
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid measurement of hemoglobin-oxygen dissociation by leveraging Bohr effect and Soret band bathochromic shift†
Zoe
Sekyonda
 a,
Ran
An
b,
Utku
Goreke
b,
Yuncheng
Man
b,
Karamoja
Monchamp
bc,
Allison
Bode
bc,
Qiaochu
Zhang
b,
Yasmin
El-Gammal
d,
Cissy
Kityo
e,
Theodosia A.
Kalfa
df,
Ozan
Akkus
abg and
Umut A.
Gurkan
*abgh
a,
Ran
An
b,
Utku
Goreke
b,
Yuncheng
Man
b,
Karamoja
Monchamp
bc,
Allison
Bode
bc,
Qiaochu
Zhang
b,
Yasmin
El-Gammal
d,
Cissy
Kityo
e,
Theodosia A.
Kalfa
df,
Ozan
Akkus
abg and
Umut A.
Gurkan
*abgh
aDepartment of Biomedical Engineering, Case Western Reserve University, Cleveland, OH, USA
bDepartment of Mechanical and Aerospace Engineering, Case Western Reserve University, 10900 Euclid Ave., Glennan Building 616B, Cleveland, OH 44106, USA. E-mail: umut@case.edu; Tel: +1 216 368 6447
cDivision of Hematology and Oncology, University Hospitals Cleveland Medical Center, Cleveland, OH, USA
dDivision of Hematology, Cancer and Blood Diseases Institute, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA
eThe Joint Clinical Research Center, Kampala, Uganda
fDepartment of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA
gDepartment of Orthopedics, Case Western Reserve University, Cleveland, OH, USA
hCase Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH, USA
First published on 7th March 2024
Abstract
Oxygen (O2) binds to hemoglobin (Hb) in the lungs and is then released (dissociated) in the tissues. The Bohr effect is a physiological mechanism that governs the affinity of Hb for O2 based on pH, where a lower pH results in a lower Hb-O2 affinity and higher Hb-O2 dissociation. Hb-O2 affinity and dissociation are crucial for maintaining aerobic metabolism in cells and tissues. Despite its vital role in human physiology, Hb-O2 dissociation measurement is underutilized in basic research and in clinical laboratories, primarily due to the technical complexity and limited throughput of existing methods. We present a rapid Hb-O2 dissociation measurement approach by leveraging the Bohr effect and detecting the optical shift in the Soret band that corresponds to the light absorption by the heme group in Hb. This new method reduces Hb-O2 dissociation measurement time from hours to minutes. We show that Hb deoxygenation can be accelerated chemically at the optimal pH of 6.9. We show that time and pH-controlled deoxygenation of Hb results in rapid and distinct conformational changes in its tertiary structure. These molecular conformational changes are manifested as significant, detectable shifts in Hb's optical absorption spectrum, particularly in the characteristic Soret band (414 nm). We extensively validated the method by testing human blood samples containing normal Hb and Hb variants. We show that rapid Hb-O2 dissociation can be used to screen for and detect Hb-O2 affinity disorders and to evaluate the function and efficacy of Hb-modifying therapies. The ubiquity of optical absorption spectrophotometers positions this approach as an accessible, rapid, and accurate Hb-O2 dissociation measurement method for basic research and clinical use. We anticipate this method's broad adoption will democratize the diagnosis and prognosis of Hb disorders, such as sickle cell disease. Further, this method has the potential to transform the research and development of new targeted and genome-editing-based therapies that aim to modify or improve Hb-O2 affinity.
Introduction
Oxygen (O2) binds to hemoglobin (Hb) in the red blood cells (RBCs) and is primarily transported via the bloodstream to various tissues, where it is released for cellular metabolism. O2 release is strongly dependent on blood pH, and this relationship is explained by the Bohr effect.1 As blood flows through the lungs, pH rises, increasing Hb's affinity for oxygen, and assisting O2 loading onto Hb. In contrast, the blood pH drops in tissue capillaries, facilitating O2 dissociation (O2 offloading).2 The fraction of O2 that dissociates from the Hb is of paramount importance in evaluating tissue oxygenation, cellular oxygen availability, assessment of Hb-oxygen affinity (Hb-O2 affinity), prevention of hypoxia, and Hb variants that affect O2 dissociation.3–5Optical absorption spectroscopic techniques have found extensive applications in the determination of Hb-O2 dissociation,5,6 RBCs biophysical properties,7 and blood physiological analyses.8 These methods leverage the multifaceted properties of Hb and its interaction with different regions of electromagnetic radiation. Hb exists in the dynamic equilibrium between the oxygen-bound oxygenated form (oxyHb) and the unbound deoxygenated form (deoxyHb).3 Each of those forms, oxyHb, and deoxyHb exhibit distinct optical characteristics, and by analyzing the different optical patterns using various methods such as circular dichroism (CD), optical density ratios, and peak wavelength intensity of Hb's (Soret (400–450 nm) and Q (500–600 nm) bands) have been studied.9–13
Determination of Hb-O2 dissociation has faced challenges due to limitations in throughput and technical complexity of the measurement techniques.14,15 Typically, O2 dissociation is assessed by measuring optical intensity at a specific wavelength while systematically exposing the blood sample to nitrogen gas, and constructing an oxygen dissociation curve (ODC). This curve illustrates the relationship between O2 tension (oxygen partial pressure, pO2) and the fraction of Hb binding sites occupied by O2 (oxygen saturation, sO2). The Hb-O2 dissociation curve can be simplified as a p50 reading, which indicates the oxygen tension at 50% Hb-O2 saturation.
Although the ODC measurement method is robust and precise, it is complex, time-consuming, and requires specialized equipment, and a dedicated gas supply. Only a small number of laboratories possess the necessary Hemox analyzer (TSC Scientific Corp., New Hope, PA) to perform this measurement.6,14 For example, a typical ODC measurement using a Hemox analyzer takes more than 30 minutes, which includes diluting a blood sample in a Hemox buffer at pH 7.4 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 ratio followed by adding the sample in a custom cuvette for optical readout while nitrogen gas is bubbled to slowly deoxygenate the hemoglobin.6 Other methods for Hb-O2 binding and O2 dissociation have been developed as alternatives.16,17 These methods rely on changes in absorbance spectra intensity in the Soret and Q bands of Hb, using samples in a pH 7.4 buffer, and nitrogen gas equilibration to deoxygenate hemoglobin. These techniques require gas supply lines or gas tanks and involve recording spectra intensity changes over 2 hours as the hemoglobin deoxygenates gradually.16–18 Moreover, these assays require purified Hb samples, which may not accurately represent O2 dissociation since O2 dissociation from Hb is higher in intact RBCs compared to purified Hb in a solution.19–21 Intracellular organic phosphates such as 2,3-diphosphoglycerate (2–3 DPG) serve as regulators of Hb-O2 dissociation and deformability within intact RBCs.22–24
:1000 ratio followed by adding the sample in a custom cuvette for optical readout while nitrogen gas is bubbled to slowly deoxygenate the hemoglobin.6 Other methods for Hb-O2 binding and O2 dissociation have been developed as alternatives.16,17 These methods rely on changes in absorbance spectra intensity in the Soret and Q bands of Hb, using samples in a pH 7.4 buffer, and nitrogen gas equilibration to deoxygenate hemoglobin. These techniques require gas supply lines or gas tanks and involve recording spectra intensity changes over 2 hours as the hemoglobin deoxygenates gradually.16–18 Moreover, these assays require purified Hb samples, which may not accurately represent O2 dissociation since O2 dissociation from Hb is higher in intact RBCs compared to purified Hb in a solution.19–21 Intracellular organic phosphates such as 2,3-diphosphoglycerate (2–3 DPG) serve as regulators of Hb-O2 dissociation and deformability within intact RBCs.22–24
Herein, we take advantage of the unique optical shift of Hb's Soret band and combine it with partial chemical deoxygenation to optically measure Hb-O2 dissociation under physiologically acidic conditions at pH 6.9. First, we harnessed the acidic pH environment to facilitate rapid chemically induced partial deoxygenation of Hb (increased Bohr effect),1,25 to reduce Hb deoxygenation time down to 90 seconds. We have shown that, during this process, the conformational changes in Hb's tertiary structure result in significant shifts in the optical absorption spectra which concede with Circular Dichroism (CD) changes.26,27 This new rapid Hb-O2 dissociation assay can be completed within 2 minutes with the following two steps: (1) mixing 1 part of the sample (whole blood, RBCs, or pure Hb) with 300 parts of 6.9 pH buffer and 0.052 M sodium metabisulfite (Na2S2O5) which takes approximately 90 seconds per sample, and (2) adding the sample into a cuvette or a 96-well microplate for a light absorbance readout using a standard spectrophotometer (i.e., plate reader) in the visible range of 390 nm to 450 nm that includes the characteristic Hb Soret band, which takes approximately 30 seconds per sample.
