
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Generation and reactivity of the fragment ion [B12I8S(CN)]− in the gas phase and on surfaces†
Sebastian
Kawa
 a,
Jaskiran
Kaur
b,
Harald
Knorke
a,
Ziyan
Warneke
a,
Myriam
Wadsack
a,
Markus
Rohdenburg
a,
Marc
Nierstenhöfer‡
c,
Carsten
Jenne
c,
Hilkka
Kenttämaa
b and
Jonas
Warneke
*ad
a,
Jaskiran
Kaur
b,
Harald
Knorke
a,
Ziyan
Warneke
a,
Myriam
Wadsack
a,
Markus
Rohdenburg
a,
Marc
Nierstenhöfer‡
c,
Carsten
Jenne
c,
Hilkka
Kenttämaa
b and
Jonas
Warneke
*ad
aWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstr. 2, 04103, Leipzig, Germany. E-mail: jonas.warneke@uni-leipzig.de
bDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA
cAnorganische Chemie, Fakultät für Mathematik und Naturwissenschaften, Bergische Universität Wuppertal, Gaußstr. 20, 42119, Wuppertal, Germany
dLeibniz Institute of Surface Engineering (IOM), Permoserstr. 15, 04318, Leipzig, Germany
First published on 29th February 2024
Abstract
Gaseous fragment ions generated in mass spectrometers may be employed as “building blocks” for the synthesis of novel molecules on surfaces using ion soft-landing. A fundamental understanding of the reactivity of the fragment ions is required to control bond formation of deposited fragments in surface layers. The fragment ion [B12X11]− (X = halogen) is formed by collision-induced dissociation (CID) from the precursor [B12X12]2− dianion. [B12X11]− is highly reactive and ion soft-landing experiments have shown that this ion binds to the alkyl chains of organic molecules on surfaces. In this work we investigate whether specific modifications of the precursor ion affect the chemical properties of the fragment ions to such an extent that attachment to functional groups of organic molecules on surfaces occurs and binding of alkyl chains is prevented. Therefore, a halogen substituent was replaced by a thiocyanate substituent. CID of the precursor [B12I11(SCN)]2− ion preferentially yields the fragment ion [B12I8S(CN)]−, which shows significantly altered reactivity compared to the fragment ions of [B12I12]2−. [B12I8S(CN)]− has a previously unknown structural element, wherein a sulfur atom bridges three boron atoms. Gas-phase reactions with different neutral reactants (cyclohexane, dimethyl sulfide, and dimethyl amine) accompanied by theoretical studies indicate that [B12I8S(CN)]− binds with higher selectivity to functional groups of organic molecules than fragment ions of [B12I12]2− (e.g., [B12I11]− and [B12I9]−). These findings were further confirmed by ion soft-landing experiments, which showed that [B12I8S(CN)]− ions attacked ester groups of adipates and phthalates, whereas [B12I11]− ions only bound to alkyl chains of the same reagents.
1 Introduction
Gas-phase fragmentation reactions in mass spectrometers frequently yield reactive ionic intermediates, which cannot be isolated in the condensed phases.1,2 Fragment ions (e.g., those generated by collision-induced dissociation (CID)) are of great analytical value but have attracted little attention as “building blocks” for the synthesis of new molecules. Gaseous ions are usually separated based on their mass-to-charge ratio, detected, and eventually discarded without further usage in an analytical mass spectrometer. However, exploiting such mass-selected ions for preparative purposes is enabled by the ion soft-landing technique.3–7Electrospray ionization (ESI)-coupled ion soft-landing can be utilized to deposit ionized, complex, non-volatile molecules or pre-charged ions onto surfaces.6–9 This approach was previously used for the deposition of, for example, intact quasi-molecular ions of biomolecules and nanoribbons.10–15 Pre-charged ions have been deposited in order to generate special electrode–electrolyte model interfaces,16 to prepare model-catalysts,17 to study self-organization processes in deposited layers,18 and resistive switching applications in molecular electronics.19 Within the last decade, new advanced ESI sources and improved ion soft-landing instruments have been developed,20–24 which significantly increased the usually low ion currents of CID-generated fragment ions.17 In recent years, experiments have involved deposition of fragment ions that then were bound to organic molecules on surfaces and were used to form covalent bonds between cluster ions of same polarity.25–28 Very recently, the reaction product of a fragment ion and gaseous N2 was accumulated on surfaces in amounts that were sufficient for conventional characterization of the new compound using infrared (IR) and nuclear magnetic resonance (NMR) spectroscopy.29 These recent developments extent the value of fragment ions beyond mere analytical applications towards small-scale chemical synthesis, in which fragment ions act as “building blocks” for molecules that are not accessible via conventional bulk- or solution-based methods. A catalogue of readily available fragment ions with well-understood reactivities and which can be obtained in high ion currents would distinctly broaden the possibilities of this unconventional synthetical approach that still is in its early infancy. The influence of precursor ion design on fragment ion generation and reactivity is essential to enable the rational selection of a fragment ion for tailored bond formation.
We have systematically explored fragmentation reactions of closo-borate anions [BnXn]2− (n = 6,10,11,12; X = halogen, CN) and the reactivity of their fragment ions both in the gas phase and after deposition on surfaces.25,27–40 Fragments of [BnXn]2− anions show exceptional binding properties: for example, [B12X11]− ions are highly electrophilic and bind noble gases even at room temperature.38 Also, [B6X5]− ions exhibit pronounced π-backbonding to reagents like N2, similar to transition metal complexes.39 In particular, CID of [B12I12]2− has been intensively explored and found to enable the formation of the full series of [B12In]− (n = 1–11) ions.30,35,41 Ion spectroscopy in the gas phase and ion soft-landing were employed to study the structures and the reactivities of these ions.25,27–29,36,38–40
The goal of the current study is to investigate whether and how the substitution of a single substituent X in the precursor ion [B12X12]2− will induce changes in the fragmentation behavior and reactivity of the fragment ions. Purposeful modifications of the precursor ions may allow control of chemical properties of the fragment ions, e.g., selectivity towards different reagents or functional groups. For this proof-of-concept study, we choose X = I, because multiple losses of iodine substituents (I˙ and [I]−) can be initiated by fragmentation without the decomposition of the B12 moiety. Therefore, studying the binding properties of [B12Xn]− (X = I) ions with different substitution levels n is possible. The precursor ion [B12I11(SCN)]2− was synthesized to study the chemical reactivity of the main fragment ions formed during CID compared to those of [B12I12]2−. The results show that the substitution of one iodine substituent by a thiocyanate group (–SCN) leads to the generation of previously unknown fragment ions with significantly different reactivities than those formed upon CID of [B12I12]2−. CID at moderate collision energies applied to [B12I12]2− leads to [B12I11]− that is highly reactive even towards alkanes. In contrast, CID at similar collision energies applied to [B12I11(SCN)]2− result in the formation of [B12I8SCN]− that demonstrates a more selective binding with functional groups of organic molecules. The reactivity of [B12I8SCN]− is further compared with the reactivity of [B12I9]− because both ions are formed by the loss of one [I]− and two I˙ from their respective precursors. Hybrid compounds containing closo-dodecaborate moieties and organic residues may find applications in medicine (boron neutron capture therapy,42 radiopharmaeuticals43) and materials sciences (non-linear optics, ionic liquids).44 The new “building blocks” investigated herein may be useful for the small-scale synthesis of new closo-dodecaborate-containing ions by using the fragment ion soft-landing approach.
2 Method section
The reagents iodine (99.8%, Roth), iodine monochloride (Alfa Aesar), tetraphenyl phosphonium chloride (abcr GmbH) and 1H,1H,2H,2H-perfluorodecanethiol (Sigma Aldrich) were purchased and used as received. Reagents for gas-phase reactions (cyclohexane, dimethyl sulfide, and dimethyl amine) were purchased from Sigma Aldrich and used as received.2.1 Synthetic details
K2[B12I12] was prepared from K2[B12H12] by a published method.45,46 The synthesis of the previously unknown Cs2[B12I11(SCN)] followed a related procedure. Dried Cs2[B12H11(SCN)] and iodine were dissolved in 1,2-dichloroethane (Alfa Aesar) in a pressure tube. The tube was closed, and the mixture was stirred for one hour at room temperature. Subsequently, the temperature was raised to 80 °C and the mixture was stirred for 90 min. After cooling to room temperature, the tube was carefully opened and an excess of iodine monochloride was added. The tube was closed again and heated to 180 °C for three days. The mixture was slowly cooled to room temperature. The residue was transferred onto a fine frit, washed with hexane (VWR), and dried in vacuo, which gave pink crude Cs2[B12I11(SCN)] suitable for gas-phase experiments. Full synthetic and spectroscopic details are given in the ESI section 1.† The molecular structure of [Cs2(NCCH3)2][B12I11(SCN)] was determined by single crystal X-ray diffraction. Details of the crystallographic data are summarized ESI section 1.† The supplementary crystallographic data for this structure has been deposited with the Cambridge Crystallographic Data Centre (CCDC number 2308756†).2.2 Mass spectrometry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ms and the kinetics of adduct formation were qualitatively compared for different reactant anions. The number of individual mass spectra averaged for the mass spectra was chosen dependent on the reaction time.
000 ms and the kinetics of adduct formation were qualitatively compared for different reactant anions. The number of individual mass spectra averaged for the mass spectra was chosen dependent on the reaction time.
Higher-energy collision-induced dissociation (HCD) experiments were performed using an Orbitrap Exploris 480 mass spectrometer equipped with an ESI source and a quadrupole mass filter and operated in the negative ion mode (Thermo Scientific, Bremen, Germany). K2[B12I12] and Cs2[B12I11(SCN)] salts were dissolved in acetonitrile (Chromasolv™ LC-MS, ≥99.9%, Honeywell) at a concentration of 10−7 to 10−8 mol L−1. The solutions were injected into the inlet by using a syringe pump at a flow rate of 5 μL min−1. An ESI spray voltage of −2.6 kV and an ion transfer tube temperature of 320 °C were used. Ions were mass-selected in a quadrupole mass filter using an isolation width of m/z 10, transferred into the HCD cell via a C-trap, and subjected to collisions with N2 gas in the HCD cell. Normalized collision energies are shown in manufacturer-specified arbitrary units. After dissociation, all ions were transferred back into the C-trap and then injected into the Orbitrap mass analyzer for detection. The scan range of the mass spectrometer was adjusted according to the mass of the fragments that were between m/z 40 to m/z 1700. All mass spectra acquired were an average of at least 10 individual mass spectra.
