
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitivity-improved blocking agent-free fluorescence polarization assay through surface modification using polyethylene glycol†
Hao
Liu
 ab,
Mao
Fukuyama
*a,
Yu
Ogura
a,
Motohiro
Kasuya
c,
Sho
Onose
d,
Ayuko
Imai
d,
Koji
Shigemura
d,
Manabu
Tokeshi
e and
Akihide
Hibara
*f
ab,
Mao
Fukuyama
*a,
Yu
Ogura
a,
Motohiro
Kasuya
c,
Sho
Onose
d,
Ayuko
Imai
d,
Koji
Shigemura
d,
Manabu
Tokeshi
e and
Akihide
Hibara
*f
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: maofukuyama@tohoku.ac.jp
bSchool of Science, Tohoku University, 6-3, Aramaki Aza-Aoba, Aoba-ku, Sendai 980-8578, Japan
cFaculty of Production Systems Engineering and Sciences, Komatsu University, Nu 1-3 Shicho-machi, Komatsu, Ishikawa 923-8511, Japan
dTianma Japan, Ltd., Shin-Kawasaki Mitsui Building West Tower 28F 1-1-2, Kashimada, Saiwai-ku, Kawasaki, Kanagawa 212-0058, Japan
eDivision of Applied Chemistry, Hokkaido University, Kita 13 Nishi 8, Kita-ku, Sapporo 060-8628, Japan
fDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: hibara.a.aa@m.titech.ac.jp
First published on 3rd September 2024
Abstract
Fluorescence polarization (FP) assays are widely used to quantify biomolecules, and their combination with microfluidic devices has the potential for application in onsite analysis. However, the hydrophobic surface of polydimethylsiloxane (PDMS)-based microfluidic devices and the amphiphilicity of the blocking agents can cause the nonspecific adsorption of biomolecules, which in turn reduces the sensitivity of the FP assay. To address this, we demonstrated an FP assay with improved sensitivity in microfluidic devices using a polyethylene glycol-based surface modification to avoid the use of blocking agents. We evaluated the effectiveness of the modification in inhibiting nonspecific protein adsorption and demonstrated the improved sensitivity of the FP immunoassay (FPIA). Our study addressed the lack of sensitivity of FP assays in microfluidic devices, particularly for the quantification of low-abundance analytes.
Introduction
Fluorescence polarization (FP), which reflects the rotational diffusion of fluorescent molecules during their lifetime (Fig. 1a), is a versatile property for investigating interactions between molecules in solutions.1,2 The FP assay can be used to measure changes in the size of fluorescent molecules without additional processes such as washing. Therefore, it is widely used as a one-step homogeneous assay and is applied to the quantification of various biomolecules in many research fields, such as drug discovery,3–5 food safety,6,7 and disease diagnosis.8 | ||
| Fig. 1 Principle of PEG-PDMS to improve sensitivity of FP assays in microfluidic devices. (a) Principle of FP assays. I∥ and I⊥ are the fluorescence intensities of the vertical and horizontal components. (b) Schematic diagram of nonspecific binding between tracer and BSA blocking agent and its standard curve. (c) Schematic diagram of the absence of nonspecific binding in PEG-PDMS and its standard curve. PDMS, polydimethylsiloxane; PEG, polyethylene glycol; FP, fluorescence polarization; BSA, bovine serum albumin. | ||
The combination of FP and microfluidic devices reduces reagent consumption, increases analysis throughput, and miniaturizes the system.9–18 High-throughput screening methods examine biomolecular interactions in a time-efficient manner, which is important for understanding biological functions and for drug discovery.19,20 Cheow et al. developed a high-throughput protein–ligand binding assay based on FP on a microfluidic platform that could simultaneously interrogate over 2300 interactions.11 The organ-on-a-chip technology simulates the functions of human organs in a controlled environment. Integrating this with the FP assay provides accurate, real-time molecular analysis, which is critical for biomedical research.21 A. L. Glieberman et al. designed an “islet on a chip” for continuous, automated insulin quantification through FP assay, enabling high-resolution dynamic analysis.22 Recently, we developed a portable FP analyzer using a polydimethylsiloxane (PDMS)-based microfluidic device for on-site analysis.23 Using this system, we report the application of an FP immunoassay (FPIA) for various samples such as mycotoxins in wheat, exosomes, and protein biomarkers in serum.24–26
