
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved reversibility of lithium deposition and stripping with high areal capacity under practical conditions through enhanced wettability of the polyolefin separator to highly concentrated electrolytes†
Yosuke
Ugata
 ab,
Chihaya
Motoki
a,
Satoshi
Nishikawa
c and
Naoaki
Yabuuchi
*ab
ab,
Chihaya
Motoki
a,
Satoshi
Nishikawa
c and
Naoaki
Yabuuchi
*ab
aDepartment of Chemistry and Life Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: yabuuchi-naoaki-pw@ynu.ac.jp
bAdvanced Chemical Energy Research Center (ACERC), Institute of Advanced Sciences, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
cBattery Materials Business Department, Teijin Limited, 2-1 Hinode-cho, Iwakuni, Yamaguchi 740-8511, Japan
First published on 27th February 2023
Abstract
Highly concentrated electrolytes (HCEs) have attracted great interest as electrolyte candidates for Li metal batteries because of their functionalities in improving the reversibility and cycling performance of the Li metal negative electrode. However, the poor wettability of conventional polyolefin separators toward HCEs with high viscosity remains a critical issue to be addressed. Although porous glass fiber filters are often used as separators for HCEs, Li dendrites easily penetrate the separator during repeated Li deposition/stripping under practical conditions, leading to an internal short circuit. Here, we report that the use of a meta-aramid-coated polyolefin separator improves the wettability with HCEs owing to its polar surface functional groups and enables stable and dendrite-free Li deposition/stripping with high coulombic efficiency of ∼98% at practical areal capacity (2 mA h cm−2) for 100 cycles. A combined strategy utilizing HCEs and functional separators provides a promising possibility for the development of practical Li metal batteries.
Li metal is considered to be one of the most attractive negative electrode materials for high energy density rechargeable batteries because of its high theoretical capacity (3860 mA h g−1) and low standard electrode potential (−3.04 V vs. SHE).1–5 However, its practical application has been hampered by the growth of Li dendrites during repeated Li deposition/stripping and the low coulombic efficiency due to the side reactions between Li metal and the electrolyte. The solid electrolyte interphase (SEI) formed by the reductive decomposition of the electrolyte on the Li metal surface, which is a Li+-ion-conductive but electronically insulating passivation layer, is one of the most critical factors that strongly affects the morphology and reversibility of the Li metal electrode. In conventional carbonate-based electrolytes used in Li-ion batteries, the structural uniformity and mechanical stability of the SEI are not sufficient to prevent the Li dendrite formation and the parasitic reaction of Li metal with the electrolyte during the Li deposition process. Therefore, the development of novel electrolytes that form a uniform and stable SEI is crucially important for improving the Li metal electrode performance.
In recent years, long-term and highly reversible Li deposition/stripping cycling has been achieved using highly concentrated electrolytes (HCEs) composed of LiN(SO2F)2 (LiFSA) and aprotic solvents.6–9 Qian et al. firstly reported that a Li/Cu cell using 4 mol dm−3 LiFSA in 1,2-dimethoxyethane electrolyte can be stably cycled for more than 1000 cycles with a high average coulombic efficiency of 98.4% and non-dendritic Li metal deposition.6 Subsequently, several research groups found similar stable Li deposition/stripping cycling in the LiFSA-based HCEs containing other aprotic solvents such as carbonate,7,8,10 sulfone,9,11,12 and phosphate.13,14 The excellent performance of the Li metal electrode in these electrolytes can be attributed to the superior SEI-forming ability of the FSA− anions. In the LiFSA-based HCEs, FSA− anions are preferentially reduced instead of the solvent on the Li metal surface, and its reductive decomposition products form an inorganic-rich robust SEI layer, which leads to uniform Li deposition and suppresses the reductive decomposition of the electrolytes.7,15,16
However, poor wettability of conventional polyolefin separators to HCEs remains a critical issue that should be addressed. Compared with conventional and non-concentrated carbonate-based electrolytes, the viscosity and surface tension of HCEs is much higher due to the strong intermolecular interaction between the Li+ ion and ligands (solvent and anion); therefore, the non-polar polyolefin separator with low porosity is hardly wetted by HCEs. Instead, a porous glass fiber filter has been widely used as a separator for HCEs in laboratory-scale research,17–21 but its large pores cause non-uniform current flow and thus dendritic Li growth during Li deposition/stripping cycling, resulting in an internal short circuit when the areal capacities are increased to >1.0 mA h cm−2. Therefore, the areal capacity of metallic Li deposition/stripping with HCEs is generally limited to 1.0 mA h cm−2,6,7,22 which is small for practical battery applications.23 Dilution of HCEs with non-coordinating solvents is an another solution to the wettability issue, but compromises the intrinsic advantages of HCEs, such as their high Li+ ion transference number and low volatility.8,9,24–26 Therefore, to overcome these challenges, an alternative separator with low porosity and strong affinity to HCEs is needed. In this context, we herein focus on an aramid-coated polyolefin separator. Surface coating with aramid resin is an effective strategy for improving the electrolyte wettability of the polyolefin separator.27,28 In addition, the aramid-coated polyolefin separator provides uniform current flow inside the separator, leading to uniform Li deposition without the Li dendrite growth in non-concentrated electrolyte.29,30 To the best of our knowledge, the use of an aramid-coated polyolefin separator with HCEs has not been reported elsewhere. Here, we report that the aramid-coated polyolefin separator exhibits an excellent wetting ability to HCEs and enables stable Li deposition/stripping cycling even at high areal capacity (up to 4 mA h cm−2).