In addition to significantly expediting the Hb-O2 dissociation measurement, this new assay allowed for a systematic comparison of healthy Hb (HbAA) and sickle Hb (HbSS), revealing previously unknown variations in the absorption spectra of these two variants in partially deoxygenated states. We examined spectral differences in purified forms of HbSS and HbAA, as well as in intact RBCs and whole blood samples. Our findings show that the absorption spectra of HbAA and HbSS were identical in the oxygenated state and at near-neutral pH. However, under specific, time-controlled deoxygenation levels and pH conditions, the Soret band (at 414 nm wavelength) of HbAA and HbSS exhibited distinct behaviors. This distinction allowed us to differentiate between the two Hb variants based on their O2 dissociation. HbSS displayed a significantly more rapid and pronounced shift of the Soret peak wavelength to the right (bathochromic shift) compared to HbAA. We assessed the utility of this assay as an optical indicator of treatment status by comparing it with existing clinical laboratory tests for Sickle Cell Disease (SCD) and by evaluating the impact of treatments (hydroxyurea, exchange transfusion) that modify hemoglobin composition in blood as well as Hb-O2 oxygen affinity and dissociation.
Leveraging this newly discovered approach, we evaluated bathochromic shift as an optical marker for O2 dissociation and compared it with p50 values obtained using a Hemox analyzer on the same samples. As with p50 values, the extent of the bathochromic shift in HbSS notably correlated with the percentage of sickle hemoglobin in the blood samples. We demonstrated the practical application of this technique in screening for sickle hemoglobin (HbS) in whole blood samples from individuals with SCD and healthy individuals. Additionally, this method allowed us to differentiate between various SCD genotypes and patients undergoing different therapies. This technique is poised to democratize basic and translational research on Hb and red cell disorders and transform the development and clinical testing of Hb-O2 affinity-modifying drugs and gene-based therapies.
Materials and methods
Materials
Phosphate buffer solutions of pH 6.9, 7.2, 7.4, 8.0, and 10.0 were purchased from Fisher Scientific (Pittsburgh, PA). All buffer solutions were stored at room temperature. Sodium metabisulfite (Na2S2O5) was purchased from Sigma Aldrich (St. Louis, MO). Nunc polystyrene 90 microwell plates with nontreated surface and a flat bottom and lid were purchased from Thermo Fisher Scientific (Waltham, MA).Blood samples and human subjects
Blood samples were collected from healthy donors and individuals with SCD as part of standard clinical care. We received ethical approval from the University Hospitals of Cleveland Medical Center's Institutional Review Board (IRB) and Joint Clinical Research Ethics Committee in Kampala, Uganda, for the Naïve SCD group. All participants in the study provided written informed consent. When collected, blood samples were de-identified, kept at 4 degrees Celsius, and processed within 6 hours of receipt. Samples were collected in EDTA-containing vacutainer tubes and separated into two groups: HbAA from healthy donors, HbSS samples from individuals with SCD, and HbAS blood samples with individuals that have heterozygous sickle cell trait (HbAS). Hemoglobin profiles of samples were verified using the reference standard high performance liquid chromatography (HPLC,VARIANT™ II, Bio-Rad Laboratories, Inc., Hercules, California). The results were presented as a percentage of HbS, A2, and F; the remainder was considered HbA since the values were normalized to 100%. Intracellular hemoglobin concentration for all blood samples was measured under normoxia with complete blood count (CBC, Hemavet 950FS, Hematology System, Draw Scientific Inc.; Miami, Florida).Sample preparation
Three independent sample preparations were performed to test different hypotheses. (1) Whole blood, (2) isolated intact red blood cells (RBC), and (3) purified hemoglobin (lysed samples).For whole blood preparation, 75 μl of whole blood was mixed with pH 6.9 buffers at a ratio of 1:300 and incubated for 10 minutes at room temperature before deoxygenation.
For intact RBC sample preparation, whole blood was centrifuged at 500g for 10 minutes in a microcentrifuge (model 21r; Thermo Scientific, Waltham, MA). The plasma and buffy coat were removed via aspiration. The RBCs were washed thrice with phosphate-buffered saline (PBS) at pH 7.4 (Gibco, Thermo Scientific, Waltham, MA). Next, 45 μL of the washed RBC suspension was diluted with a pH 6.9 buffer solution until the baseline hematocrit of 0.2% was achieved. Before deoxygenation, RBCs were incubated for 10 minutes in a pH 6.9 buffer solution at room temperature.
For purified hemoglobin preparation; lysed samples were prepared according to Andrade et al.'s28 protocol with few modifications. Hemolysis was carried out by sonication for 30 seconds (Hemex Health; Portland, OR). Following hemolysis, the lysed cell suspension was added to various pH buffer solutions and centrifuged at 2000g for 1 hour. Three hundred μl of the cell lysate was removed, and the supernatant was discarded. Cell lysates were diluted in 700 μl of the specific pH buffer solution and centrifuged at 2000g for 1 hour. The 1 ml of pH buffer solution and cell lysate were filtered through a 0.02 μm Millipore membrane. The hemoglobin concentration in the lysed sample was determined using the hemoglobin cyanide (HiCN) method and measured with a cuvette spectrophotometer at 540 nm.29 Each purified lysed sample was diluted with different pH solutions (pH 6.9, 7.2, 7.4, 8.0, and 10.0) to achieve a hemoglobin concentration of 2.8 mg dl−1, determined as the baseline for lysed sample analysis. Methemoglobin was evaluated in blood samples, and if the optical spectra included a 635 nm band (methemoglobin peak), the sample was not included in our data analysis.