An external manifold for introducing reagent gases into the quadrupole ion trap via the helium line was attached to the LQIT mass spectrometer.48 The reagents were introduced into the manifold by using a syringe pump operating at a flow rate of 3–5 μL h−1. The reagent was diluted with helium at a flow rate of 500 μL min−1. A fraction of the diluted reagent-helium mixture (2 μL min−1) was introduced into the ion trap via a leak valve. The remaining fraction was directed into the exhaust. The nominal pressure within the ion trap region was 2.7 mbar, which corresponds primarily to the helium buffer gas. Using an isolation width of m/z 2 and a q value of 0.25, analyte ions were isolated in the ion trap by ejecting all unwanted ions from the trap. The isolated ions were allowed to react with the reagent for 30 ms at a q value of 0.25. For different analyte ions, the respective partial pressure of the reagent was kept constant. For CID experiments, the advanced scan feature of the LTQ Tune software interface was used to isolate the desired ion-molecule reaction product ions with isolation width varying between m/z 0.5 and m/z 2. The isolated ions were then subjected to CID at collision energies of 5–30 arbitrary units for 30 ms by using helium buffer gas as the collision gas. The detection mass range was from m/z 500 up to 2000. All mass spectra acquired were an average of at least 20 individual mass spectra.
2.3 Ion soft-landing experiments
:1 v/v) and subjected to a subsequent chip-based nano-ESI MS analysis (Spray voltage: −1.3 kV).
2.4 Computational chemistry
Geometric and electronic structure calculations, vibrational frequency analysis, 0 K attachment enthalpy calculations, and natural population analysis were performed using the Gaussian 16, rev. C.0150 software package using density functional theory (DFT) (B3LYP51–53/def2-TZVPP54,55 with GD3BJ dispersion correction56,57). 0 K attachment enthalpies (ΔH0 K) were calculated as the differences between electronic energies of the product anion ([B12X11Y]−) and the separated reagents ([B12X11]− and Y). They were corrected for zero-point energy (ZPE) and basis set superposition error (BSSE) by using the counterpoise method.58,59
3 Results and discussion
3.1 Synthesis of Cs2[B12I11(SCN)]
The non-halogenated precursor [B12H11(SCN)]2− was reported for the first time by Preetz in 1984.60 They attached the thiocyanate group onto the boron cluster [B12H12]2− by the reaction with dirhodan (SCN)2.60 Later on, a new synthesis avoiding the difficult handling of (SCN)2 was developed.61 The reagent dirhodan was generated in situ using potassium thiocyanate and copper(II)chloride in water as the solvent to give [B12H11(SCN)]2− and [B12H10(SCN)2]2− in good yields and purities.61 In this work, we used the latter method and removed the twofold-substituted side product [B12H10(SCN)2]2− by fractional crystallization from hot ethanol. Since halogenated [B12I11(SCN)]2− was needed for the gas-phase experiments, we decided to perform an iodination reaction, applying reaction conditions also suitable for the synthesis of [B12I12]2−. Indeed, iodination with a mixture of iodine and iodine monochloride in 1,2-dichloroethane at higher temperature and pressure led to the replacement of all hydrogen atoms by iodine atoms. The thiocyanate group (–SCN) was mainly retained but partly oxidized to the novel thioiodine group (–SI), which could not be separated from the main product. However, this impurity was acceptable as the desired ions were isolated from all other ions in the following gas-phase experiments. Cs2[B12I11(SCN)] was characterized by heteronuclear NMR spectroscopy, vibrational spectroscopy (IR and Raman), mass spectrometry, and single crystal X-Ray diffraction. Full details are presented in the ESI section 1.†3.2 Comparing CID of [B12I12]2− and [B12I11(SCN)]2− in the gas phase
The two pre-charged precursors [B12I12]2− and [B12I11(SCN)]2− were transferred into the instrument via ESI, mass-selected, and fragmented using CID in an HCD cell. The collision energy-dependent CID MS2 spectra are compared in Fig. 1. As it has been observed in previous studies for [B12I12]2−,30 successive losses of iodine substituents occurred. [B12I11(SCN)]2− showed the same losses of I˙ and [I]− as observed for [B12I12]2−, but the relative abundances of the fragment ions were very different as shown in Fig. 1a and b. The fragment ion [B12I8SCN]− dominated the CID MS2 spectra of [B12I11(SCN)]2− over a wide range of collision energies and was detected in high abundance. Therefore, this ion may be available in high ion currents for preparative mass spectrometry experiments. The ion [B12I9]−, formed in an analogous manner by the loss of two I˙ and one [I]− from [B12I12]2−, was also observed. However, the normalized collision energy required to form [B12I9]− was higher than that required to form [B12I8SCN]−. Several other ions with similar abundances were also detected in these mass spectra, either precursor ions (such as [B12I10]˙2−/−) at lower normalized collision energies or smaller fragment ions (such as [B12I8]˙− and [B12I7]−) at higher normalized collision energies. The selective formation of [B12I8SCN]− ions over a wide range of normalized collision energies suggests that [B12I8SCN]− ions constitute a special “energetic sink” in the sequential loss of iodine substituents from [B12I11(SCN)]− ions due to the presence of the SCN group.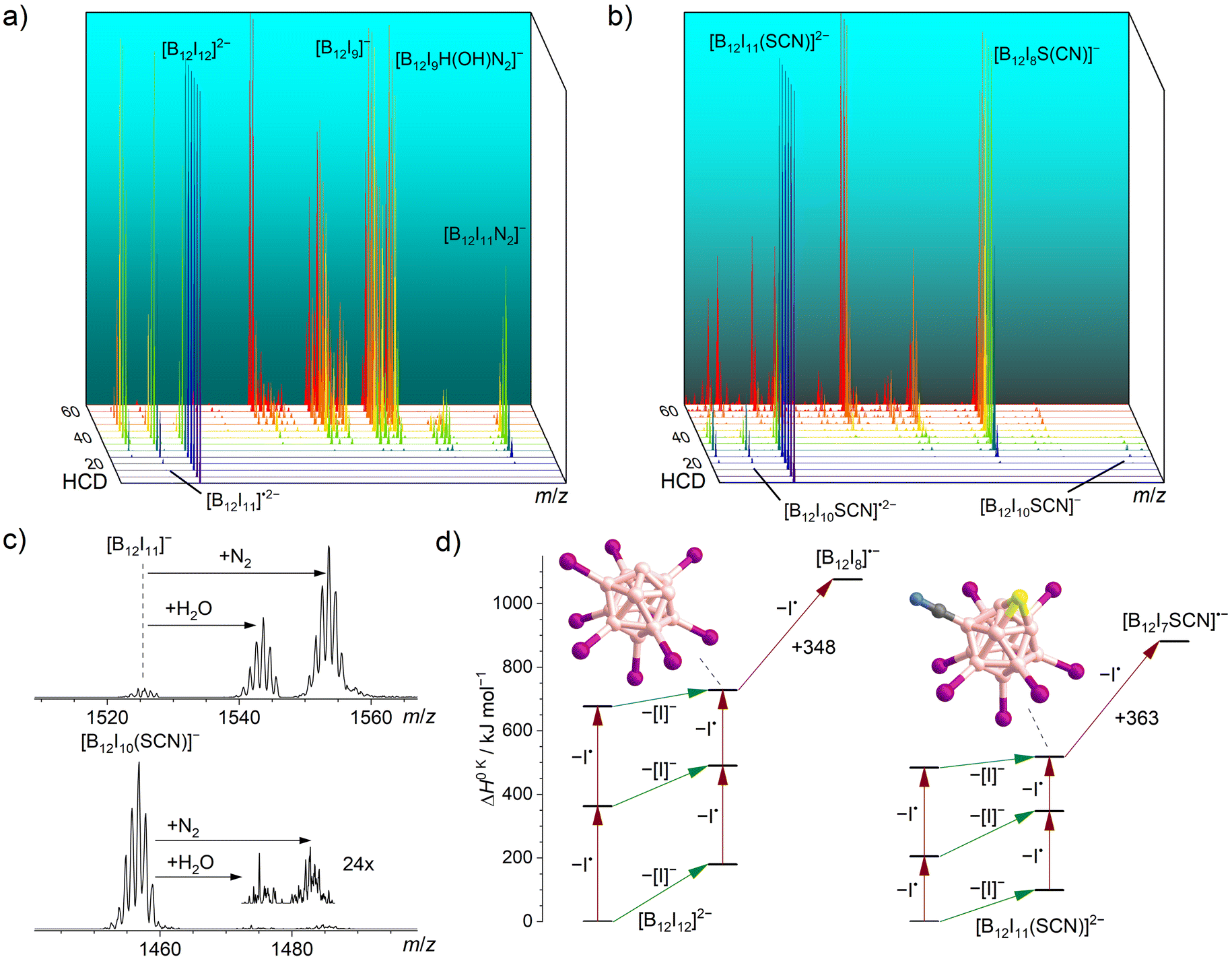 | ||
| Fig. 1 HCD mass spectra of isolated (a) [B12I12]2− (m/z 826) and (b) [B12I11(SCN)]2− (m/z 792) ions at different normalized collision energies (arbitrary units). Energy-resolved relative abundances of the generated fragment ions are shown in the ESI Fig. S9.† (c) Isolation of [B12I11]− (top) and [B12I10SCN]− (bottom) in an ion trap in which N2 and H2O are present as background gases (isolation time: 60 ms). (d) Calculated relative 0 K enthalpies ΔH0 K for stripping of I˙ (red arrows) or [I]− (green arrows) from [B12I12]2− (left) and [B12I11(SCN)]2− (right) and the proposed structures of [B12I9]− (left) and [B12I8S(CN)]− (right). | ||
In order to shed more light on the differences between the formation of the [B12I9]− and [B12I8SCN]− ions, we evaluated the individual I˙ and [I]− losses from the precursors. [B12I11]− ions possess one vacant boron atom and react with residual N2 in the HCD cell. The product [B12I11(N2)]− was observed as the ion with the largest m/z ratio in the CID mass spectra of [B12I12]2− ions. Calculated 0 K enthalpies (ΔH0 K) of N2 attachment towards [B12I11]− and [B12I10SCN]− ions are almost equal. (Table S4†). However, the abundance ratio [B12I10(SCN)(N2)]−/[B12I10SCN]− was much smaller than the abundance ratio [B12I11(N2)]−/[B12I11]−.