The wettability of the HCE to pristine and aramid-coated polyolefin separators was tested as shown in Fig. 1a. Herein, a composition of LiFSA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 1:1.1 (mol:mol) was used as the HCE, which possesses a wide electrochemical window to enable the stable operation of 5 V-class Li-ion batteries.31 A droplet of LiFSA:DMC = 1:1.1 electrolyte remained intact on the pristine polyolefin separator with low porosity due to high viscosity (238.9 mPa s at 30 °C),31 whereas the HCE easily spreads and soaks through the aramid-coated polyolefin separator. The preparation of the aramid-coated polyolefin separator is described in the ESI.† The difference in the wettability with these two separators originates from the difference in the surface functional groups of the separator. Fig. 1b shows the attenuated total reflection Fourier transform infrared (ATR-FTIR) spectra for different separators. Peaks at 2952, 2920, 2871, 2841, 1456, and 1375 cm−1 in the spectrum of the pristine polyolefin separator correspond to the vibrational modes of CH2 and CH3 groups (see the detailed peak assignment shown in ESI,† Table S1),32 indicating the non-polarity on the pristine polyolefin separator surface. In the spectrum of the aramid-coated separator, peaks at 1656 and 1538 cm−1 appear, which are assigned to the vibrational modes of C
:DMC = 1:1.1 (mol:mol) was used as the HCE, which possesses a wide electrochemical window to enable the stable operation of 5 V-class Li-ion batteries.31 A droplet of LiFSA:DMC = 1:1.1 electrolyte remained intact on the pristine polyolefin separator with low porosity due to high viscosity (238.9 mPa s at 30 °C),31 whereas the HCE easily spreads and soaks through the aramid-coated polyolefin separator. The preparation of the aramid-coated polyolefin separator is described in the ESI.† The difference in the wettability with these two separators originates from the difference in the surface functional groups of the separator. Fig. 1b shows the attenuated total reflection Fourier transform infrared (ATR-FTIR) spectra for different separators. Peaks at 2952, 2920, 2871, 2841, 1456, and 1375 cm−1 in the spectrum of the pristine polyolefin separator correspond to the vibrational modes of CH2 and CH3 groups (see the detailed peak assignment shown in ESI,† Table S1),32 indicating the non-polarity on the pristine polyolefin separator surface. In the spectrum of the aramid-coated separator, peaks at 1656 and 1538 cm−1 appear, which are assigned to the vibrational modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N groups (ESI,† Table S2) contained in meta-aramid used for the surface coating layer (Fig. 1c).33,34 Note that a uniform distribution of N and O is evidenced from scanning electron microscopy (SEM) coupled with elemental mapping by energy dispersive X-ray spectroscopy (EDX), as shown in ESI,† Fig. S1. Although nanosized pores are found in the aramid-coated polyolefin separator (Fig. 1d), the polar functional groups of the aramid coating layer would enhance the affinity of the polyolefin separator toward HCE, resulting in the superior electrolyte wettability.
O and C–N groups (ESI,† Table S2) contained in meta-aramid used for the surface coating layer (Fig. 1c).33,34 Note that a uniform distribution of N and O is evidenced from scanning electron microscopy (SEM) coupled with elemental mapping by energy dispersive X-ray spectroscopy (EDX), as shown in ESI,† Fig. S1. Although nanosized pores are found in the aramid-coated polyolefin separator (Fig. 1d), the polar functional groups of the aramid coating layer would enhance the affinity of the polyolefin separator toward HCE, resulting in the superior electrolyte wettability.