pH measurements for the buffer and samples
The pH of the buffer solutions was checked before and after in addition to the prepared samples with an acumen AE150 pH meter (Fisher Scientific, Waltham, MA). Using the same pH meter, we also measured the pH of a mixture of buffer and whole blood samples before and after adding Na2S2O5.Oxygenation and deoxygenation of the samples
All samples were initially exposed to ambient air for 600 seconds. The oxygen partial pressure (pO2), 21% of atmospheric pressure (773 mmHg), was then normalized to 162 mmHg PO2 during oxygenation (Fig. 1A). Sodium metabisulphite (Na2S2O5) was used to deoxygenate the samples chemically (Fig. 1B). Predetermined amounts of sodium metabisulphite were mixed with 2 ml of the prepared samples for 90 seconds using a vortexer (Scientific Industries, Bohemia, NY). The concentrations of Na2S2O5 used with purified hemoglobin were 0.039 M, 0.046 M, 0.053 M, 0.059 M, 0.066 M, 0.079 M, and 0.082 M, which corresponded to a gradual reduction in the oxygen partial pressure in the samples. For whole blood and RBC analyses, we used 0.053 M of Na2S2O (17-fold more molecules than Hb molecules in solution) to achieve partial deoxygenation (Table S1†). A blood gas analyzer (Nova Starter Pro, Nova BioMed, Boston, Massachusetts) was used to confirm the deoxygenation levels at those Na2S2O5 concentrations.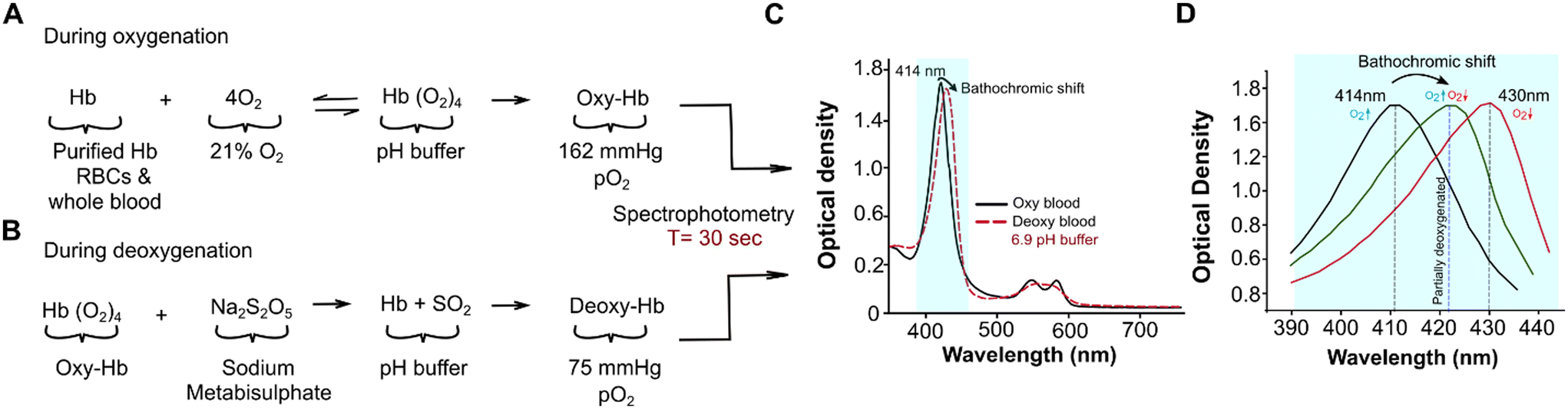 | ||
| Fig. 1 Rapid measurement of hemoglobin-oxygen dissociation by leveraging the Bohr effect and the Soret band bathochromic shift. (A) Sample analytes, purified hemoglobin (Hb), red blood cells (RBCs), or whole blood are prepared in a buffer with controlled pH. The analyte is then oxygenated at room air (162 mmHg) to form oxygenated Hb (Oxy-Hb). (B) Oxy-Hb analytes are mixed with sodium metabisulphite (Na2S2O5) for 90 seconds to form partially deoxygenated Hb (Deoxy-Hb). (C) The optical absorption spectrum of a representative blood sample from an individual with sickle cell disease, readout acquired in the 350–750 nm range. The bathochromic shift from the 414 nm Soret band (light-blue highlighted) is measured. (D) The Soret band peak shifts from 414 nm to 430 nm peak upon full deoxygenation but halfway on partial deoxygenation. The bathochromic shift was systematically evaluated with partial deoxygenation and optical readout in the 390 nm to 450 nm range, which takes 30 seconds to acquire. | ||
Hemoglobin-oxygen dissociation assay
During testing with either oxygenation or deoxygenation, samples were analyzed in three microplate wells using a spectroscopy microplate reader Spectramax M2e (Molecular Devices, San Jose, CA), over a spectral range of 350–750 nm, with a wavelength resolution of 2 nm and at a customized microplate well reading setting at room temperature. Each acquisition lasted 30 seconds, for each microplate well for a spectral range of 380–450 nm and 90 seconds for 350–750 nm. The sequence of the assay is as follows: (1) initially, the fully oxygenated samples were analyzed to obtain reference signatures that included a Soret band (390–450 nm) and two peaks in Q-band (560–580 nm). All oxygenated samples had their highest oxygenated absorption peak at 414 nm (Fig. 1C). (2) The deoxygenated samples were analyzed across the same spectral range as the oxygenated samples but with Na2S2O5 concentration at 0.053 M. The concentration of Na2S2O5 used corresponded to a decrease from 75 mmHg of pO2 in samples. The spectra of deoxygenated samples yielded two distinct peaks with pO2 reduction (Fig. 1C). The most prominent Hb peak at 414 nm in the oxygenated spectral range shifted, and the bathochromic shift varied depending on the type of sample under analysis and the pH buffer added, while the two peaks in the Q band range converged into a single peak at 560 nm (Fig. 1C). At pO2 reduction of 75 mmHg in the sample, the spectral analysis yielded three different peaks; the bathochromic shift from 414 nm and the two peaks at 540 nm and 570 nm with a reduced gap between while the hump still present. (3) For each sample and at concentration of 0.053 M Na2S2O5, the bathochromic wavelength shift and intensity were assessed for further analysis (Fig. 1D).Oxygenated and deoxygenated absorption spectra
Oxygenated and deoxygenated spectra were obtained and processed in SoftMax Pro 6.3. Individual peaks, intensities, and peak wavelength shifts were identified from the spectra through an automated peak and intensity search (Fig. 1D). Using SpectraGryph 1.2, the Soret peak was baselined, smoothed, and examined. The area under the peak (380–460 nm) and full width at half maximum (FWHM) were then calculated from the normalized Soret peak (Fig. 1D).Circular dichroism spectra of purified hemoglobin at pH 6.9
The circular dichroism (CD) spectra, like the absorption spectra in the Soret and visible range, reveal the molecular structure of the hemoglobin globin and heme groups. To confirm the effect observed in our assay, we performed circular dichroism on oxygenated purified hemoglobin (OxyHbS, OxyHbA) and deoxygenated purified hemoglobin (DeoxyHbS, DeoxyHbA) at 75 mmHg and at pH 6.9 for HbAA and HbSS samples. CD spectra were collected between 190 and 650 nm wavelength on a Jasco J-1500 Spectropolarimeter with a 1 mm quartz cuvette and a scan rate of 4 seconds. The recorded spectra were background subtracted, accounting for a pH 6.9 buffer. The data was baselined and smoothed in the Spectra Manager software (Release 2018; Version 2.15.01).Statistical analysis