Experiments were performed in the ion trap to gain more insight into the reactivity of [B12I11]− ions compared to that of [B12I10SCN]− ions. The reaction time of the ions in the ion trap was systematically varied to observe their time-dependent reactions with residual gases. The kinetics of the reactions of [B12I11]− ions with H2O and N2 showed a typical (quasi-)first order reaction kinetics (Fig. S10†). At a reaction time of 60 ms, most of the [B12I11]− ions had already reacted with H2O and N2 (Fig. 1c) and longer isolation times led to an almost complete depletion of the signal of the unbound [B12I11]− ions. In contrast, only a small fraction of the [B12I10SCN]− ions had bound N2 at a reaction time of 60 ms (Fig. 1c), and increasing the isolation time even up to seconds did not result in a significant depletion of the [B12I10SCN]− abundace. Therefore, only a minor fraction of the [B12I10SCN]− ions contain a vacant boron atom as in [B12I11]−. DFT results indicate that [B12I10SCN]− ions can be stabilized by an intrinsic rearrangement of the SCN substituent in which the vacant boron atom becomes occupied, explaining their lack of reactivity towards the background gases. The most stable isomer found in the computational investigations is formed by splitting of the SCN group (Scheme 1) into a sulfur and a CN substituent. Therefore, we denote the fragment ion in the following as [B12I10S(CN)]−. Note that we cannot rule out the presence of other isomers in the experiment. Table S5† lists all identified isomers and their calculated relative energies. The exothermic intramolecular reaction following the loss of an [I]− increases the internal energy of the ion vibrational modes, which promotes secondary fragmentation and explains the overall low abundance of [B12I10S(CN)]−. Further stripping of I˙ in the next fragmentation steps results in [B12I9S(CN)]˙− and [B12I8S(CN)]− ions containing a sulfur atom that bridges two and three boron atoms, respectively (Scheme 1 and Fig. 1d). Other, less stable isomers containing eight iodine substituents are shown in Table S6.†
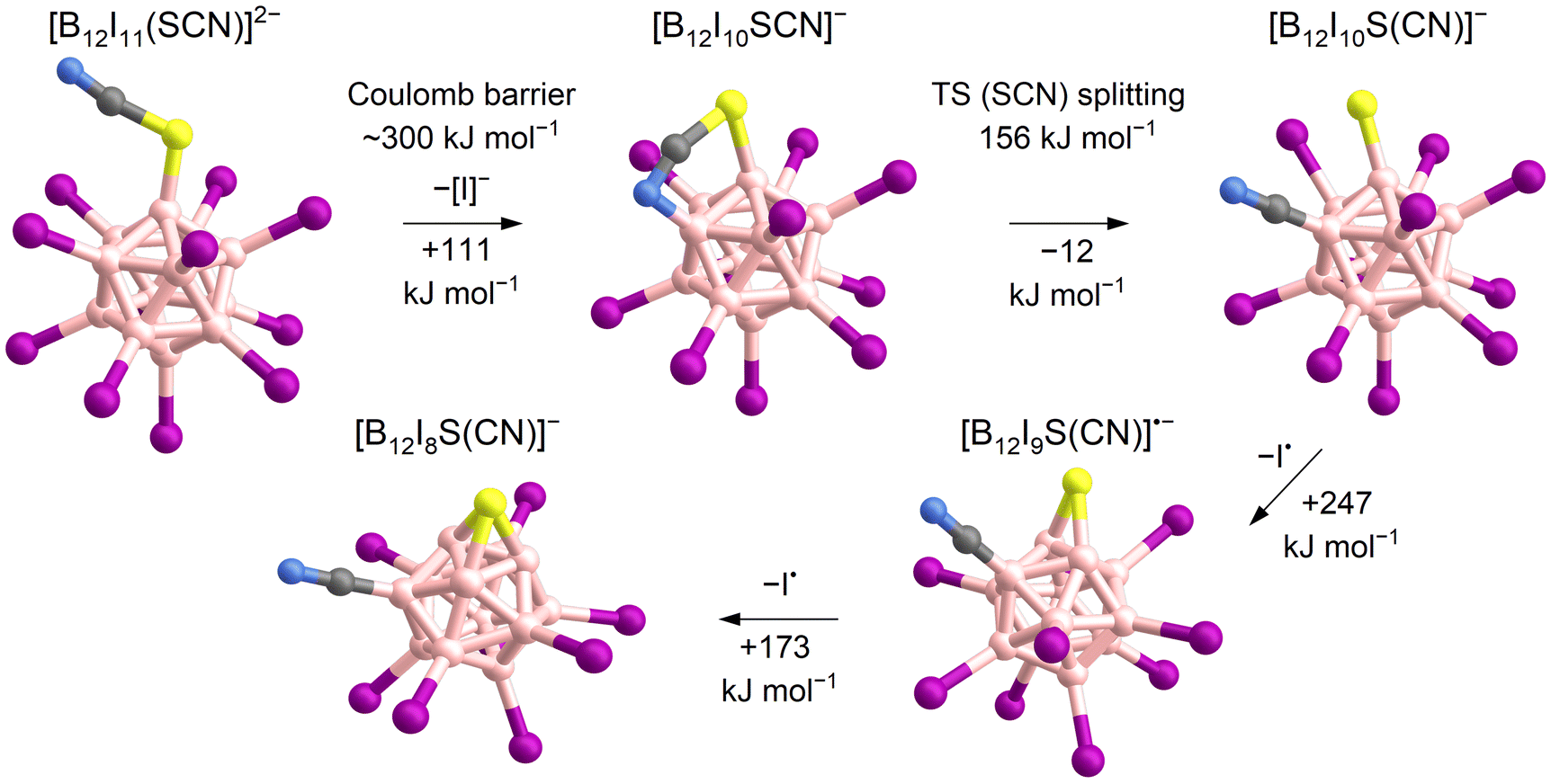 | ||
| Scheme 1 Proposed mechanism for the formation of [B12I8S(CN)]− ions based on calculated ΔH0 K. The loss of [I]− requires overcoming the Coulomb barrier (first step).62,63 A transition state (TS) was found for the splitting of the SCN-group (second step). Note that other isomers of [B12I10SCN]− also exist, as shown in Table S5.† | ||
Similar to the loss of [I]− in the first fragmentation step described above, a rearrangement of the SCN group is also expected when I˙ is eliminated from [B12I11SCN]2−. Therefore, [B12I11]˙2− and [B12I10SCN]˙2− ions were accumulated in the ion trap to examine their reactions with residual O2. Reactions with O2 were previously identified to be a characteristic reaction of dodecaborate radical anions with one vacant boron atom.37 While prolonged reaction times resulted in an almost complete conversion of [B12I11]˙2− to [B12I11(O2)]˙2−, only some of the [B12I10SCN]˙2− ions reacted with O2, pointing towards an intramolecular rearrangement in this radical anion, similar to [B12I10SCN]− (Fig. S10†).
Fig. 1d shows the calculated DFT electronic energies of the energetically lowest lying isomers of the observed fragment ions formed by the first three I˙/[I]− losses. Note that the competition of I˙ and [I]− loss in the first fragmentation steps cannot be estimated from the enthalpy differences shown in Fig. 1d (green vs. red arrows), because a repulsive Coulomb barrier is present for the loss of the [I]− from a dianion.62,63 The formation of [B12I8S(CN)]− ions require less ΔH0 K than the formation of [B12I9]− ions (518 kJ mol−1vs. 728 kJ mol−1) and further fragmentation by I˙ loss requires more ΔH0 K than in the case of [B12I9]− ions (363 kJ mol−1vs. 348 kJ mol−1), rationalizing the observed differences for the formation and further fragmentation of [B12I9]− and [B12I8S(CN)]− ions (Fig. 1a and b).
The proposed structure of [B12I8S(CN)]− shown in Fig. 1d and Scheme 1 constitutes a fully substituted B12 unit without vacant boron atoms. The proposed structure is in agreement with the reactivity of this ion: in contrast to [B12I9]−, no reactions with background gases were observed in MSn experiments in ion traps. Results of a study with other reagents than N2, H2O and O2 are shown in section 3.3 (vide infra). Further excitation of [B12I8S(CN)]− ions results in the formation of the radical anion [B12I7S(CN)]˙− ions, which spontaneously bind O2 in the ion trap (Fig. S11†). Since the spontaneous binding of O2 is characteristic for dodecaborate radical anions with one vacant boron atom, this observation indirectly supports the full boron substitution level proposed for [B12I8S(CN)]−. In contrast, [B12I8]˙− ions form multiple adducts with H2O (Fig. S12†).