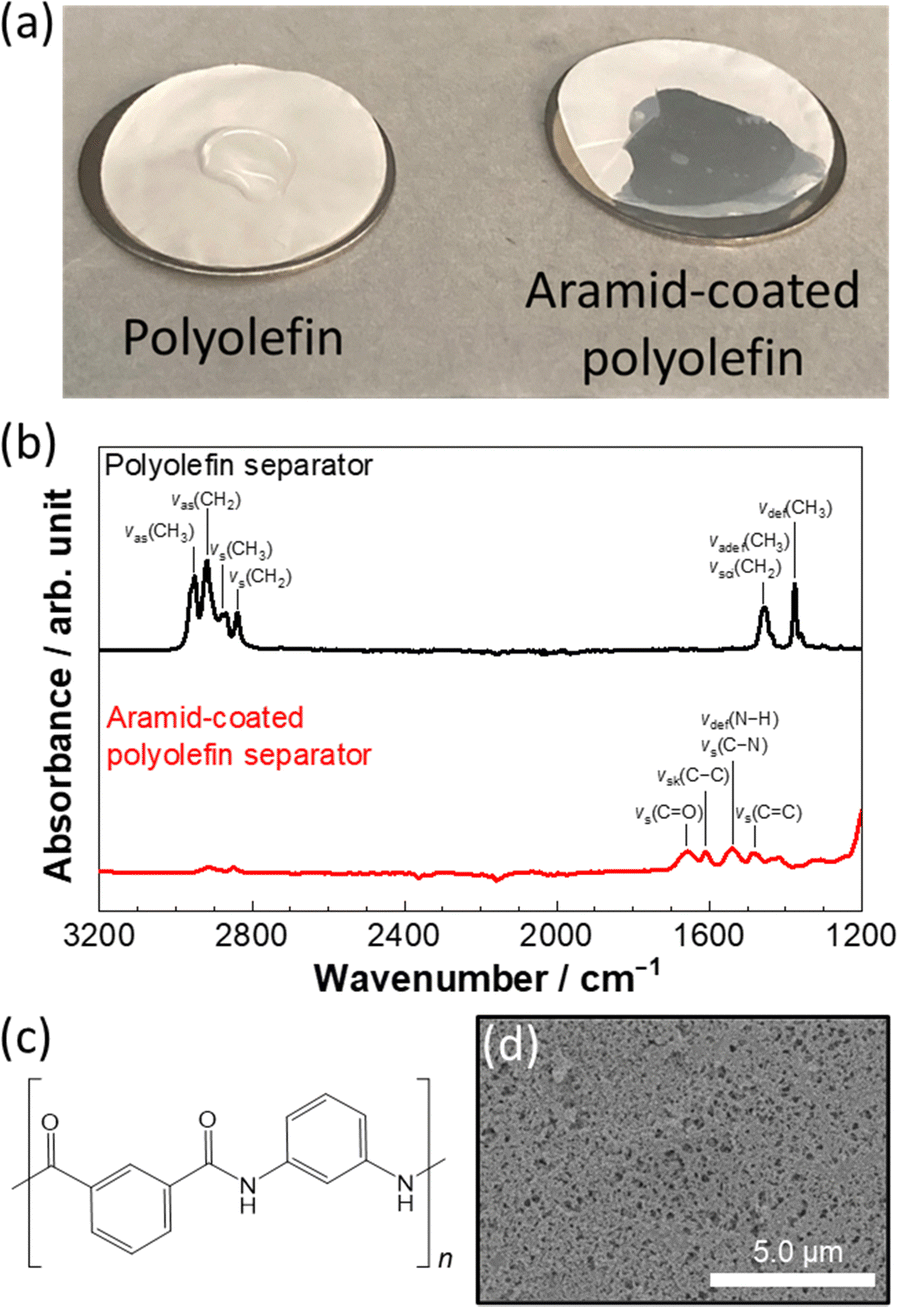 | ||
| Fig. 1 (a) Photographs of wettability of a polyolefin separator (left) and aramid-coated polyolefin separator (right) to the highly concentrated electrolyte (LiFSA:DMC = 1:1.1 in a molar ratio), and (b) ATR-FTIR spectra of the separators. (c) Chemical structural formula of meta-aramid used for coating. (d) A SEM image of the aramid-coated polyolefin separator. | ||
To examine the effect of the aramid-coated polyolefin separator on the electrochemical performance of Li metal electrodes in LiFSA:DMC = 1:1.1 electrolyte, chronopotentiometry was conducted using Li/Cu cells. For comparison, the electrochemical performance of a cell with a glass fiber filter was also studied. A schematic illustration of the used Li/Cu cell is shown in Fig. 2a. Fig. 2b shows the chronopotentiometric curves of the Li/Cu cells with a glass fiber filter and aramid-coated polyolefin separator measured at various Li deposition capacities ranging from 0.7 to 4.2 mA h cm−2 with fixed current density at a rate of 0.35 mA cm−2. At the first cycle, the Li deposition capacity is 0.7 mA h cm−2, whereas the Li stripping capacity in the cell with the glass fiber filter was approximately 0.56 mA h cm−2. This irreversible capacity is possibly due to the initial SEI formation on the Cu electrode surface. After the first cycle, the obtained Li stripping capacity is increased and the coulombic efficiency of the Li deposition/stripping is improved to >90% owing to the stabilization of the SEI layer (Fig. 2c). As shown in Fig. 2b, the cell with the glass fiber filter showed a stable cycling with low polarization of ∼50 mV at the deposition capacity of 0.7 mA h cm−2. However, with increasing the deposition capacity to 1.4 mA h cm−2, the cell voltage suddenly dropped to almost zero during Li deposition at the 7th cycle. This behavior is due to the internal short circuit caused by the dendritic growth of Li metal. Indeed, the voltage of the cell does not reach 0.5 V on the stripping process from the 7th cycle, leading to coulombic efficiencies of >100% in the following cycles (Fig. 2c). This fact indicates the direct contact between Cu and Li electrodes associated with dendritic Li deposition. The glass fiber filter possesses large pores, 5–10 μm (inset in Fig. 2b), and thus dendritic Li metal formation results in the internal short circuit at high areal capacity (>1 mA h cm−2) for Li deposition. In contrast, for the cell with the aramid-coated polyolefin separator, a highly reversible Li deposition and stripping without short circuiting and superior coulombic efficiency of ∼98% are observed even when the Li deposition areal capacity was increased to 4.2 mA h cm−2 (Fig. 2b). Such large amount of Li deposition/stripping is sufficient to realize practical high energy density in Li metal batteries.23,35 Furthermore, the cell with the aramid-coated polyolefin separator can be stably cycled with increasing the current density, ∼1.0 mA cm−2 (ESI,† Fig. S2). When the deposition capacity is increased to 4.9 mA h cm−2, the reversibility of Li deposition/stripping is gradually lost upon cycling (ESI,† Fig. S3). The aramid-coated polyolefin separator has nanosized pores (Fig. 2d), which effectively suppresses the penetration of dendritic Li metal through the separator and enables reversible cycling of Li metal even at high areal capacity.
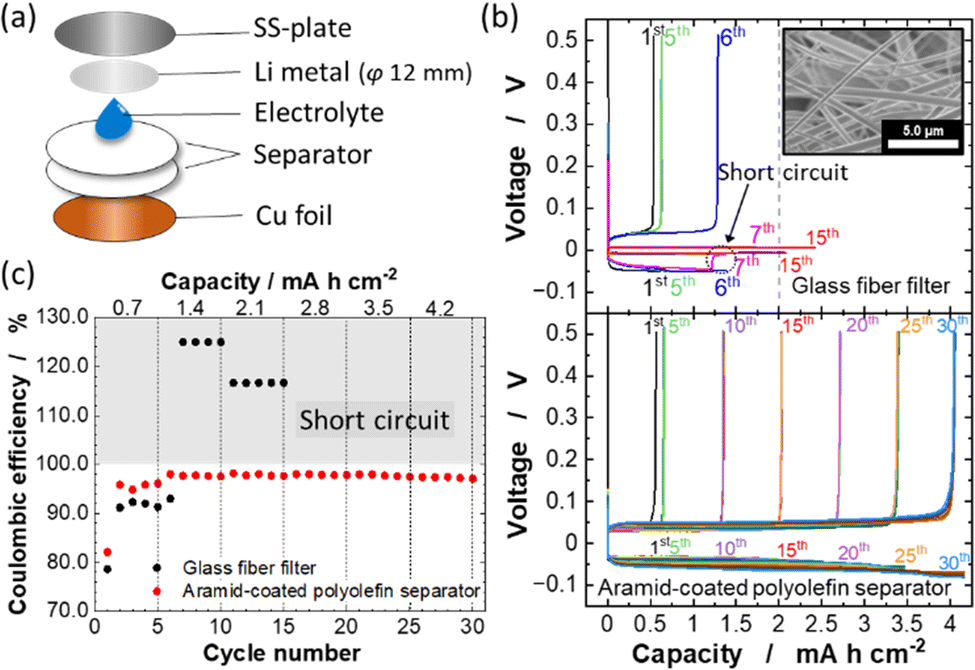 | ||
| Fig. 2 (a) A schematic illustration of a Li/Cu cell used in this study. LiFSA:DMC = 1:1.1 was used as the electrolyte. (b) Chronopotentiometric curves of Li deposition/stripping in Li/Cu cells with a glass fiber filter (top) and aramid-coated separator (bottom) measured at various Li deposition capacities. The current density was fixed at 0.35 mA cm−2. A SEM image of the glass fiber filter is also shown in the inset. (c) Coulombic efficiency of Li deposition/stripping in each cell. | ||
The cycling stabilities of Li deposition/stripping in conventional electrolyte (1 M LiPF6 in EC/DMC) with and without 2 vol% fluoroethylene carbonate (FEC) and highly concentrated LiFSA/DMC electrolyte were compared using Li/Cu cells (Fig. 3). The separators used for the low and high concentration electrolytes are conventional polyolefin and aramid-coated polyolefin membranes, respectively. For the cycling test, the current density was set to 0.35 mA cm−2 and the Li deposition areal capacity to 2.1 mA h cm−2. As shown in Fig. 3a, polarization for Li deposition/stripping processes in the cell with the conventional 1 M LiPF6 in EC/DMC electrolyte gradually increased during 100 continuous cycles. The coulombic efficiency remains 95–96% for the initial 40 cycles and then gradually dropped to ∼91% (Fig. 3d). These results suggest that the irreversible reductive decomposition of the electrolyte continuously occurs in the conventional electrolyte during the Li deposition in each cycle. In contrast, the increase in the polarization with cycling was greatly suppressed and the average coulombic efficiency was significantly improved in the cell with 1 M LiPF6 in EC/DMC + 2 vol% FEC (Fig. 3b and d). This observation is probably because the addition of a small amount of FEC in the conventional electrolyte forms a stable LiF-rich SEI on the Cu electrode surface, which suppresses the reductive decomposition of the electrolyte.36,37 More importantly, the cell with the LiFSA:DMC = 1:1.1 exhibits lower irreversible capacity (Fig. 3c) and higher coulombic efficiencies (>98%) for 100 cycles (Fig. 3d) compared to the cells containing LiPF6-based electrolytes, despite the polarization being larger on 100 continuous cycles, probably associated with lower ionic conductivity.31 In the highly concentrated LiFSA-based electrolyte, the FSA-derived SEI is considered to be formed on the deposited Li metal surface and minimizes the irreversible reaction between the Li metal and the electrolyte.7,9,10 It is noted that the use of aramid-coated polyolefin separator is also responsible for the superior cycling performance for the cell with LiFSA:DMC = 1:1.1. High electrolyte wettability of the aramid-coated separator (Fig. 1) induces the uniform Li deposition (vide infra), thereby enabling dendrite-free Li deposition/stripping reactions for 100 cycles. The insets of Fig. 3a–c show the SEM images of Li metal deposited on the Cu substrate. In the 1 M LiPF6 in EC/DMC electrolyte, needle-like Li deposits with a large surface are formed (Fig. 3a), which accelerates the parasitic reactions between the Li metal and electrolyte, resulting in the poor cycling stability (Fig. 3a and d). In contrast, granular Li deposits were formed uniformly in the 1 M LiPF6 in EC/DMC + 2 vol% FFC and LiFSA:DMC = 1:1.1 electrolytes (Fig. 3b and c), which may contribute to the high coulombic efficiency. The grain sizes of the Li deposits in LiFSA:DMC = 1:1.1 are clearly larger compared with those in the LiPF6 in EC/DMC + 2 vol% FEC, leading to the reduced surface areas of the Li deposits.6–8 As such, the parasitic reactions could be minimized more effectively in the highly concentrated LiFSA/DMC electrolyte, which improves the reversibility of Li deposition/stripping with practical areal capacity.
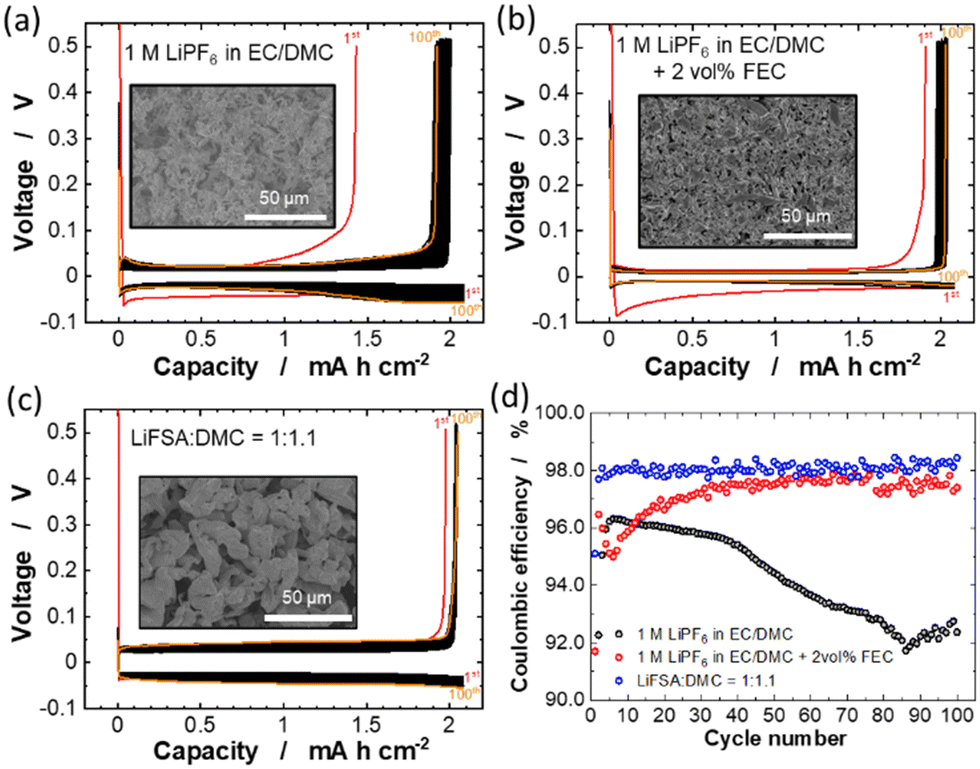 | ||
| Fig. 3 Chronopotentiometric curves of Li deposition/stripping in Li/Cu cells with (a) 1 M LiPF6 in EC/DMC, (b) 1 M LiPF6 in EC/DMC + 2 vol% FEC and (c) LiFSA:DMC = 1:1.1 electrolyte solutions at a current density of 0.35 mA cm−2. SEM images of Li metal deposited on the Cu electrode at a current density of 0.35 mA cm−2 for 6 h are also shown in the inset of (a–c). (d) Coulombic efficiency of Li deposition/stripping in each cell. | ||
In summary, the use of an aramid-coated polyolefin separator solves the wetting issue of conventional polyolefin separators to HCEs. This is because the polar surface functional group on the aramid-coating layer provides strong affinity of the polyolefin separator to the electrolyte. Furthermore, the aramid-coated separator enables stable Li deposition/stripping reactions in LiFSA/DMC = 1:1.1 at practical areal capacity (2 mA h cm−2) for 100 cycles. Unlike in the conventional LiPF6/EC + DMC-based electrolyte, a high coulombic efficiency over 98% for Li deposition/stripping can be achieved in the highly concentrated LiFSA/DMC electrolyte even without the addition of functional additives such as FEC. Thus, the combined use of highly concentrated electrolytes and functional separators offers the promising possibility for practical applications of Li metal batteries.
Author contributions
Yosuke Ugata: investigation, validation, writing – original draft. Chihaya Motoki: investigation. Satoshi Nishikawa: validation. Naoaki Yabuuchi: conceptualization, validation, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NY acknowledges the partial support from JSPS, Grant-in-Aid for Scientific Research (Grant Numbers 19H05816 and 21H04698). This work was partially supported by JST, CREST Grant Number JPMJCR21O6, Japan.Notes and references
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- S. K. Sharma, G. Sharma, A. Gaur, A. Arya, F. S. Mirsafi, R. Abolhassani, H.-G. Rubahn, J.-S. Yu and Y. K. Mishra, Energy Adv., 2022, 1, 457–510 RSC.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Chem, 2017, 4, 174–185 Search PubMed.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J. G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- Y. Maeyoshi, D. Ding, M. Kubota, H. Ueda, K. Abe, K. Kanamura and H. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 25833–25843 CrossRef CAS PubMed.
- Y. Ugata, R. Tatara, T. Mandai, K. Ueno, M. Watanabe and K. Dokko, ACS Appl. Energy Mater., 2021, 4, 1851–1859 CrossRef CAS.
- Z. Zeng, V. Murugesan, K. S. Han, X. Jiang, Y. Cao, L. Xiao, X. Ai, H. Yang, J.-G. Zhang, M. L. Sushko and J. Liu, Nat. Energy, 2018, 3, 674–681 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- K. Sodeyama, Y. Yamada, K. Aikawa, A. Yamada and Y. Tateyama, J. Phys. Chem. C, 2014, 118, 14091–14097 CrossRef CAS.
- Y. Ugata, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2019, 21, 9759–9768 RSC.
- S. Kondou, K. Dokko, M. Watanabe and K. Ueno, Electrochemistry, 2021, 89, 389–394 CrossRef CAS.
- N. Takeda, I. Ikeuchi, R. Natsui, K. Nakura and N. Yabuuchi, ACS Appl. Energy Mater., 2019, 2, 1629–1633 CrossRef CAS.
- J. Yun, R. Sagehashi, Y. Sato, T. Masuda, S. Hoshino, H. B. Rajendra, K. Okuno, A. Hosoe, A. S. Bandarenka and N. Yabuuchi, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024969118 CrossRef CAS PubMed.
- R. Qi, B. D. L. CampÉOn, I. Konuma, Y. Sato, Y. Kaneda, M. Kondo and N. Yabuuchi, Electrochemistry, 2022, 90, 037005 CrossRef CAS.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217–1224 CrossRef CAS.
- S. Chen, C. Niu, H. Lee, Q. Li, L. Yu, W. Xu, J.-G. Zhang, E. J. Dufek, M. S. Whittingham, S. Meng, J. Xiao and J. Liu, Joule, 2019, 3, 1094–1105 CrossRef CAS.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- A. Nakanishi, K. Ueno, D. Watanabe, Y. Ugata, Y. Matsumae, J. Liu, M. L. Thomas, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2019, 123, 14229–14238 CrossRef CAS.
- M. Yanagi, K. Ueno, A. Ando, S. Li, Y. Matsumae, J. Liu, K. Dokko and M. Watanabe, J. Electrochem. Soc., 2020, 167, 070531 CrossRef CAS.
- B. Yang, L. Wang, M. Zhang, W. Li, Q. Zhou and L. Zhong, J. Mater. Chem. A, 2021, 9, 12923–12946 RSC.
- S. Hu, S. Lin, Y. Tu, J. Hu, Y. Wu, G. Liu, F. Li, F. Yu and T. Jiang, J. Mater. Chem. A, 2016, 4, 3513–3526 RSC.
- I. Arise, Y. Miyahara, K. Miyazaki and T. Abe, J. Electrochem. Soc., 2022, 169, 010536 CrossRef.
- I. Arise, Y. Miyahara, K. Miyazaki and T. Abe, J. Electrochem. Soc., 2022, 169, 020546 CrossRef.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- R. Morent, N. De Geyter, C. Leys, L. Gengembre and E. Payen, Surf. Interface Anal., 2008, 40, 597–600 CrossRef CAS.
- C. H. Do, E. M. Pearce, B. J. Bulkin and H. K. Reimschuessel, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 2409–2424 CrossRef CAS.
- L. Yao, C. Lee and J. Kim, Fibers Polym., 2010, 11, 1032–1040 CrossRef CAS.
- M. Ue, K. Sakaushi and K. Uosaki, Mater. Horiz., 2020, 7, 1937–1954 RSC.
- J. Heine, P. Hilbig, X. Qi, P. Niehoff, M. Winter and P. Bieker, J. Electrochem. Soc., 2015, 162, A1094–A1101 CrossRef CAS.
- X.-Q. Zhang, X.-B. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00359g |
| This journal is © The Royal Society of Chemistry 2023 |