However, although the use of microfluidic devices reduces the sample and reagent volumes, it also limits the sensitivity of FP owing to the nonspecific adsorption of the analyte and tracer molecules to the solid–liquid interface in the device. Although PDMS is a widely used material for fabricating microfluidic devices,27,28 hydrophobic and amphiphilic molecules, including proteins and lipids, are non-specifically adsorbed onto the interfaces because PDMS is hydrophobic.29 While hydrophilic surface modification and non-adsorptive materials have been used in both microplates30,31 and in microfluidic devices,32,33 the use of blocking agents still remains a more common practice because it is simpler to implement. In microfluidic bioanalytical applications, blocking agents such as bovine serum albumin (BSA) are typically used to prevent nonspecific adsorption. However, even when blocking agents were used, interference occurred (Fig. 1b). Blocking agents can interfere with immunoreactivity,34,35 such as through nonspecific binding of amphiphilic blocking agents with tracers and analytes.36,37 In addition, the blocking agent cannot completely cover the solid surface and there is still room for a tracer.38 In this case, the polarization degree (P) change does not reflect the concentration of the analyte because tracer complexation to the blocking agent and its adsorption to the surface increase P and potentially hinder the P increase by analyte-tracer bonding. Although the number of molecules bound to the blocking agents was negligible when the tracer concentration was high, the amount of binding was significant when the concentration was low. Because the tracer concentration must be low to achieve high sensitivity39 (details are explained in Fig. 4a), the use of blocking agents can be critical for FP assay sensitivity.
In this study, we demonstrate an improvement in the sensitivity of the FP assay through surface modification. Using our previously reported polyethylene glycol (PEG)-based modification method, the hydrophobic PDMS surface was modified into a hydrophilic surface by simple one-step mixing of PDMS with PEG-functionalized silicone.40 This method requires fewer modifications than other methods that involve multiple coating modifications after PDMS fabrication.41,42 The PEG group contains long chains that exhibit steric repulsion and inhibit protein adsorption.43 This PDMS-PEG copolymer can effectively suppress the nonspecific adsorption of the tracer without the need for blocking agents (Fig. 1c), allowing P to accurately reflect the sample concentration and thus improve sensitivity. We used positively and negatively charged proteins to evaluate the ability and versatility of this modification method to inhibit nonspecific protein adsorption. Sensitivity improvement was demonstrated by FPIA in a PEG-PDMS device with a portable FP analyzer (Fig. 2a).44
 | ||
| Fig. 2 Portable fluorescence polarization analyzer and microfluidic device. (a) Picture of the portable fluorescence polarization analyzer. (b) Photo and schematic diagram of the microfluidic device. | ||
Experimental
Chemicals
A Sylgard 184 silicone elastomer kit for the PDMS microdevice fabrication and PEG-functionalized silicone (501 W additive) consisting of heptamethyl-3-(propyl(polyethylene oxide)methyl)trisiloxane (74–90% w/w) and allyloxypolyethylene glycol methyl ether (14–20% w/w) were purchased from Dow Corning Toray (Japan). The PDMS included black silicon rubber to reduce background noise. Alexa Fluor® 488 AffiniPure Fab Fragment Goat Anti-Rabbit IgG (H + L), Alexa Fluor® 647 AffiniPure Fab Fragment Goat Anti-Rabbit IgG (H + L), and bovine serum albumin (BSA) were purchased from Jackson Immuno Research Laboratories Inc. (United States). Phosphate-buffered saline (PBS), fluorescein isothiocyanate (FITC) isomer I, dimethyl sulfoxide (DMSO), and lysozyme were purchased from Fujifilm Wako Pure Chemical Corporation (Osaka, Japan). Rabbit IgG and FITC-BSA conjugates were purchased from Sigma-Aldrich (St Louis, MO, USA).Microfluidic device fabrication
For PEG-PDMS devices (Fig. 2b), the PDMS prepolymer with black silicon rubber was mixed with PEG functionalized silicone and curing agent at a ratio of 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:100. It was then cured at 100 °C for 30 min to obtain PEG-PDMS sheets. The cured PEG-PDMS sheets and glass were plasma treated using a CUTE-1MP/R vacuum plasma system (Femto Science, Gwangju, Korea). The treatment time was fixed at 20 s and the power was set to 10 W. Oxygen or air was used as the process gas. Subsequently, the PEG-PDMS sheets were bonded to the glass and heated using a 75 °C hot plate to obtain PEG-PDMS devices.