We used Minitab software (Release 2021, Version 20; Minitab) for the statistical analysis. Mann Whitney U-test was performed to compare spectral variables between the HbAA and HbSS groups. Pearson's correlation analysis assessed the correlation between spectral variables; P < 0.05 was considered statistically significant. Unless otherwise noted, results were expressed as means ± standard error of the mean (SEM). Four optical variables (peak wavelength shift, intensity, FWHM, and area under the peak) data from HbAA, HbSS, and HbAS blood samples were pooled and subjected to two-dimensional principal components analysis (PCA) using R Studio (Release 2021; 4.1.1) independently. The first and second principal components were PC1 and PC2, respectively. The two components were clustered and visualized to represent the HbSS, HbAS and HbAA samples.Results
Sodium metabisulphite (Na2S2O5) induces Hb-O2 dissociation and oxygen partial pressure (pO2) decrease in a dose- and pH-dependent manner
We used sodium metabisulphite (Na2S2O5) to deoxygenate purified hemoglobin, intact RBCs, or whole blood (Fig. 1A & B). To quantify the effect of Na2S2O5 on deoxygenation, we measured the oxygen partial pressure (pO2) in both HbAA and HbSS whole blood samples reconstituted with a range of Na2S2O5 concentrations from 0 to 0.08 M at pH values of 6.9, 7.2, 7.4, 8.0, and 10.0. At all pH levels, the pO2 levels of both HbAA and HbSS whole blood samples were determined to be 162 mmHg before the addition of Na2S2O5 (Fig. 2). The pO2 levels of both HbAA and HbSS whole blood samples decreased with increasing Na2S2O5 concentration from 0.039 M to 0.059 M (Fig. 2A & B). Under all Na2S2O5 concentrations, the pO2 of both HbAA and HbSS whole blood decreased with a decrease in pH (Fig. 2A & B). HbSS whole blood demonstrated lower pO2 levels compared to HbAA whole blood at the same Na2S2O5 concentration and pH value, except at zero Na2S2O5 concentration (max. pO2 = 162 mmHg) and 0.053 M (min. pO2 around 10 mmHg) (Fig. 2B). The 0.053 M concentration of Na2S2O5 leads to partial deoxygenation of hemoglobin, where the number of Na2S2O5 molecules are more than 17-fold of available oxygen molecules in solution (Table S1†). Na2S2O5 scavenges soluble oxygen in an aqueous buffer but does not fully deoxygenate oxyhemoglobin. At elevated concentrations of Na2S2O5, corresponding to high levels of deoxygenation or pO2 close to 30 mmHg, the curve plateaus (Fig. 2). This occurs because, at these concentrations, both HbAA and HbSS exhibit similar maximum oxygen dissociation behaviors in these conditions. Together, these results demonstrate that: (1) increase in the concentration of Na2S2O5 and decrease in pH together lower pO2 levels in blood samples, and (2) pO2 levels in HbSS whole blood samples are subjected to a greater magnitude of decrease than HbAA whole blood samples when treated with the same concentration of Na2S2O5.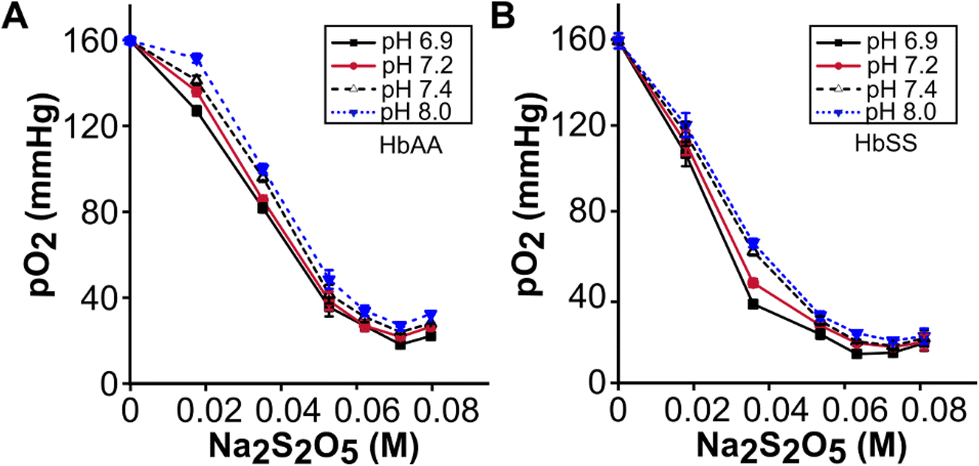 | ||
| Fig. 2 pO2 reduction in whole blood depends on the Na2S2O5 concentration and pH. pO2 measured after 4 minutes in HbAA (A), and HbSS (B), (n = 5) whole blood at pH of 6.9, 7.2, 7.4, and 8.0, at different independent Na2S2O5 concentrations ranging from 0–0.08 M. At all pH values, pO2 was at 160–162 mmHg without Na2S2O5 and decreased to a range of 20–40 mmHg with increased Na2S2O5 concentration. | ||
The bathochromic shift of hemoglobin with oxygen dissociation depends on the pH
At all pH values, fully oxygenated (162 mmHg) HbAA and HbSS had the same peak wavelengths of 414 nm (Fig. 3). Decreasing the pO2 from 162 to 20 mmHg induced peak wavelength shifts in both HbAA and HbSS purified hemoglobin at all pH values (Fig. 3A–D) except pH of 10.0 (Fig. 3E). At partial deoxygenation (pO2 = 75 mmHg), significant differences in peak wavelength shifts between HbAA and HbSS were found at pH = 6.9 (Fig. 3F) (p < 0.001) and pH = 8 (p < 0.001). At pH = 7.2 (Fig. 3F) (p > 0.05) and pH > 7.4 (Fig. 3F) (p = 0.72), there were no significant changes in peak wavelength shifts between HbAA and HbSS. At pH 6.9, RBCs and whole blood followed the same trend as purified Hb (Fig. 3G & H). These results demonstrate that pH influenced the bathochromic shift's magnitude for HbAA and HbSS purified hemoglobin. For HbAA and HbSS purified hemoglobin, the highest magnitude of bathochromic shift was obtained at pH 6.9, followed by pH 8.0. At all pH values except pH 10, HbSS had a greater magnitude of bathochromic shift compared to that of HbAA.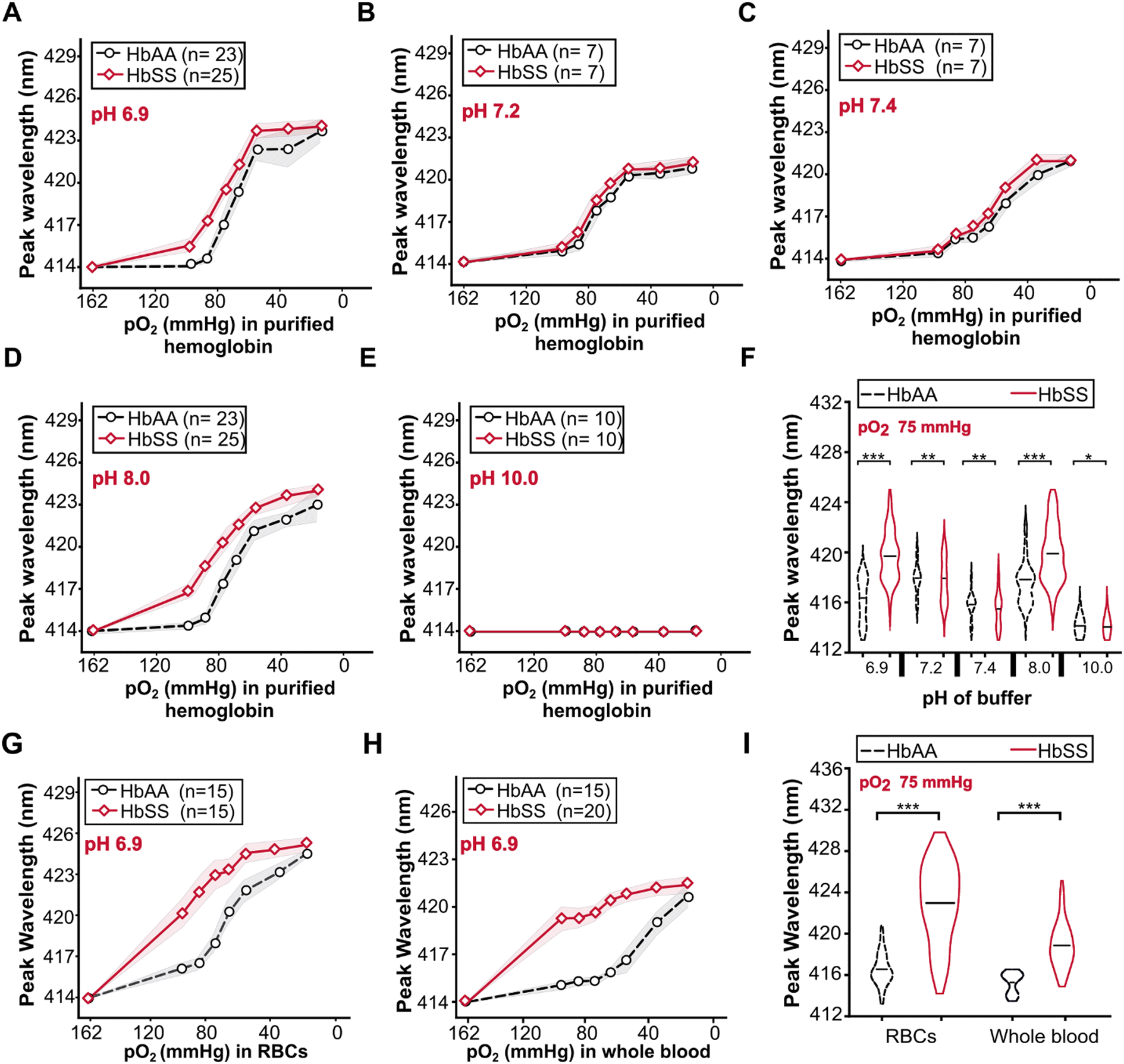 | ||
| Fig. 3 Peak shifts with deoxygenation for HbAA and HbSS differ with pH changes. Bathochromic shifts of purified HbAA and HbSS at pO2 from 162–10 mmHg and pH of 6.9 (A), 7.2 (B), 7.4 (C), 8.0 (D), and 10.0 (E). (F) At 75 mmHg pO2, HbAA vs. HbSS. (G) RBCs at pH 6.9. (H) Whole blood at pH 6.9. (I) At 75 mmHg pO2, RBCs, and whole blood (***p < 0.001), (**p > 0.05) and (*p > 0.05). | ||
For the RBCs and whole blood samples, we focused our analyses on pH 6.9 because the highest peak wavelength difference between HbAA and HbSS purified hemoglobin was obtained at this pH value. The variations of bathochromic shift shown for purified hemoglobin were also evident for RBCs (Fig. 3G) and whole blood (Fig. 3H). At 75 mmHg pO2, bathochromic shifts were significantly different between HbAA and HbSS whole blood samples (Fig. 3I) (***p < 0.001) and HbAA and HbSS whole blood samples (Fig. 3I) (***p < 0.001) were significantly different. Compared to the bathochromic shift of purified hemoglobin for HbAA and HbSS at pH 6.9, RBCs had the largest magnitude of the bathochromic shift, followed by purified hemoglobin and whole blood samples. Bathochromic shift summaries are shown in Table S2.† HbSS RBCs and whole blood had a larger bathochromic shift than normal RBCs and whole blood. The findings demonstrate that the bathochromic shift difference shown by purified hemoglobin HbAA and HbSS is also observable when using RBCs and whole blood analytes.