3.3 Reactivity of [B12I8S(CN)]− compared to [B12I11]− and [B12I9]− in the gas phase
Considering that [B12I8S(CN)]− does not react with background gases, consistent with the proposed structure, we also expected its reactivity to differ from that of [B12I11]− and [B12I9]− towards other reagents. Although both the [B12I8S(CN)]− and [B12I9]− ions are the result of a loss of two I˙ and one [I]− from their respective precursor dianions, [B12I9]− contains three vacant boron atoms, while the proposed structure of [B12I8S(CN)]− does not contain any vacant boron atoms. A sulfur atom that bridges three boron atoms is a previously unreported functional group and, therefore, its reactivity needs to be evaluated. Atomic charges derived from natural population analysis (NPA) are negative for the boron atoms bridged by the sulfur atom in [B12I8S(CN)]− but positive for the vacant boron atoms in [B12I11]− and [B12I9]− (Table S7†). Consistently, the lowest unoccupied molecular orbital (LUMO) is higher in energy for [B12I8S(CN)]− than for [B12I11]− and [B12I9]− (Fig. S6†). These computational results suggest that [B12I8S(CN)]− should have reduced electrophilicity compared to the other investigated ions. The reduced electrophilicity may enhance the selectivity of this ion towards different functional groups. Consequently, the ions [B12I8S(CN)]−, [B12I9]− and [B12I11]− were isolated in the ion trap and their reactivity towards residual gas and selected neutral reagents was probed.The reagents cyclohexane (84.1 Da), dimethyl sulfide (62.0 Da), and dimethyl amine (45.1 Da) were selected to investigate the reactivity of the ions of interest towards alkanes with no functionalities and a few functional groups typically found in organic compounds (–SR and –NR2). Mass spectra measured after the introduction of cyclohexane, dimethyl sulfide, and dimethyl amine into the ion trap containing [B12I11]−, [B12I9]−, and [B12I8S(CN)]−, respectively, are shown in Fig. 2 (cyclohexane), Fig. 3 (dimethyl sulfide), and Fig. S22† (dimethyl amine). Reaction products with the background gases that were also observed without the introduction of an additional neutral reagent are marked with orange color, and the products formed upon reactions with the introduced neutral reagent are marked with blue color. As mentioned above, [B12I11]− and [B12I9]− ions are highly reactive toward residual gases within mass spectrometers. Despite the introduction of the selected reagents into the ion trap, reactions between these ions and H2O and N2 were observed (Fig. 2 and 3). It should be noted that binding of residual N2 is characteristic for [B12X11]− anions that possess only one vacant boron atom (see literature on “superelectrophilic” anions).38 Binding of residual N2 was not observed for the other ions in the [B12In]− series (n = 1–10).30 Consistently, [B12I9]− ions bind to H2O but not to N2 (Fig. 2b). The bound H2O molecule dissociates into a hydrogen and an OH substituent, each of which occupies a vacant boron site, resulting in [B12I9(OH)H]− ([B12X11]− configuration) with only one vacant boron site.30 Subsequently, this ion binds to H2O and N2. The reaction enthalpy of the first H2O attachment to [B12I9]− is sufficient to overcome the barrier for the elimination of HI. A substantial part of the [B12I9(OH)H]− ions fragment in this manner, resulting in [B12I8(OH)]−. This ion again contains three vacant boron atoms and undergoes the same reaction sequence as observed for [B12I9]− (Fig. S24†).
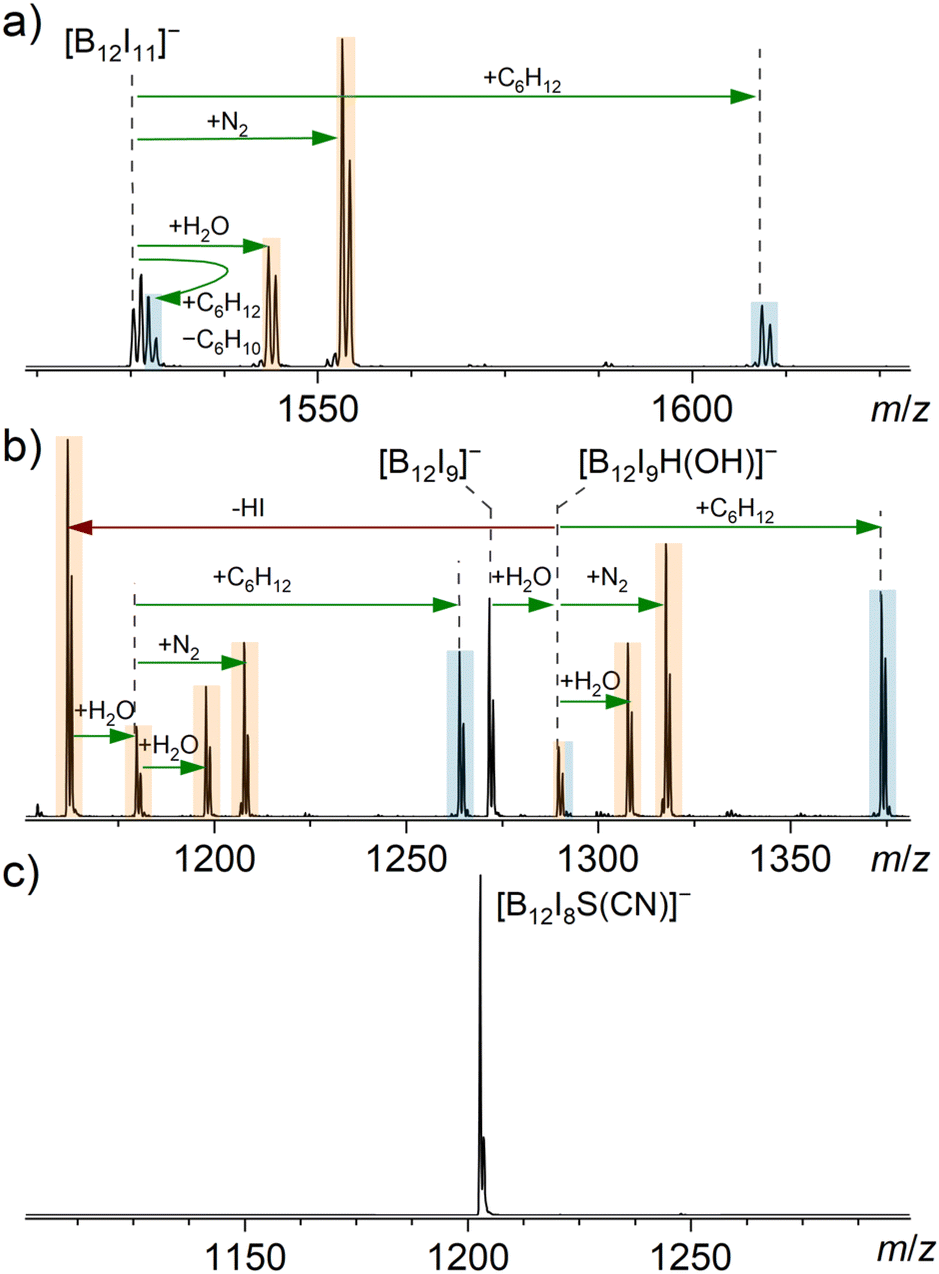 | ||
| Fig. 2 MS2 spectra obtained for the isolated (a) [B12I11]−, (b) [B12I9]− and (c) [B12I8S(CN)]− anions (isolation width m/z 2) after 30 ms reactions with cyclohexane in an ion trap. The ion trap also contained the ubiquitous residual gases H2O and N2. Binding of a reagent/residual gas (green arrows) and fragmentation reactions (red arrows) were individually verified by MSn experiments (Fig. S13–S16†). Signals assigned to reaction products resulting from reactions with cyclohexane are highlighted in blue, while signals assigned to reaction products resulting from reactions with residual gas are highlighted in orange. | ||
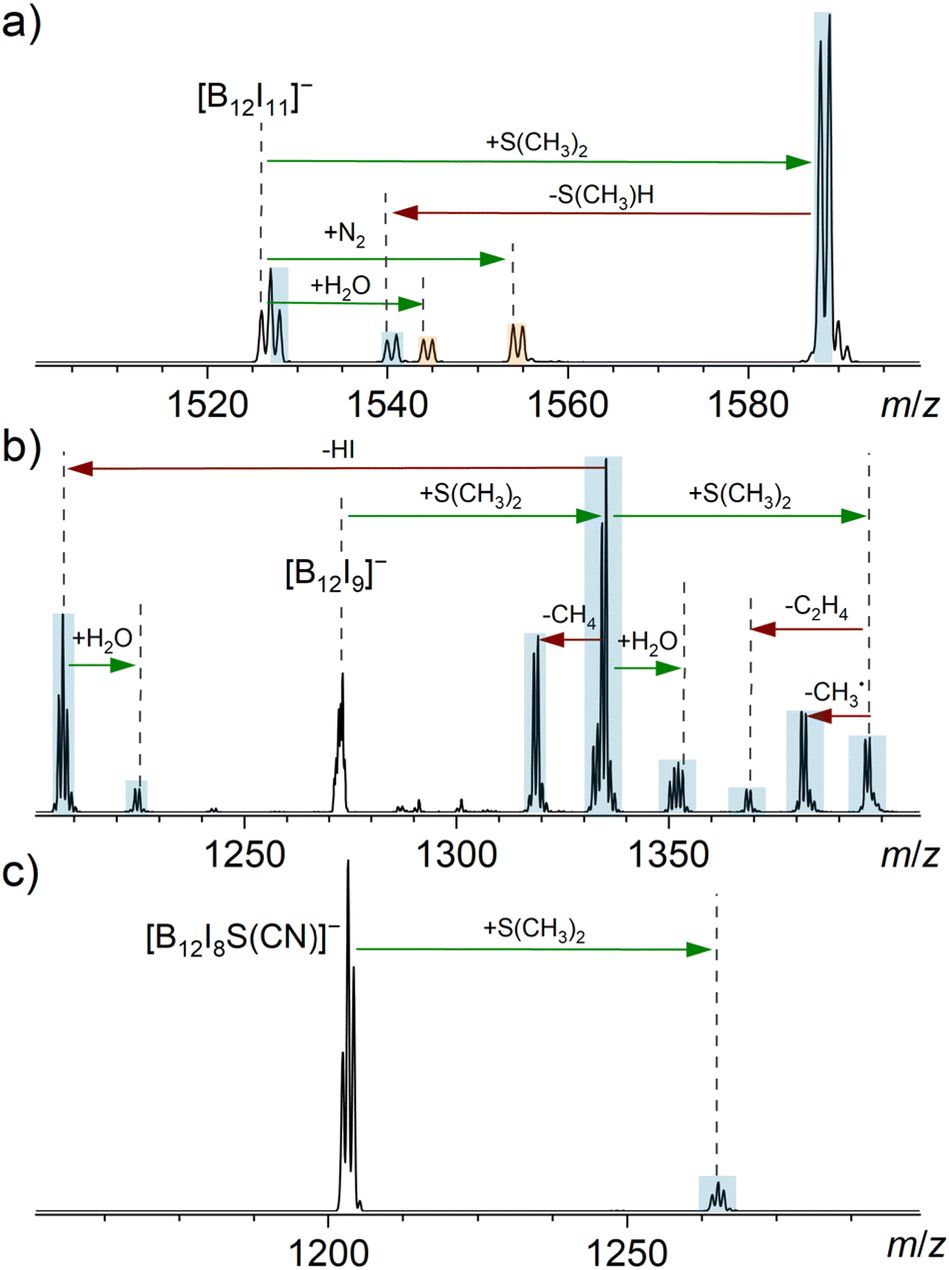 | ||
| Fig. 3 MS2 spectra obtained for the isolated anions (a) [B12I11]−, (b) [B12I9]−, and (c) [B12I8S(CN)]− (isolation width m/z 2) after 30 ms reactions with dimethyl sulfide in an ion trap. The ion trap also contained the ubiquitous residual gases H2O and N2. Binding of a reagent/residual gas (green arrows) and fragmentation reactions (red arrows) were individually verified by MSn experiments (Fig. S17–S20†). Signals assigned to reaction products resulting from reactions with cyclohexane are highlighted in blue, while signals assigned to reaction products resulting from reactions with residual gas are highlighted in orange. Note that [B12I9]− had completely reacted in (b) and the doubly charged isotopic pattern of [B24I18]2− became visible. [B24I18]2− likely originates from a synthesis side product or a fragment thereof. | ||
In addition to the reaction products described above, we have previously reported that [B12X11]− ions (X = Cl, Br) react with alkanes.28,40 Experimental evidence for binding to cyclohexane (C6H12) for X = I is shown in Fig. 2a. The stable adduct [B12I11(C6H12)]− is predominantly formed by B–C bond formation.40 The second product [B12X11H]2−[H]+, observed two m/z units above the isolated precursor ion, is the result of a two-step reaction: (i) hydride abstraction from cyclohexane yields [B12I11H]2−[C6H11]+ containing a carbocation and (ii) deprotonation of the cation and elimination of cyclohexene yields [B12I11H]2−[H]+. Formation of [B12X11H]2−[H]+ ions has been observed for reactions of [B12X11]− with different alkanes and has also been observed for other neutral reagents (vide infra).40 The relative abundance of this product ion may be strongly affected by the stability of the cation formed upon hydride abstraction from the neutral reagent, and the ability of the formed cation to further transfer a proton to the anion.