:5:100. It was then cured at 100 °C for 30 min to obtain PEG-PDMS sheets. The cured PEG-PDMS sheets and glass were plasma treated using a CUTE-1MP/R vacuum plasma system (Femto Science, Gwangju, Korea). The treatment time was fixed at 20 s and the power was set to 10 W. Oxygen or air was used as the process gas. Subsequently, the PEG-PDMS sheets were bonded to the glass and heated using a 75 °C hot plate to obtain PEG-PDMS devices.
The PDMS microfluidic devices were fabricated as described before.45 The PDMS prepolymer with black silicone rubber was mixed with a curing agent in a ratio of 10:1. It was then cured at 80 °C for 60 min to obtain PDMS sheets. The PDMS sheets were then pasted onto the glass substrates without plasma treatment.
This device was designed with nine channels (Fig. 1c) to enable simultaneous analysis of nine groups of samples. The width of the channel was 200 μm and the depth was 900 μm.
Preparation of FITC-conjugated lysozyme (FITC-lysozyme)
FITC-lysozyme was synthesized according to a previous report.46 A total of 2 mg lysozyme was weighed and dissolved in 1 mL carbonate buffer (0.1 M pH = 9); 1 mg FITC was dissolved in 1 mL dry DMSO; 100 μL FITC solution was slowly added to 1 mL of lysozyme solution; the lysozyme solution was gently mixed as the FITC was added. The mixed solution was then incubated for 8 h at 4 °C. The crude product was purified on a PD MidiTrap G-25 column. The concentration of purified FITC-lysozyme was determined by measuring the absorbance using a BioSpectrometer (Eppendorf, Japan).Evaluation of the adsorption of FITC-BSA and FITC-lysozyme to PDMS and PEG-PDMS microfluidic devices
Three sets of 20 μL FITC-BSA and FITC-lysozyme solutions with 10−7, 10−7.5, and 10−8 M were injected with a 30 min-interval period. After the final set of solutions was injected, the device was kept in the dark for 30 min. Fluorescence images were obtained using a portable analyzer. Afterwards, all channels were washed with 100 μL PBS and the fluorescence images were obtained in the same manner. The fluorescence intensity was quantified using ImageJ software.Demonstration of nonspecific adsorption between tracer and BSA
Tracer Alexa Fluor® 488-Fab (1.0 and 0.1 nM) were mixed with 0.1–0.7 wt% BSA and injected into the PDMS channel. Changes in fluorescence intensity and P with BSA concentration were measured using a portable analyzer. After 1 h of incubation, the channel was washed with 200 μL PBS, and the fluorescence intensity was measured again.Demonstration of FPIA in PDMS and PEG-PDMS microfluidic devices
For FPIA in PDMS microfluidic devices, a mixed solution of 0.007 nM–108 nM rabbit IgG (analyte/antigen), 0.1 nM or 1 nM Alexa Fluor 488-conjugated anti-Rabbit IgG Fab fragment (tracer/antibody) and 0.1 wt% blocking agent (BSA) was prepared. The mixed solution was incubated in the dark at 24 °C for 60 min (1 nM tracer) or overnight (0.1 nM tracer). Subsequently, 20 μL of the sample was injected into the PDMS device.For FPIA in PEG-PDMS microfluidic devices, a mixed solution of 0.007 nM–108 nM rabbit IgG (analyte/antigen), 0.1 nM or 1 nM Alexa Fluor 488-conjugated anti-Rabbit IgG Fab fragment (tracer/antibody) was prepared. BSA was not added to the solution. The mixed solution was incubated in the dark at room temperature for 60 min (1 nM tracer) or overnight (0.1 nM tracer). Subsequently, 20 μL of the sample was injected into the PDMS device.
