 1 mM hexaammineruthenium(III) chloride (Ru-Hex) in PBS were carried out using the following parameters: CV voltage was scanned between 0.1 and −0.6 V at 100 mV s−1 with a step of 0.02 V. SWV voltage was scanned between 0.1 and −0.6 V with an amplitude of 0.1 V, step of 0.01 V and frequency of 20 Hz. EIS was performed at E1/2 (calculated as −0.25 V after initial CV investigation) between 10000.0 and 5.0 Hz (10 steps per decade) with 10 mV sine wave stimulation.
Dopamine
CV measurement was carried out using 0.25 mM dopamine hydrochloride in PBS. Voltage was scanned between −1 and 1.5 V at increasing scan rates of 100, 300, 500, 700, 900, 1400 and 3000 mV s−1 with a step of 0.2 V. To gauge the oxidation voltage of dopamine SWV, measurements were then performed on a range of dopamine hydrochloride concentrations between −0.1 and 1.1 V with an amplitude of 0.1 V, step of 0.01 V and frequency of 20 Hz. A similar experiment was performed using chronoamperometric measurement at 1 V, measurements were taken at 0.1 s intervals for 60 s. The common interferents ascorbic acid, uric acid and serotonin hydrochloride were also tested via the SWV detailed above at a concentration of 0.5 mM.ELISA protocol
IL-6 ELISA was performed as instructed by the duoset ELISA kit. In summary, 32 wells of a standard 96 well plate were successively incubated with 100 μL of capture antibody (2 μg ml−1), 300 μL of reagent diluent (1× diluted from kit stock), 100 μL of IL-6 standard added to 10% human serum in PBS (1200–9.38 pg ml−1), 100 μL of biotinylated detection antibody (50 ng ml−1) and 100 μL of streptavidin-tagged enzyme (40 fold dilution from kit standard). Between incubations, wells were washed via three successive washes consisting of filling and emptying wells with 300 μL of wash buffer (diluted from kit). This produced an antibody ‘sandwich’ as seen in Fig. 1 tipped with HRP. 100 μL of HRP substrate was then added to these wells and incubated for 20 minutes before 50 μL of stop solution was added. Absorbance and chronoamperometry were then measured to reflect the concentration of IL-6 added to each well.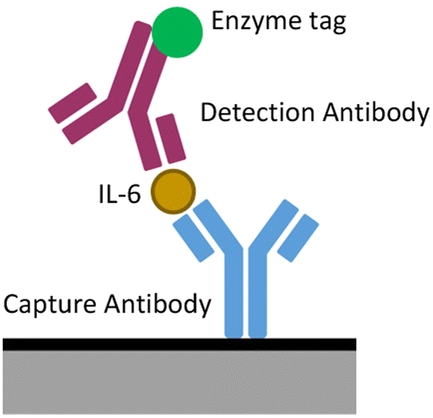 | ||
| Fig. 1 Schematic of a sandwich ELISA. | ||
Limit of detection calculation
Limit of detection (LoD) was calculated from concentration curves as shown in literature.39 Firstly, the limit of the blank (LoB), the highest apparent concentration found from blank measurements, was calculated using eqn (2). Eqn (3) was then used to calculate the LoD of the performed experiment.Calculation of the limit of the blank:
| LoB = Mean(blank) + 1.645(STD)(blank). | (2) |
Calculation of the limit of detection:
| LoD = LoB + 1.645(STD)(Sample). | (3) |
To relate this LoD to the real measurements taken, an experimental LoD (ELoD) will be taken as the closest measured concentration point above the LoD to represent the lowest analyte concentration able to be detected consistently.
K 0 Calculation
The heterogenous electron transport rate constant (K0) is of great interest in the characterisation of electrochemical systems, indicating the rate of electron transfer between an electrode surface and electroactive substrate. In this investigation, it was calculated as shown by Lavagnini, Antiochia and Magno (2003).40,41 This approach is based on two equations, both centred around the dimensionless parameter, ψ.| ψ = (0.0021ΔEp − 0.6288)/(1 − 0.017ΔEp), | (4) |
| ψ = K0[πDnFv/RT]−1/2, | (5) |
The measurement of a defined electroactive species was measured at several scan speeds using CV. Peak separation was extracted and eqn (4) used to obtain ψ. Based on eqn (5), the gradient obtained from fitting ψ plotted against [πDnFv/RT]1/2 therefore represents K0.
Results and discussion
Electrode manufacturing
Fig. 2 shows the produced electrode base (Fig. 2a) and well (Fig. 2b) which were filled with a 50:50 parrafin:graphite paste mixture to produce the electrode format (Fig. 2c). In general, each chip consisted of 10 channels into which heated paste was pushed before excess was removed with a flat spatula end. There were 8 linear working electrodes and two large L shaped electrodes, one of which operated as a carbon paste counter and one of which had a Ag/AgCl wire inserted into it to act as a reference. This produced chips with a geometric working electrode area of 4.77 mm2 and a counter electrode with an area of 44.25 mm2 with a similar layout to other designs utilised in literature.41 The estimated cost of goods to produce one chip is £0.023 (excluding the reusable Ag wire that is regenerated between uses) and manufacturing including printing required around half an hour of work.