No reaction of cyclohexane with [B12I9]− was observed. Apparently, [B12I9]− is not electrophilic enough to abstract a hydride ion from an alkane, as also indicated by the less positive NPA charges of the three vacant boron atoms compared to the vacant boron in [B12I11]− (Table S7†). However, after binding the first H2O molecule, the [B12I9H(OH)]− ion reacts with cyclohexane, as expected for a [B12X11]− ion (Fig. 2b). In contrast, [B12I8S(CN)]− did not undergo any reactions with residual gases or cyclohexane (Fig. 2c), even when the ions were stored in the ion trap for up to ten seconds.
Introducing dimethyl sulfide (S(CH3)2) into the ion trap showed that [B12I11]− binds this molecule, forming [B12I11(S(CH3)2)]− (Fig. 3a). Also [B12I11H]˙− was observed, which may be explained by hydride abstraction from S(CH3)2 and a subsequent electron transfer, resulting in the elimination of CH3–S–CH2˙ radical. The loss of this radical is energetically preferred over the deprotonation and subsequent elimination of C2H4S as shown by DFT calculations (Fig. S7†), which explains the difference to the cyclohexane experiment.
Dimethyl sulfide is a more nucleophilic reagent than cyclohexane. In contrast to cyclohexane, S(CH3)2 binds directly to [B12I9]− and the addition of a second sulfide molecule was observed. This second addition indicates that the B12-unit was not fully substituted after the first sulfide addition. Product ions formed by binding dimethyl sulfide were observed to fragment by elimination of, for example, CH4 and HI.
The addition of only one S(CH3)2 molecule was observed for [B12I8S(CN)]−. This product formation demonstrates that the proposed, novel sulfur-bridging functional group in [B12I8S(CN)]− is not inert towards the sulfide. The reaction product is stable and contains most likely a fully substituted B12-unit, because no further addition reactions were observed. The binding of S(CH3)2 is much slower for [B12I8S(CN)]− than in the case of [B12I11]−. Experimental data with longer reaction times are shown in Fig. S21.†
It should be noted that the examination of the structures of the dimethyl sulfide adducts of [B12I8S(CN)]− and [B12I9]− is beyond the scope of the present study, which aims to show the difference in the reactivity of these ions. The variety of ways in which dimethyl sulfide can bind to multiple boron atoms (and a sulfur atom in the case of [B12I8S(CN)]−) and different possibilities to dissociate after binding makes the computational search for the lowest-lying isomers of these products challenging.
The introduction of dimethyl amine (NH(CH3)2) into the ion trap resulted in a different reaction behavior. Direct addition of dimethyl amine without fragmentation was found to be slower than the competing B–H bond formation based on the measured product ion mass spectra for both [B12I11]− and [B12I9]−. For [B12I11]−, the formation of [B12I11H]2−[H]+ dominates the spectrum. The high tendency towards hydride abstraction may be explained by the high stability of the resulting iminium ion [CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH–CH3]+, which can transfer a proton to yield [B12I11H]2−[H]+. [B12I11H]2−[H]+ acts as a Brønsted acid.64 Due to the comparatively high basicity of dimethyl amine, protonation of the amine occurs to form the adduct [B12I11H]2−[H2N(CH3)2]+ (Fig. S22a†). While [B12I11N2]− does not undergo any further reactions, the Brønsted acidity of [B12I11(H2O)]− enables binding to an amine molecule. The hydride abstraction/protonation reaction sequence is investigated by DFT calculations (Fig. S8†). For [B12I9]−, in addition to direct binding, the formation of [B12I9H3]2−[H]+ was observed in the presence of the amine. This observation is contrary to the results obtained for the reactions of cyclohexane and dimethyl sulfide with [B12I9]− (Fig. S25†). The greater stability of the [CH2NH–CH3]+ iminium ion compared to that of [C6H11]+ or [CH2SCH3]+ apparently enables the hydride abstraction from the amine. A subsequent proton transfer results in the formation of [B12I9H2]− ([B12X11]− configuration). The reaction with a second amine molecule leads to the formation of [B12I9H3]2−[H]+ ([B12X12]2−[H]+ configuration). Due to the overlapping isotopic patterns of reaction products of various [B12X9]−, [B12X11]−, and [B12X12]2−[H]+ ions (X = I, H, OH) with residual gas and with the amine, the mass spectra are very complex (Fig. S22b†). In contrast, selective binding to NH(CH3)2 without any side reactions was observed in the case of [B12I8S(CN)]− (Fig. S22c†). The reduced electrophilicity of [B12I8S(CN)]− compared to [B12I9]− and [B12I11]− is likely responsible for binding solely to the nucleophilic nitrogen of the amine.
NH–CH3]+, which can transfer a proton to yield [B12I11H]2−[H]+. [B12I11H]2−[H]+ acts as a Brønsted acid.64 Due to the comparatively high basicity of dimethyl amine, protonation of the amine occurs to form the adduct [B12I11H]2−[H2N(CH3)2]+ (Fig. S22a†). While [B12I11N2]− does not undergo any further reactions, the Brønsted acidity of [B12I11(H2O)]− enables binding to an amine molecule. The hydride abstraction/protonation reaction sequence is investigated by DFT calculations (Fig. S8†). For [B12I9]−, in addition to direct binding, the formation of [B12I9H3]2−[H]+ was observed in the presence of the amine. This observation is contrary to the results obtained for the reactions of cyclohexane and dimethyl sulfide with [B12I9]− (Fig. S25†). The greater stability of the [CH2NH–CH3]+ iminium ion compared to that of [C6H11]+ or [CH2SCH3]+ apparently enables the hydride abstraction from the amine. A subsequent proton transfer results in the formation of [B12I9H2]− ([B12X11]− configuration). The reaction with a second amine molecule leads to the formation of [B12I9H3]2−[H]+ ([B12X12]2−[H]+ configuration). Due to the overlapping isotopic patterns of reaction products of various [B12X9]−, [B12X11]−, and [B12X12]2−[H]+ ions (X = I, H, OH) with residual gas and with the amine, the mass spectra are very complex (Fig. S22b†). In contrast, selective binding to NH(CH3)2 without any side reactions was observed in the case of [B12I8S(CN)]− (Fig. S22c†). The reduced electrophilicity of [B12I8S(CN)]− compared to [B12I9]− and [B12I11]− is likely responsible for binding solely to the nucleophilic nitrogen of the amine.
3.4 Reactivity of [B12I8S(CN)]− in molecular surface layers compared to [B12I11]− and [B12I9]−
Previous studies investigated the deposition of [B12X11]− ions in the presence of phthalates, common plasticizers that are known to be present in the background of high vacuum instruments.18,25,28 Although phthalates do not adsorb in significant amounts on the highly repellent FSAM surfaces used in this study, these molecules have been found to accumulate on the surface in significant amounts with pre-charged anions during ion soft-landing experiments,18,28 if no charge balancing counterions were provided.29 Surprisingly, it has been found that [B12X11]− ions react with the phthalate alkyl chains, although binding to the ester groups is thermochemically preferred.28 It has been hypothesized that the organic molecules orient at the layer-vacuum interface, exposing the non-polar alkyl groups towards the vacuum. Highly reactive [B12X11]− ions are able to bind these alkyl chains and may directly react upon landing on the surface. Our gas-phase experiments described in section 3.3 indicate that different reaction behavior can be expected for [B12I9]− and [B12I8S(CN)]−. Since phthalate molecules contain ester groups and alkyl chains, their presence on the surface enables us to test whether the conclusions of section 3.3 also apply to reactions on the surface in the condensed phase. [B12I9]− and [B12I8S(CN)]− ions may react preferentially with the ester groups because they should be unreactive towards the hydrocarbon chains. Based on our gas-phase experiments, non-specific reactions of [B12I9]− are expected. In contrast, a more selective binding of the functional groups is expected in the case of [B12I8S(CN)]−. Ion soft-landing experiments were performed at background pressures of 10−6 mbar and therefore background gases including H2O were present and an adsorbate layer was formed on the surface. An ion soft-landing experiment with [B12I9]− was reported previously, but the full product spectrum was not analyzed and only the occupation of vacant boron atoms with hydrogen and OH substituents was discussed.27[B12I9]−, [B12I8S(CN)]−, and for comparison, [B12I11]− ions, were deposited via ion soft-landing in amounts of 52 pmol (3.1 × 1013 ions), 155 pmol (9.3 × 1013 ions), and 52 pmol, respectively, on gold surfaces covered with a fluorinated self-assembled monolayer (FSAM) (section 2.3). The deposited material was analyzed using LESA-MS. Sections of the resulting mass spectra are shown in Fig. 4. In all spectra, multiply charged ions were observed. In this instrument, the organic molecules which accumulate with pre-charged anions at the surface were identified with LESA-MS in the positive-ion mode. Abundant signals for ions of m/z 441.3 and m/z 393.3 can be assigned to sodium adducts of dinonyl phthalate (C6H4(COOC9H19)2) and dioctyl adipate (C4H8(COOC8H17)2), respectively, or isomers thereof (ESI Fig. S26†).