In the FPIA measurement, P is expressed by the following equation:23

The measured P was fitted using four-parameter logistic models47 to obtain a standard curve f(x) and the fitting equation can be expressed as
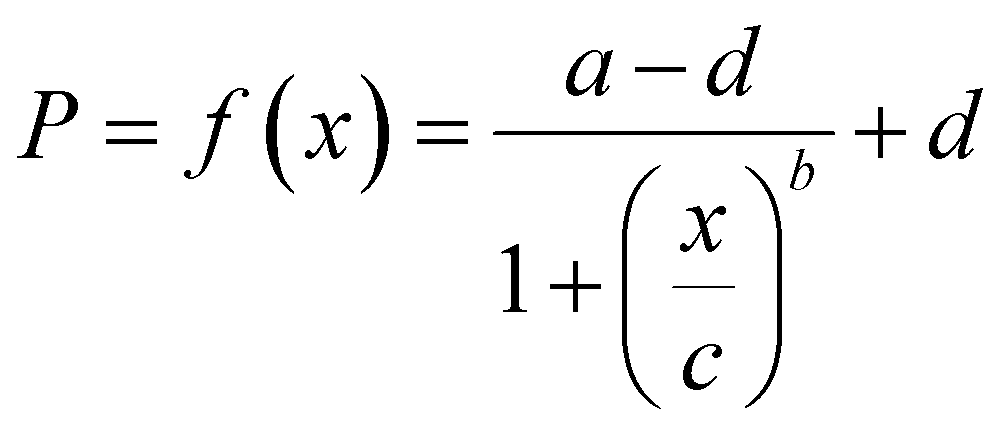
The limit of detection (LOD) of the standard curve was calculated as
| LOD = f−1(P0 + 3σ) |
Results and discussion
Investigation of protein adsorption to PEG-PDMS device
PEG-PDMS microfluidic devices were fabricated by the same procedure except for the bonding process as PDMS. In the plasma treatment method, air plasma was used to avoid damage to the PEG functional groups (Fig. S1†). After plasma treatment, PEG-PDMS was heated at 75 °C for more than 19 h for strong bonding (Table S1†).Subsequently, the protein adsorption to the microfluidic channel walls in PDMS and PEG-PDMS devices was investigated by using fluorescent-dye-conjugated proteins. Fig. 3a shows fluorescent images of the microfluidic channels loaded with FITC-BSA (pI = 5.1–5.5, negatively charged in pH 7.4) solutions before and after the washing process. In the PDMS device, the FITC-BSA fluorescence was retained after washing. In Fig. 3b, the fluorescence intensities for immersion times of 30–90 min are plotted as a function of concentration. The result indicates that 38–79% of FITC-BSA adsorbed to the PDMS channel walls. On the other hand, in the PEG-PDMS device, the fluorescence of FITC-BSA drastically decreased after the washing process. This indicates that FITC-BSA was not adsorbed onto the PEG-PDMS channel walls. Higher molecular weight PEG effectively inhibits protein adsorption.43 As protein adsorption was not observed in this experiment, the molecular weight of PEG in the PEG-functionalized silicone was considered sufficiently high to perform highly sensitive FPIA.
 | ||
| Fig. 3 Evaluation of the inhibitory adsorption effect. (a) Fluorescence images and (b) fluorescence intensity plot of FITC-BSA in PDMS and PEG-PDMS devices. PDMS, polydimethylsiloxane; PEG, polyethylene glycol. | ||
To confirm the versatility of the inhibition of protein adsorption onto PEG-PDMS, we examined the adsorption of FITC-lysozyme (pI = 11.4; positively charged at pH 7.4). As shown in Fig. S2,† FITC-lysozyme did not absorb to PEG-PDMS channel walls while it adsorbed to PDMS ones. These results indicated that PEG-PDMS devices inhibited the nonspecific adsorption of proteins with both positive and negative charges. Therefore, we concluded that the FP assay could be conducted without blocking agents in the PEG-PDMS devices.