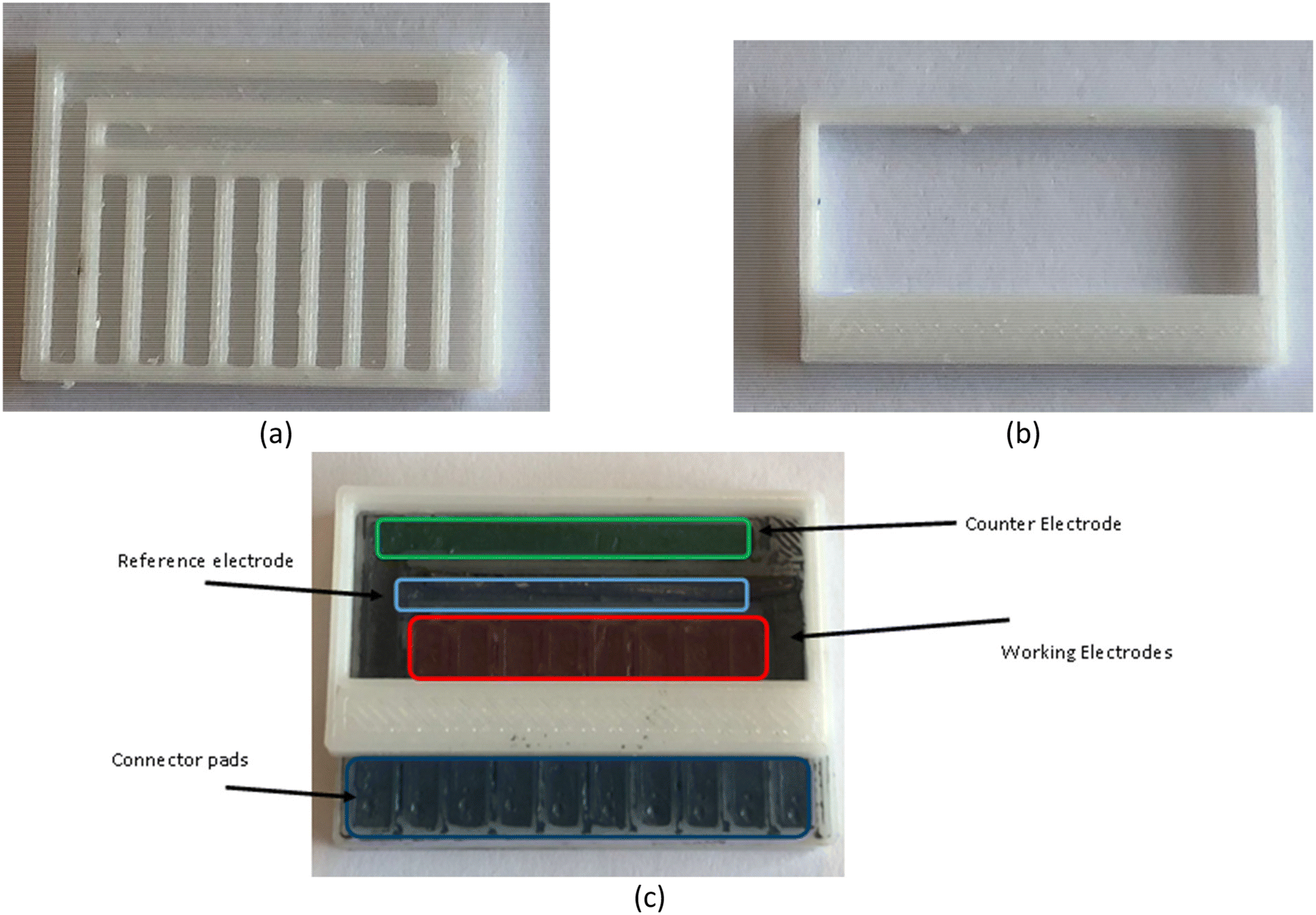 | ||
| Fig. 2 Paste electrodes in a multiplexed format (a) PETG base produced via 3D printing. (b) PETG well produced via 3D printing. (c) The 8 WE carbon paste chip produced and used during this investigation with attached well. | ||
To characterise the CPEs performance with different redox mediators and enzymes, it was necessary to perform a series of investigations involving CV, amperometry and SWV while increasing the complexity of the species being a simple outer sphere redox mediator (hexaammineruthenium(III) chloride), to the small biological molecule dopamine with it well known complex electrochemistry to the inflammatory biomarker IL-6 (23 kDa). The following sections outline the results generated with the newly fabricated devices shown here.