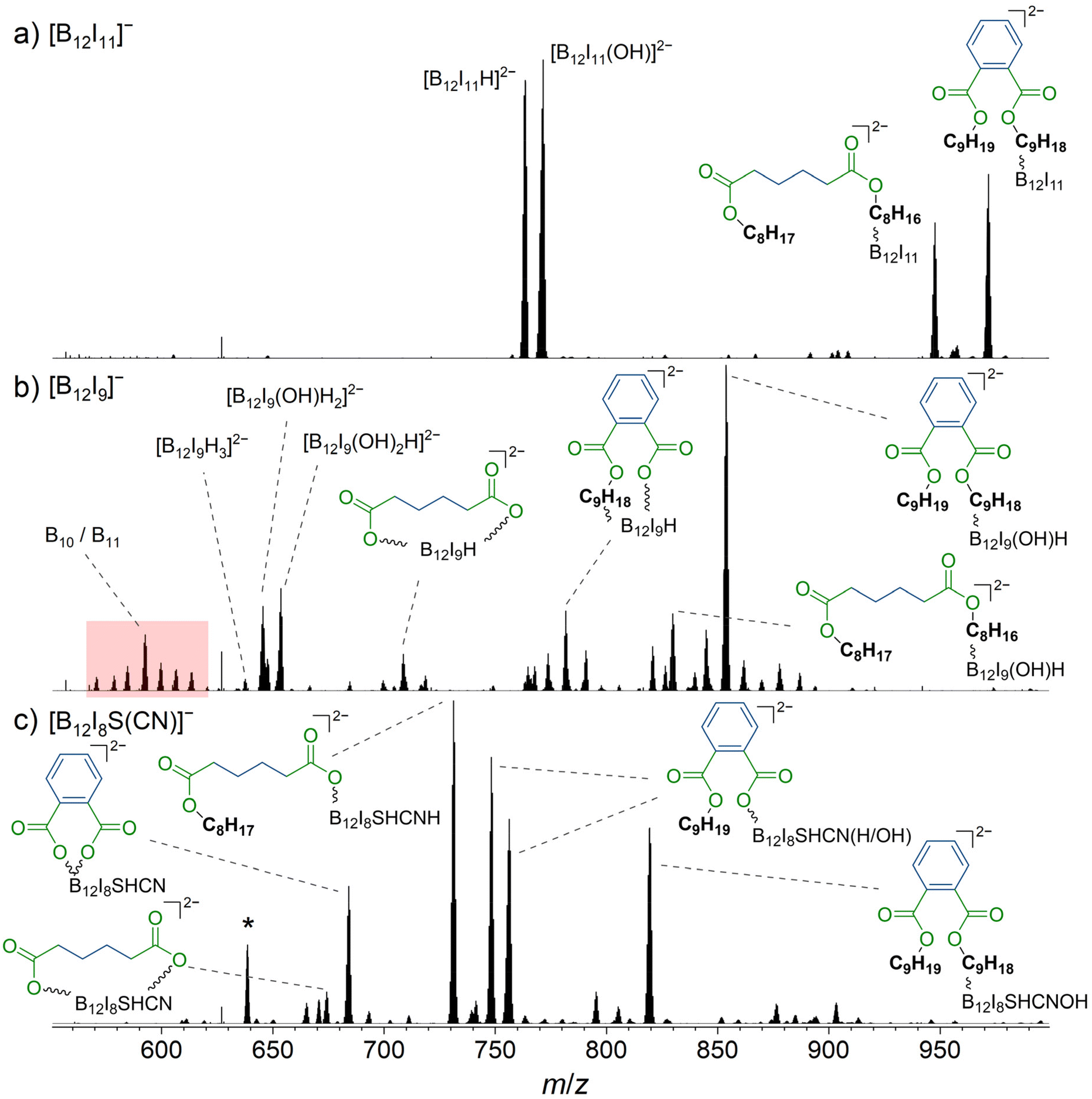 | ||
| Fig. 4 Mass spectra obtained by LESA-MS after the deposition of (a) [B12I11]−, (b) [B12I9]−, and (c) [B12I8S(CN)]− on gold covered with FSAM. Reaction products with dinonyl phthalate and dioctyl adipate are marked with a blue organic backbone. The ester moiety is shown in green. In (b), signals assigned to fragments with m/z values smaller than that of [B12I9H3]2− are highlighted in red, indicating degradation of the B12-scaffold. The isotopic pattern of the fourfold negatively charged [(B12I8(SH)(CN)H)2(C4H8(COO)2)]4− is marked with an asterisk and additionally shown enlarged in Fig. 5. More detailed signal assignments are shown in Table S8.† | ||
The deposition of [B12I11]− (Fig. 4a) led to the formation of previously reported [B12I11H]2− (m/z 763.5),27 likely formed by hydride abstraction from an organic background molecule, and [B12I11(OH)]2− (m/z 771.5) formed by the reaction with H2O. [B12I11(C4H8(COOC8H17)(COOC8H16))]2− (m/z 947.7) and [B12I11(C6H4(COOC9H19)(COOC9H18))]2− (m/z 971.7) were formed by the reaction with dioctyl adipate and dinonyl phthalate, respectively, which are known to be organic background molecules in the instrument used.27 In order to understand why the observed products were doubly charged although the reactants were a singly charged [B12I11]− ion and a neutral organic molecule, it is noteworthy that the highly electrophilic, partially positively charged, vacant boron atom in [B12X11]− reacts by substituting a proton in organic C–H bonds.18,28,40 Substituting a [H]+ in an organic molecule with an anionic unit (formally bond formation between the carbanion formed by deprotonation and the electrophilic vacant boron atom) results in the formation of a dianion with a stable configuration of [B12X12]2− as a closo-structure. The results of CID MS2 experiments support the conclusion that binding occurs via the alkyl chain for both dioctyl adipate and dinonyl phthalate (Fig. S27 and S28†).
In contrast to [B12I11]−, occupying the vacant boron sites with hydrogen substituents is not a major process for [B12I9]− on the surface. [B12I9H3]2− (m/z 637.6) was only observed in low abundance in the LESA mass spectrum (Fig. 4b) after the deposition of [B12I9]−. Instead, [B12I9]− reacts with H2O to form [B12I9(OH)H]−, which then reacts similarly to [B12I11]− as previously observed in gas-phase experiments.30 [B12I9(OH)H]− binds dioctyl adipate and dinonyl phthalate via the alkyl chain, leading to the reaction products [B12I9(OH)H(C4H8(COOC8H17)(COOC8H16))]2− (m/z 829.8) and [B12I9(OH)H(C6H4(COOC9H19)(COOC9H18))]2− (m/z 853.8) (see Fig. S29 and S30† for CID MS2 experiments). Occupation of the vacant boron sites with hydrogen and OH substituents and subsequent binding of the organic molecules by substitution of a [H]+ constitutes the main reaction on surfaces for [B12I9]−. However, [B12I9]− also showed reaction products that apparently form by direct binding to the functional groups (ester groups) of the organic molecules and therefore result by the loss of alkyl chains and formation of B–O bonds, as in the case of [B12I9H(C4H8(COO)2)]2− (m/z 708.7) (for CID MS2 see Fig. S31†). Here, both ester groups were cleaved. Also, the cleavage of one ester group and the preservation of the other was observed, as for [B12I9H(C6H4(COO)(COOC9H18))]2− (m/z 781.7) (CID MS2 experiments are shown in Fig. S32†). To conclude, while [B12I11]− shows a strong selectivity towards binding the alkyl chains of the organic molecules accumulated on the surface, [B12I9]− reacts predominantly with H2O in the first step but can also attack the ester groups. This reactivity is in agreement with the observed reactivity in the gas phase discussed in section 3.3. A significant difference from the gas-phase results can be noted for the ions with smaller m/z values observed in the spectra. The dominant HI loss after binding H2O and S(CH3)2 observed in (Fig. 2b and 3b) was not observed on the surface (Fig. 4b). Instead, boron cage fragmentation was observed, as demonstrated by the formation of [B11I8(OH)3]2− (m/z 592.7) and [B10I8(OH)2]2− (m/z 578.7). The signals corresponding to these fragmentation reactions are marked in red in Fig. 4b. The kinetic energy of the landed ions was very similar for [B12I11]− and [B12I9]−, see ESI section 8† for details. Since boron cage fragmentation was observed only after deposition of [B12I9]− but not after deposition of [B12I11]−, we assume that [B12I11]− ions are more stable against “crash-landing” than [B12I9]− ions and that reaction products of deposited [B12I11]− ions are more resistant to subsequent fragmentation than those of deposited [B12I9]− ions. Fig. 4c shows the LESA mass spectrum obtained after the soft-landing of [B12I8S(CN)]−. The most abundant signals correspond to reaction products in which at least one alkyl chain of dioctyl adipate and dinonyl phthalate is cleaved from the ester group by binding [B12I8S(CN)]−. This observation demonstrates preferred binding at the only functional group present in these molecules – the ester groups – in agreement with the conclusions drawn from the gas-phase experiments. In [B12I8(SH)(CN)(C6H4(COO)2)]2− (m/z 684.2), the phthalate backbone is bound via the ester groups to [B12I8S(CN)]−. We hypothesize that during the reaction on the surface the bridging sulfur atom transforms to a singly-bound SH group and two new B–O bonds are formed. In [B12I8(SH)(CN)H(C6H4(COOC9H19)(COO))]2− (m/z 748.2) and [B12I8(SH)(CN)(OH)(C6H4(COOC9H19)(COO))]2− (m/z 756.2), binding occurs via one ester group, while one alkyl chain of the phthalate is preserved and one previously vacant boron atom is occupied by a hydrogen or OH substituent. The main reaction product with dioctyl adipate [B12I8(SH)(CN)H(C4H8(COOC8H17)(COO))]2− is also the result of an attack on the ester group by [B12I8S(CN)]−. Remarkably, the product [(B12I8(SH)(CN)H)2(C4H8(COO)2)]4− (m/z 638.7) is formed in significant abundance (Fig. 5). This product ion results from the attack of two [B12I8S(CN)]− fragment ions at the two ester groups of one adipate molecule. Reaction product formation by the attack of two fragment ions on one organic molecule is very much less abundant in the case of [B12I11]− and [B12I9]− ions. From these observations, we conclude that [B12I8S(CN)]− ions exhibit higher selectivity towards ester groups in comparison to [B12I11]− and [B12I9]− ions. CID MS2 experiments on the above-mentioned reaction products support the assigned binding motif (ESI Fig. S33–S37†).