Demonstration of sensitivity enhancement in FPIA by using PEG-PDMS microfluidic device
In FPIA, tracer concentration is a key factor that limits sensitivity. Fig. 4a shows the theoretical curves of FPIA48 composed of small tracers and large analytes at different tracer concentrations with KD = 0.2 nM. As the tracer concentration decreases, the dynamic range shifts to lower analyte concentrations. This indicated that lower tracer concentrations were more suitable for analyzing low-abundance analytes and provided a lower LOD. | ||
| Fig. 4 Sensitivity improvement of FPIA in the PEG-PDMS microfluidic device. (a) Theoretical FPIA standard curve with different tracer concentration. Dissociation constant (Kd) of tracer and analyte is 0.2 nM. FPIA standard curves in (b) PDMS devices with 0.1 wt% BSA and in (c) PEG-PDMS devices without BSA. PDMS, polydimethylsiloxane; PEG, polyethylene glycol; FPIA, fluorescence polarization immunoassay; BSA, bovine serum albumin. | ||
Fig. 4b shows the standard curve of FPIA in PDMS devices with blocking agents (0.1% BSA). Although sigmoidal P increased with increasing analyte concentration, which is a typical response of FPIA, it was observed with a 1 nM tracer but not with a 0.1 nM tracer. With a 0.1 nM tracer, the P value increased with the decrease in analyte concentration in the lower concentration range (0.0016–0.026 nM). In addition, the P of all data points increased. This may result from two types of interference: nonspecific adsorption of the tracer to the blocking agent and the PDMS surface, which is insufficiently covered by the blocking agent. Further experiments (Fig. S4†) indicated that nonspecific adsorption of the tracer to the blocking agent and BSA autofluorescence caused the increase of P. However, the adsorption of the tracer on the PDMS surface was not significant. As a result, the standard curve at a 0.1 nM tracer concentration could not be fitted by the four-parameter logistic function.
Fig. 4c shows the standard curve for FPIA using PEG-PDMS devices. A sigmoidal P increase was observed with both 0.1 nM and 1 nM tracers. Compared to the results for the PDMS device, there was no significant increase in the baseline of P for the 0.1 nM concentration of the tracer concentration. This also supports the notion that nonspecific adsorption to the surface was suppressed by PEG-PDMS. FPIA cannot be demonstrated in PDMS devices using a 0.1 nM tracer but can be successfully demonstrated using PEG-PDMS devices. Using a PEG-PDMS microfluidic device with a 0.1 nM tracer, we obtained an LOD of 0.25 nM which is lower than the LOD in a PDMS device (0.81 nM) and lower than or comparable to a previous report of FP assays in microfluidic devices (Table 1). These results demonstrate that the use of PEG-PDMS devices and the removal of blocking agents improve the sensitivity of FPIA and increase the applicability of FPIA to analyze biomarkers at low cut-off values.
Conclusions
In this study, we report an improvement in the sensitivity of an FP assay using PEG surface modification. The PEG-PDMS devices obtained through this simple modification method effectively inhibited the nonspecific adsorption of proteins. Using these PEG-PDMS devices, it is possible to demonstrate the FP assay at low tracer concentrations without the use of blocking agents and achieve a higher sensitivity.This study has significant implications for the quantitative analysis of low-abundance samples using FP assays. The improved sensitivity increases the potential of the FP assay for analyzing biomarkers at low cut-off values. This expands its application in clinical diagnostics and further enhances its importance for on-site analysis.
Author contributions
M.F. and A.H. conceived and designed the study. L.H. contributed to the data collection and analysis. M.F., A.H., and L.H. participated in discussions and provided constructive suggestions. M.F., A.H., and L.H. prepared initial drafts of the manuscript. Y.O., M.K., S.O., I.A., K.S., and M.T. critically reviewed and revised the manuscript. All the authors approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKEN-HI (Kiban A, 18H03912), the Strategic International Collaborative Research project promoted by the Ministry of Agriculture, Forestry and Fisheries, Tokyo, Japan, and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices (MEXT)”. Liu Hao acknowledges the GP-Chem program of Tohoku University.References
- D. M. Jameson and J. A. Ross, Chem. Rev., 2010, 110, 2685–2708 CrossRef CAS.
- M. C. Gutierrez, A. Gomez-Hens and D. Perez-Bendito, Talanta, 1989, 36, 1187–1201 CrossRef CAS.
- M. D. Hall, A. Yasgar, T. Peryea, J. C. Braisted, A. Jadhav, A. Simeonov and N. P. Coussens, Methods Appl. Fluoresc., 2016, 4, 022001 CrossRef.
- C. Vinegoni, P. F. Feruglio, I. Gryczynski, R. Mazitschek and R. Weissleder, Adv. Drug Delivery Rev., 2019, 151–152, 262–288 CrossRef CAS.
- W. A. Lea and A. Simeonov, Expert Opin. Drug Discovery, 2011, 6, 17–32 CrossRef CAS.
- G. Seymour Shephard, Chem. Soc. Rev., 2008, 37, 2468–2477 RSC.
- H. Zhang, S. Yang, K. De Ruyck, N. Beloglazova, S. A. Eremin, S. De Saeger, S. Zhang, J. Shen and Z. Wang, TrAC, Trends Anal. Chem., 2019, 114, 293–313 CrossRef CAS.
- C. Y. Lee, I. Degani, J. Cheong, R. Weissleder, J. H. Lee, J. Cheon and H. Lee, Acc. Chem. Res., 2021, 54, 3991–4000 CrossRef CAS.
- J. E. Adablah, Y. Wang, M. Donohue and M. G. Roper, Anal. Chem., 2020, 92, 8464–8471 CrossRef CAS.
- A. M. Schrell, N. Mukhitov and M. G. Roper, Anal. Chem., 2016, 88, 7910–7915 CrossRef CAS PubMed.
- L. F. Cheow, R. Viswanathan, C. S. Chin, N. Jennifer, R. C. Jones, E. Guccione, S. R. Quake and W. F. Burkholder, Anal. Chem., 2014, 86, 9901–9908 CrossRef CAS.
- S. Okada, Y. Muto, B. Zhu, H. Ueda and H. Nakamura, Anal. Chem., 2023, 95, 6198–6202 CrossRef CAS PubMed.
- F. Gelen, M. Butz, E. J. Rees, M. Erdelyi, T. Moschetti, M. Hyvonen, J. B. Edel, C. F. Kaminski and F. Hollfelder, Anal. Chem., 2017, 89, 1092–1101 CrossRef PubMed.
- J. W. Choi, D. K. Kang, H. Park, A. J. Demello and S. I. Chang, Anal. Chem., 2012, 84, 3849–3854 CrossRef CAS PubMed.
- X. Liu, H. Li, W. Jia, Z. Chen and D. Xu, Lab Chip, 2017, 17, 178–185 RSC.
- Y. Wang, D. I. Adeoye, Y. J. Wang and M. G. Roper, Anal. Chim. Acta, 2022, 1212, 339942 CrossRef CAS.
- V. K. Yadavalli and M. V. Pishko, Anal. Chim. Acta, 2004, 507, 123–128 CrossRef CAS.
- J. H. Kim, H. J. Shin, H. Cho, S. M. Kwak, H. Cho, T. S. Kim, J. Y. Kang and E. G. Yang, Anal. Chim. Acta, 2006, 577, 171–177 CrossRef CAS.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio De Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. Macmillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- J. Bajorath, Nat. Rev. Drug Discovery, 2002, 1, 882–894 CrossRef CAS.
- D. E. Ingber, Nat. Rev. Genet., 2022, 23, 467–491 CrossRef CAS PubMed.
- A. L. Glieberman, B. D. Pope, J. F. Zimmerman, Q. Liu, J. P. Ferrier, J. H. R. Kenty, A. M. Schrell, N. Mukhitov, K. L. Shores, A. B. Tepole, D. A. Melton, M. G. Roper and K. K. Parker, Lab Chip, 2019, 19, 2993–3010 RSC.
- O. Wakao, Y. Fujii, M. Maeki, A. Ishida, H. Tani, A. Hibara and M. Tokeshi, Anal. Chem., 2015, 87, 9647–9652 CrossRef CAS PubMed.
- A. Nakamura, M. Aoyagi, M. Fukuyama, M. Maeki, A. Ishida, H. Tani, K. Shigemura, A. Hibara and M. Tokeshi, ACS Food Sci. Technol., 2021, 1, 1623–1628 CrossRef CAS.
- K. Takahashi, S. Chida, T. Suwatthanarak, M. Iida, M. Zhang, M. Fukuyama, M. Maeki, A. Ishida, H. Tani, T. Yasui, Y. Baba, A. Hibara, M. Okochi and M. Tokeshi, Lab Chip, 2022, 22, 2971–2977 RSC.
- K. Nishiyama, M. Fukuyama, M. Maeki, A. Ishida, H. Tani, A. Hibara and M. Tokeshi, Sens. Actuators, B, 2021, 326, 128982 CrossRef CAS.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563–3576 CrossRef CAS PubMed.
- J. Zhou, D. A. Khodakov, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2012, 33, 89–104 CrossRef CAS PubMed.
- A. Fiene, Y. Baqi, J. Lecka, J. Sévigny and C. E. Müller, Analyst, 2015, 140, 140–148 RSC.
- E. A. Kumar, C. D. Charvet, G. L. Lokesh and A. Natarajan, Anal. Biochem., 2011, 411, 254–260 CrossRef CAS.
- C. I. Rogers, J. V. Pagaduan, G. P. Nordin and A. T. Woolley, Anal. Chem., 2011, 83, 6418–6425 CrossRef CAS.
- J. Liu, X. Sun and M. L. Lee, Anal. Chem., 2007, 79, 1926–1931 CrossRef CAS PubMed.
- M. Á. Sentandreu, L. Aubry, F. Toldrá and A. Ouali, Eur. Food Res. Technol., 2007, 224, 623–628 CrossRef CAS.
- W. Y. Craig, S. E. Poulin, M. F. Collins, T. B. Ledue and R. F. Ritchie, J. Immunol. Methods, 1993, 158, 67–76 CrossRef CAS PubMed.
- Y. L. Jeyachandran, E. Mielczarski, B. Rai and J. A. Mielczarski, Langmuir, 2009, 25, 11614–11620 CrossRef CAS PubMed.
- J. N. Klatt, T. Hutzenlaub, T. Subkowski, T. Müller, S. Hennig, R. Zengerle and N. Paust, ACS Appl. Polym. Mater., 2021, 3, 3278–3286 CrossRef CAS.
- Y. L. Jeyachandran, J. A. Mielczarski, E. Mielczarski and B. Rai, J. Colloid Interface Sci., 2010, 341, 136–142 CrossRef CAS PubMed.
- S. R. J. Hoare, SLAS Discovery, 2021, 26, 835–850 CrossRef CAS.
- M. Fukuyama, M. Tokeshi, M. A. Proskurnin and A. Hibara, Lab Chip, 2018, 18, 356–361 RSC.
- S. Sugiura, J. Edahiro, K. Sumaru and T. Kanamori, Colloids Surf., B, 2008, 63, 301–305 CrossRef CAS.
- H. P. Long, C. C. Lai and C. K. Chung, Surf. Coat. Technol., 2017, 320, 315–319 CrossRef CAS.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. De Gennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- O. Wakao, K. Satou, A. Nakamura, P. A. Galkina, K. Nishiyama, K. Sumiyoshi, F. Kurosawa, M. Maeki, A. Ishida, H. Tani, M. A. Proskurnin, K. Shigemura, A. Hibara and M. Tokeshi, Lab Chip, 2019, 19, 2581–2588 RSC.
- K. Nishiyama, Y. Takeda, M. Maeki, A. Ishida, H. Tani, K. Shigemura, A. Hibara, Y. Yonezawa, K. Imai, H. Ogawa and M. Tokeshi, Sens. Actuators, B, 2020, 316, 128160 CrossRef CAS PubMed.
- M. Takano, Y. Koyama, H. Nishikawa, T. Murakami and R. Yumoto, Eur. J. Pharmacol., 2004, 502, 149–155 CrossRef CAS.
- S. K. Kim, S. H. Hwang and H. B. Oh, BioChip J., 2016, 10, 346–353 CrossRef CAS.
- D. S. Smith and S. A. Eremin, Anal. Bioanal. Chem., 2008, 391, 1499–1507 CrossRef CAS PubMed.
- K. Nishiyama, K. Takahashi, M. Fukuyama, M. Kasuya, A. Imai, T. Usukura, N. Maishi, M. Maeki, A. Ishida, H. Tani, K. Hida, K. Shigemura, A. Hibara and M. Tokeshi, Biosens. Bioelectron., 2021, 190, 113414 CrossRef CAS PubMed.
- M. Fukuyama, A. Nakamura, K. Nishiyama, A. Imai, M. Tokeshi, K. Shigemura and A. Hibara, Anal. Chem., 2020, 92, 14393–14397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00569d |
| This journal is © The Royal Society of Chemistry 2024 |