Hexaammineruthenium(III) chloride measurement
Hexaammineruthenium(III) chloride (RuHex) is a standard redox mediator often used to gauge the basic effectiveness of electrodes. Due to the outer sphere nature of this redox couple, it is often used with carbon materials. Outer sphere reactions occur when electrons are transferred from outer electron spheres easily via weak interactions. This can happen without the need of specific adsorption and so kinetics are less dependent on electrode surfaces.42A 1 mM RuHex solution was made in PBS and used to investigate the consistency of electrode manufacturing. Three freshly manufactured and cleaned chips were characterised in 1 mM RuHex using SWV. Fig. 3 demonstrated that responses obtained were within a similar order of magnitude (82 ± 12, 122 ± 16, 104 ± 25 μA). The produced CPEs would therefore be suitable for measurements which do not require ‘high precision’, since deviation both between individual electrodes on a chip and between chips was noticed, even after surface conditioning. This could be expected given their handmade nature and the potentially random generation of conductive paths in the bulk material, to be further improved.
 | ||
| Fig. 3 SWV peaks taken from measurements of three separate chips to highlight manufacturing consistency, four electrodes from each chip were measured in 1 mM RuHex in PBS (n = 4). | ||
Ruhex was also measured through CV and EIS methods to estimate the basic electrochemical ability of CPEs (Fig. 4). As can be seen clear oxidation and reduction peaks are present, indicating suitable electrode performance.
 | ||
| Fig. 4 (a): CV measurements of 1 mM RuHex in PBS between −0.6 and 0.1 V at a scan speed of 100 mV s−1. (b) Nyquist plot of EIS characterisation of electrode in 1 mM RuHex in PBS, performed at E1/2 between frequencies of between 10000.0 and 5.0 Hz with 10 mV sine wave stimulation. (c) Bode plot of the same EIS characterisation. | ||
On inspection of the CV response (Fig. 4a), a few key points can be seen. The Randles–Ševčík equation (inputting the geometric area of the electrode surface) predicts a reduction current of 12.88 μA. The mean value of 8.70 μA ± a standard deviation of 4.38 μA is in reasonable agreement with the equation, with the discrepancy explained by electrode roughness and slight variations in area arising from the fabrication process.
A peak separation of 79 mV was recorded (Fig. 4a), higher compared to the theoretical separation of 59 mV expected. In literature a breadth of performances has been reported, with paste electrodes from Fanjul-Bolado et al. (2008) capable of peak separation as low as 63 mV,43 while Wang et al. (2001) CPEs produced much more similar peak separations of 77 mV.44 This range highlights both the variation in paste composition in literature and the effect of the paraffin binder on typical performance.
Differences in the oxidation and reduction peak amplitudes were also observed (10.56 (±0.83) μA vs. 15.45 (±2.15) μA). This indicates a quasi-reversible behaviour of RuHex on the electrodes as expected from reports of other untreated paste surfaces in literature.45 To confirm this quasi-reversibility, CV measurements of 1 mM RuHex in PBS were then taken at scan speeds of 100, 300, 500, 700 and 900 mV s−1. Peak separations were then recovered and the K0 calculated using a reported diffusion coefficient of 8.4 × 10−6 cm2 s−1.46 The obtained K0 value of 0.055 cm s−1 is comprised within the range of a quasi-reversible system (10−1–10−5 cm s−1), where currents are constrained both by charge transfer and mass transport.47
EIS measurements displayed in Fig. 4b and c showed a highly resistive system. This was first evidenced in the Nyquist plot (Fig. 4b), which although difficult to fit to standard circuits (Randles/simplified Randles equivalents) most closely resembles a single Warburg element, albeit at a much shallower angle than usual. This indicates a system under control of mass transport or ‘semi-infinite’ diffusion rather than any electrode effects. The Bode representation (Fig. 4c) also showed a system dominated by resistance.48 Both SEM and FTIR measurements respectively shown in Fig. S2 and S3 (ESI†) may offer some explanations for this electrochemical activity. In particular, the ‘solid dispersion’49 structure of the paste shown both in this investigation and other pastes made in both solid50 and liquid binders51–53 where pastes consist of carbon particles coated in binder. This produced an uneven structure with a surface composed of high and low conductivity areas, shown by bright and dark patches in SEM images (Fig. S2†) and through the presence of both hydrocarbon and pure carbon bonds in FTIR (Fig. S3†).
This dispersion produced conductive paths and therefore performed as an electrode. However, much of the bulk of the material can still be considered as ‘inactive’, either completely non-conducting paraffin or isolated graphite. This supports the behaviour observed in EIS measurements, governed by the ability of analytes to find conductive areas on the electrodes surface as well as in CV measurements, where high ‘distributed resistance’ has been shown to spread CV peaks.54
Dopamine measurement with CPEs
A 250 μM dopamine solution was made in PBS and measured by CV using several scan rates. Oxidation peaks showing the formation of dopamine-o-quinone (DOQ) were clear and although reduction peaks are not visible at slower rates due to the rapid desorption of this product from the electrode surface,55 faster scan rates record DOQ before desorption can take place.Inspecting the CV responses, scan speed had minor impact on peak potentials, indicating a reasonably reversible system for the parameters tested. However, when plotting peak heights vs. the square root of scan rate, a poor linear fit is seen (see Fig. 5b), especially at higher scan rates where currents deviate from the expected significantly, producing lower than anticipated peak heights. This highlights the complex nature of dopamine electrochemistry and may indicate the point at which diffusion becomes a limiting factor in the redox system, or potentially the point at which the oxidative production of polydopamine seen commonly in literature56 begins to interfere with measurement.
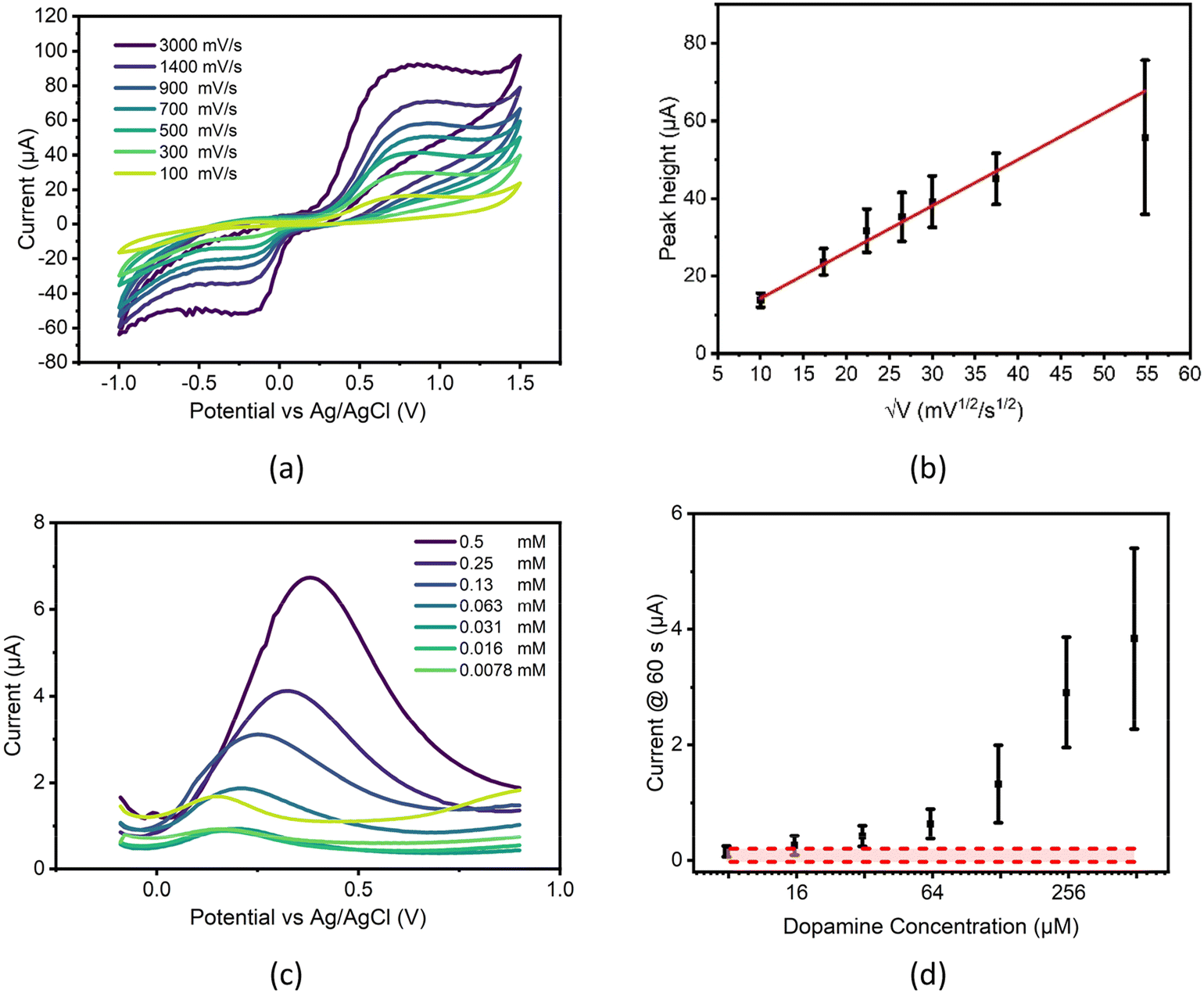 | ||
| Fig. 5 (a): Measurements of 0.25 mM dopamine in PBS at increasing scan rates (mV s−1) showing stability of the system. One channel of 8 WE chip shown for clarity. (b): Measurements of 0.25 mM dopamine in PBS at increasing scan rates of 100, 300, 500, 700, 900, 1400 and 3000 mV s−1, oxidation peak heights against the square root of the scan speed plotted (n = 8) (error bars plotted as ± one sample standard deviation) (c): SWV characterization of dopamine on the carbon paste sensor, example from a single channel over multiple concentrations of dopamine in PBS (mM) (d): chronoamperometric measurements of dopamine concentrations seen in (c), measured using all electrodes on a single chip, compared to PBS baseline measurements (red zone) (n = 8) (error bars plotted as ± one sample standard deviation). | ||
When different concentrations of dopamine are tested using SWV, concentration dependent differences are seen in peak currents down as low as 62.5 μM, however this plot also shows potential drift in measurements (see Fig. 5c), with higher concentrations of dopamine requiring higher potentials for full oxidation, one possible cause of this drift may be due to the effect of iR drop within the chip. iR drop is the difference between the expected potential applied at the working electrode and the actual potential that is applied and is influenced by several factors including electrode and electrolyte conductivity, distance between working and reference electrodes, working electrode roughness and double layer thickness57 and can lead to the shifting of electrochemical peaks on a plot. The increasing concentrations of dopamine measured may potentially affect this drop in two major ways, firstly by increasing the solution resistance and secondly through the altering of electrode conductivity through the binding of dopamine polymers to electrode surfaces.
These same dopamine solutions were then measured chronoamperometrically along with a PBS baseline to ascertain the electrode's ability to detect dopamine, producing the concentration curve seen in Fig. 5d. When analysed, this data produced an ELoD of 62.5 μM of dopamine, although standard deviation error bars indicated that distinguishing between concentrations above this point may be difficult.
Although this LoD is much higher than those seen in literature (Table 1) and is not suitable for direct dopamine detection from any biological fluid, it does show the general utility of the produced chips for the direct measurement of biological molecules. In its current format, the chip may be useful for detecting biomarkers which have higher clinically relevant concentrations, or with modifications like those seen in literature may be capable of relevant dopamine detection.
SWV testing was also performed to gauge how well the chip may potentially fare in a more clinical setting with the potential impact from the common interferents ascorbic acid, uric acid58 and serotonin hydrochloride which are often detected simultaneously.59 SWV measurements of 0.5 mM solutions in PBS were performed both separately and with dopamine hydrochloride using the same electrode chip. Results of these tests are shown in the supplementary (Fig. S5†).
The chip produced performed poorly in these mixed solutions, showing that use of the bare chip in real world samples will lead to interference from ascorbic and uric acid which both have overlapping SWV peak positions. Interestingly, combinations of these interferents and dopamine were able to produce large single peaks and further work will be performed to ascertain if these are related to interferent or dopamine concentration. In the meantime, the detection of dopamine on these unmodified carbon sensors should be carried out with experimental removal of interferents before electrochemical testing, as suggested in literature.60
Detection of electrochemically active enzymatic products
A sandwich ELISA tagged with HRP (see Fig. 1) was performed to gauge the ability of the electrode chip to detect the products of a more general measurement system. Interleukin 6 (IL-6) was chosen as a suitable biomarker for initial detection. It is diluted in 10% human serum, the liquid component of blood with both cells and clotting factors removed. This was chosen as a suitable medium for detection as it does not contain anti coagulants as plasma does, is stable and still contains many of the markers required for biosensing.67 Importantly for the development of POC systems, it is routinely sampled from patients and so should indicate the chip's likely performance in a clinically relevant sample.
Results of this experiment are shown in Fig. 6. Raw chronoamperometric plots (Fig. 6a) contain some noise, potentially to be expected from a graphite-based sensor at low voltage.68 While not able to seriously affect results, this noise should be watched closely and potentially dealt with using commonly employed data processing techniques such as ‘spike removal’ if deemed necessary for other applications.
 | ||
| Fig. 6 (a) Example chronoamperometric data taken from one chipo during measurement of ELISA, measured at 0.2 V vs. Ag/AgCl for 60 s. (b) Absorbance of IL-6 sandwich ELISA performed with 1200, 600, 300, 150, 75, 37.5 and 18.75 pg ml−1 standards of IL-6 diluted in 10% human serum, four wells of ELISA were performed for each concentration and plotted with zero line (error bars plotted as ± one sample standard deviation) (n = 4). (c) Chronoamperometric measurement of well solutions pipetted onto chip, measured at 0.2 V over 60 s using all 8 electrodes of a single chip (n = 8), compared to PBS baseline measurements (red zone) (n = 8) (error bars plotted as ± one sample standard deviation). | ||
Colourimetric measurement of the assay performed well (Fig. 6b), with results showing a logarithmic trend that did not visually intersect the zero line in the y axis at all and a very low ELoD of 18.75 pg ml−1. Clear visual difference between discrete concentration points also showed that colourimetric measurement was not only able to detect the presence of IL-6 but is also able to discern small differences in its concentration. Although a similar logarithmic trend was seen in chronoamperometric results (Fig. 6c), a higher ELoD of 75 pg ml−1 was required to distinguish between control and IL-6 solutions. Large standard deviations prevent signal differentiation at low IL-6 concentrations. Even if the electrochemical LoD obtained was higher compared to optical ELISA results, this value proved much more comparable to literature (Table 2). Using the indirect detection strategy, CPEs performed at an equivalent level to literature reports and were in the useful and clinically relevant range for the biomarker tested (IL-6). This was achieved without further modification of these low-cost carbon paste electrodes indicating potential for use as a low-cost ‘point of need’ decision support tool. Advancing the system into an ‘in the field’ detection platform could provide a clinician an inflammatory response snapshot in terms of elevation and likelihood of associated illnesses, e.g. sepsis.69
Further work will focus on the development of the CPE electrodes into an even more convenient and reproducible format through development of working electrode surface areas and reference electrode stability studies to give the improvement in precision necessary to push electrochemical ELISA sensitivity higher.
Conclusions
A low-cost carbon paste material has been produced which is easy to deploy in various electrode formats. For an assay format, electrode chips were obtained in 0.5 hours, costing £0.023 each. This format focused on being produced using widely accessible equipment with little specific knowledge or expertise required by users in the field. Chips were capable of detection of biomarkers of interest both directly (dopamine) and indirectly (IL-6) with an enzyme-tagged ELISA format. The system presently does not produce clear, quantitative data at the low abundance biomarker concentration (sub 100 pg ml−1) but is able to detect clinically relevant elevations. In its current form, it may be more suitable for qualitative sensing or producing YES/NO diagnostic results at diagnostically useful thresholds.Author contributions
Author contributions are tracked using the appropriate CRediT system categories: conceptualization – DC (equal), SM (equal); data curation – SM (lead); formal analysis – SM (lead); funding acquisition – DC (lead); investigation – SM (lead); methodology – DC (equal), SM (equal); project administration – SM (lead); resources – DC (lead); supervision – DC (lead); validation – SM (lead); writing – SM (lead), DC (supporting); writing – review & editing – SM (equal), PL (equal).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
SM thanks the EPSRC Doctoral Training Partnership EP/T517938/1 for his scholarship.References
- H. Dellweg, J. W. Engels, J. L. Fox, L. M. Gierasch, R. P. Gregson, B. Heinritz, H. G. W. Leuenberger, M. Moo-Young, A. Moser, B. Nagel, L. Nyeste, L. Pénasse, G. B. Petersen, M. Van Montagu and Y. Yamada, Pure Appl. Chem., 1992, 64, 143–168 CrossRef.
- F. Ricci, G. Adornetto and G. Palleschi, Electrochim. Acta, 2012, 84, 74–83 CrossRef CAS.
- H. Abu-Ali, O. Cansu, F. Davis, N. Walch and A. Nabok, Chemosensors, 2020, 8, 1 CrossRef.
- Z. Lin, G. Wu, L. Zhao and K. W. C. Lai, IEEE Nanotechnol. Mag., 2019, 13(5), 4–14 Search PubMed.
- O. Fernando and E. Magner, Sensors, 2020, 20, 1 Search PubMed.
- K. Jacobs, S. F. Wolf, L. Haines, J. Fisch, J. N. Kremsky, J. P. Dougherty, R. H. Symons, N. Habili, J. L. Mclnnes, G. Gentilomi, E. Ferri, S. Girotti, Y. W. Kan, F. F. Chehab, J. S. Sevall, H. Prince, G. Garratty, W. A. O′brien, J. A. Zack, K. M. Millan, S. R. Mikkelsen, K. Hashimoto, K. Miwa, Y. Ishimori, K. Ito, A. J. Bard and T. Cater, Anal. Chem., 1994, 66, 3830 CrossRef PubMed.
- L. Fritea, M. Tertis, R. Sandulescu and C. Cristea, Methods Enzymol., 2018, 609, 293–333 CAS.
- Z. Taleat, A. Khoshroo and M. Mazloum-Ardakani, Microchim. Acta, 2014, 181, 865–891 CrossRef CAS.
- S. Lee, W. J. Kim and M. Chung, Analyst, 2021, 146, 5236–5244 RSC.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- S. A. Wring and J. P. Hart, Analyst, 1992, 117, 1215 RSC.
- D. Bellido-Milla, L. M. Cubillana-Aguilera, M. El Kaoutit, M. P. Hernández-Artiga, J. L. Hidalgo-Hidalgo De Cisneros, I. Naranjo-Rodríguez and J. M. Palacios-Santander, Anal. Bioanal. Chem., 2013, 405, 3525 CrossRef CAS PubMed.
- S. Tajik, H. Beitollahi, F. G. Nejad, M. Safaei, K. Zhang, Q. Van Le, R. S. Varma, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 21561–21581 RSC.
- P. Kartikay, A. Yella and S. Mallick, Mater. Chem. Phys., 2020, 256, 1–9 CrossRef.
- R. N. Adams, Sci. Commun., 1958, 1576 CAS.
- I. Švancara, K. Vytřas, K. Kalcher, A. Walcarius and J. Wang, Electroanalysis, 2009, 21, 7 CrossRef.
- S. Suresh, A. K. Gupta, V. K. Rao, O. Kumar and R. Vijayaraghavan, Talanta, 2010, 81, 703 CrossRef CAS PubMed.
- V. Rajendran, E. Csöregi, Y. Okamoto and L. Gorton, Anal. Chim. Acta, 1998, 373, 241 CrossRef CAS.
- K. Sugawara, T. Takano, H. Fukushi, S. Hoshi, K. Akatsuka, H. Kuramitz and S. Tanaka, J. Electroanal. Chem., 2000, 482, 81 CrossRef CAS.
- S. H. Zuo, L. F. Zhang, H. H. Yuan, M. B. Lan, G. A. Lawrance and G. Wei, Bioelectrochemistry, 2009, 74, 223 CrossRef CAS PubMed.
- M. O. Klein, D. S. Battagello, A. R. Cardoso, D. N. Hauser, J. C. Bittencourt and R. G. Correa, Cell. Mol. Neurobiol., 2019, 39, 31 CrossRef PubMed.
- S. J. Li, D. H. Deng, Q. Shi and S. R. Liu, Microchim. Acta, 2012, 177, 325 CrossRef CAS.
- M. Hasanzadeh, N. Shadjou and M. de la Guardia, TrAC, Trends Anal. Chem., 2017, 86, 107–121 CrossRef CAS.
- D. J. Wiedemann, K. T. Kawagoe, R. T. Kennedy, E. L. Ciolkowski and R. M. Wightman, Anal. Chem., 1991, 63, 2965 CrossRef CAS PubMed.
- A. R. Macdonald, F. Charlton and D. K. Corrigan, Anal. Bioanal. Chem., 2023, 415, 1137 CrossRef CAS PubMed.
- L. Yu, L. Feng, Z. Wei, S. Wang, Y. Feng, Y. Shen, J. Cai, J. Wu and Y. Xiao, Adv. Funct. Mater., 2023, 33, 1–11 Search PubMed.
- X. Ouyang, Y. Wu, L. Guo, L. Li, M. Zhou, X. Li, T. Liu, Y. Ding, H. Bu, G. Xie, J. Shen, C. Fan and L. Wang, Angew. Chem., 2023, 62, 1–8 Search PubMed.
- M. A. Dayton, A. G. Ewing and R. M. Wightman, Anal. Chem., 1980, 52(14), 2392–2396 CrossRef CAS.
- S. Hosseini, P. V.-V. Marco, R.-P. Sergio and O. Martinez-Chapa, Enzyme - linked Immunosorbent Assay (ELISA) From A to Z, Springer Nature, 2018, vol. 1 Search PubMed.
- Z. Yin, Y. Liu, L. P. Jiang and J. J. Zhu, Biosens. Bioelectron., 2011, 26, 1890–1894 CrossRef CAS PubMed.
- R. R. Mustafa, R. Sukor, S. Eissa, A. N. Shahrom, N. Saari and S. M. M. Nor, Sens. Actuators, B, 2021, 345, 1–10 CrossRef.
- M. A. Khan and M. Mujahid, Sensors, 2020, 20, 1–27 Search PubMed.
- S. Kibe, K. Adams and G. Barlow, J. Antimicrob. Chemother., 2011, 66, 33–40 CrossRef PubMed.
- S. Kaur, Y. Bansal, R. Kumar and G. Bansal, Bioorg. Med. Chem., 2020, 28, 115327 CrossRef CAS PubMed.
- C. Deng, F. Qu, H. Sun and M. Yang, Sens. Actuators, B, 2011, 160, 471–474 CrossRef CAS.
- G. Wang, X. He, L. Chen, Y. Zhu and X. Zhang, Colloids Surf., B, 2014, 116, 714–719 CrossRef CAS PubMed.
- R. Kapoor and C. W. Wang, Biosens. Bioelectron., 2009, 24, 2696–2701 CrossRef CAS PubMed.
- A. E. Ferreira Oliveira, A. C. Pereira, M. A. Campos de Resende and L. F. Ferreira, Electroanalysis, 2022, 34, 809–819 CrossRef CAS.
- D. A. Armbruster and T. Pry, Clin. Biochem. Rev., 2008, 29, 49–52 Search PubMed.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505–506 CrossRef CAS.
- R. Sánchez-Salcedo, R. Miranda-Castro, N. de-los-Santos-Álvarez, M. J. Lobo-Castañón and D. K. Corrigan, Anal. Bioanal. Chem., 2023, 415, 7035–7045 CrossRef PubMed.
- C. Banks, Understanding Voltammetry, Imperial College Press, 2nd edn, 2011, vol. 1 Search PubMed.
- P. Fanjul-Bolado, D. Hernández-Santos, P. J. Lamas-Ardisana, A. Martín-Pernía and A. Costa-García, Electrochim. Acta, 2008, 53, 3635–3642 CrossRef CAS.
- J. Wang, Ü. A. Kirgöz, J.-W. Mo, J. Lu, A. N. Kawde and A. Muck, Electrochem. Commun., 2001, 3, 203–208 CrossRef CAS.
- G. Cui, J. H. Yoo, J. Su Lee, J. Yoo, J. H. Uhm, G. S. Cha and H. Nam, Analyst, 2001, 126, 1399–1403 RSC.
- J. Moldenhauer, M. Meier and D. W. Paul, J. Electrochem. Soc., 2016, 163, H672–H678 CrossRef CAS.
- J. Wang, in Analytical Electrochemistry, Wiley, 3rd edn, 2006, pp. 34–35 Search PubMed.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed.
- Y. M. Issa, H. M. Abdel-Fattah, O. R. Shehab and N. B. Mohamed, Electroanalysis, 2017, 29, 2541–2550 CrossRef CAS.
- C. Laghlimi, Y. Ziat, A. Moutcine, M. Hammi, Z. Zarhri, O. Ifguis and A. Chtaini, Chem. Data Collect., 2021, 31, 1–17 Search PubMed.
- E. Y. Frag, M. E. B. Mohamed and H. S. Salem, J. Iran. Chem. Soc., 2017, 14, 2355–2365 CrossRef CAS.
- I. Švancara, M. Hvízdalová, K. Vytřas, K. Kalcher and R. Novotný, Electroanalysis, 1996, 8, 61–65 CrossRef.
- T. Mikysek, I. Švancara, K. Kalcher, M. Bartoš, K. Vytřas and J. Ludvík, Anal. Chem., 2009, 81, 6327–6333 CrossRef CAS PubMed.
- R. G. Keil, Electrochem. Sci. Technol., 1986, 133, 1375–1379 CrossRef CAS.
- B. J. Venton and Q. Cao, Analyst, 2020, 145, 1158–1168 RSC.
- S. Schindler and T. Bechtold, J. Electroanal. Chem., 2019, 836, 94–101 CrossRef CAS.
- S. Anantharaj and S. Noda, J. Mater. Chem. A, 2022, 10, 9348–9354 RSC.
- S. Pruneanu, A. R. Biris, F. Pogacean, C. Socaci, M. Coros, M. C. Rosu, F. Watanabe and A. S. Biris, Electrochim. Acta, 2015, 154, 197–204 CrossRef CAS.
- M. Kundys-Siedlecka, E. Baczyńska and M. Jönsson-Niedziółka, Anal. Chem., 2019, 91, 10908–10913 CrossRef CAS PubMed.
- S. Rantataro, L. Ferrer Pascual and T. Laurila, Sci. Rep., 2022, 12, 1–9 CrossRef PubMed.
- Y. Chang and B. J. Venton, Anal. Methods, 2020, 12, 2893–2902 RSC.
- M. Ni, J. Chen, C. Wang, Y. Wang, L. Huang, W. Xiong, P. Zhao, Y. Xie and J. Fei, Microchem. J., 2022, 178, 1–11 CrossRef.
- S. E. Elugoke, O. E. Fayemi, A. S. Adekunle, B. B. Mamba, T. T. I. Nkambule and E. E. Ebenso, FlatChem, 2022, 33, 1–13 CrossRef.
- A. Rahman, R. K. Pal, N. Islam, R. Freeman, F. Berthiaume, A. Mazzeo and A. Ashraf, Sensors, 2023, 23, 1–17 Search PubMed.
- K. Gjesing Welinder, FEBS Lett., 1976, 72, 19–23 CrossRef PubMed.
- S. Gu, S. Risse, Y. Lu and M. Ballauff, ChemPhysChem, 2020, 21, 450–458 CrossRef CAS PubMed.
- C.-M. José-Luis, MS-Based Proteomic Analysis of Serum and Plasma: Problem of High Abundant Components and Lights and Shadows of Albumin Removal, Springer, 2019 Search PubMed.
- S. S. Ghoreishizadeh, G. Nanda, S. Carrara and G. De Micheli, 5th International Workshop on Advances in Sensors and Interface (IWASI), 2013, 1(1), 36–39 Search PubMed.
- C. Russell, A. C. Ward, V. Vezza, P. Hoskisson, D. Alcorn, D. P. Steenson and D. K. Corrigan, Biosens. Bioelectron., 2019, 126, 806–814 CrossRef CAS PubMed.
- X. Zhou, P. Li, X. Wu, X. Lin, L. Zhao, H. Huang, J. Wu, H. Cai, M. Xu, H. Zhou and P. Sun, Biosens. Bioelectron., 2022, 210, 1–10 CrossRef PubMed.
- D. Huang, H. Ying, D. Jiang, F. Liu, Y. Tian, C. Du, L. Zhang and X. Pu, Anal. Biochem., 2020, 588, 1–6 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00392f |
| This journal is © The Royal Society of Chemistry 2024 |