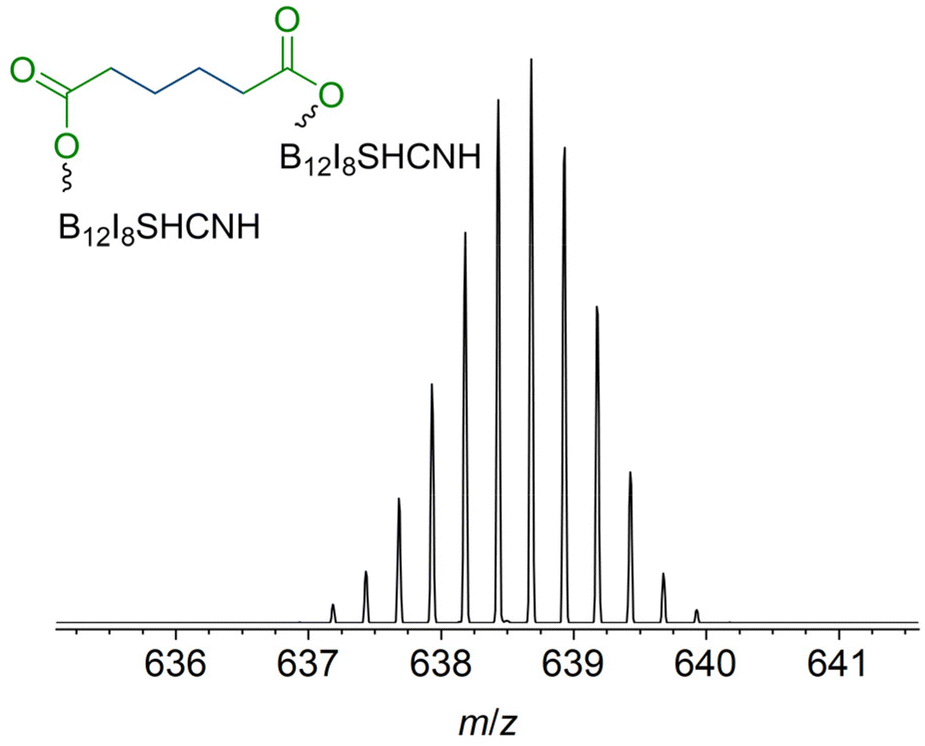 | ||
| Fig. 5 Enlarged section of the LESA-mass spectrum shown in Fig. 4c marked with an asterisk. Isotopic pattern was assigned to the fourfold negatively charged [(B12I8(SH)(CN)H)2(C4H8(COO)2)]4− anion. Two fragment ions are bound to the adipate backbone. | ||
Although [B12I8S(CN)]− was unreactive towards residual H2O in the gas-phase experiments, we associate the ion of m/z 819.3 with such a reaction on the surface. Binding to H2O results in [B12I8(CN)(SH)(OH)]−, which constitutes a [B12X11]− substitution level. Therefore, the typical binding of the intact organic background molecules via electrophilic substitution at the alkyl chain is observed, yielding [B12I8(SH)(CN)(OH)(C6H4(COOC9H19)(COOC9H18))]2−. The increased tendency to bind H2O in the fragment ion deposition experiments compared to the gas-phase experiments may be due to a layer of surface-adsorbed H2O at the deposition target. Therefore, to avoid the binding of alkyl chains of co-accumulated organic molecules and a highly selective binding solely via functional groups may require a reduced amount of water in the deposition chamber.
4 Conclusions
With the synthesis of Cs2[B12I11(SCN)], a purposeful modification of [B12I12]2− precursor dianions becomes accessible for gas-phase ion chemistry and ion soft-landing experiments. Upon CID, [B12I11(SCN)]2− ions yield fragment ions with a more selective reactivity than the fragment ions of [B12I12]2−. Similar to the previously studied [B12I12]2− ions,30 [B12I11(SCN)]2− ions fragment upon CID by loss of [I]− and sequential loss of I˙. However, in the case of [B12I11(SCN)]2−, a pronounced tendency to form the fragment ion [B12I8SCN]− was observed. This fragment ion is substantially less reactive than its analogue [B12I9]−. Experimental and computational investigations indicate that the [B12I8SCN]− ion does not exhibit three vacant boron atoms but instead the SCN group splits up to form a boron-bound CN substituent and a sulfur substituent bridging three boron atoms. Therefore, the ion is denoted as [B12I8S(CN)]−. The chemistry of this highly unusual functional group toward different organic molecules was probed in the gas phase and in ion soft-landing experiments. [B12I8S(CN)]− ions were found to react differently with organic molecules compared to [B12I11]− and [B12I9]− ions. While [B12I11]− ions react with alkyl groups and [B12I9]− ions show a high affinity to water in order to occupy their vacant boron atoms, [B12I8S(CN)]− ions show an increased selectivity towards functional groups in organic molecules, such as –SR, –NR2, and RCOOR′ in dimethyl sulfide, dimethyl amine, and phthalates, respectively. Therefore, the selective substitution of different substituents in [B12X12]2− ions enables the formation of fragment ions with previously unknown functional groups (here a sulfur bridging three boron atoms) and unique reactivity and selectivity that may be exploited for small-scale synthesis of new substances by using preparative mass spectrometry. So far, only reactions with background molecules were probed in ion soft-landing experiments, but future work will explore defined bond formations of [B12I8S(CN)]− and [B12I11]− ions with selected reagents on the surface. These experiments are expected to enable functionalization of organic molecules with closo-dodecaborate ions at either their alkyl chains ([B12I11]−) or functional groups ([B12I8S(CN)]−), respectively.Author contributions
SK analyzed experimental data and computational results, prepared the figures for the main text and the ESI,† and performed the ion soft-landing and LESA experiments. JK performed the ion molecule reaction and CID experiments, analyzed data and participated in data discussion. HK performed a significant part of computational investigations and prepared graphical material for the ESI.† ZW performed CID and HCD experiments. MW participated in the CID experiments, evaluated the mass spectra, and prepared graphical material for the ESI.† MR performed computational investigations and discussed the manuscript, in particular, the CID and LESA results. MN performed the synthesis of the precursor ion, provided synthetic details and characterization data, discussed the manuscript, and prepared graphical material for the ESI.† CJ designed the synthesis and discussed the manuscript. HIK designed ion-molecule reaction and CID experiments and participated in manuscript discussion. JW designed and coordinated the study. SK and JW wrote the manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JW is grateful to the Volkswagen foundation for a Freigeist fellowship. MN is grateful for a Kekulé fellowship from the Fonds der Chemischen Industrie. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB TRR 102 (Polymers under multiple constraints: restricted and controlled molecular order and mobility) and Project number 498397108. The authors are grateful to Lauren Blaudeau and Hugo Y. Samayoa-Oviedo (Purdue University) for carefully proofreading the manuscript. Computations for this work were done with resources of Leipzig University Computing Center.References
- J. P. Max, X. Ma, R. R. Kotha, D. Ding, J. Milton, J. J. Nash and H. I. Kenttämaa, Int. J. Mass Spectrom., 2019, 435, 280–290 CrossRef CAS.
- M. G. Leeming, G. N. Khairallah, G. Da Silva and R. A. J. O'Hair, Organometallics, 2011, 30, 4297–4307 CrossRef CAS.
- V. Franchetti, B. H. Solka, W. E. Baitinger, J. W. Amy and R. G. Cooks, Int. J. Mass Spectrom. Ion Phys., 1977, 23, 29–35 CrossRef CAS.
- S. A. Miller, H. Luo, S. J. Pachuta and R. G. Cooks, Science, 1997, 275, 1447–1450 CrossRef CAS.
- G. E. Johnson, Q. Hu and J. Laskin, Annu. Rev. Anal. Chem., 2011, 4, 83–104 CrossRef CAS PubMed.
- J. Laskin, G. E. Johnson, J. Warneke and V. Prabhakaran, Angew. Chem., Int. Ed., 2018, 57, 16270–16284 CrossRef CAS PubMed.
- S. Rauschenbach, M. Ternes, L. Harnau and K. Kern, Annu. Rev. Anal. Chem., 2016, 9, 473–498 CrossRef CAS PubMed.
- Z. Ouyang, Z. Takáts, T. A. Blake, B. Gologan, A. J. Guymon, J. M. Wiseman, J. C. Oliver, V. J. Davisson and R. G. Cooks, Science, 2003, 301, 1351–1354 CrossRef CAS PubMed.
- G. E. Johnson, D. Gunaratne and J. Laskin, Mass Spectrom. Rev., 2016, 35, 439–479 CrossRef CAS PubMed.
- P. Wang and J. Laskin, Angew. Chem., Int. Ed., 2008, 47, 6678–6680 CrossRef CAS PubMed.
- Z. Deng, N. Thontasen, N. Malinowski, G. Rinke, L. Harnau, S. Rauschenbach and K. Kern, Nano Lett., 2012, 12, 2452–2458 CrossRef CAS PubMed.
- G. Rinke, S. Rauschenbach, L. Harnau, A. Albarghash, M. Pauly and K. Kern, Nano Lett., 2014, 14, 5609–5615 CrossRef CAS PubMed.