Whole blood optical absorption parameters demonstrate a correlation with hemoglobin oxygen affinity (p50), sickle hemoglobin fraction, and hemoglobin concentration
Aside from obtaining the measurable optical variables and examining their confounding effects, it was also critical to study the correlation between bathochromic shift and absorption intensity with p50, HbS fraction, and Hb concentration (Fig. 4). HbAA and HbSS whole blood bathochromic shifts were associated with the changes in hemoglobin oxygen affinity as measured by p50 value (Fig. 4A & B) from the ODCs obtained using Hemox analyzer for each whole blood sample at pH 7.681.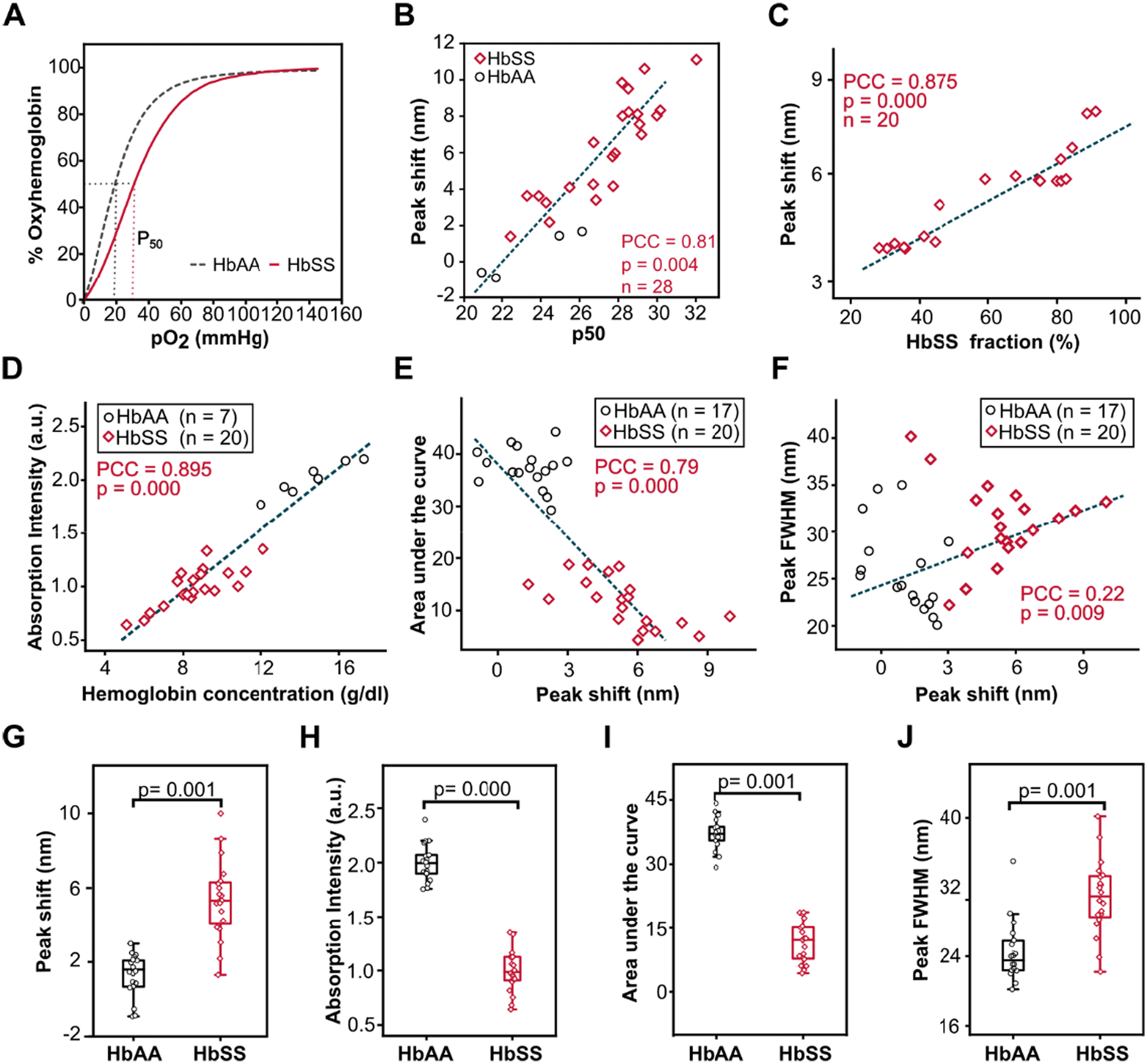 | ||
| Fig. 4 Optical absorption variables correlate with p50 (mmHg), sickle hemoglobin fraction (% HbSS), and Hb concentration (g dl−1) at 75 mmHg pO2 at pH 6.9. (A) Representative Hb-O2 dissociation curves. (B) Bathochromic shift correlates with p50. (C) HbSS bathochromic shift associates with sickle hemoglobin fraction in the sample. (D) Absorption intensity correlates with Hb concentration. (E) The area under the curve is negatively associated with peak wavelength, and (F) positively associated with FWHM. (G) HbSS vs. HbAA peak wavelength shift, (H) absorption intensity (I), the area under the curve, and peak FWHM (J). | ||
HbSS whole blood bathochromic shifts at pH 6.9 and pO2 of 75 mmHg were highly associated with the changes in the percentage of hemoglobin S (HbS%) obtained using High-Performance Liquid Chromatography (HPLC) for each whole blood sample at pH 6.9 and pO2 75 mmHg (Fig. 4C) (PCC = 0.875, p = 0.000, n = 20). The absorption intensity for whole blood samples at pH 6.9 and 28 mmHg pO2 (Fig. 4D) was strongly and positively associated with the concentration of hemoglobin (g dl−1) obtained by the standard Complete Blood Count (CBC) for both HbAA and HbSS whole blood samples (PCC = 0.89, p = 0.000, HbSS (n = 20), HbAA (n = 7)). At 75 mmHg pO2 (Fig. 4E), the area under the shifted Soret peak was significantly and inversely associated with a bathochromic shift in both HbAA and HbSS whole blood (PCC = −0.79, p = 0.000, HbSS (n = 20), HbAA (n = 17)) (Fig. 4G). Full width at half max (FWHM) were significantly but weakly linked with peak wavelength shift for both HbAA and HbSS whole blood samples (PCC = 0.22, p = 0.009, HbSS (n = 20), HbAA (n = 17)). Additional correlations between the variables and physiological importance are shown in Table S3.†
Comparisons of the optical variables between HbAA and HbSS whole blood indicated that HbSS whole blood had a significantly higher magnitude of bathochromic shift and FWHM compared to HbAA whole blood (HbSS; 420.6 ± 0.44, HbAA; 415.3.0 ± 0.21), (p = 0.001) (Fig. 4G), and (HbSS; 30.9 ± 0.84, HbAA; 24.3 ± 0.82) (p = 0.001) (Fig. 4J) respectively. HbAA whole blood had a significantly higher peak intensity and area under the peak compared to HbSS whole blood (HbSS: 0.99 ± 0.04, HbAA: 2.0 ± 0.03), (p = 0.000), (Fig. 4H) and (HbSS: 11.6 ± 0.69, HbAA: 37.2 ± 0.8) (p = 0.001) (Fig. 4I) respectively. These results revealed that the magnitude of bathochromic shift could be used to predict the hemoglobin oxygen affinity (p50) and percentage of HbS in SCD patient samples, absorption intensity for determination of the concentration of hemoglobin, and anemia status and FWHM to assess the homogeneity of the sample. When compared to HbAA samples, the homogeneity of HbSS whole blood was less consistent, as shown by greater standard deviation, since HbSS patients were on different therapies, leading to varying fractions of HbS.
Optical peak wavelength shifts and other optical variable differences are driven by differences in Hb-O2 dissociation of HbAA, HbAS, HbSS and sickle cell patients based on treatment
Principal component analysis (PCA) included optical variables (bathochromic shift, peak intensity, FWHM, and AUC data set constructed from HbAA, HbSS, and heterozygous sickle cell traits (HbAS) whole blood samples (Fig. 5). PCA resolved the differences in our data set. Initially, two components accounted for 94.5% variability in the optical parameters data set, with the first component accounting for 77.96% of the total variation between HbAA and HbSS whole blood samples (n = 20) (Fig. 5A). Secondly, two principal components accounted for 98.5% variability in the optical parameters data set, with the first component accounting for 86.56% of the total variation between heterozygous sickle traits (HbAS) = 9 and HbSS patients on various treatments: hydroxyurea (Hu HbSS = 15), exchange transfusion (TF HbSS = 8) and those not on any treatment (treatment Naïve HbSS = 20) (Fig. 5B). We evaluated the HbAA, HbAS, and HbSS patient whole blood samples stratified in the context of treatment by only peak wavelength variable under both oxygenated and deoxygenated conditions at 75 mmHg at pH 6.9. Bathochromic shift displayed significant differences for HbAA, HbAS, and HbSS patient whole blood samples and stratified based on treatment (hydroxyurea, transfusion) (Fig. 5C). No significant differences were seen for peak wavelength shift for oxygenated HbSS, HbAA, Hu HbSS, TF HbSS, and HbAS except for Naïve HbSS p = 0.02 (Fig. 5C). The PCA clearly showed grouping when using the four variables which diverged from the peak wavelength. We conclude that optical absorption variables or the bathochromic shift can be used to screen Hb-O2 affinity disorders and evaluate the efficacy of treatments that modify Hb-O2 affinity and dissociation.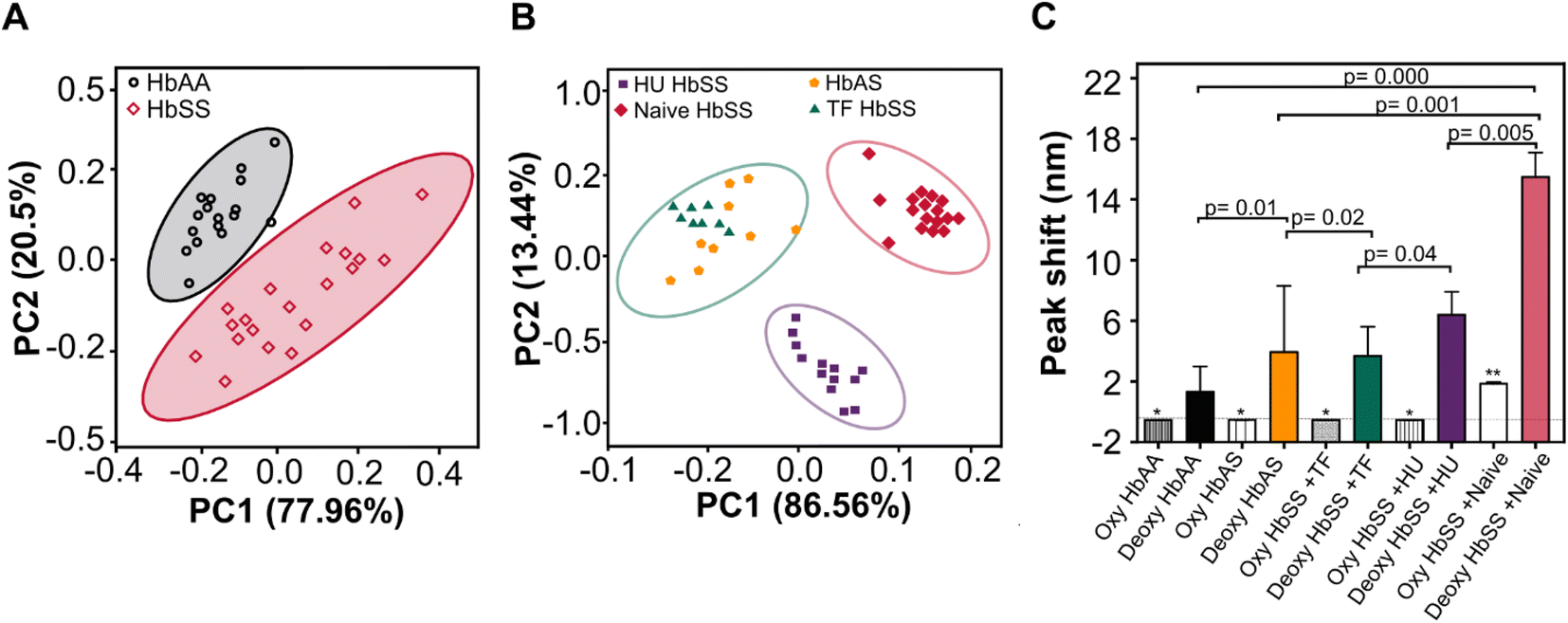 | ||
| Fig. 5 Optical absorption variables and peak wavelength shifts characterize Hb-O2 dissociation differences. (A) Principal component analysis (PCA) of the absorption peak wavelength data were used to distinguish HbAA and HbSS, (B) PCA of the absorption peak wavelength data used to categorize sickle cell trait (HbAS = 9), homozygous sickle cell disease (HbSS) on various treatments, hydroxyurea (Hu) HbSS = 15, exchange transfusion (TF) HbSS = 8, and treatment Naïve HbSS = 20. (C) Peak wavelength shift for whole blood was used as a single variable to characterize HbAA, Hu HbSS, TF HbSS, Naïve HbSS, and HbAS. | ||
CD spectra of purified hemoglobin at pH 6.9 reveals differences in the heme–heme interaction for deoxygenated HbAA and HbSS
To understand the fundamental mechanism, behold the observed differences in the peak wavelength shift of HbAA and HbSS, the CD spectra of purified hemoglobin HbAA and HbSS were conducted under oxygenated and partially deoxygenated states (Fig. 6). Under Hb oxygenation, the heme iron atom rotates to become planar with the porphyrin rings, pulling the histidine and creating a broader-scale structural change in the protein (Fig. 6A). Under Hb deoxygenation, the iron atom is non-planar with the porphyrin rings due to its association with a histidine side chain (Fig. 6B). Heme–heme interaction arises from the transition between the Hb oxygenated (Fig. 6A), and Hb deoxygenated (Fig. 6B), which differ in the tertiary and quaternary structures. In the spectral region below 250 nm, which reveals quaternary conformations of Hb, Oxy HbAA and Oxy HbSS show identical spectra (Fig. 6C). Similar CD spectra were observed with deoxy HbAA and deoxy HbSS in that wavelength range (Fig. 6D). In the spectral range above 300 nm, the CD spectral results from the heme environment are caused by heme interactions between α and β chains of protein and with the aromatic residues of the globin. Slight differences were observed in the Oxy HbAA and Oxy HbSS (Fig. 6E & G). Still, more significant differences were observed with Deoxy HbAA and Deoxy HbSS, particularly in the 260–470 nm wavelength range, which is close to the Soret band range (Fig. 6F & H). The CD spectral differences between Deoxy HbAA and Deoxy HbSS in the Soret band corresponded to the differences we observed with peak wavelength shift. The Soret peak of deoxy HbSS was at 431 nm, while that of deoxy HbAA was 425 at partial deoxygenation. Those results provide a clear fundamental understanding of peak wavelength shift; the spectral differences we report for HbSS results from its unique heme environment as demonstrated with the shifted CD Soret band induced by heme interactions between α and β chains. The peak wavelength shift at partial deoxygenation and at pH 6.9 provide molecular conformational characterization of the tertiary Hb structure, reflecting variations in Hb-O2 affinity.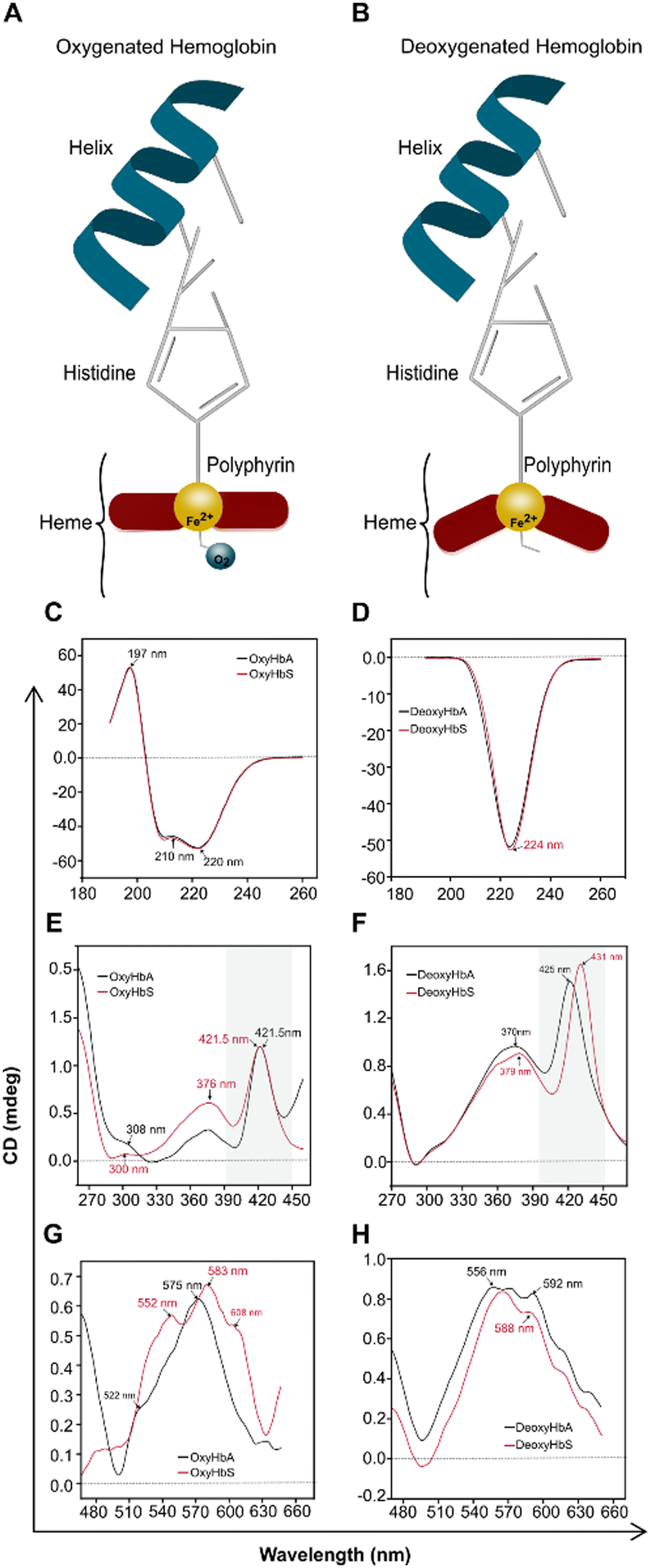 | ||
| Fig. 6 Circular Dichroism (CD) spectra of purified HbAA differ from that of HbSS at pH 6.9 and coincide with the peak wavelength shift differences. (A) Schematic structure of Oxy-Hb showing the position of heme. (B) Structural change of Deoxy Hb's heme porphyrin position. (C) CD spectra of Oxy HbAA and Oxy HbSS in 190–260 nm range. (D) CD spectra of Deoxy HbAA and Deoxy HbSS in 190–260 nm range. (E) CD spectra of Oxy HbAA and Oxy HbSS in 270–460 nm range. (F) CD spectra of Deoxy HbAA and Deoxy HbSS in 270–460 nm range. (G) CD spectra of Oxy HbAA and Oxy HbSS in 480–650 nm range. (H) CD spectra of Deoxy HbAA and Deoxy HbSS 480–650 nm range. | ||
Discussion
We describe a new optical assay approach to quickly measure Hb-O2 dissociation. This method enabled us to detect low oxygen affinity HbSS, stratify SCD patients based on treatment status, and measure Hb concentration for whole blood samples. We have demonstrated that, this new approachcorrelated with the reference standard p50 value, which is a clinical indicator of Hb-O2 affinity. In clinical practice, Hb's oxygen affinity is one of the most underutilized blood parameters, despite its critical significance in assessing blood oxygen availability and congenital disorders of erythrocytosis. However, the interest in p50 measurement has been heightened with the accelerating development of Hb-O2 affinity-modifying therapeutics for inherited Hb disorders. One of the major reasons why Hb oxygen affinity is frequently underutilized might be the unavailability of an alternative facile and accurate p50 measurement method.14,30 We believe that the method presented here will significantly streamline the Hb-O2 affinity assessment and improve its accesibility.To our knowledge, this is the first time that the optical absorption spectra of HbSS's Soret band were shown to be significantly different from HbAA under specific deoxygenation and pH conditions, based on actual spectral bathochromic shifts. We have shown that the bathochromic shifts of HbAA and HbSS purified Hb were significantly different only at pH 6.9. No bathochromic shift was observable at pH 10.0. At higher pH, we noticed that HbSS has a similar O2 dissociation to HbAA regardless of the hypoxia (pO2) level at those conditions. We may counter that there is no bathochromic shift at alkali conditions, implying that O2 is not rapidly released from the HbSS. Earlier attempts by researchers to alleviate SCD crises by alkalizing the blood can be deemed reasonable in this context.31,32
SCD samples had a greater bathochromic shift magnitude under deoxygenation at all pH levels than normal healthy blood. Furthermore, the higher the magnitude of the wavelength shift, the greater the O2 release and the lower the O2 affinity, as seen with the p50 correlation. Importantly, the bathochromic shift's magnitude in SCD samples significantly correlated to the percentage of HbSS. The dependence of HbSS O2 affinity on its concentration is consistent with earlier research.33 From a diagnostic standpoint, this finding proves that our assay may be utilized to measure Hb-O2 affinity and the percentage of HbSS, which are clinically important in monitoring SCD.
Both HbAA and HbSS variants displayed a reduction in oxygen content with partial deoxygenation, which also manifested as a bathochromic shift in the wavelength of the Soret peak. However, the magnitude of the bathochromic shift was significantly greater for HbSS than for HbAA. There are various possible explanations for this dichotomy. The absorption wavelength of hemoglobin is largely determined by the tertiary and quaternary structures of the protein.34 Notably, the visible absorption wavelength (350–650 nm) spectra of Hb are determined by the tertiary structure changes which explicitly include heme–globin environment changes (Fig. 6). In reported experiments, multiple effectors can distort protein conformations. Changing solvent–solute interactions due to increased H+ ions in the solution with increased acidity is a potential contributor that may affect both HbAA and HbSS. Increased H+ affects the Hb conformation, facilitating O2 release through the heme–globin interaction. Typically, the bathochromic shift both for HbAA and HbSS can easily be obtained in the acidic condition, at those conditions, the heme ring becomes protonated, which leads to the release of O2. Furthermore, the localization of sulphite ions to oxygen binding sites may also affect the protein conformation, resulting in the bathochromic shift for HbAA and HbSS. We speculate that the key effector that defines the greater magnitude of bathochromic shift for HbSS is its unique heme environment at lower pH, which otherwise differs from HbAA (Fig. 6). We observed more significant differences in the CD spectra Soret band and aromatic bands of HbSS from those of HbAA in the deoxygenated state at 6.9 pH. Changes in the CD deoxygenated Soret band have been correlated with the heme–globin interaction environment, which directly reflects the oxygen affinity.35,36 HbSS has been reported with slightly different β aromatic residue chains, which may contribute to its distinguished heme environment.
In this method, the purified hemoglobin concentration used is 2.8 mg dl−1 (Fig. S1†), more than 10000 times less than the critical intracellular concentration of ∼34 g dl−1 for HbSS polymerization.37 Therefore, we don't anticipate the polymerization of purified HbSS upon deoxygenation in this assay. We also observed a notably greater bathochromic shift in RBCs for both sickle and normal compared to purified Hb (Table S2) & (Fig. S3).† This result could be due to intracellular differences in pH and 2,3-DPG, which are known to affect Hb-O2 dissociation. A complementary investigation is needed to explain the possible mechanisms contributing to the differences in the heme environment of HbSS at lower pH as it happens physiologically. It is not straightforward to study these variables of pH, solvent, SO3–O2 exchange, heme–heme interaction, and polymerization contributions individually because these effectors are correlated. Optical absorption may be a valuable tool for future studies to study these mechanisms. Recent studies report the effects of 2,3-DPG on improving O2 retention of HbSS but also lowered intracellular pH;38–41 thus, the bathochromic shift can also be studied to understand the mechanisms by which 2,3-DPG plays into O2 retention and polymerization of HbSS.
The full absorption spectrum (350–750 nm) and the four optical variables (bathochromic shift, peak wavelength, AUC, and FWHM) data sets were used to detect HbSS and HbAA variants using the principal component analysis (PCA) algorithm, an unsupervised dimensional reduction tool. Our study highlights the diagnostic potential of using this method to screen for HbSS and HbAS according to the significant cluster differences between HbSS and HbAA variants that PCA revealed and to stratify SCD patients based on treatment such as transfusion, hydroxyurea and treatment-Naïve SCD patients. We believe that our assay can screen other Hb oxygen affinity abnormalities in addition to HbSS. Robustness and repeatability of the assay support this finding (Fig. S2†).
Sodium metabisulfite is a chemically stable antioxidant food additive,42 which has been used to deoxygenate blood components.43 Sodium metabisulfite based deoxygenation has been used to induce sickling of RBCs from individuals with SCD.43,44 Due to its stability and well-established use in the literature, we used sodium metabisulphite in buffer with a pH of 6.9 to controllably deoxygenate purified hemoglobin, intact RBCs, and whole blood. Sodium metabisulfite dissolves in aqueous buffer to produce sodium bisulfate: Na2S2O5 + H2O → 2Na+ + 2HSO3−. Oxyhemoglobin releases oxygen to result in deoxyhemoglobin and dissolved oxygen: HbO2 → Hb + O2. Sodium bisulfate is a commonly used reducing agent that readily reacts with dissolved oxygen and is converted into sodium hydrogen sulfate:42 2Na+ + 2HSO3− + O2 → 2NaHSO4. Scavenging of dissolved oxygen by sodium bisulfate leads to increased deoxygenation of oxyhemoglobin in the sample. Deoxygenation reaction of sodium metabisulfite is controllable by adjusting its concentration and time, which allows partial deoxygenation of blood components, as we have shown in this study.
The method presented here is designed to assess Hb-O2 dissociation and eventually determine Hb-O2 affinity based on the magnitude of Soret band shift but not designed to allow recordings of ODCs over the whole pO2 range. Future development of this method would include further quantification of the Soret band with the goal to provide absolute quantification of SO2 and pO2 for each sample to construct the ODCs. This study also lays the foundation for understanding optical absorption parameters that could differentiate Hb variants with abnormal oxygen affinity and motivates investigation to understand HbSS deoxygenation response dynamics and cellular oxygen availability assessment, which is critical for emerging pharmaceutical and genome editing-based therapies for SCD. In addition, we report evidence that could be used to improve the diagnostic accuracy of systems that use light absorption spectrophotometry, which may be affected by Hb variants and Hb-O2 affinity abnormalities.
Human subjects statement
This study involved human subjects as research participants. All experiments in the United States were performed in accordance with the Guidelines of the United States Food and Drug Administration (US FDA) and Declaration of Helsinki and approved by the ethics committee at the University Hospitals Cleveland Medical Center (UHCMC IRB# 05-14-07C). In Uganda, all experiments were performed in accordance with the Guidelines of the Uganda National Council for Science and Technology (UNCST) based on international standards and approved by the ethics committee at the Joint Clinical Research Centre in Kampala, Uganda (JCRC IRB# JC1122). Informed consents were obtained from all human participants of the study.Data availability
The corresponding author of this publication will fulfill all reasonable requests for materials and data.Author contributions
ZS, UAG, and OA conceptualized the idea, ZS, UAG, and OA contributed to the proof-of-concept experiments and method development. KM, AB, YM, QZ, RA, YE, TAK, and CMK assisted with the planning and execution of clinical sample acquisition and testing, including human subject research protocol development, subject recruitment, and blood sample collection. ZS, RA, UG, YM, OA, and UAG performed the experiments, investigation, data analysis and visualization. UAG and OA supervised the work. UAG acquired funding and administrated the project. ZS drafted the original manuscript, and all authors reviewed and edited the manuscript.Conflicts of interest
RA, UAG, and Case Western Reserve University have financial interests in Hemex Health Inc. UAG and Case Western Reserve University have financial interests in BioChip Labs Inc. UAG and Case Western Reserve University have financial interests in Xatek Inc. UAG has financial interests in DxNow Inc. Financial interests include licensed intellectual property, stock ownership, research funding, employment, and consulting. Hemex Health Inc. offers point-of-care diagnostics for hemoglobin disorders, anemia, and infectious diseases. BioChip Labs Inc. offers commercial clinical microfluidic biomarker assays for inherited or acquired blood disorders. Xatek Inc. offers point-of-care global assays to evaluate the hemostatic process. DxNow Inc. offers microfluidic and bio-imaging technologies for in vitro fertilization, forensics, and diagnostics. The competing interests of Case Western Reserve University employees are overseen and managed by the Conflict of Interests Committee according to a Conflict-of-Interest Management Plan.Acknowledgements
Authors acknowledge and thank all individuals and patients who participated in this study. The authors acknowledge the following funding sources: National Heart Lung and Blood Institute awards: R41HL172662, R42HL162214, R44HL140739, K25HL159358, R56HL165946, and U01AI176469; National Institute of Diabetes and Digestive and Kidney Diseases awards: R42DK119048, and U54DK126108; and Case-Coulter Translational Research Partnership pilot award. Zoe Sekyonda acknowledges American Society of Hematology (ASH) Graduate Hematology Award and National Institute of Biomedical Imaging and Bioengineering (NIBIB) RADx® Tech fellowship. Ozan Akkus acknowledges Kent Hale Smith Professorship at Case Western Reserve University. Umut Gurkan acknowledges Wilbert J. Austin Professorship at Case Western Reserve University. This article's contents are solely the authors’ responsibility and do not necessarily represent the official views of the National Institutes of Health.References
- F. B. Jensen, Acta Physiol. Scand., 2004, 182, 215–227 CrossRef CAS PubMed.
- H. Malte, G. Lykkeboe and T. Wang, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2021, 254, 110880 CrossRef CAS PubMed.
- M. K. Safo, M. H. Ahmed, M. S. Ghatge and T. Boyiri, Biochim. Biophys. Acta, 2011, 1814, 797–809 CrossRef CAS PubMed.
- H. Mairbäurl and R. E. Weber, Compr. Physiol., 2012, 2, 1463–1489 Search PubMed.
- G. Di Caprio, C. Stokes, J. M. Higgins and E. Schonbrun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9984–9989 CrossRef CAS PubMed.
- R. Guarnone, E. Centenara and G. Barosi, Haematologica, 1995, 80, 426–430 CAS.
- M. Nagai, N. Mizusawa, T. Kitagawa and S. Nagatomo, Biophys. Rev., 2018, 10, 271–284 CrossRef CAS PubMed.
- E. D. Chan, M. M. Chan and M. M. Chan, Respir. Med., 2013, 107, 789–799 CrossRef PubMed.
- X. D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- M. Nagai, Y. Sugita and Y. Yoneyama, J. Biol. Chem., 1969, 244, 1651–1653 CrossRef CAS.
- B. R. Wood, L. Hammer, L. Davis and D. McNaughton, J. Biomed. Opt., 2005, 10, 14005 CrossRef PubMed.
- M. R. Dayer, A. A. Moosavi-Movahedi and M. S. Dayer, Protein Pept. Lett., 2010, 17, 473–479 CrossRef CAS PubMed.
- A. Cruz-Landeira, M. J. Bal, O. Quintela and M. López-Rivadulla, J. Anal. Toxicol., 2002, 26, 67–72 CrossRef CAS PubMed.
- B. Balcerek, M. Steinach, J. Lichti, M. A. Maggioni, P. N. Becker, R. Labes, H. C. Gunga, P. B. Persson and M. Fähling, Sci. Rep., 2020, 10, 16920 CrossRef CAS PubMed.
- T. J. Morgan, Z. H. Endre, D. M. Kanowski, L. I. Worthley and R. D. Jones, J. Lab. Clin. Med., 1995, 126, 365–372 CAS.
- M. P. Patel, V. Siu, A. Silva-Garcia, Q. Xu, Z. Li and D. Oksenberg, Drug Des., Dev. Ther., 2018, 12, 1599–1607 CrossRef CAS PubMed.
- A. Nakagawa, F. E. Lui, D. Wassaf, R. Yefidoff-Freedman, D. Casalena, M. A. Palmer, J. Meadows, A. Mozzarelli, L. Ronda, O. Abdulmalik, K. D. Bloch, M. K. Safo and W. M. Zapol, ACS Chem. Biol., 2014, 9, 2318–2325 CrossRef CAS PubMed.
- S. Woyke, M. Ströhle, H. Brugger, G. Strapazzon, H. Gatterer, N. Mair and T. Haller, Physiol. Rep., 2021, 9, e14995 CAS.
- R. Benesch, R. E. Benesch and C. I. Yu, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 526–532 CrossRef CAS PubMed.
- M. Seakins, W. N. Gibbs, P. F. Milner and J. F. Bertles, J. Clin. Invest., 1973, 52, 422–432 CrossRef CAS PubMed.
- P. W. Trembath, E. A. Taylor, P. Turner, M. Roberts, P. Cole and J. Amess, Br. J. Clin. Pharmacol., 1981, 11, 19–24 CrossRef CAS PubMed.
- H. F. Bunn and J. H. Jandl, N. Engl. J. Med., 1970, 282, 1414–1421 CrossRef CAS PubMed.
- Y. Suzuki, T. Nakajima, T. Shiga and N. Maeda, Biochim. Biophys. Acta, 1990, 1029, 85–90 CrossRef CAS PubMed.
- Y. Man, R. An, K. Monchamp, Z. Sekyonda, E. Kucukal, C. Federici, W. J. Wulftange, U. Goreke, A. Bode, V. A. Sheehan and U. A. Gurkan, Front. Physiol., 2022, 13, 954106 CrossRef PubMed.
- Z. Sekyonda, C. Kityo, Y. Elgammal, T. A. Kalfa, O. Akkus and U. A. Gurkan, Blood, 2023, 142, 2270 CrossRef.
- M. F. Perutz, Nature, 1972, 237, 495–499 CrossRef CAS PubMed.
- Y. Sugita, M. Nagai and Y. Yoneyama, J. Biol. Chem., 1971, 246, 383–388 CrossRef CAS.
- C. T. Andrade, L. A. Barros, M. C. Lima and E. G. Azero, Int. J. Biol. Macromol., 2004, 34, 233–240 CrossRef CAS PubMed.
- A. Zwart, O. W. van Assendelft, B. S. Bull, J. M. England, S. M. Lewis and W. G. Zijlstra, J. Clin. Pathol., 1996, 49, 271–274 CrossRef CAS PubMed.
- A. J. Srinivasan, C. Morkane, D. S. Martin and I. J. Welsby, Expert Rev. Hematol., 2017, 10, 449–458 CrossRef CAS PubMed.
- Cooperative Urea Trials Group, J. Am. Med. Assoc., 1974, 228, 1129–1131 CrossRef.
- K. Hugh-Jones, H. Lehmann and J. M. McAlister, Br. Med. J., 1964, 2, 226–229 CrossRef CAS PubMed.
- A. May and E. R. Huehns, Br. J. Haematol., 1975, 30, 317–335 CrossRef CAS PubMed.
- L. Ronda, S. Bruno, C. Viappiani, S. Abbruzzetti, A. Mozzarelli, K. C. Lowe and S. Bettati, Protein Sci., 2006, 15, 1961–1967 CrossRef CAS PubMed.
- M. Perutz, Nature, 1972, 237, 495–499 CrossRef CAS PubMed.
- C. Zentz, S. Pin and B. Alpert, Methods Enzymol., 1994, 232, 247–266 CAS.
- C. T. Noguchi, D. A. Torchia and A. N. Schechter, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 5487–5491 CrossRef CAS PubMed.
- W. N. Poillon and B. C. Kim, Blood, 1990, 76, 1028–1036 CrossRef CAS.
- E. F. McCarthy, J. Physiol., 1943, 102, 55–61 CrossRef CAS PubMed.
- J. F. Tisdale, S. L. Thein and W. A. Eaton, Science, 2020, 367, 1198–1199 CrossRef CAS PubMed.
- W. A. Eaton, Am. J. Hematol., 2020, 95, 205–211 CrossRef PubMed.
- H. K. Trivedi and M. C. Patel, Sci. Pharm., 2011, 79, 909–920 CrossRef CAS PubMed.
- K. Torabian, D. Lezzar, N. Z. Piety, A. George and S. S. Shevkoplyas, Biosensors, 2017, 7(3), 39 CrossRef PubMed.
- P. C. Chikezie, Pharmacogn. Mag., 2011, 7, 126–132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an02071a |
| This journal is © The Royal Society of Chemistry 2024 |