- P. Fremdling, T. K. Esser, B. Saha, A. A. Makarov, K. L. Fort, M. Reinhardt-Szyba, J. Gault and S. Rauschenbach, ACS Nano, 2022, 16, 14443–14455 CrossRef CAS PubMed.
- H. Ochner, S. Szilagyi, M. Edte, T. K. Esser, S. Rauschenbach, L. Malavolti and K. Kern, Sci. Rep., 2023, 13, 10241 CrossRef CAS PubMed.
- W. Ran, A. Walz, K. Stoiber, P. Knecht, H. Xu, A. C. Papageorgiou, A. Huettig, D. Cortizo-Lacalle, J. P. Mora-Fuentes, A. Mateo-Alonso, H. Schlichting, J. Reichert and J. V. Barth, Angew. Chem., Int. Ed., 2022, 61, e202111816 CrossRef CAS PubMed.
- V. Prabhakaran, B. L. Mehdi, J. J. Ditto, M. H. Engelhard, B. Wang, K. D. D. Gunaratne, D. C. Johnson, N. D. Browning, G. E. Johnson and J. Laskin, Nat. Commun., 2016, 7, 11399 CrossRef CAS PubMed.
- G. E. Johnson and J. Laskin, Chem. – Eur. J., 2010, 16, 14433–14438 CrossRef CAS PubMed.
- J. Warneke, M. E. McBriarty, S. L. Riechers, S. China, M. H. Engelhard, E. Aprà, R. P. Young, N. M. Washton, C. Jenne, G. E. Johnson and J. Laskin, Nat. Commun., 2018, 9, 1889 CrossRef PubMed.
- F. Yang, M. Moors, D. A. Hoang, S. Schmitz, M. Rohdenburg, H. Knorke, A. Charvat, X.-B. Wang, K. Y. Monakhov and J. Warneke, ACS Appl. Nano Mater., 2022, 5, 14216–14220 CrossRef CAS.
- K. D. D. Gunaratne, V. Prabhakaran, Y. M. Ibrahim, R. V. Norheim, G. E. Johnson and J. Laskin, Analyst, 2015, 140, 2957–2963 RSC.
- P. Su, X. Chen, A. J. Smith, M. F. Espenship, H. Y. Samayoa-Oviedo, S. M. Wilson, H. Gholipour-Ranjbar, C. Larriba-Andaluz and J. Laskin, Anal. Chem., 2021, 93, 11576–11584 CrossRef CAS PubMed.
- P. Su, H. Hu, J. Warneke, M. E. Belov, G. A. Anderson and J. Laskin, Anal. Chem., 2019, 91, 5904–5912 CrossRef CAS PubMed.
- M. Pauly, M. Sroka, J. Reiss, G. Rinke, A. Albarghash, R. Vogelgesang, H. Hahne, B. Kuster, J. Sesterhenn, K. Kern and S. Rauschenbach, Analyst, 2014, 139, 1856–1867 RSC.
- L. Bernier, H. Pinfold, M. Pauly, S. Rauschenbach and J. Reiss, J. Am. Soc. Mass Spectrom., 2018, 29, 761–773 CrossRef CAS PubMed.
- F. Yang, K. A. Behrend, H. Knorke, M. Rohdenburg, A. Charvat, C. Jenne, B. Abel and J. Warneke, Angew. Chem., Int. Ed., 2021, 60, 24910–24914 CrossRef CAS PubMed.
- H. Gholipour-Ranjbar, H. Y. Samayoa-Oviedo and J. Laskin, ACS Nano, 2023, 17, 17427–17435 CrossRef CAS PubMed.
- H. Y. Samayoa-Oviedo, K.-A. Behrend, S. Kawa, H. Knorke, P. Su, M. E. Belov, G. Anderson, J. Warneke and J. Laskin, Anal. Chem., 2021, 93, 14489–14496 CrossRef CAS PubMed.
- J. Warneke, M. Mayer, M. Rohdenburg, X. Ma, J. K. Y. Liu, M. Grellmann, S. Debnath, V. A. Azov, E. Apra, R. P. Young, C. Jenne, G. E. Johnson, H. I. Kenttämaa, K. R. Asmis and J. Laskin, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 23374–23379 CrossRef CAS PubMed.
- M. Rohdenburg, Z. Warneke, H. Knorke, M. Icker and J. Warneke, Angew. Chem., Int. Ed., 2023, 62, e202308600 CrossRef CAS PubMed.
- J. Warneke, T. Dülcks, C. Knapp and D. Gabel, Phys. Chem. Chem. Phys., 2011, 13, 5712–5721 RSC.
- M. Rohdenburg, Z. Yang, P. Su, E. Bernhardt, Q. Yuan, E. Apra, S. Grabowsky, J. Laskin, C. Jenne, X.-B. Wang and J. Warneke, Phys. Chem. Chem. Phys., 2020, 22, 17713–17724 RSC.
- J. Warneke, S. Z. Konieczka, G.-L. Hou, E. Aprà, C. Kerpen, F. Keppner, T. C. Schäfer, M. Deckert, Z. Yang, E. J. Bylaska, G. E. Johnson, J. Laskin, S. S. Xantheas, X.-B. Wang and M. Finze, Phys. Chem. Chem. Phys., 2019, 21, 5903–5915 RSC.
- M. Mayer, M. Rohdenburg, V. van Lessen, M. C. Nierstenhöfer, E. Aprà, S. Grabowsky, K. R. Asmis, C. Jenne and J. Warneke, Chem. Commun., 2020, 56, 4591–4594 RSC.
- M. Mayer, V. van Lessen, M. Rohdenburg, G.-L. Hou, Z. Yang, R. M. Exner, E. Aprà, V. A. Azov, S. Grabowsky, S. S. Xantheas, K. R. Asmis, X.-B. Wang, C. Jenne and J. Warneke, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8167–8172 CrossRef CAS PubMed.
- M. R. Fagiani, L. L. Zeonjuk, T. K. Esser, D. Gabel, T. Heine, K. R. Asmis and J. Warneke, Chem. Phys. Lett., 2015, 625, 48–52 CrossRef CAS.
- M. Rohdenburg, M. Mayer, M. Grellmann, C. Jenne, T. Borrmann, F. Kleemiss, V. A. Azov, K. R. Asmis, S. Grabowsky and J. Warneke, Angew. Chem., Int. Ed., 2017, 56, 7980–7985 CrossRef CAS PubMed.
- J. Warneke, M. Rohdenburg, J. K. Liu, E. Johnson, X. Ma, R. Kumar, P. Su, E. Aprà, X.-B. Wang, C. Jenne, M. Finze, H. I. Kenttämaa and J. Laskin, Int. J. Mass Spectrom., 2019, 436, 71–78 CrossRef CAS.
- M. Mayer, M. Rohdenburg, S. Kawa, F. Horn, H. Knorke, C. Jenne, R. Tonner, K. R. Asmis and J. Warneke, Chem. – Eur. J., 2021, 27, 10274–10281 CrossRef CAS PubMed.
- S. Kawa, H. Knorke, J. Jin, M. Rohdenburg, K. R. Asmis, R. Tonner-Zech, E. Bernhardt, C. Jenne, M. Finze and J. Warneke, Chem. – Eur. J., 2023, e202302247 CrossRef CAS PubMed.
- X. Ma, M. Rohdenburg, H. Knorke, S. Kawa, J. K.-Y. Liu, E. Aprà, K. R. Asmis, V. A. Azov, J. Laskin, C. Jenne, H. I. Kenttämaa and J. Warneke, Phys. Chem. Chem. Phys., 2022, 24, 21759–21772 RSC.
- P. Farràs, N. Vankova, L. L. Zeonjuk, J. Warneke, T. Dülcks, T. Heine, C. Viñas, F. Teixidor and D. Gabel, Chem. – Eur. J., 2012, 18, 13208–13212 CrossRef PubMed.
- M. A. Dymova, S. Y. Taskaev, V. A. Richter and E. V. Kuligina, Cancer Commun., 2020, 40, 406–421 CrossRef PubMed.
- D. S. Wilbur, M.-K. Chyan, D. K. Hamlin and M. A. Perry, Bioconjugate Chem., 2009, 20, 591–602 CrossRef CAS PubMed.
- N. S. Hosmane, Boron Science New Technologies and Applications, CRC Press, Boca Raton, FL, USA, 2016 Search PubMed.
- V. Geis, K. Guttsche, C. Knapp, H. Scherer and R. Uzun, Dalton Trans., 2009, 2687–2694 RSC.
- W. H. Knoth, H. C. Miller, J. C. Sauer, J. H. Balthis, Y. T. Chia and E. L. Muetterties, Inorg. Chem., 1964, 3, 159–167 CrossRef CAS.
- J. F. Parcher, M. Wang, A. G. Chittiboyina and I. A. Khan, Drug Test. Anal., 2018, 10, 28–36 CrossRef CAS PubMed.
- S. C. Habicht, N. R. Vinueza, E. F. Archibold, P. Duan and H. I. Kenttämaa, Anal. Chem., 2008, 80, 3416–3421 CrossRef CAS PubMed.
- V. Kertesz and G. J. van Berkel, J. Mass Spectrom., 2010, 45, 252–260 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- H.-G. Srebny and W. Preetz, Z. Anorg. Allg. Chem., 1984, 513, 7–14 CrossRef CAS.
- M. Lepsík, M. Srnec, J. Plesek, M. Budesínský, B. Klepetárová, D. Hnyk, B. Grüner and L. Rulísek, Inorg. Chem., 2010, 49, 5040–5048 CrossRef PubMed.
- M. K. Scheller, R. N. Compton and L. S. Cederbaum, Science, 1995, 270, 1160–1166 CrossRef CAS.
- J. Warneke and X.-B. Wang, J. Phys. Chem. A, 2021, 125, 6653–6661 CrossRef CAS PubMed.
- C. Jenne, M. Keßler and J. Warneke, Chem. – Eur. J., 2015, 21, 5887–5891 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an02175k |
| ‡ Current address: Chemische Sicherheit und Abwehrender Brandschutz, Fakultät für Maschinenbau und Sicherheitstechnik, Bergische Universität Wuppertal, Gaußstr. 20, 42119, Wuppertal, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |