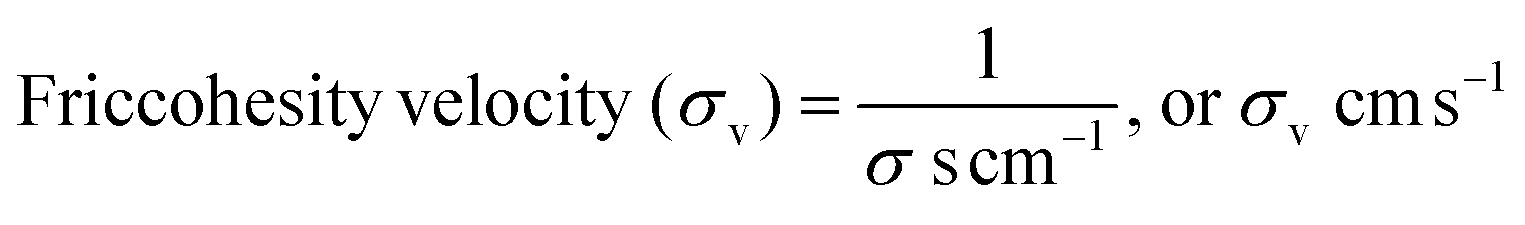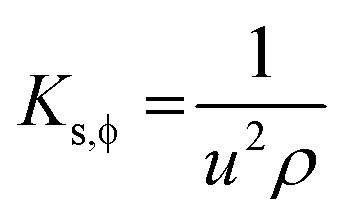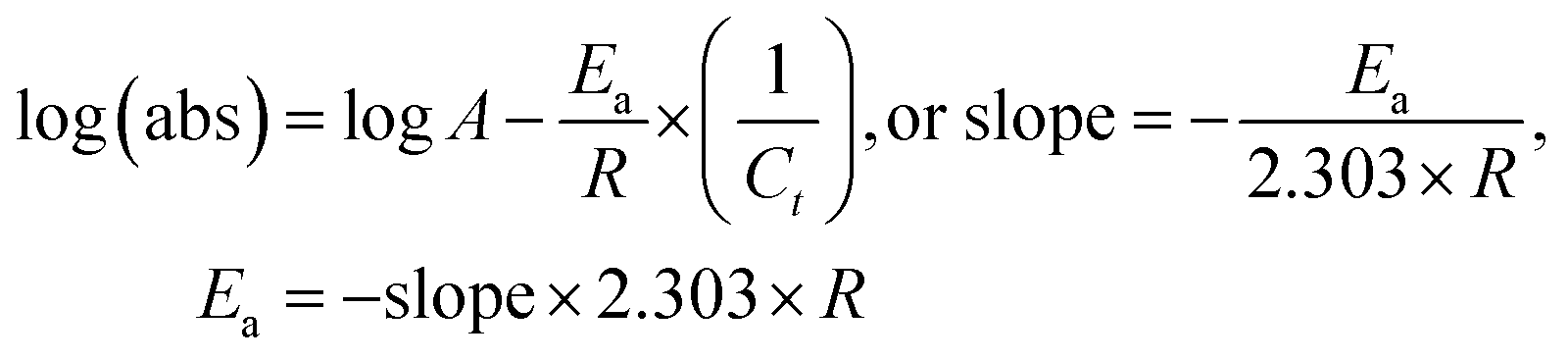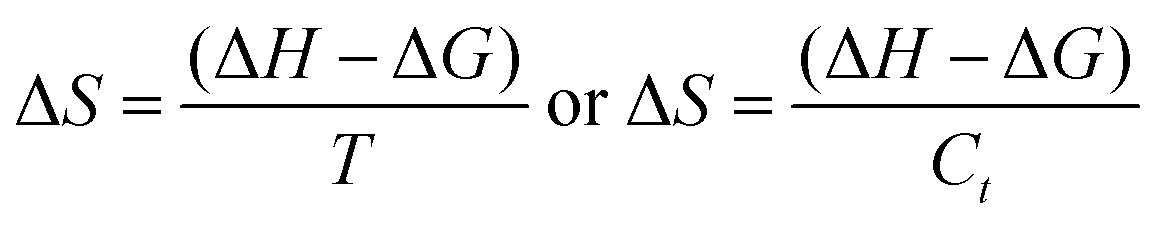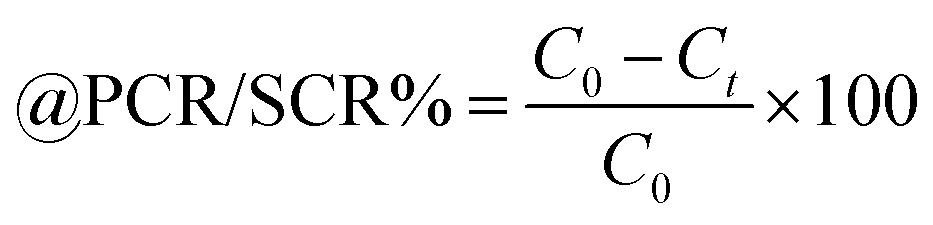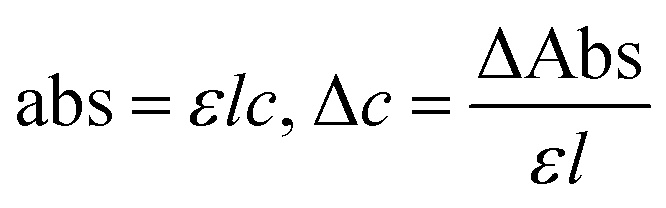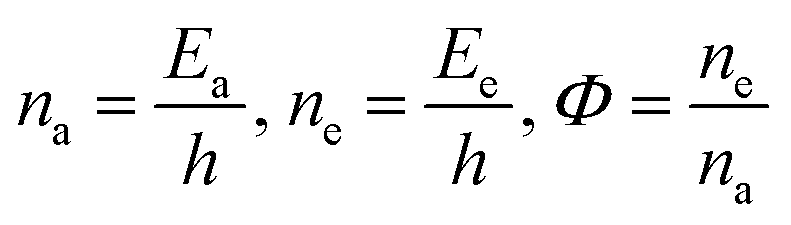DOI:
10.1039/D2YA00274D
(Paper)
Energy Adv., 2023,
2, 198-225
LSNR:GO template for BSA interaction, photo and sonocatalytic reductions of fluorescent dyes in aqueous solutions†
Received
7th October 2022
, Accepted 9th December 2022
First published on 9th December 2022
Abstract
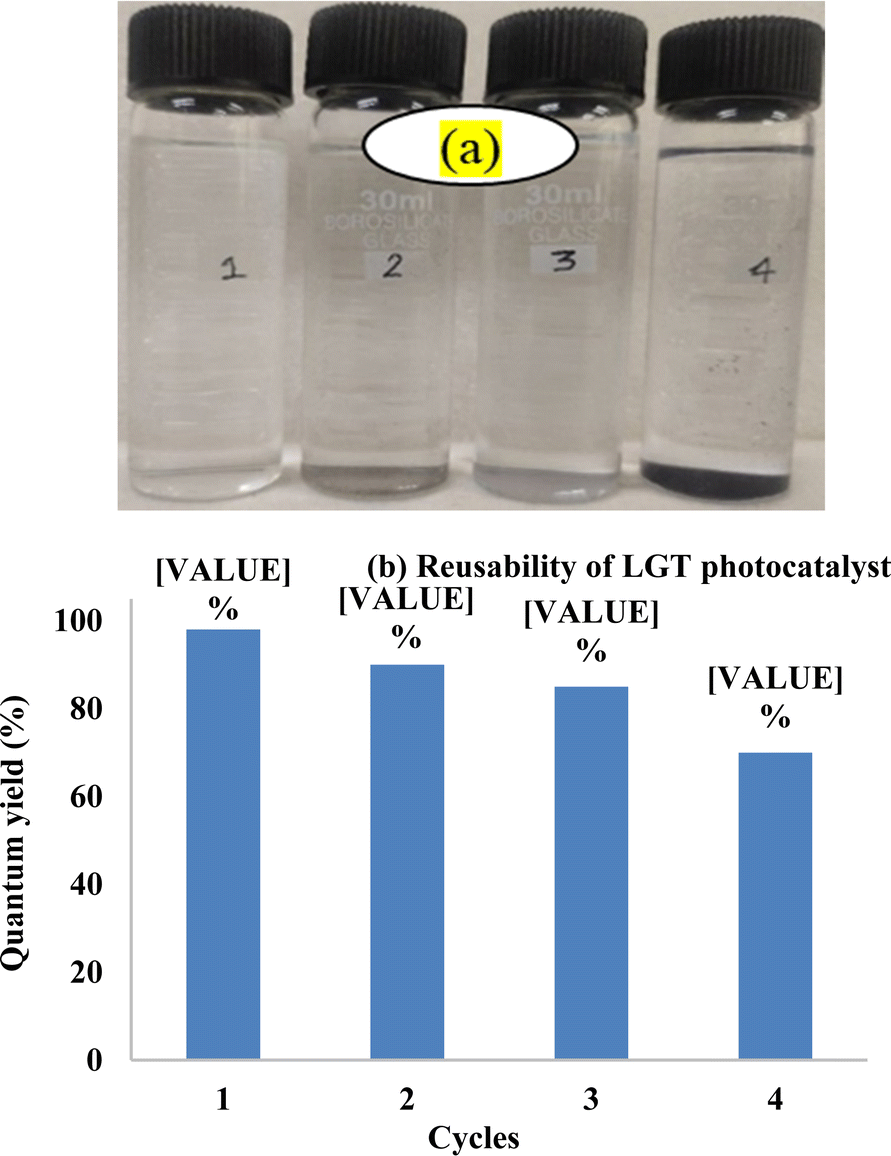
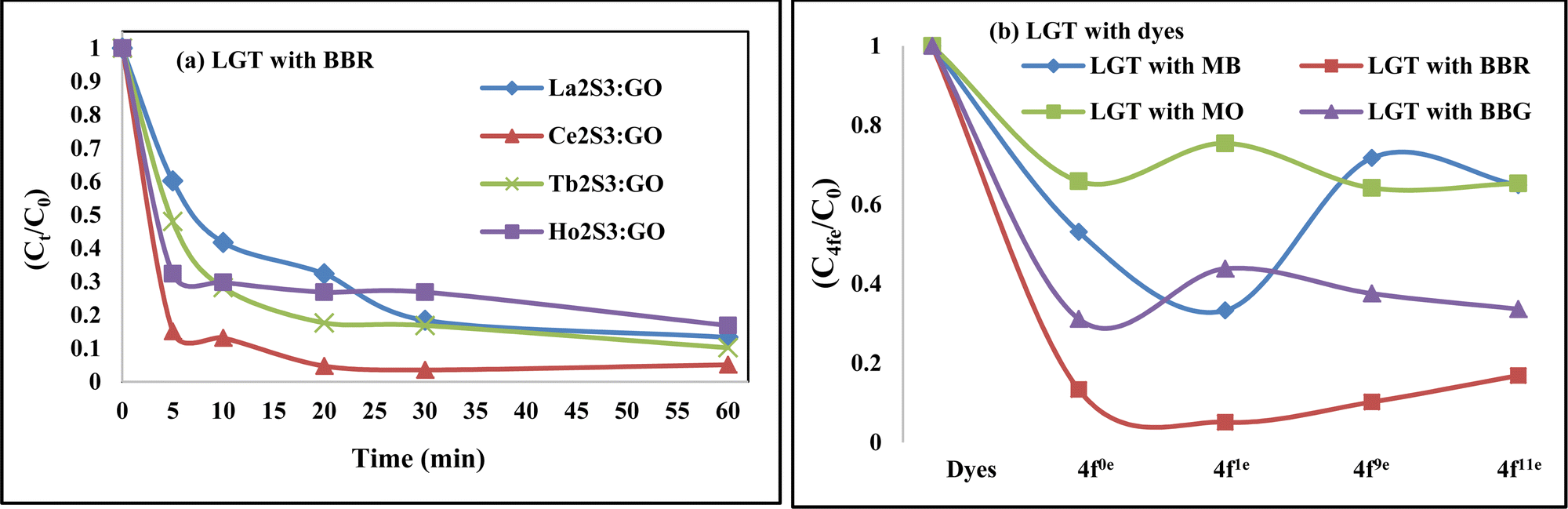
Lanthanide sulfide nanorods (LSNRs) were synthesized by fusing sodium (Na) metal with hexahydrate chloride salts of lanthanum (La), cerium (Ce), terbium (Tb), and holmium (Ho) (LnCl3·6H2O) with 4f0, 4f1, 4f9, and 4f11 electrons (4f#e), respectively. Na reduced LnCl3·6H2O to Ln(OH)3 + 3H2 as a crash reaction methodology (CRM). Aq-Ln(OH)3 with H2S developed La2S3, Ce2S3, Tb2S3, Ho2S3, LSNRs, which were doped with graphene oxide (GO) at ∼60 for an LSNR:GO template (LGT), characterized with XRD, FT-IR, TGA/DTG, SEM, DLS, FESEM + EDS, C, H, N/S elemental analysis, Raman spectroscopy and survismetry. The 0.010 g% aq-LSNR and LGT were studied separately and with 0.010 g% (w/v) bovine serum albumin (BSA) from 283.15 to 315.15 K, and 0.002–0.010 g% @0.004 g% LSNR and LGT with 0.010% aq-BSA at 298.15 K. Their densities (ρ, g cm−3), viscosity (η, mPa s), sound velocity (μ, m s−1), surface tension (γ, mN m−1), friccohesity (σ, s cm−1), isentropic compressibility (Ks,ϕ, cm s2 g−1), activation energy (Ea, J mol−1), and the thermodynamic parameters enthalpy (ΔH, kJ mol−1), Gibbs free energy (ΔG, kJ mol−1), and entropy (ΔS, J mol−1) imply interactive activities with globular protein. LSNRs and LGT photocatalytically (PCR) and sonocatalytically (SCR) reduced methylene blue (MB), brilliant blue red (BBR), brilliant blue green (BBG) and methyl orange (MO). LGT recycled wastewater containing fluorescent dyes in ∼60 min while GO did it in 120 min and LSNRs in 24 h under sunlight (SL). Sonicated dyes with aq-LGT at 28 kHz for 5, 10, 20, 30, 60 min were reduced with 40–90% Φ compared to 98% with PCR in 1st order kinetics. The physicochemical properties (PCPs) of LSNR and LGT predicted their structural activities with reusability.
1. Introduction
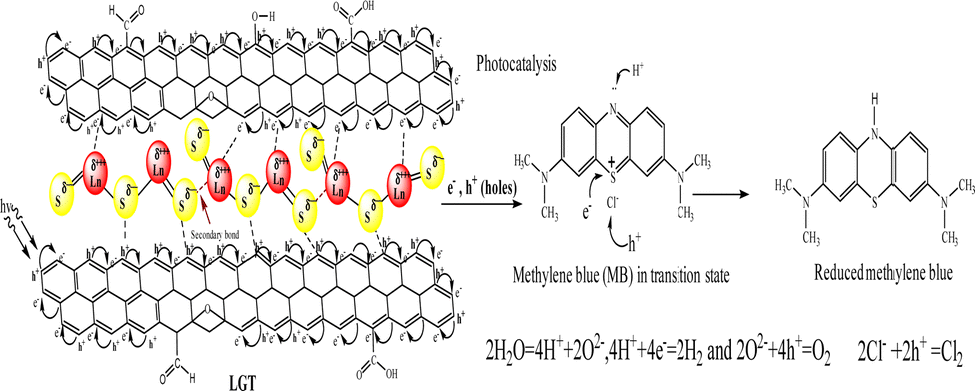
Lanthanide and GO individually have been widely used for research and industrial applications, but their interfaces have not yet been reported.1,2 Therefore, developing their LGT has been a novel research project and it was found to be relentlessly stable, reusable, and efficient photocatalyst to split water expeditiously compared to GO and LSNRs alone.3,4 Synthesizing new photocatalyzing nanomaterials for reducing wastewater effluents in sunlight is a biological and natural requirement for recycling. PCR splits wastewater into H2 and O2 gases, and also recycles its fluorescent effluents and transitional metals to collect usable water.5 For decades, the focus has been on developing photocatalyzing metallic organic frameworks, metallic biocomplexes, and bimetallic complexes using transitional metals, as reported in previous studies.5,6 Transition metals are comparatively easy to handle, and their crystalline water molecules are removed at NTP unlike the 800–1300 for lanthanide metals that hinders chemical processes.7 The crystalline 6H2O of LnCl3·6H2O is not detached by dissolving it in water at NTP, though, its Cl− dissociate and respond to AgNO3 but are unable to form Ln2S3. As a result, researchers have been heating LnCl3·6H2O up to 800–1300 to prepare Ln2S3 without understanding the contraction effect of 4f# electrons8,9 (Table 1). The 6s electrons shield a nuclear charge that affects the activities of 4f#e and the water dipoles are strongly bound with the nuclear charge, restricting lanthanides interacting with H2S for LSNR preparation and applications in the areas of photocatalysts, and adsorbents. The problem of preparing Ln2S3 by heating to detach a crystalline 6H2O from LnCl3·6H2O due to the energy barrier for electronic reduction of 6H2O is resolved by allowing a reaction with sodium metal at 50 with LnCl3·6H2O. The 3s1 electron of sodium metal reduces 6H2O to H2 and Ln(OH)3 (Fig. 3). The idea of synthesizing LSNRs and LGT for use as photocatalysts and adsorbents has never been initiated, despite huge industrial potential in areas of effluent disposal of infectious and hazardous biomedicals, and also, for solving the most tedious and hazardous chemical processes. LSNRs and LGT act as renewable energy storage systems and dopants with pharmaceuticals, digital inks, textiles, integrated circuits (ICs), and heat sensors.5,6,10,11 PCPs, PCR, and SCR predict their structural abilities to photocatalyzed and solubilize the biomolecules as BSA LSNR and LGT nanomaterials could also be doped and blended with a fine powder of waste plastics to functionalize and recycle them.12 LSNRs and LGT could structurally align GO and polymer composites for wider applications as supercapacitors and metallic nanocatalysts.8,9,13 The quantized interface (4f#e ∼ FE) could split light and water due to mutually resonating energy transfer for separating light.14 Thereby, LGT depicted as 4f#e ∼ FE could replace the expensive prisms used for splitting light for the faster photocatalysis of industrial effluents.15 LSNRs could also be doped with proteins, chitosan, carbohydrates/cellulose as biotemplates with a 4f#e ∼ FE interface for biophysical applications.16 LGT with 4f#e ∼ FE as an energy simulator could trap solar radiation for bio and piezoelectric sensors and minimize light scattering, and quantum confinements within optoelectronics.3,17 Unlike MOFs of transition metals, the lanthanide organic framework (LOF) could fill a need for applications with higher frequency of recycling and energy storage.18 The functional edges (FE) and π conjugations of GO with photons (hν) generates a wavefunction (ψ) in different phases (φ), but the 4f#e ∼ FE interface photocatalyzed a dye in a shorter time with an intensified redox cycle (ROC).15–17 LSNRs and LGT could affect salt bridges, electrostatics, and the peptide bonds of a protein by generating thermodynamic parameters tuned by 4f#e electrons.18,19 GO as an hν receptor has been a key factor in the fluorescence and photoluminescence of LGT.20,21 The 4f#e align GO with the lowest permittivity (ε) and quantum energy barrier (QEB) without causing electron–electron collisions. LGT with a highly resonating frequency of 4f#e and GO together attain outstanding optical, luminescence, and magnetic properties with extensive screening for diagnostic applications in the biological domain.22 LGT with balanced hydrophobicity and hydrophilicity could boost drug delivery, fluorescent probes, bioimaging, biosensors, and contrast agents in MRI scanning interaction with globular protein, but no study has been reported yet.22–26 The 4f#e ∼ FE interface as a new framework is aimed at separating out hazardous materials from biomedical waste effluents. PCPs of LGT suggest intensification of molecular interactions to recycle wastewater via hydrophobic and hydrophilic activities. The morphology, crystallinity, and redox activities of LSNRs have not yet been reported.26–28 The hydrophobic and hydrophilic interactions of Ln(NO3)2·6H2O with BSA were extended to LSNRs and LGT in previous studies.29 LSNRs and LGT could not unfold aq-BSA except by solubilising by using partial surface charges to interact with LGT. Aq-LSNR and LGT solutions align the dipoles of water that affects the adherence and cohesion of a solution with higher PCR and SCR activities interfaced with PCPs for understanding the QEB. The multifunctional LGT gets fitted with electrostatic dipoles, gaining a photon trapping ability for PCR and SCR activities. BSA interactions with partially soluble LSNRs and LGT could tune the activities of hormones and enzymes due to S2− with temperature.30 The shear stress implies a suspended BSA aligned in solution, so survismetry explores their alignments.31 The aligned dipoles in aqueous medium catalyse the reduction of dyes via photonic and sonication energies. Therefore, the rheology, surface energy, and thermodynamic parameters have been determined as a prerequisite database for PCR and SCR activities.
Table 1 Comparative study of lanthanide sulfide preparation with temperature
| Sr. no. |
Temp. (°C) |
Method |
Ref. |
| This study |
50–60 |
Crash reaction methodology |
|
| 1 |
800–1000 |
Hydrothermal process |
26 and 32 |
| 2 |
650–750 |
Solid phase reaction |
8 and 33 |
| 3 |
500–650 |
Solvothermal |
34 and 35 |
| 4 |
100–500 |
Hydrothermal reaction |
21 and 36 |
| 5 |
500–1000 |
Solvothermal |
37 and 38 |
2. Experimental section
2.1. Materials
Gt, H2SO4 (99.9%), H3PO4 (85%), KmnO4, H2O2, HCl, chloride hexahydrate salts of lanthanum, cerium, terbium, holmium, sodium metal, BSA, iron sulfide, methylene blue, brilliant blue R, brilliant blue G, and methylene orange were procured from Sigma Aldrich; petroleum ether (40–60) from Sisco Research Laboratory (SRL) India, and absolute alcohol from SCVUS Mandli India, and the solutions were prepared in Milli-Q water weighed using a ±1 × 10−6 balance.
2.2. Preparation of GO and GtO from Gt
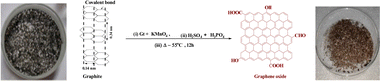
Graphite oxide (GtO) and GO were synthesized from Gt flakes with KmnO4 and H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H3PO4 in 8.5:1.5 ratio poured dropwise into an RB flask placed on an ice bath containing Gt and KmnO4 in 1:6 ratio. These were refluxed at 55 @650 rpm in an oil bath for 12 h to reach RT. To obtain pure GtO, ice-cold water was mixed with a reaction mixture on adding 3 mL H2O2 (30%) and stirred for 10 min to cease a reaction.5 The resultant product was centrifuged at 8000 rpm for 10 min and washed with Milli-Q water, HCl and absolute alcohol until the supernatant attained pH 7. AgNO3 and BaCl2 tests ensured complete removal of NaCl and Na2SO4 after 5 washings, and a brownish sediment was coagulated with petroleum ether. It was extracted to a Petri dish for drying at 60 for 24 h, a brown-colour solid GtO was obtained with good reproducibility and feasibility after repeating the procedure several times. GtO was exfoliated by sonicating at 28 kHz for 3 h to disrupt the ABAB sheet stacking of the Gt layers. GtO disperses brownish GO homogeneously39 (Fig. 1 and 2). The reaction possibly occurs as follows:
:H3PO4 in 8.5:1.5 ratio poured dropwise into an RB flask placed on an ice bath containing Gt and KmnO4 in 1:6 ratio. These were refluxed at 55 @650 rpm in an oil bath for 12 h to reach RT. To obtain pure GtO, ice-cold water was mixed with a reaction mixture on adding 3 mL H2O2 (30%) and stirred for 10 min to cease a reaction.5 The resultant product was centrifuged at 8000 rpm for 10 min and washed with Milli-Q water, HCl and absolute alcohol until the supernatant attained pH 7. AgNO3 and BaCl2 tests ensured complete removal of NaCl and Na2SO4 after 5 washings, and a brownish sediment was coagulated with petroleum ether. It was extracted to a Petri dish for drying at 60 for 24 h, a brown-colour solid GtO was obtained with good reproducibility and feasibility after repeating the procedure several times. GtO was exfoliated by sonicating at 28 kHz for 3 h to disrupt the ABAB sheet stacking of the Gt layers. GtO disperses brownish GO homogeneously39 (Fig. 1 and 2). The reaction possibly occurs as follows:
 |
| | Fig. 1 GO preparation from Gt flakes. | |
 |
| | Fig. 2 LSNR preparation. | |
GO functionalization
| 6KMnO4 + 3H2SO4 + 4H3PO4 → 3K2SO4 + 2Mn3(PO4)2 + 9H2O + 15[O] |
and
| KMnO4 → K+ + Mn7+ + 4O2−, |
2.3. Crash reaction methodology (CRM) for LSNR synthesis
Crushed Na metal taken in a boiling tube was heated to 50 °C for ∼1 min, then LnCl3·6H2O(s) was added in 1:6 ratio. It sparked the production of H2 gas and Ln(OH)3 within nanoseconds, and the mechanism is denoted CRM.40 To solubilize Ln(OH)3, water in 1:4 ratio was added to homogenize the solution (Fig. 1). An excess of H2S gas as a source of S2− was passed (10 min) to react with Ln3+ to produce Ln2S3. The H2 gas, NaCl and NaOH were confirmed with gas chromatography, AgNO3 and phenolphthalein, respectively (Fig. 3). The 3s1 Na reacted with 6H2O to disrupt its dipolar linkage with Ln3+, generating H+ that received 3s1 as 2H+ + 2e → H2 (Fig. 3) as follows:
| LnCl3·6H2O + 6Na → Ln(OH)3 + 3NaCl + 3NaOH + 3H2↑ |
| 2Ln(OH)3 + 3H2S → Ln2S3 + 6H2O (LSNR) |
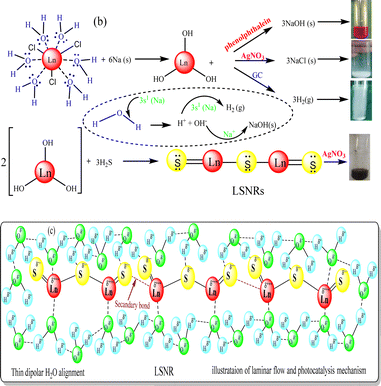 |
| | Fig. 3 (a) CRM with evolution of H2 gas, (b) LSNR confirmatory test and (c) alignment. | |
A temperature of 50 °C ignited 3s1 to gain adequate oscillations which simultaneously interfaced with H2O to dissociate it. The product was washed with Milli-Q water 4/5 times to remove NaCl and NaOH. The whitish to greenish colour confirmed LSNRs which with AgNO3 had formed black precipitate implying S2− (Fig. 3). Ln2S3 was washed 2–3 times and dried at 50 °C for 2 h in a vacuum oven.40
La2S3 (S2− = Ln3+–Ln3+ = S2−) with 4f0e have developed cohesive forces and with less kinetic energy have aligned to form linear rods (Fig. 4), although the white patches imply thick nanorods (NRs) due to linearly aligned S2− and Ln3+. The S2− oscillate to align LSNRs and 4f#e in different φ. The double bonds of S2− = Ln3+– and –Ln3+ = S2− and both double bonds of S2− = Ln3+–S2− = S2− align linearly with the spatial lattice as NRs. The oscillation of the double bonds of S2− = Ln3+–S2−–S2− affects the electronic activities of Ln3+ and S2− due to electrostatic interactions at the terminals. There are no possibilities of charge oscillations as their ψ2 is attained and Ce2S3 (4f1e) has created a cage-like structure compared to La2S3 with maximum oscillations in different φ. Additionally, Tb2S3 with 4f9e shows a finer distribution for developing a sheet-like arrangement. Ho2S3 with 4f11e shows perfect NRs on aligning the S2− with the conduction band (CB) (Fig. 4). The LSNR has one dimension (1D) due to having a wavefunction of the unoccupied state of the lone pair of electrons of the sulfur atom including its negatively charged domain facilitated by 4f#e (Fig. 4a1). The secondary bond of Ln3+ and S2− static ions help to equipartition their residual electronic charges through quantizing their wavefunctions, generating communication for their spatial alignment as a state. Thus, their eigenenergy and eigenfunctions generate a ψ(x, t) × ψ*(x, t) density of energy state (DOS), which was elucidated with elemental diffraction X-ray spectroscopy and CHNS/O analysis. Our previous study of GO, Pr2S3–GO, Sm2S3–GO, Eu2S3–GO, and Gd2S3–GO used HR-TEM images with 20 nm range to infer 1D nanorods.40 Additionally, the HR-TEM analysis of Gd2O3 doped with Eu3+ and capped with PVP functionalised with HSA maintained the nanorod dimension reported in our earlier study.41 A similar methodology to that of LSNR and LGT was extended to the present study where the 4f#e of LSNR and FE of GO have been coordinated through their respective ψ#4fe and ψFE to initiate the ψ#4fe(x1, t) × ψFE(x2, t) DOS. The x1 and x2 positions of the 4f#e and FE, respectively, differ infinitesimally to generate (q#4fe, qFE)/4πεr2, as the intermixing distance r is approachable. These highly inclusive electronic states, as per the Born–Oppenheimer approximation, optimize and simulate the electronic and nuclear charges, leading to coordination via the S2− ions of an LSNR with consecutive LSNRs. Thereby, the changes occur at nanometre distance, causing stacking LSNRs to stack linearly, as in Fig. 4a–d. SEM images of La2S3, Ce2S3, Tb2S3, and Ho2S3 imply linear rods. However, they have formed a bundled state on combining close to each other, but the dimension remains broadly 1D.
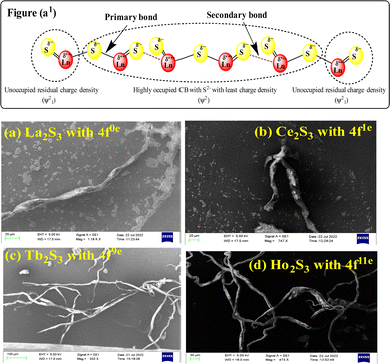 |
| | Fig. 4 (a1) Preferred interaction for LSNR formation and SEM images (a) La2S3 (b) Ce2S3 (c) Tb2S3 (d) Ho2S3. | |
2.4. LGT synthesis via sonothermal process
GO (mg mL−1) was sonicated for 3 h to disperse the sheets in water to generate FE (Fig. 5a). LSNR powder in GO solution with 1:5 ratio (dopant:matrix) was sonicated for 30 min at 60 and cooled to RT (Fig. 5). GO sheets with a floppy structure engaged with LSNRs for doping. (Fig. 5d). After the LGT was washed with H2O to remove LSNRs weakly bonded with GO, it was put through a centrifuge for 8 min at 8000 rpm. The LFT was dried in a vacuum oven for 2 h at 50 as shown below:
| GO + ≊ → GO*, GO* + Ln2S3 → Ln2S3:GO (LGT) |
Spatial alignments and the geometrical appearance of LGT are shown in Fig. 5.
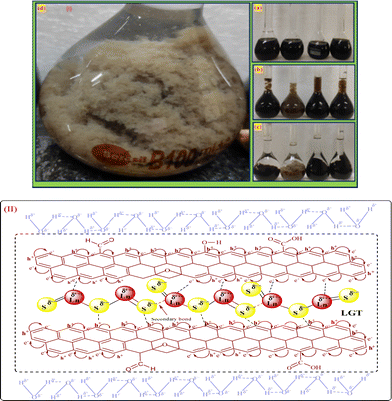 |
| | Fig. 5 (I) LGT preparation and (II) alignment. | |
2.5. Characterizations
UV/Vis spectra were recorded with a Shimadzu UV1800 double beam spectrophotometer and X-ray diffraction (XR) with a Bruker Advance-D8 diffractometer with Cu Kα radiation (λ = 0.15406 nm, 40 kV, 40 mA). TGA was conducted in an N2 environment on an EXSTAR TG/DTA 7300 to analyze thermal stability. C, H, N, and S analysis was undertaken with a Euro-vector EURO EA 3000, and FESEM + EDS with a JEOL JSM 7100F. The Raman spectra were recorded with a Horiba HR 800 instrument with a 480 nm Ar laser. Functional groups (FGs) were analysed with an FT-IR spectrometer (PerkinElmer, Spectrum 65), H2 evolution with a gas chromatograph (PerkinElmer, Clarus 680), and PCPs and density with a Mansingh survismeter (Cal.06070582/1.01/C – 0395,NPL,GOI) and Anton par Densimeter-5000, respectively.
3. Results and discussion
LnCl3·6H2O with a stronger nuclear charge can hold 6H2O and detach 3Cl− in aqueous solution in response to AgNO3. Na+Cl− with H2S gas developed Na2S but was unable to develop Ln2S3. Heating LnCl3·6H2O could not thermally induce electronic spin to overcome a nuclear charge being applied on 6H2O. LnCl3·6H2O reacted directly with H2S gas but did not form LSNRs. The NaOH added in aq-LnCl3·6H2O gives NaCl and free S2− to capture Ln3+ by weakening their inner hydration sphere (IHS). The weaker IHS of LnCl3·6H2O attracts free S2− and forms a cloudy cluster around Ln3+ (Fig. 6).
 |
| | Fig. 6 Direct sulfurization with aq-NaCl and LnCl3·6H2O. | |
Previous researchers could not analyse the electronic bonding of 6H2O with LnCl3 in areas of lanthanide chemistry (Table 1). LGT with 4f#e with a resonating energy tuned the biophysical and biochemical activities of BSA for an interfacial study extendable to haemoglobin via the holding/releasing ability of O2 and CO2 gases. Solute and solvent interaction affect various activities, where PCPs of LSNRs and LGT imply alignment to enhance the PCR and SCR. Survismetry of GO, LSNRs, and LGT in aqueous solutions through a uniform capillary has elucidated their incompressible dipoles interfaced with PCR and SCR activities. The aligned dipoles of LGT could have minimized the localized collisions of e− and h+ holes by avoiding their recombination due to a robust friccohesity resonating energy transfer (FRET). In the reaction LnCl3·6H2O → Ln3+ + 3Cl−, in addition to the 6H2O molecules, infinite dipoles of H2O align with Ln3+, developing the strongest electrostatic interactions. These activities spontaneously favour the generation of electron-deficient ψLn3+ and ψOH2 clouds. These clouds are highly stable even when dissolved in H2O, though the 3Cl− anions get dissociated, which further exceptionally reorients the H2O dipole to optimize the negatively charged clouds of H2O with maximum hybridization. The Cl− had hurdled the alignments of H2O to LnCl3·6H2O, but after the removal of the 3Cl−, an infinite number of H2O had surrounded the Ln3+, which interfered with the bonding the S2− supplied on passing H2S. These activities are supported with 4f#e of lanthanide due to 5d and 6s suborbitals sharing a nuclear charge that weakens the attraction of the 4f#e for a nuclear charge. The dissociated Cl− gave a positive test with AgNO3 as a white AgCl cloud while it was noted that strongly hydrated Ln3+ could not respond to H2S or Na2 (Fig. 6). The chemical species Na+ + S2− and H+ + S2− obtained on dissociation of the Na2S and H2S respectively could not attract the hydrating H2O dipoles. As the dipoles were electronically bonded with Ln3+H2O–Ln3+–6H2O and confined to quantum dots (QD–Ln–NHS) highly intensified with a highest energy. This energy acted as non-permissible reaction so the S2− could not penetrate the QD–Ln–NHS. Hence, the large S2− could not penetrate QD–Ln–NHS, so aq-Na2S could not develop Ln2S3. The S2− could not cause adequate spin, rotational, vibrational, or translational movement compared to a 3s1 electron due to the least size that could enter the QD–Ln–NHS to reduce the H2O (Fig. 3a). The 2H+(2H+ + 2e− → H2) + 2OH− and Na+ expeditiously develop NaOH. Another negative factor in using Na2S is that it is dangerous for the H2O molecule to develop a cationic hydration sphere. The Na metal with a 3s1 electron is not a danger, but rather is a highly favourable mechanism where the spin of the 3s1 electron gains a robust ψ to overcome a quantum tunnelling effect to separate out the hydrating H2O dipoles. Hence, there are no electronic or vibrational energies to facilitate the Na-3s1 electron reaching the QD–Ln–NHS unlike S2−. The energy which is expeditiously released during the reaction excites the electron of the hydrating H2O molecule of LnCl3·6H2O and separates it out. A precaution which is maintained to synthesize LSNRs using Na-3s1 is that small amounts of both the reagents were taken. Additionally, Na2S acquires energy from the reacting system to dissociate the 2Na+ + S2−, which attracts the cation (CHS) and anion (AHS) hydration spheres, weakening the activities of the H2O medium to affect the alignment of the CHS and AHS around QD–Ln–NHS, to not only immobilize the structural activities of the H2O medium but also remove the chemical potential (μ) to develop Ln2S3 as a = eμ/RT activities. Several reactions with Na2S, H2S, K2S, Ca2S, and others were conducted which could not produce Ln2S3. The Na-3s1 electron is easily accessible to the Hδ+ population and could not reduce Ln3+ to Ln0 metal, avoiding the Ln3+ + 3s1 → Ln0 reaction and instead consuming the Hδ+ charges. The present mechanism is safer as almost all the H2O molecules are either strongly hydrogen bonded where a major chunk of H2O is hydrating Ln3+ and 3Cl−. Hence, the Na metal was warmed up to 50–55 to react with Ln3+. Had there been free H2O, there would have been a stronger reaction. Detailed studies of the CRM were reported in our previous study.40 Lots of trials were made to synthesize the LSNRs by avoiding a direct heating to 800–1300, as reported in earlier studies (Fig. 6 and Table 1). Developing the infrastructure for generating 800–1300 is very difficult and dangerous, unlike using Na metal. The flame shown in Fig. 3a belongs to the H2 gas which was produced during the reaction. Of course, by developing pressure within a few seconds it is diffused. The H2 gas was intensively confined to a flame as we wanted to confirm it with a popping sound and a yellowish flame. However, provision was made to generate Ln3+ by allowing contact with a small amount of Na metal by passing it through a conveyor belt to the reaction chamber and attracting the resultant Ln3+ to H2S gas to develop Ln2S3 + H2. Hence, the conveyor belt system is highly scalable.
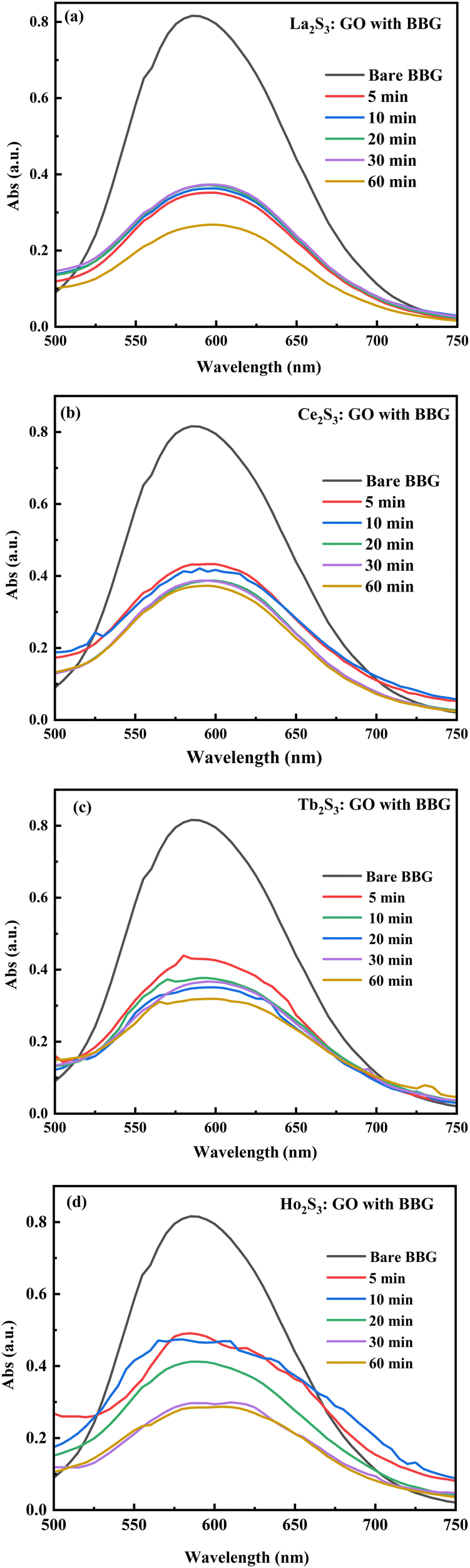
LGT and LSNRs with a smaller surface area induce stronger interactions between the δ− and δ+ aligned dipoles of H2O. The 4f#e of LGT oscillates with ψ and minimized ε for expeditious PCR and SCR activities of dyes, causing the uniform alignment shown with PCPs. The electrostatic dipoles of H2O interact with monodispersed aq-LSNR and aq-LGT, so a laminar flow was studied from 0.002 to 0.10 g% @ 0.004 g% to align them (Fig. 13). The and values do not vary on increasing the g% of LSN and LG, as they could act in ordered alignment as a supercapacitor (Tables 5 and 6). The water dipoles and the aligned LSNR:GO develop a pendant drop number (pdn) saturated at 298.15 K with new surfaces (Fig. 14b). LGT formed a large size drop with increasing g% due to alignment with a more strongly adhered solution and multiple nanohydration spheres (NHSs) that isolated their cohesive forces (Fig. 6). Aq-LGT adheres to a solid glass surface by developing a pdn in continuous flow, cohesively holding a solution with an adequate cohesive force of adhered water to LGT and surrounding water molecules. This action develops the size and length of a pdn due to stronger adhesive and cohesive forces of H2O to LGT and with itself. The lower surface area of a drop implies H2O adhered to LGT forming an NHS around the LGT, weakening the H2O–H2O electrostatic interactions (Fig. 7). GO, LSNR, and LGT act as competent photocatalysts to PCR and SCR fluorescent dyes in aqueous solution. On receiving hν, the surface area and surface energy, with their realignment and reorientation in the photocatalyzing liquid mixture, have become the most critical interfaces. The photocatalysts on receiving the hν haphazardly collide, increasing the probability of holes recombining, so the hν may not generate effective quantum dots to cause hν to excite the electron. Hence surface enhanced Raman spectroscopy (SERS) highlights the surface activities reported in our previous study.3,40 Additionally, due to the surface energies, the molecules develop NHSs, which could combine via the cohesive force that develops pdns of different sizes, affecting the photocatalysis. Thereby, the compositions of GO, LSNRs, and LGT generate the surface activities which influence the width of the pdn with a lower surface area. Fig. 7 shows that the 0.002, 0.004, 0.006, and 0.010 g% LSNRs respectively increase the size of the pdn. The 0.010% sample has a comparatively longer pdn due to stronger cohesive forces with slightly milder adhesive forces with a lower tip of the hanging capillary with the controlled pressure unit (Fig. 7). This mechanism of milder adhesion, with either a concentrated photocatalyst or a dye with comparatively stronger cohesive forces, by itself increases the QEB to hamper the photocatalysis. The pdn decreases as the cohesive forces are strengthened with increasing composition, which could reduce the quantum yield (Fig. 7).
 |
| | Fig. 7 Increasing size of drop with @g% due to lower surface area. | |
GO has been synthesized and widely characterized with XPS, XRD, TGA, HR-TEM, SEM, and EDX reported in our previous studies.42 Widely and vividly reported studies suggested that GO has a 2D sheet structure with hexagonal units with moderate VWF. GO has been functionalised by generating oxygenated FGs, which further realigned the 2D GO sheet with slightly different intersheet VWF causing the lowest relaxation time. These structures have been extensively studied with stretching frequency and bending frequency (Fig. 9 and Fig. S2.9, ESI†). The FT-IR analysis for LSNRs and LGT individually elaborated the spatial geometry with specific rotational, vibrational, and electronic motions integrated as stretching and bending frequencies. The C–C–C atomic domains of LGT for 1900–3000 cm−1 are almost linear compared to a GO matrix, which indicates the activities of the FGs. These activities were minimized and FGs were used in doping the LSNRs. But dopants with specific doping activities to tune the C–C–C atomic domains are lacking for individual LSNRs (Fig. S1.1, ESI†). The S–S atomic oscillations are depicted with the pattern of split of stretching and bending frequencies where the integrated 2D GO sheet with highly stable C–C–C atom domain that could not split the stretching and bending frequencies. These fundamental patterns exclusively elucidate the structure of GO, LSNRs, and LGT. Intrinsic friccohesity correlates the stretching and bending frequencies of GO, LSNRs, and LGT as the intrinsic electronic configurations of each adjoining atom rotate, vibrate, and translate with specific oscillations. These generate specific patterns of electron–electron repulsion, electronic collision, and mean free path, as per Lorentz activities. These oscillations generate a specific pattern in TGA, DTG, and DSC (Fig. 10). GO generates a single sharp line reflecting its single lattice structural pattern. However, the single sharp line is transformed into different patterns with the LSNR spatial lattice, implying that the 4f#e of LSNRs seem to coordinate for mutual optimization. Hence the specific patterns of the LGT differ from those of GO with faster photocatalyzing activities. These changes imply the participation of the FGs of GO and the 4f#e of the LSNRs due to their structural alignments. FT-IR and TGA analyses have extensively elaborated the originality and functionality of their structures, developing specific patterns of wavevector and lattice, as depicted by the XRD pattern (Fig. 8). The specific characteristics of GO and the lattice are depicted with XRD. The specific features express the GO skeleton doped with LSNRs, as depicted with an XRD pattern for each LGT (Fig. 8). The XRD pattern of GO differs from that of the LGT which authentically indicates the doping mechanism due to the interaction and optimization activities of the FGs and 4f#e of LSNRs. Thereby, FT-IR, TGA, and XRD depict the structural lattice and features expressed as an energy spectrum denoted as DOS. These specific structural patterns supported by specific patterns of their morphology are depicted with SEM. The DLS data and distribution patterns of GO and the LGT in a most controlled pattern indicate the structural morphology and topography, which further synchronize their interacting PCP profiles. Structural illustrations focussing on almost each and every atom and their linkages in GO, LSNRs, and LGT indicate the stability of their structural frameworks despite subjecting them to photons. The elemental analysis further gives their structural constituents and their ratios to predict their presence in the structural lattice (Table 2). EDX spectroscopy was run on the samples, which elucidated their structural frameworks with their elemental composition (Table 3). Reports of our previous study with LSNRs and LGT of Pr2S3, Sm2S3, Eu2S3, and Gd2S3 included XPS that confirmed the elemental composition and participation of each constituent stabilizing their resultant lattices.40
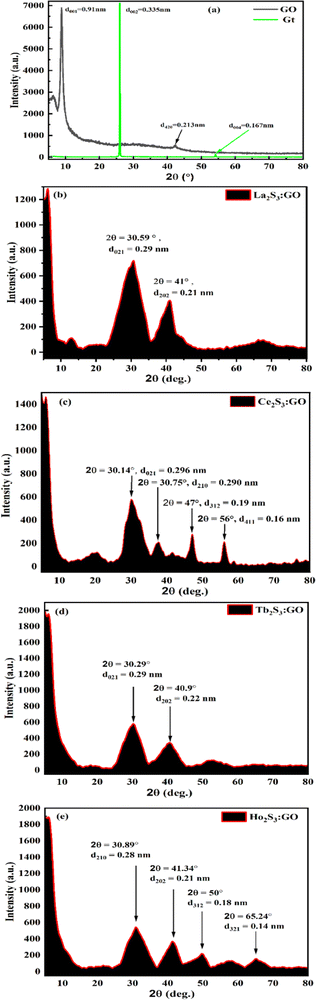 |
| | Fig. 8 XRD of (a) bare GO, (b) La2S3:GO, (c) Ce2S3:GO, (d) Tb2S3:GO, (e) Ho2S3:GO. | |
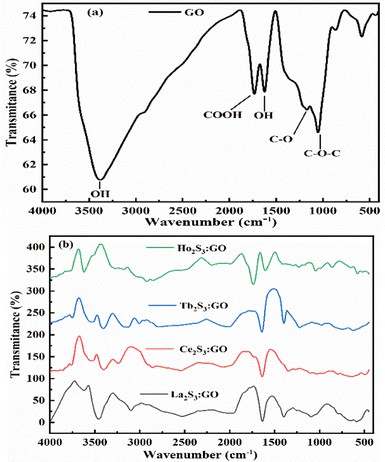 |
| | Fig. 9 FT-IR spectra analysis: (a) GO, (b) LGT. | |
Table 2 Elemental composition analysis for LSNR, GO, and LGT
| Sr. no. |
Name |
C (%) |
H (%) |
S (%) |
Weight (mg) |
| 1 |
GO |
60.558 |
3.306 |
— |
1.0752 |
| 2 |
La2S3:GO |
18.782 |
2.669 |
2.134 |
1.0756 |
| 3 |
Ce2S3:GO |
20.915 |
1.799 |
2.642 |
1.0995 |
| 4 |
Tb2S3:GO |
19.264 |
2.480 |
1.764 |
1.0299 |
| 5 |
Ho2S3:GO |
25.024 |
2.475 |
1.964 |
1.0295 |
3.1. X-ray diffraction analysis
The single sharp GO peak at 2θ = 8.83°, despite treatment with Gt, means a stronger oxidising mixture could not disrupt the covalent bond between C atoms, but it weakened the van der Waals forces (VWF) binding the GO sheets. The GO matrix accommodated a lesser amount of LSNR dopant due to 4f#e in the doping43 (Fig. 8a–e). The Gt plane at (002) is shifted to (001) in GO due to the intersheet distance (Fig. 8a, green line). LSNRs with LGT has reduced the intensity of minor peaks from 7000 for GO to 2200 for LGT8. The LGT generates a new intensity due to ROC of h+ and e− holes resulting in lower intensity (Fig. 8a–e). XRD suggests there is no dissociation, with stronger interatomic forces generating prominent peaks at 2θ = 30.59° and 41° for La2S3:GO with 0.29 and 0.21 nm d-spacings, respectively.27 While Ce2S3:GO has peaks at 2θ = 20°, 30.14°, 30.75°, 47° and 57° with 0.443, 0.296, 0.290, 0.190 and 0.160 nm d-spacings of the lattice plane, respectively.44 This has direct implications for PCPs, as a lattice plane develops different NHSs according to the d spacing. This indicates the relevance of PCPs to PCR and SCR, as the alignment of water affects electronic transitions. Tb2S3:GO with five half-filled electrons from the valence band (VB) to the conduction band (CB) causes the maximum number of collisions, unlike Ce2S3:GO, which generates sharper peaks at 2θ = 30.29° and 40.9° with d-spacings of 0.29 and 0.22 nm. Ho2S3:GO has peaks at 2θ = 30.89°, 41.34°, 50° and 65.24° with 0.28, 0.21, 0.18 and 0.14 nm d-spacings, respectively, with 4f11e with four CBs that split into multiple peaks after with robust collisions, while the GO sheet remains structurally intact. LGT aligned with different peaks as GO is aligned at 2θ = 8.83° with 6900 a.u. The FG synchronizes with a continuous electronic plane to interface the 4f#e ∼ FE collisions. The electronic configurations with higher d spacings for GO suggest stronger reducing activities as the FG might have pushed the LSNRs into the GO intersheet due to a higher mean free path (Fig. 8).
3.2. FT-IR analysis
The bands at 3375, 1726, 1167 1051 and 1625 cm−1 for the stretching of –OH, –COOH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O–C and OH, respectively, come GO alone (Fig. 9a). These FGs are aligned with different intensities around the various paths developing different lattices (Fig. 8). Their geometries influence the shear and surface energy for a PCP sensor. The bending vibrations of –OH, epoxide (–O–), and the skeletal rings of GO occur at 1624 cm−1 with a small stretching vibration hump at 1372 cm−1 from tertiary C–OH. The PCPs have repeated their state functions for conversion into the resulting product. The LGT has aligned the GO sheet to quench undesirable π → π* transitions of FGs and residual potential charges. FE exists with Ce2S3:GO and d-spacing with the restrictions, unlike Tb2S3:GO and Ho2S3:GO with 4f9e and 4f11e, respectively. A new band at 1633 cm−1 indicates LSNR:GO optimization by FGs and 4f#e transitions. The stretching depends on electron–electron repulsion of 4f#e with nuclear charge.36 The Born–Oppenheimer approximation optimizes LSNRs via 4f#e ∼ FE clouds on the metal surfaces. The S2− of LGT might not have stretched except for a minor simulation, as stretching from 2000 to 3000 cm−1 indicates the C–C, C–H, CC, and aromatic ring of LGT (Fig. 9). The stable GO sheet with prominent peaks decreases the stretching intensity. Multiple splits in stretching indicate the collision of 4f#e on interacting with an electronic cloud of FE (Fig. 9). The stretching of an FG with milder peaks within 500–1000 cm−1 for La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO indicates GO doping with LSNR dopants (Fig. S2.9, ESI†). C–C is stabilized as FGs attached to the C atom are aligned to accommodate the dopants.40
O, C–O–C and OH, respectively, come GO alone (Fig. 9a). These FGs are aligned with different intensities around the various paths developing different lattices (Fig. 8). Their geometries influence the shear and surface energy for a PCP sensor. The bending vibrations of –OH, epoxide (–O–), and the skeletal rings of GO occur at 1624 cm−1 with a small stretching vibration hump at 1372 cm−1 from tertiary C–OH. The PCPs have repeated their state functions for conversion into the resulting product. The LGT has aligned the GO sheet to quench undesirable π → π* transitions of FGs and residual potential charges. FE exists with Ce2S3:GO and d-spacing with the restrictions, unlike Tb2S3:GO and Ho2S3:GO with 4f9e and 4f11e, respectively. A new band at 1633 cm−1 indicates LSNR:GO optimization by FGs and 4f#e transitions. The stretching depends on electron–electron repulsion of 4f#e with nuclear charge.36 The Born–Oppenheimer approximation optimizes LSNRs via 4f#e ∼ FE clouds on the metal surfaces. The S2− of LGT might not have stretched except for a minor simulation, as stretching from 2000 to 3000 cm−1 indicates the C–C, C–H, CC, and aromatic ring of LGT (Fig. 9). The stable GO sheet with prominent peaks decreases the stretching intensity. Multiple splits in stretching indicate the collision of 4f#e on interacting with an electronic cloud of FE (Fig. 9). The stretching of an FG with milder peaks within 500–1000 cm−1 for La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO indicates GO doping with LSNR dopants (Fig. S2.9, ESI†). C–C is stabilized as FGs attached to the C atom are aligned to accommodate the dopants.40
3.3. C, H, N/S analysis
The structural changes are supported by the spatial arrangement of the constituent elements in the highest reproducible primitive lattice as a uniform wavevector of C, H, and S detected with a C, H, N, and S analyser, while the constituents of LSNRs and LGT are detected with elemental diffraction spectroscopy (Tables 2 and 3).
Table 3 Elemental composition via EDS for LSNRs, GO, and LGT
| System |
Element |
Weight (%) |
Atomic weight (%) |
| Tb2S3 |
Tb |
88.24 |
60.23 |
| S |
11.76 |
39.77 |
|
|
| Ho2S3 |
Ho |
97.17 |
86.97 |
| S |
2.83 |
13.03 |
|
|
| GO |
C |
69.57 |
75.28 |
| O |
30.43 |
24.72 |
|
|
| Tb2S3–GO |
C |
55.90 |
64.10 |
| O |
40.42 |
34.65 |
| S |
1.04 |
1.02 |
| Tb |
2.64 |
0.23 |
|
|
| Ho2S3–GO |
C |
56.19 |
63.84 |
| O |
41.23 |
36.01 |
| S |
1.01 |
0.06 |
| Ho |
1.57 |
0.13 |
3.4. FESEM with EDS analysis
The morphologies of GO, LSNRs, and LGT have already been explained with SEM (Fig. 14). There is a further explicit focus on morphology with FESEM including elemental diffraction X-ray spectroscopy to explore their nucleating frameworks, elucidating their most primitive alignments (Fig. 10). Their morphologies elucidated their surface profiles at 1 μm, 10 μm, and 100 nm for GO, LSNRs, and LGT. The LSNR morphology suggests workable structural frameworks that constitute the specific atomic bonding and the sheet arrangements (Fig. 10B and D). GO constitutes the most symmetric and synchronized 2D sheet, closely interfaced with moderate VWF or Lennard Jones potential, as per the Prigogine intrinsic entropies and the Born–Oppenheimer approximation. The morphology which was further elucidated with a 1 μm highly expanded view of the 2D GO sheet could be defined as top-to-bottom and bottom-up with a few areas and edges sharply diffracting laser light. Hence, this is a bottom-up tentropic surface with comparatively localised and ordered surface entropies within the closest interlink to resonate their energies for mutual transfer with single-valued ψ, enabling a PCR. The front view of the 1 μm scale morphology further indicates synchronized 2D GO sheets with the most symmetric upridges noted as basement spaced 2D GO sheets able to sense each minimum temperature and pressure fluctuation. These have been highly resolved at a scale of 100 nm; however, the longer 2D GO sheet with a structure similar to a carbon-fibre-like structure also reflects laser light (Fig. 10A). One side of the 2D GO fibrous sheets are anchored or adhered to a longer with few layered sheets. These sheets are highly and constantly fixed with their C atoms obtaining the 100% hybridization of potential energy via |ψc−1|, |ψc−2|, |ψc−3|…|ψc−n| = 100% hybridization. The index n indicates the number of C atoms. Hence, 2D GO fibrous sheets slightly differ in an equipartitioning of energy, where their residual energies seem to facilitate a well-defined ordered tentropy with explicit friccohesity chemistry by balancing the PE of the basement sheet and the KE of fibrous sheets developing a remarkable interfacial φ to equilibrate the φFibrous × φBasement matrix to balance a higher stability with definite FEs and hexagonal sheets. Therefore, |φFibrous| ≈ |φBasement| develops an energetic way of sticking to the higher surface area that could act as an anticoagulant, obeying autocatalytic friccohesity interfacial restrictions.
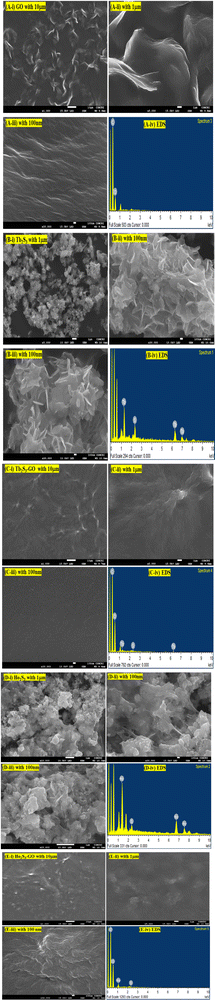 |
| | Fig. 10 The morphologies of GO, LSNRs and LGT obtained with an X-ray spectrometer (A, i–iv) GO, (B, i–iv) Tb2S3, (C, i–iv) Tb2S3:GO, (D, i–iv) Ho2S3, (E, i–iv) Ho2S3:GO. | |
The morphology of the GO differs from LSNRs, as LSNRs could not act as 2D sheets, but rather act as one-dimensional entities with more residual energies at the terminal 3S2− and 2La3+. These spatial DOS could align them in a 3D energetic expression of LSNRs due to residual or electron-rich and charged defect-rich domains (Fig. 4a1). These defect-rich domains occupy the spatial dimension with ψS2− ≈ ψ2La3+ within the energy clouds of S2− and La3+. These develop a cloud-like DOS with specific virtual wavevectors where the real nanorod alignments of these LSNR entities stabilize them (Fig. 10B and D) expressed as the Schrödinger equation:
| | | Eψ = [p2/2m + V(x, t)]ψ | (1) |
E is total energy of 4f
#e and
ψ is the wavefunction,
p is momentum,
m is mass, and
V is the potential energy (PE) at position
x at time
t. The position
x at time
t remains synchronized to symmetrize the alignments of the LSNRs. Positions
x1,
x2,
x3,…,
xn align symmetrically with virtual boundaries. Hence, Tb
3+ with 4f
8e probably strengthens the bonding with S
2− (S
16 = 1s
2, 2s
2, 2p
6, 3s
2, 3p
4) where 3p
4 has UNPE 3p
2x, 3p
1y, 3p
1z. Hence, the electronic activities of 3p
4e (S) and 4f
8e (Tb) lead to the development of very functional interfacial adherence with highly optimized energies from 10 μm and 100 nm (
Fig. 10B). The most novel alignment of the
ψ is
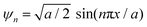
, where the
ψ could have π/2, π, and 2π coordinates of the wavevector. Hence, these hardcore wave mechanics laid down a function of interfacial alignment of the LSNRs and 2D GO sheets (
Fig. 10). Therefore, the LGT attained a matrix with the most QDs, highly integrated with the uniformly distributed lattice of the LSNR ≈ GO energy plane without disrupting the basic structural framework of the LSNRs and GO. In fact, at 100 nm scale highly uniform energetic ODs are seen without energy spreading. Hence, the
ψLGT ≈ [∂
p2/2
m ±
V(
x,
t)]
ψ integrate with the most synchronized arrangement depicted with the sharpest single light order.
These are compared with Ho2S3:GO having 4f10e without QD rates, where the most symmetric sin waves are depicted (Fig. 10E). Hence, the electronic cloud of 4f10e influences the QDs of GO. The elemental compositions of GO, LSNRs and LGT are included in Table 3.
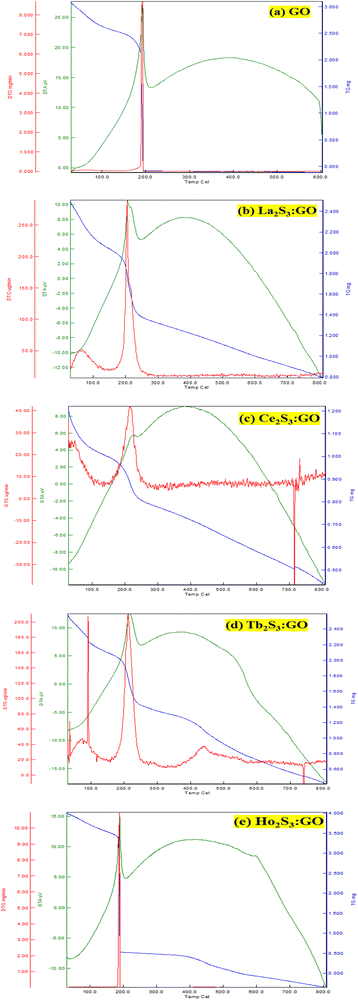
3.5. TGA, DTG and DTA/DSC analysis
Ln2S3:GO responds to a rising temperature due to oscillation of 4f#e ∼ FE interfaces: see the overlapped DTG and TGA curves of LGT with weight (wt) loss in three steps (Fig. 11a–d). La2S3:GO decomposes at 100, 221 and <800 with 20.77, 15.96 and 63.27% wt losses due to moisture, and single-bonded and double-bonded S2− with LGT surfaces, respectively. A maximum decomposition rate at 204 with a sharp exothermic peak predicts that La2S3 will interact strongly with the FEs of GO. The La2S3:GO and e−–h+ interfacial energy optimized as q+q−/4πεr2 weakens ΔH release. Ce2S3:GO at 215 and 717 lose maximum weight with sharp exothermic and endothermic peaks as 4f1e interacts with FE, causing higher stability than GO (Fig. 11a). There are 27.4, 24.74 and 47.86% wt losses of Ce2S3:GO at 138, 246 and <800, respectively (Fig. 11c). Tb2S3:GO with 20, 31.46 and 48.54% wt losses at 119, 235 and <800 has sharp peaks with maximum decomposition rate at 92 and 212, respectively. Synchronized LSNRs and GO of LGT with a few new weight losses indicate their interfacial activities. The 4f9e synchronized electronic cloud of Tb2S3:GO and Ho2S3:GO with 4f11e as a single crystal equalizes the PE to KE. Two sharp exothermic peaks indicate maximum energy release on interaction with GO due to its aligned FGs.
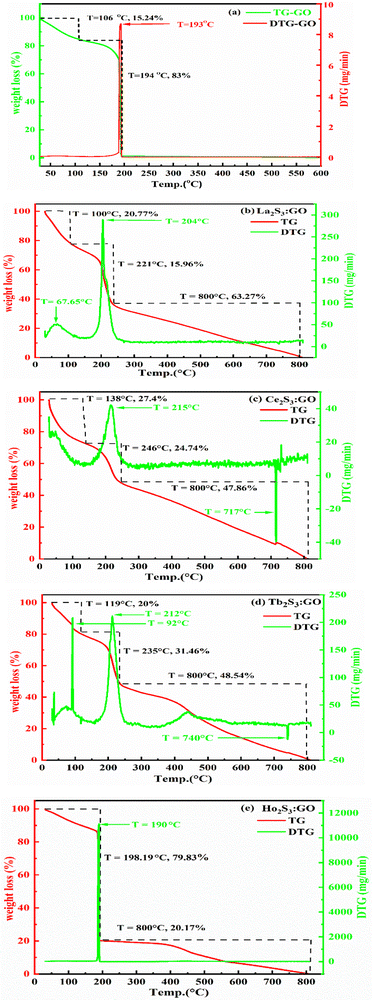 |
| | Fig. 11 TGA and DTG of GO and LGT. | |
The 4f9e of Tb2S3 with GO enhanced LGT stability (Fig. 11d). The maximum decomposition of Ho2S3:GO at 190 with a sharp exothermic peak through 79.83 and 20.17% wt loss does not suggest a crystalline structure. Ho2S3:GO shows a sharp weight loss at 198.19, as Ho3+ strongly aligns H2O dipoles as well as FE interfaces. Therefore, Ho2S3:GO reduces dyes in a shorter time by minimizing the collisions to increase the mean free path to overcome the QEB (Fig. 11e). The 4f#e enhance the thermal stability of both LSNRs and LGT by reducing the PCR time due to having a similar FE to GO (Fig. S1.0 and S1.1, ESI†). The difference in weight loss between LSNRs and LGT suggests stability due to doping. LSNRs alone lose weight in two steps due to the two strong interfacial forces of 2Ln3+ and 3S2−. One is under the control of 4f11e of Ln and the other under the control of S2− compared to LGT with thermal stability enhancing PCR and SCR activities. The LGT loses weight in three steps due to the interfacial domains of LSNRs and GO including their electronic clouds. GO which has a C–C lattice with equal heat capacities is disrupted at 193 with 82% wt loss with a sharp peak. LSNR decomposition suggests the role of 4f#e by aligning the lattices of La2S3, Ce2S3, and Ho2S3 but not Tb2S3 which has a minor simulated weight loss with increasing the 4f#e (Fig. S1.0 and S1.1, ESI†) as:
| | 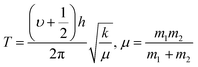 | (2) |
here
υ ∝
T,
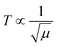
and
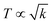
, and frequency depends on temperature and oscillations as per Hooke's law, and for the bond constant
k,
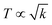
.
The basic structural units of LSNRs and LGT have been expressed on by subjecting them to the XRD and TGA, as their electronic DOS respond to the laser as per according to their wavevectors. The weight of a well-defined lattice also responds to the thermal energy, which induces the oscillation to transfer the oscillating energies via FRET with a 1st first-order weight loss transfer rate. Thus, the TGA and DTA also define the energy changes based on a the nature of the electron–electron collision (EEC) and electron–electron repulsion (EER) that interfere with the development in the same φ in the same φ (Fig. 12, green line). Thermal energy excites the unpaired electron within the wavevector with the patterns of the DOS of the DTA peak with LSNRs with comparatively sharper apex DTA along with differential heat flow, −7.0 to 10, −0.4 to 14, −7.0 to 12, and −0.4 to 13 μV for La2S3, Ce2S3, Tb2S3 and Ho2S3, respectively, at 400 °C apex temperature (Fig. S3.5, ESI†). Thus, the lattice of the LSNRs that responded at 400 °C remained fairly much the same that responded at 400 °C. However, the sharper DTA apex for an LGT is broader without any sharper peak-like shoulder. The DTA apex is broader than that of the LGT, which proves that the LSNRs have been fairly doped with into the 2D GO 2D sheet. These structural changes have fastened an energy flow within the LGT lattice on receiving the hν without causing the EEC and or EER by avoiding the recombination of the holes. Therefore, no the lack of a transitional peak in the DTA of LSNRs, GO, and LGT have inferred indicates smaller heat energy flow as equipartitioning the energy is equipartitioned (Fig. 12, green line). Initially LGT gains heat energy to get begin to oscillated and become ordered, where almost all the lattice points acquire a relaxation time. The unit becomes ordered within the coordinates of the initial state except the excited electronic state or the DOS. There is heat energy transfer without a loss and the PCR done is enacted as the gained heat excites an electron with a similar pattern where the hν could also push the electron within the a similar framework with to ψNor,T → ψRef,T. The DTA analysis for LSNRs is included in the ESI† (Fig. S3.5).
 |
| | Fig. 12 Differential thermal analyses: (a) GO, (b–e) LGT. | |
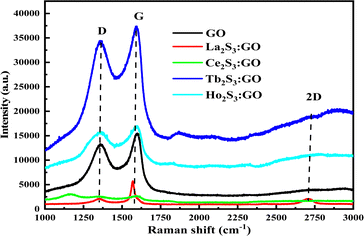
3.6. Raman spectra analysis
The laser light interacts with the molecular vibrations in GO as well in LSNR:GO that inferred from two sharper peaks in the same range of Raman shift. The ratio of D and G bands infer indicates the participation of LSNRs in the GO sheet, and creating disorder with the FGs and electrostatic moieties within the matrix. The ID/IG < 1 shows symmetric deformation on the GO sheet due to packing of LSNRs (Tables 1–3).
The D, G and 2D band peaks at 1350, 1580, and 2690 cm−1 respectively indicate sp2–sp3 and C–C stretching, respectively, and the intensity ratio shows the disorder of the GO sheet. The modulated bump on the 2D band at 2728 cm−1 is caused by minor reorientation, and the higher intensity of the G band indicates there are more defects in the GO sheets.5 The shift in energy bands is caused by the direct interaction of molecules with laser light. However, for the LGT, Raman spectra and intensities indicate the lattice and energy transfer behaviour of the LGT photocatalysts during the PCR. The Raman spectra have excellently developed similar patterns of the defunct activity of the 2D GO sheet doped with LSNRs in a most ordered manner (Fig. 13), as the D and G bands for GO, La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO are developed at 1350, 1580 cm−1 with variable intensities. Thus, the basic matrix remained the same with a well-defined wavevector. The fixed wavevector of a geometry with highly ordered entropy allows the increasing 4f#e to equilibrate with the FE of the GO, developing a single-valued ψLGT with negligible EEC and EER, leading to an efficient ∼100% FRET to dye for the PCR of a dye (Table 4).
 |
| | Fig. 13 Raman spectroscopy with D, G and 2D bands for GO and LSNR@GO. | |
Table 4 Raman shifts with D and G, bands with their ratio for GO and LGT
| LSNR@GO |
D-shift (cm−1) |
G-shift (cm−1) |
I
D/IG |
| GO |
13180 |
15152 |
0.87 |
| La2S3@GO |
1827 |
5400 |
0.33 |
| Ce2S3@GO |
2501 |
2609 |
0.95 |
| Tb2S3@GO |
34021 |
37340 |
0.91 |
| Ho2S3@GO |
15465 |
16812 |
0.92 |
3.7. Scanning electron microscopy (SEM)
Micrographs of GO indicate a lace-like morphology of a thin sheet of C atoms in 2D at 20 m resolution with a single distribution pattern due to synchronized FGs illustrated with PCPs that focus the alignments. A single sharp peak in the GO thermogram indicates a single crystal structure (Fig. 11a). The increasing 4f#e alter the topography and FE distribution in LGT depicted with the patterns of XRD, FT-IR, TGA/DTG and DLS (Fig. 8, 9 and 15). The La2S3:GO fragments have reoriented due to VWF to hold La2S3 within sheets, creating intersheet interfaces. The LGT fragments of smaller sizes enhance surface charges with higher increasing rate of PCR and SCR compared to LSNRs and GO alone (Fig. 14a and b). Ce2S3 with 4f1 having a maximum number of CB reoriented the GO sheets with the ψ in different φ and the reorienting energies spatially aligned the sheets. The upgraded ridges and furrows indicate interconnected nanorods (Fig. 4a1). Tb2S3 with 4f9 having ψ with a highly intensified RE unlike  unpaired electronic states with [ψ1 ≈ ψ2 ≈ ψ3]2 probability. Subscripts 1, 2, 3 on ψ denote electron transitions from 1 → 2 → 3 energy states of the 4f#e suborbital. Critical analysis of the micrographs of GO, La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO indicate topographical differences at 4f#e (Fig. 14). The changes are caused by the splitting energy of Ln2S3:GO with GO due to 4f#e. LSNR dopants have influenced the GO morphology due to 4f#e ∼ FE on aligning a GO matrix vis-à-vis LGT. La2S3:GO, Tb2S3:GO, and Ho2S3:GO produced almost equally distributed energy patterns, unlike Ce2S3:GO. Equal distribution of similar lacings of GO sheets are split into equal distributions as each GO sheet participates in LGT formation. The unpaired electron of Tb3+ symmetrises a GO topography, accepting more hν for PCR and SCR with Tb2S3:GO (Fig. 14d). Ho2S3 with 4f11e develops ψ4f11 with the least QEB, inducing a symmetric topography RE of FE. The ψ4f11 and ψFE in the same φ have developed a single crystal with a sharp peak, as depicted with TDA/DTG (Fig. 11e).
unpaired electronic states with [ψ1 ≈ ψ2 ≈ ψ3]2 probability. Subscripts 1, 2, 3 on ψ denote electron transitions from 1 → 2 → 3 energy states of the 4f#e suborbital. Critical analysis of the micrographs of GO, La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO indicate topographical differences at 4f#e (Fig. 14). The changes are caused by the splitting energy of Ln2S3:GO with GO due to 4f#e. LSNR dopants have influenced the GO morphology due to 4f#e ∼ FE on aligning a GO matrix vis-à-vis LGT. La2S3:GO, Tb2S3:GO, and Ho2S3:GO produced almost equally distributed energy patterns, unlike Ce2S3:GO. Equal distribution of similar lacings of GO sheets are split into equal distributions as each GO sheet participates in LGT formation. The unpaired electron of Tb3+ symmetrises a GO topography, accepting more hν for PCR and SCR with Tb2S3:GO (Fig. 14d). Ho2S3 with 4f11e develops ψ4f11 with the least QEB, inducing a symmetric topography RE of FE. The ψ4f11 and ψFE in the same φ have developed a single crystal with a sharp peak, as depicted with TDA/DTG (Fig. 11e).
 |
| | Fig. 14 SEM for surface morphology: (a) GO and (b–e) LSNR:GO. | |
Tb2S3:GO and Ho2S3:GO as robust photocatalysts and adsorbents receive maximum hν almost in the same φ as the GO sheets for uninterrupted PCR and SCR.
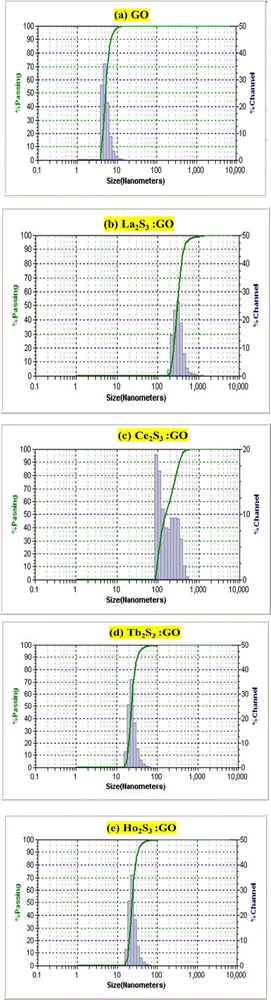
3.8. Dynamic light scattering (DLS)
The ζ > 0 of GO indicates aligned h+ holes on a surface of LGT and LSNRs either from S2− or Ln3+ charges. LGTs receive hν with different capacities. A broader La2S3 distribution pattern with GO sheets indicates weaker 4f0e interactions with FEs. The oxidation potential of La3+ and reduction potential of S2− are unable to generate a caging of water (Fig. 15). La2S3 is actively distributed with less passage while on doping with GO there is maximum capping and a decrease in the passage of population on interaction of LSNRs and FEs. Ce2S3 with 4f1e develops prominent collisions with a stoichiometric ratio of GO and LSNRs for LGT with a 4f0e → 4f11e interface The conductivities of LSNRs and LGT are in the range 17 to 43 μS cm−1 compared to the 50 μS cm−1 of GO alone, so the 4f#e and FE interactions might have occupied the surface charges and mildly responded to an electrical potential with a lower mobility (Table 5). The distribution of La2S3 is scattered while that of La2S3:GO is narrow, as GO has an ordered lattice due to similar GO sheets. There is a narrower range of distribution between 100 and 1000 nm at −0.49 mV compared to that at 22.24 mV, as the La3+ develops Coulombic interaction with negative holes that confine C atoms (Fig. S3.4, ESI†). The redistributing electronic clouds equipartition the electronic energy of S2− to interact with the h+ holes or electron-deficient sites of GO. Their interactions have narrowed down the distribution range of La2S3:GO (Fig. 14). These changes are supplemented by the TGA of GO and LGT, with major structural disruption of La2S3 at 365 and of GO at 194, but La2S3:GO has elevated the GO stability of La2S3:GO with a major structural disruption at 221 (Fig. 11). These disruptive activities indicate the doping mechanism and pattern of GO with La2S3. The 3e− + La3+ local –C–C–C– network is simultaneously accompanied by 3h+ + 3S2−. The interfaces of La2S3 and GO narrow down the distribution range with an active state due to FRET. A similar pattern is noticed with Tb2S3 and Tb2S3:GO, as their distribution range remains almost the same with a sharp lattice disruption at 235 (Fig. 1d). Thus, the Tb3+–3e− and S2−–2h+ electronic strings are influenced by 4f9e due to their oscillations. However, the Ln3+–3e− and S2−–2h+ loops are quantized and their localized quantum dots may be equally distributed in Ln2S3 and Ln2S3:GO. The FGs of GO could opt to interact with Ln3+–3e− and S2−–2h+ electronic strings as Ln2S3:GO. Surprisingly Ce2S3 and Ho2S3 with their templates produced a fine distribution in the 10 to 1000 nm range. Ce3+–3e− and Ho3+–3e− with S2−–2h+ are simulated with 4f1e and 4f11e of Ce3+ and Ho3+, respectively, which are duly supplemented with the TGA pattern (Fig. 11c and e). Since these electronic strings substantially influence their solubilizing activities, so their PCPs indicate the thermodynamic state function. The Ln3+–3e−(GO) and S2−–2h+(GO) interfaces are highly functional and localized cohesively as per their respective charges. So, solvent adherence could cause further alignment or reorientation despite their cohesiveness, and their friccohesity index could reveal a unique interaction mechanism. Ln2S3 and Ln2S3:GO have developed new interaction centres unlike the others. Their hν receiving and phonon generating activities have been found to be directly interconnected with their PCPs. Therefore, the photocatalysts were solubilized for PCP, PCR, and SCR studies. Their electronic interactions could be influenced by sonication to induce some electronic arrays in contact with 2Ln3+–3S2− as S = Ln–S–Ln = S nanorods. Thus, the PCPs and DLS profile elucidate applications to reduce and recycle wastewater. Ho2S3 has shown a 5.35 PDI which was reduced to 1.369 with Ho2S3:GO, as the GO capped the Ho2S3 around its surface. These are understood with TGA, in which it has developed multiple disruptive temperatures, but on doping developed a single crystal lattice resulting in a sharper single line at 198 against the 194 of GO (Fig. 11a and e). Ho2S3 has elevated the GO disruption temperature by 4. Thus, the PDI and lattice disruption via TGA could have a unique relationship. The diameter of La2S3 is 457 nm, the highest among the series, which is also explained by TGA due to the Gaussian distribution pattern of heat (Fig. 15 and Fig. S3.4, ESI†).
 |
| | Fig. 15 Size (nm) of (a) GO and (b–e) LGT analysed by DLS. | |
Except for Ce2S3:GO and Ho2S3:GO, their mobilities are almost in the same range. La2S3:GO and Tb2S3:GO have negligible ζ values, which may be due to the negative holes of GO. Their conductivity remains within 50 μS cm−1 and responds to electrode charges on subjection to the probe (Table 5). A detailed study of DLS for GO, LSNR and LGT is incorporated in Fig. S3.4 (ESI†).
4. LSNR and LGT solutions for PCPs, PCR and SCR
The NHSs of LSNRs and LGT with BSA could have been deformed irreversibly on acquiring unequal activation energy due to the selective charge clouds of Ln3+–3e− and S2−–2h+ electronic strings with the LGT. The highly sensitive survismetry technique measures viscous flow time (vft) and pdn at 283.15 to 315.15 K.45 Lifting a sample from a reservoir to a buffer bulb under a reduced pressure within capillaries avoided the mechanical shock that could have disarrayed the FE. A sample was equilibrated in the buffer bulb for 3 min before allowing for a backward flow to a CPU (controlled pressure unit) with a hassle-free capillary flow in survismetry.46 PCPs for 0.002–0.010% @0.004% of LSNRs, LGT alone and with 0.010% aq-BSA at 298.15 K and 0.010% with Milli-Q water at 283.15 to 315.15 K indicate a monodispersion and structural alignment of the solution (Fig. 16a–d). LSNRs and LGT alone did not aggregate but developed a gel with BSA (Fig. 16c and d). Turbidity increases with increasing rate of g% of LSNR and LGT as 4f#e and FEs could align slightly, holding water molecules haphazardly. Their PCPs and thermodynamic parameters depict the solubilizing activity and interactions of aq-BSA with LSNRs and LGT. Viscosity elucidates the effect of Ln3+–3e− and S2−–2h+ in bulk while there is a surface tension on the surfaces. These have been used as the functional interfaces to explore the activities of LSNRs and LGT for widening their applications in areas of PCR and SCR. La2S3ΔH ≪ 0 for La2S3, and ΔH < 0 for Ce2S3, Tb2S3, and Ho2S3 indicate a milder disruption of the water structure and LSNRs and LGT could have been expeditiously realigned on releasing H. Ln3+–3e− and S2−–2h+ seem to develop stable dipoles that are explained by their photocatalyzing activities. LSNRs and LGT as a single unit with expeditious molecular motions disrupted hydrogen bonding (HB) on decreasing H and increasing S, as supported by DLS (Table 5 and Tables S1.4–S2.1, ESI†).
 |
| | Fig. 16 Aq-solutions (a) LSNR (b) LGT (c) LSNR-BSA (d) LGT-BSA. | |
4.1. Density (ρ, g cm−3)
The ρ of LSNRs and LGT alone and with aq-BSA indicate interacting activities as a function of temperature and concentration. The ρ values are in the order of aq-BSA > La2S3:GO > La2S3:GO-BSA > La2S3-BSA > La2S3 at 288.15, 298.15 and 310.15 K, predicting stronger aq-BSA and weaker aq-Ln2S3 interactions. The electrostatic salt bridges of BSA strongly attract the H2O dipoles, including Ln3+–3e− and S2−–2h+ electronic strings. Thus, La2S3 and La2S3:GO with aq-BSA decreased ρ by 0.039% and 0.009%, 0.039% and 0.0095%, and 0.048% and 0.16% at 288.15, 298.15 and 310.15 K, respectively as the BSA was weakly reoriented to optimize their interacting surfaces. The tryptophan and peptide bonds (PB) of BSA strongly interacted with water and Ce2S3:GO by occupying a majority of FEs that salted out water dipoles. BSA decreased the ρ of aq-Ce2S3 and aq-Ce2S3:GO by 0.0027% and 0.0090% at 288.15 K and 0.0026 and 0.0040% at 298.15 K, unlike the increases of 0.0029% and decreases of 0.034% at 310.15 K, respectively. Ce2S3:GO with increasing temperature could have generated vibrant oscillations, weakening BSA interactions with H2O and could attract GO. BSA decreased the ρ with Tb2S3 and Tb2S3:GO by 0.006% and 0.011%, 0.007% and 0.014%, and 0.013% and 0.039% at 288.15, 298.15 and 310.15 K, respectively, as the H2O accommodated them on holding BSA as well as Tb2S3 and Tb2S3:GO. BSA decreased the ρ of Ho2S3 and Ho2S3:GO by 0.0053% and 0.0131%, 0.0073% and 0.0160%, and 0.0073% and 0.080% at 288.15, 298.15 and 310.15 K, respectively (Tables S8.1 and S8.2, ESI†). Thus, the H2O, which was associated with BSA, Ln2S3 and Ln2S3:GO was realigned with weaker force but adequate adherence. BSA developed stronger interactions with H2O dipoles associated with LSNR:GO and LSNRs, with stronger internal pressure of FEs (Fig. 16). The H2O dipoles interact with themselves and the salt bridge, and the electrostatics of BSA could have normalized Ln2S3 and Ln2S3:GO. GO engages the LSNRs in LSNR:GO via weakly interacting H2O dipoles. The covalently bonded S2− and Ln3+ of LSNRs and LGT develop NHSs, as NH3+, COO−, >CO, and PB along with tryptophan develop stronger hydrophilic–hydrophilic interactions (HHI), aligning LSNR:GO expeditiously during a PCR.31
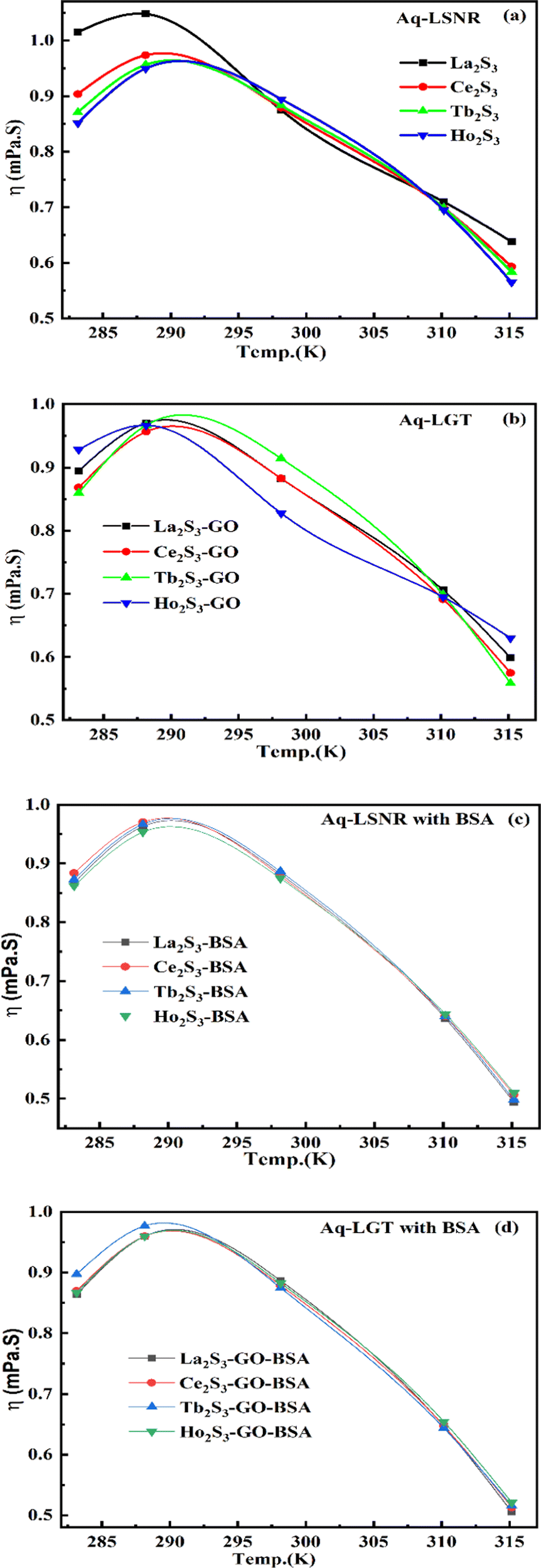
4.2. Viscosity (η, mPa s)
The η indicating the vft of Ln3+–3e− and S2−–2h+ aligned in aqueous solution is calculated with eqn (3) (Tables 6, 7 and Table S1.2, ESI†).| | 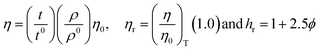 | (3) |
where ρ is the density of solution, ρ0 is the density of water, η0 is the viscosity of water, ηr is the relative viscosity, and vft is an average relaxation time (τ). Decreasing viscosities of LSNRs and LGT alone and with BSA indicate weaker interactions due to competitive adherence with S = Ln–S–Ln = S, and aq-GO, and their interfacial charges reorient and align the NHSs, generating cohesive activities (Tables 6, 7 and Fig. 17). The η values in the order La2S3 > Ce2S3 > Tb2S3 > Ho2S3 with increasing the 4f#e and temperature indicate that electron spin via electron–electron collisions weakens H2O adherence, promoting coherence. The stronger interaction of GO with Ho2S3 engages almost all the FEs, with stronger H2O adherence increasing η values. The η values for LGT decreased on the GO and LSNRs mutually interacting via their respective NHSs. The NHSs align, generating mutual cohesive forces with sharply weakening adherence of H2O, as cohesive activities induce continuity, equilibrating the adhered H2O as bulk H2O. Thereby monolayer adherence of H2O with comparatively weaker coherence enhances the η at lower temperature. These activities sensitively visualised a change compared to an uneven magnitude of γ data at higher temperature (Fig. 17c and d). The GO controls a CAC of LSNRs, but BSA has disrupted the LSNRs aggregating as colloids with a lower value due to multiple NHSs tending towards a monolayer (Fig. 17b). BSA capping of LSNRs and LGT has weakened their cohesive forces, as BSA disrupts the aligned water dipoles, forming multiple NHSs with slight monodispersion (Fig. 17b and d). The values of La2S3, Ce2S3, and Tb2S3 intersect at 0.006 g% unlike Ho2S3 with stronger adherence of H2O. The values decreased with concentration of La2S3 ∼ H2O. Additionally, Tb2S3 with aq-BSA increased the η values due to the half-filled electronic spins of 4f9e causing oscillations aligning H2O dipoles for comparatively stronger adhesion. On increasing Ho2S3, g% with aq-BSA did not produce a higher η value unlike Tb2S3 at 0.006 and 0.010 g% due to 4f9e, as an unpaired electron does not align the H2O in light of the NHS of BSA (Tables S6.3 and S6.4, ESI†). The BSA monodispersion with increasing LSNR g% develops LSNR ∼ PB interactions on solvent adherence (Fig. S1.2, ESI†). LSNRs align the surface charges of BSA to develop multiple NHSs, resulting in a weaker NHS on a continuously flickering H2O. The ordered trends for values for aq-LGT-BSA compared to aq-BSA alone and aq-LSNR-BSA redistributes and realigns the surface charges with concentration. The aq-BSA regularly adheres to LSNRs with increasing g% on engaging a solvent. Amounts of 0.002 g% for La2S3 and Ce2S3 produced almost the same values unlike the situation at 0.010 g%. The values of La2S3:GO-BSA, Ce2S3:GO-BSA, Tb2S3:GO-BSA and Ho2S3:GO-BSA depend on g% (Tables S6.3 and S6.4, ESI†). The trend in polynomial η for La2S3, Ce2S3 and Tb2S3 on increasing g% at constant temperature predicts saturation at 0.006 g% (Fig. S1.2a, ESI†). Aq-Ho2S3 mildly increased the η values, unlike the case at 0.006 g% due to hydrated LSNRs as the Ho2S3 nanorods could have aligned H2O dipoles (Fig. S1.2b, ESI†). Increasing Ho2S3 g% has mildly realigned a saturated H2O with stronger adherence, but decreases the η values due to energetically disrupting the aligned NHSs (Fig. 5b). The residual charges align the LSNR–H2O interface with Gnet = GH2O–H2O ≠ Ginterfacial as a polynomial relation. Thus, increasing the g% of Ho2S3 disrupts the NHS, which affects both the adhesive and cohesive force. Ho2S3 does not disrupt the H2O and is salted out, requiring sonication to remain suspended. La2S3, Ce2S3, and Tb2S3 decreased the η values by weakly disrupting the adhering NHS that distinguished the GH2O–H2O and Ginterfacial on sonication. LGT realigns H2O to enhance aq-LSNR solubility with higher η values due to effective adherence increasing τ. The rheology of aq-Ln2S3 with increasing temperature equalizes with their oscillation with certain ψ equipartition of the energy. The adherence of H2O to Ln2S3 is equal, however, at lower temperature and the viscosity values differ due to different oscillations and ψs. The shift from Ln2S3 to Ln2S3:GO at 298.15 K, indicates GO doping reducing energy equipartition (Fig. 17). However, BSA with Ln2S3 and Ln2S3:GO have indistinguishable viscosities due to an equal adherence pattern. Robust oscillations of electron clouds of GO with LGT influence adherence via ψs (Fig. 17a and b). Interactions of Ln2S3 and Ln2S3:GO with BSA via NHS have sandwiched Ln2S3:GO with BSA together as incompressible, that lowers the η with increasing temperature. The η of packed LSNR-BSA and LGT-BSA support expeditious resonating energy transfer for equipartition, where no energy-deficient sites are generated by varying the temperature.
 |
| | Fig. 17 Viscosity of aq-LSNR and aq-LGT with BSA at 5 T. | |
Due to its salt bridges and electrostatic sites, BSA is closely packed in between the cages or NHSs of LSNRs and LGT, lowering the η values equally (Fig. 17c and d). Tb2S3-BSA and Ho2S3-BSA interactions are weakened at 0.006 and 0.010 g% compared to Tb2S3 and Ho2S3 alone, as 4f#e ∼ PB linkages and stronger electrostatic sites of BSA affect NHS alignment (Fig. S1.2c and d, ESI†). The η values of BSA with LGT are increased on strengthening the FE of GO and electrostatic interactions of BSA that are more soluble with LGT than LSNRs, as no FE networking exists with the LSNRs on disruption of HB. The LGT-BSA biotemplate increased BSA solubility, due to expeditious charge equalization through 4f#e ∼ FE interfaces via FRET increasing the η values (Fig. S1.2d, ESI†).
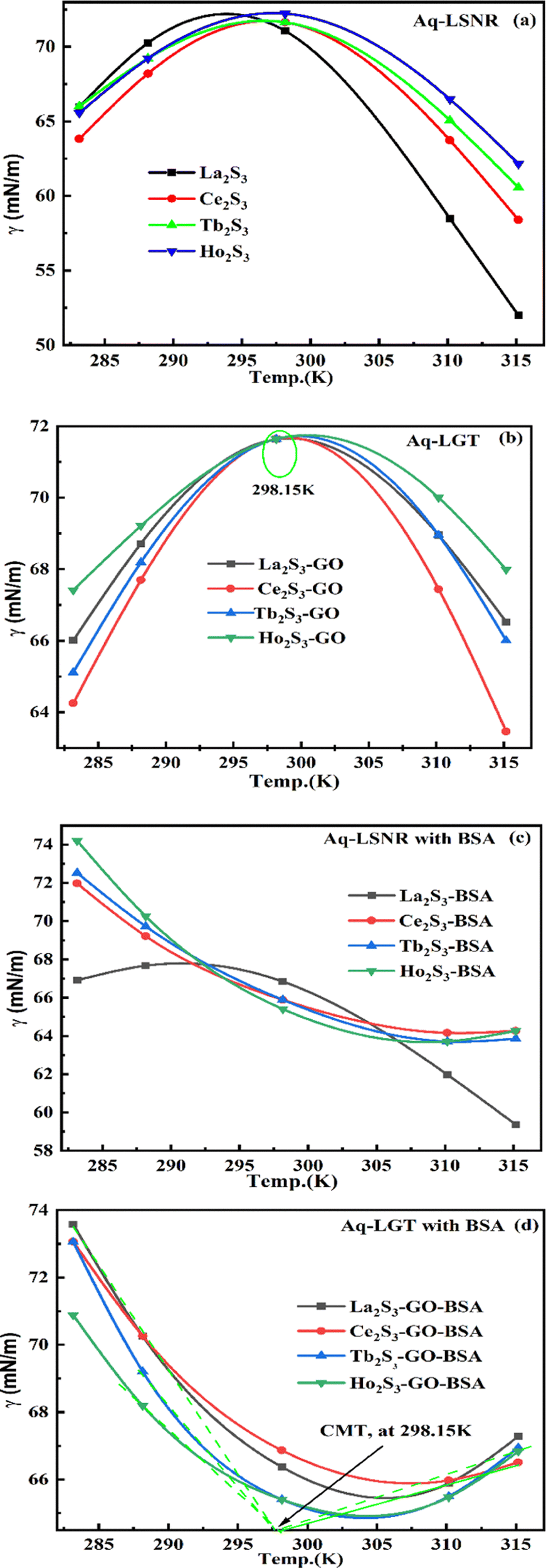
4.3. Surface tension (γ, mN m−1)
Disruption of H2O adhering to LSNRs and LGT alone and with BSA affect the cohesive force of the medium. The γ elaborates the structural interaction with the solvent and pdn indicates the cohesive force, and γ is calculated with pdn and density values (Tables 8, 9 and Table S1.1, ESI†) as:| | 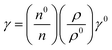 | (4) |
where γ0, n0, ρ0 and γ, n, ρ are the surface tension, pdn, and density of water and solution, respectively. The surface tension of aq-LGT with BSA is higher than aq-LSNR with BSA, aligning multiple NHSs with stronger cohesive forces (Tables 8, 9 and Fig. 18). LGT developed a critical aggregation temperature (CAT) at 298.15 K similar to a critical solution temperature.47 The H2O dipoles cap LGT to transfer a resonating energy with a moderate VWF (Fig. 18b). The CAT is controlled by 4f#e, as GO sheets adhere to LSNRs via NHSs that equalise the surface energy (SE). The FE with SE of LSNRs exchange their oscillations. Aq-LSNR does not favour CAT; thus, the 4f#e ∼ FE works to develop multiple linkages to develop CAT at ∼298.15 K (Fig. 18a and b). The GO wrapped the LGT and a CAT could not distinguish an SE while neither the LSNRs nor LGT produce a CAT with BSA. BSA increasing time binds the LSNRs via NHSs with different surface charges and heat capacities around LSNRs via cohad. On partially engaging the cohesive and adhesive forces expressed as cohad. BSA having higher SE and a lower γ with aq-LGT developed a critical micelle temperature (CMT) at 298.15 K (Fig. 18d). BSA did not initiate a CMT due to its PB salt bridges, and electrostatic dipoles that stabilize the SE of LGT at 298.15 K. The SEs of GO and BSA both mutually adhere at 298.15 K with the highly sensitive surfaces for expeditious energy transfer via VWF. The trend of change in γ values unlike the η values depicts an adherence of NHS around a core of LSNRs. The cohad infers a balances in the cohesive and adhesive forces of the solvent as well as the LSNR and LGT for the thermodynamic and kinetic stabilities of their aqueous solutions preventing their coagulation (Fig. 18). The η, γ, and σ indicating aq-Ho2S3 with three unpaired electrons are more sensitive than other LSNRs due to the same contraction effect with stable dipoles (Fig. S1.3a, ESI†). Surfaces of LSNRs are capped with GO in manifold layers to attract the electrostatic centres of PB. LSNR:GO-BSA strengthens the electrostatic interactions with dipolar water. The lower γ values for aq-Tb2S3 indicate stronger adherence with LGT and BSA with a higher (Fig. S1.3d, ESI†). The 4f#e engage the FE to get solvated or NHSs with increasing, and LSNR:GO with BSA further increases the adherence with increasing. The 4f#e ∼ FE photonic interfaces of LGT with BSA could store energy through aligned dipoles45 by preventing the recombination of holes. The electrostatic linkages with 4f#e ∼ FE could develop the functional interface with a protein of specific charge with highly stabilized dipoles from 285 to 297 K; the γ value saturates but increasing H2O drastically develops an NHS.
 |
| | Fig. 18 Surface tension of aq-LSNR and aq-LGT with BSA at ST. | |
4.4. Friccohesity (σ, S cm−1)
HB disruption aligns H2O around LSNRs and LGT separately and with BSA develops specific adherence with the cohesive force that supplements the respective FRET to Ea. It equalizes a stabilizing energy to develop σ, calculated from pdn and vft (Tables S1.1 and S1.2, ESI†):| | 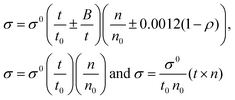 | (5) |
where σ and σ0 are the friccohesity of solution and solvent, respectively, ±B/t are kinetic and ±0.0012(1 − ρ) buoyancy corrections, other symbols are as usual. Corrections of the order of 1 × 10−7 are omitted. The common physicochemical parameters of solvents are rationalized as a Mansingh constant (Mc) that finds an interface between adhesive and cohesive forces. Mc depicts a mutual shear stress and the SE coefficient of a solvent for GO, LSNRs and LGT with BSA calculated with eqn (6):| | 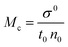 | (6) |
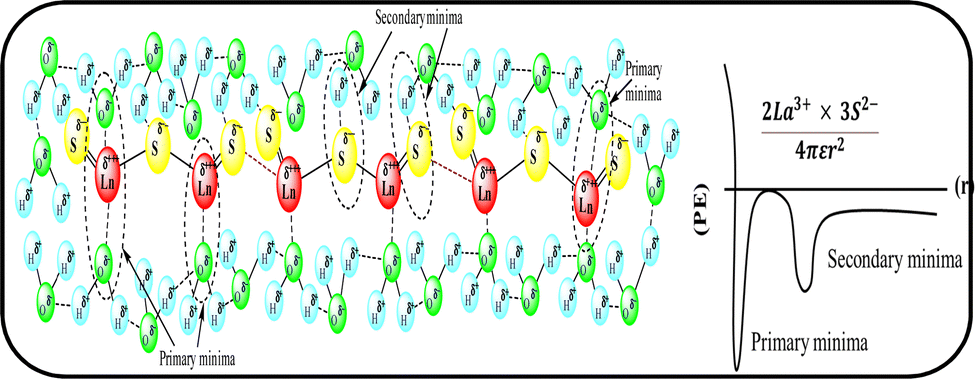
Mc as a quantizing coordinate of aligned H2O dipoles with LSNRs, LGT and BSA indicates homogenization with equipartition of their energies with a solvent via FRET. The Mc operator transforms cohesive to oscillating energies through a mutual electrostatic reorientation of solvent molecules around the LSNRs and LGT. The friccohesity of aq-LSNR, and LGT alone and with BSA indicate spontaneous solvent adherence (Tables 10, 11 and Fig. 20). The 4f#e ∼ FE interfaces of LSNRs, LGT and BSA are accessible to the internal salt bridges and electrostatic dipolar linkages of BSA to attract H2O dipoles. The H2O dipole interacts with each constituent of LSNRs, LGT and BSA via quantized negative and positive holes to adhere H2O. It avoids nanoclusters to promote PCR and SCR activities. Friccohesity generates several NHSs with different charges on surfaces that initiate the secondary global energy minima, stabilizing their NHSs for the required cohesive force. The friccohesity illustrates a mechanism and pattern of primary and secondary minima as the LSNRs, LGT and BSA interact differently through the NHS. Their NHSs mutually equalize the energy of a medium (Fig. 20).
Secondary minima attained via LSNR ∼ HS, LGT ∼ HS, and BSA ∼ HS interactions facilitate a FRET for mutual homogenization. The friccohesity for partitioning the secondary quantization of cohesive forces was transformed into oscillatory energy by reducing the QEB of an NHS (Fig. 19). The σ represents the CMT of LSNRs and LGT with BSA at 295 and 302.5 K, respectively, aligning the FEs at 302.5 K compared to η. It depicts a higher resolution incorporating the role of each constituent for NHS quantization (Fig. 20a and d). The σ values illustrate the reengineering of interactions to analyse a contribution from each charge to operate on the air–liquid interface at laminar-to-laminar flow. Friccohesity scans consolidate the contribution of surface energy from each laminar layer, causing a frictional force on maximum adherence with the least and higher η. Friccohesity decreases on increasing 4f#e from La2S3 to Ho2S3 at 283.15 and 315.15 K unlike that at 298.15 K that expeditiously exchanges via FRET, establishing a continuity to mutually exchange the alignment of H2O between adhesive and cohesive forces (Fig. 20a). LGT slightly tunes LSNR activities except for Ho2S3 with higher and the least SE. The ψPE compensates for the electronic cloud of ψUPE with the vibrant activities of FE (Fig. 20b). Friccohesity indicates LSNR and LGT systematisation on BSA capping unlike the and values with cohad on mutually equilibrating the binding and dispersing forces, respectively. The LSNRs and LGT both indicate CMT with BSA where the friccohesity mutual resonating energies transfer to normalise the cohad, the ratio of the cohesive and adhesive forces (Fig. 20c and d).
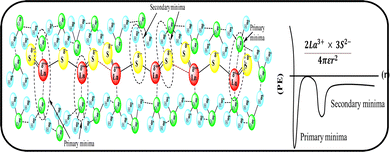 |
| | Fig. 19 Primary and secondary minima via 4f#e ∼ H2O. | |
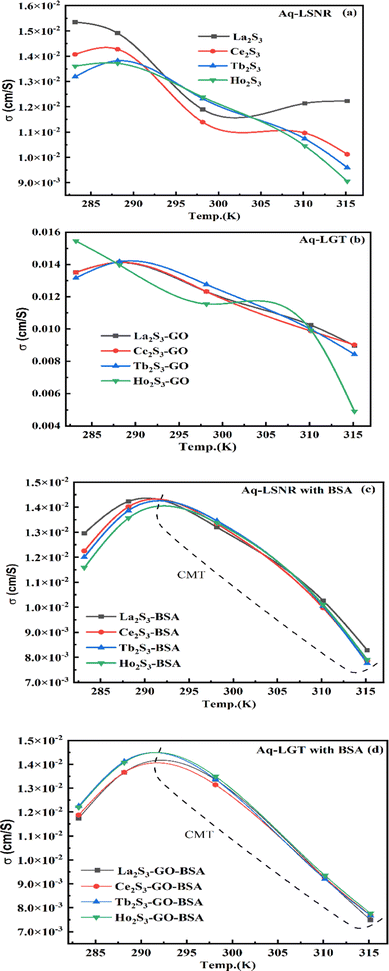 |
| | Fig. 20 Friccohesity of aq-LSNR and aq-LGT with BSA at 5 T. | |
4.5. Friccohesity velocity (σv, cm s−1)
The disruption of LSNRs, GO, and LGT alone and with BSA causes reorientation with a certain velocity that could be decisive with respect to their adhesive and cohesive forces to mutual stabilization. The solvent molecules adhere around LSNRs and LGT surfaces, forming an NHS and to suspend them needs a strengthening of the structural framework with the required adhesion supported by mutual cohesion. The spontaneity of a template with respective ψs establishes a close continuity as an ordered state. Locally aligning and adhering a solvent induce collisions for continuous flow oscillation to maintain cohesion, so σv is calculated with σ values (Tables 10 and 11).| | 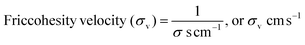 | (7) |
The σv values of LSNRs and LGT separately and with BSA at constant pH and temperature indicate alignment in capillary flow (Tables S5.9 and S6.0). The reorienting chemical species could initiate a solvent-disrupting activity to dislocate a free hydrogen-bonded state and adhere to LSNRs, GO, and LGT with specific velocity. Adhered H2O spontaneously align on the outer surface of the NHS, initiating the development of solvent–solvent coherence. The σv depicted as adherence and coherence move dynamically.
4.6. Isentropic compressibility (KS,ϕ)
LSNRs, GO, LGT, and BSA reorient to adhere H2O from the hydrogen-bonded state with a higher cohesive force, causing monodispersion. Thereby charge expression plays a key role in generating entropy on reorienting and aligning them. KS,ϕ indicates constant entropy during these activities as the species maintain their structures, except for infinitesimal changes in friccohesity, for initiating interactions producing KS,ϕ in the 5 × 10−7 cm s2 g−1 range (Tables S7.5 and S7.6, ESI†). KS,ϕ is calculated using density and sound velocity (Tables S1.3, S1.8 and S8.2, ESI†):| | 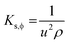 | (8) |
where u and ρ are sound velocity and density of solution. The KS,ϕ of LSNRs and LGT alone and with BSA depict their electrostatic interaction with almost constant entropy due to the ordered alignment maintained by adherence and coherence within infinitesimal changes (Tables S7.5 and S7.6, ESI†). The KS,ϕ of LSNRs and LGT gradually decrease with increasing temperature as it disrupts the ordered state. At higher temperature the ρ values are lower, as cohesive forces are weakened by oscillation of their binding energy. At lower temperature, a mild oscillation indicates a maximum KS,ϕ but increasing temperature has weakened the ε of LSNRs and LGT with BSA (Fig. S1.5 and S1.6, ESI†). The increasing temperature indicates stronger LGT ∼ BSA interaction with 4f#e ∼ FE ∼ PB linkages due to the lowest friccohesity, as adherence is dominated by the cohesive forces of a uniform NHS.
5. Thermodynamics for PCPs, PCR and SCR activities
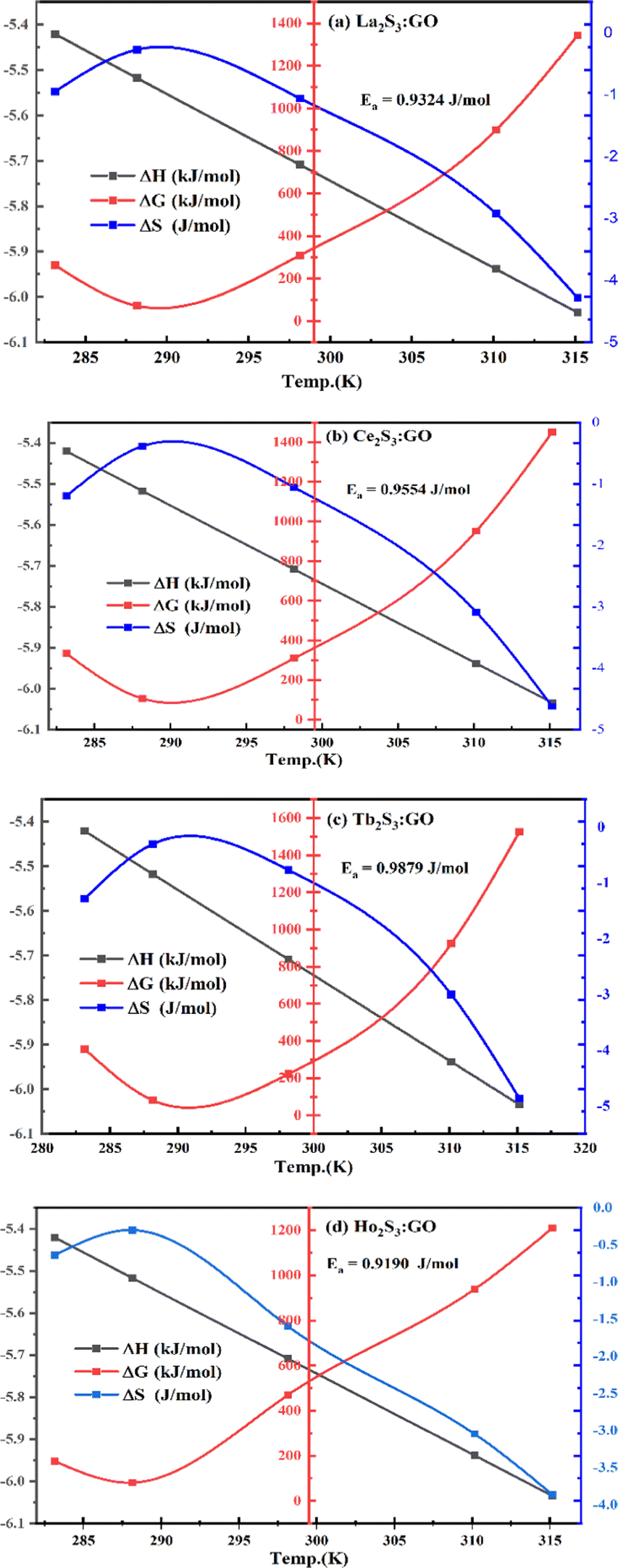
5.1.
E
a, kJ mol−1
LSNR and LGT dissolution need energy and space to disperse in H2O, so the HB disruption by weakening cohesive forces activates H2O alignment around them via electrostatic and ionic dipoles. LSNR and LGT dispersion acquire energy from water directly, indicating their interacting nature with a certain state function. The Ln3+ and S2− of LSNRs vis-à-vis the skeletal edges of GO and their spatial occupation of the LGT interface encounter BSA with an active electronic cloud. These respond to interlinking of oscillations and thermal energies on increasing temperature as:| | 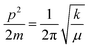 | (9) |
The acquired energy does not break bonds, but rather activates a whole structure to develop ψ that resonates energy to participate in interactions. Ea is calculated with eqn (9) and logη vs. (1/T) and log(abs) vs. (1/Ct) are plotted (Tables S1.4–S2.1, ESI†), where T is temperature and Ct is dye degradation concentration, with time t min fitted as shown below:| | 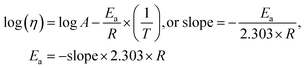 | (10) |
Ea is calculated with dye degradation concentration (Ct) on PCR and SCR.| | 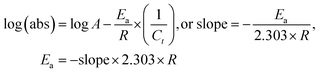 | (11) |
Ea > 0 for Ln2S3 and Ln2S3:GO indicates HB disruption realigning and reorienting H2O to flow in a streamlined way within a uniform capillary. (Ea,LSNR > Ea,LGT) > 0 indicates higher activities of the FE despite it getting doped with LSNRs. Thus, the 4f#e of LGT resonates energy to transfer to the closest FE to photocatalyze dyes in a shorter time and is proven to be an efficient photocatalyst to induce FRET. Ea calculated with PCR is 10 times higher than with PCP, as multiple and manifold collisions of 4f#e ∼ FE utilize their energies to align (Fig. 21 and Tables S1.4–S2.1, ESI†) compared to shear stress being used to align.
 |
| | Fig. 21 The thermodynamic parameters with increasing temperature of Aq-LGT for PCR. | |
5.2. Thermodynamic parameters ΔH, ΔG, and ΔS
Thermodynamic functions define the interacting activities of LSNRs, GO, LGT, and BSA in aqueous solution as state functions ΔH, ΔG, and ΔS calculated from Ea (Tables S1.4–S2.1, ESI†):| | | ΔH = Ea − 2.303RT or ΔH = Ea − 2.303RCt | (12) |
| | | ΔG = 2.303RTlog(abs) or ΔG = 2.303RCtlog(abs) | (13) |
| |  | (14) |
ΔG < 0, ΔH < 0 and ΔS > 0 with LSNRs indicate spontaneously exothermic disordered interaction activities, reducing dyes in a longer time (Tables S1.4–S2.1, ESI†). ΔH < 0, ΔG > 0 and ΔS < 0 indicate an aligned LGT with H2O dipoles where non-spontaneous endothermic activity has reduced the dyes in shorter time, acquiring a lower Ea (Tables S8.3–S8.6, ESI†). The aligned and ordered LGT with ΔG > 0 has a stronger dipolar system (ΔS < 0), as the hν absorption ability is rapid for the PCR and SCR of dyes (Tables S4.2–S5.9, ESI†). Sonication disrupts the NHS and the secondary bonds release energy, increasing the overall heat content as an exothermic process. Ea for LGT is the lowest, as the FE and 4f#e are both pre-activated with a least QEB of 4f#e and ordered FE, as 〈ψ4f#e|ψFE2〉 is a normalized quantum string on the surface. With a mutual contribution, the RE of FGs and 4f#e are partially shed to reorient and align GO around LSNRs is favoured by NHS mutually (Fig. 5b). LSNRs and LGT could not affect Ea with BSA, despite having many amino acids, tryptophan, salt bridges, and electrostatic linkages with H2O dipoles due to their structural integrity. Tb2S3 with 4f9 strongly aligns the H2O around BSA.
6. Photocatalytic reduction (PCR) of MB, MO, BBR and BBG under sunlight
The hν could interact with the electronic cloud available on the surfaces of LSNRs and LGT as an excessive cohesive force with maximum electronic sites to receive photons. Additionally, the packing of LSNRs, LGT, and GO photocatalyst causes a higher increasing rate of PCR. The monodispersion of photocatalyst generates a lower surface area with a maximum number of hν receiving sites. Therefore, the study of PCPs is noted as an essential prerequisite for monodispersion and to have thermodynamic and kinetic stability for the efficient use of active site of LSNR, GO, and LGT photocatalysts. Thus, the PCPs have been considered as preparatory ground for PCR and SCR. Fig. 22 depicts the homogeneous to heterogeneous phase; initially it dispersed in aqueous solution and after 90 min it was settled at the bottom, as these did not engage the dipoles of water without monodispersion. Thus, an adequate adherence of H2O is necessary for monodispersion for reusability, as the photon could not weaken the cohesive force for adhering H2O molecules, denoted as the secondary cohesive force. The primary cohesive forces do not monodisperse the sample and the secondary cohesive forces generate functional sites to enhance increasing rate of PCR and SCR activities. LSNRs and LGT both oscillate the electronic cloud to gain an electronic charge of dyes having quaternary nitrogen (QN) with electron-deficient sites through FRET. LSNRs and LGT both resonate their respective electronic charges to overcome the QEB to approach cycles. Their resonating energies counterbalance the respective residual charge without balancing the holes. GO holes out of hexagonal GO sheets PCR the MB, MO, BBR, and BBG with LGT. GO with a high mobility of charge carriers gains higher thermal conductivity to capture a maximum number of hν.20,35 LGT has reduced dyes in ∼30–90 min and GO in ∼120 min with Φ of 99–100 and 87%, respectively.3 The @PCR and @SCR for dyes are calculated as:| |  | (15) |
C0 is the initial dye concentration at time t = 0, and Ct is the concentration of reduced dye at t min. UV/Vis absorbance has been used to calculate the quantum yield using Einstein eqn (16):| | 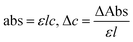 | (17) |
| | 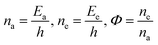 | (18) |
where m is the mass derived from eqn (17) with Δc the change in concentration, ΔAbs is the change in absorbance, c is the speed of a photon (3 × 108 m s−1) and Ea is the absorbed energy and Ee the emitted energy, na is the number of photons absorbed and ne is the number of photons emitted. The hν from sunlight generated phonons different frequencies, so a particular energy is used to calculate the number of hν. Absorption is used to calculate Ct, which directly depends on number of hν, which is then used to calculate na and ne (Fig. 24).
 |
| | Fig. 22 PCR of MB: (a) aq-GO (b) aq-LSNR under SL in 120 min and 24 h, respectively. | |
 |
| | Fig. 23 PCR of dyes under SL with LGT ∼30–90 min. | |
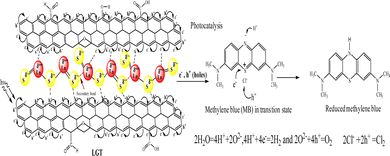 |
| | Fig. 24 LGT electronic hole generation for the PCR of dyes. | |
LGT has reduced BBG, BBR, MB and MO with 90–100, 87–97, 90–96, and 93–95% @PCR and with 94–100, 94–99, 93–97, and 93–95% Φ, respectively due to their different structures (Tables S8.3.0–S8.6, ESI†) Tb2S3:GO and Ho2S3:GO are effective photocatalysts with 4f9e and 4f11e, respectively. Photocatalysts and dyes both align mutually to exchange their electronic charges as ψ overcomes the QEB. MB with 4–CH3 causes and ER effect on absorbing hν (Fig. 25c). The active sites of dyes interact with LSNRs and LGT, but QN receives electron holes from LSNRs and LGT through FRET (Fig. 25a). LSNRs and LGT distinguished the azo group of MO despite resistance to the conjugation of electrons (Fig. 25d). MO dye is reduced in a longer time than BBG, BBR, or MB as the hydrophobicity of –CH3 influences the delocalisation of the π electrons. The binding constant (kb) of MO with LSNRs and LGT is higher with a lower reduction, indicating oscillation of MO to interact with the photocatalysts. The higher kb blocks the hν receiving sites of the photocatalysts, inducing defects on interacting with them as well as with dyes. The –NN– azo and Na+–SO3− interact, reducing the surface area to become settled at the bottom; therefore, the PCPs and PCR are directly interfaced. ERG (–CH3) intensifies their holes for reducing BBG and BBR in a shorter time than MO with LGT and LSNRs, initiating an ROC via hν. The QNs of BBG and BBR received the e− holes from LGT via ψ for the PCR (Fig. 25c and d). BBR, BBG, MB, and MO are reduced at λmax of 550, 585, 665, and 465 nm, respectively, with UV/vis, light and sunlight for disposal. The reduced dyes were phased out from wastewater for recycling (Fig. 23). The hν weakened the shared electron and FGs accompanied by certain thermodynamic parameters. ΔH < 0, ΔG > 0 and ΔS < 0 as an exothermically spontaneous reaction has fixed the @PCR and @SCR of dyes. The hν have struck FE and 4f#e on decreasing S compared to aq-LSNR to minimize the collision of holes.
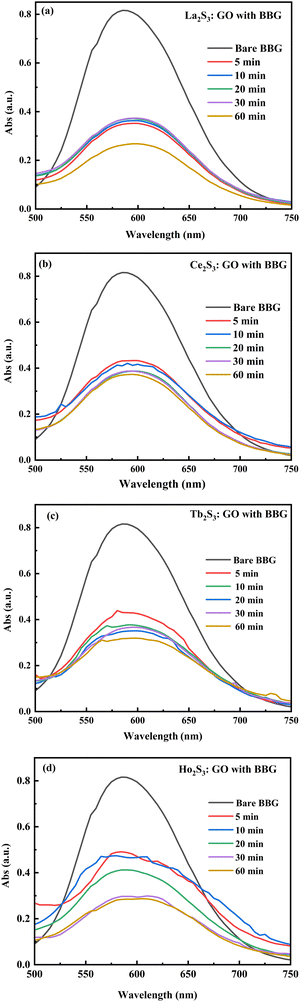 |
| | Fig. 25 Photocatalysis of (a) BBR, (b) BBG, (c) MB and (d) MO through LGT. | |
The absorbance of wastewater effluents decreases with the 4f electrons of the photocatalyzing properties of the photocatalyst. An 87–100% reduction of dyes was observed after ∼60 min. The enhanced photocatalyzing performance of LGT was noticed due to a higher amount of UV light being detained and higher photon trapping ability. The catalysing activities were observed by plotting (C4fe/C0) vs. 4f#e, to observe the role of electronic activities during PCR (Fig. 26). The @4f#e of the photocatalyst indicates the excellent photocatalyzing ability extended to determine the role of the maximum number of 4f electron in the lanthanide series.
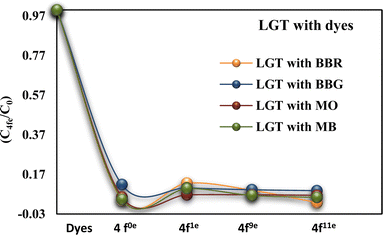 |
| | Fig. 26 Photocatalytic activity as a function of 4f electrons. | |
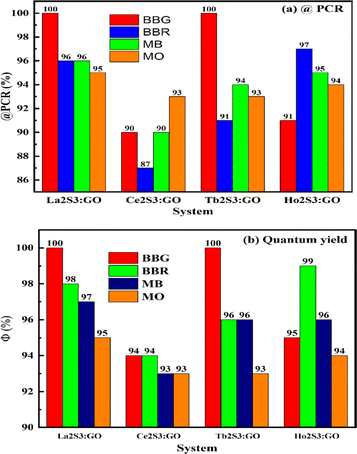 |
| | Fig. 27 (a) @PCR and (b) quantum yield for PCR under SL. | |
6.1. Reusability of LGT photocatalyst
The reusability of a photocatalyst is studied by using the same photocatalyst over 4 to 5 cycles to reduce the fresh effluents. Every cycle reduced the active sites of the photocatalyst, as the quantum yield of the dyes decreases with successive cycles. PCR performance has not significantly decreased after 4–5 cycles due to the structural stability inducing the reusability of the photocatalyst, as supported by TGA on the disruption of the lattice (Fig. 1). Reusability of photocatalysts implies a decrease in active sites in subsequent cycles and mass loss of photocatalyst over time (Fig. 28). The wastewater effluents were suspended in solution at the 4th cycle and settled at the bottom after 1 h as the active sites of the photocatalyst have been disrupted and do not respond to dyes any more (Fig. 28).
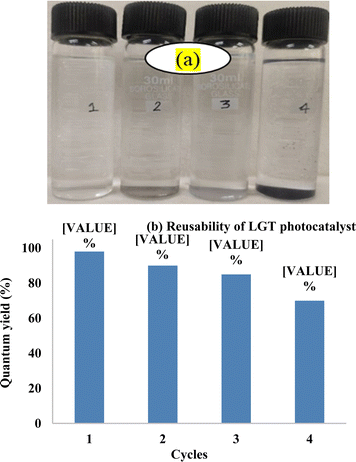 |
| | Fig. 28 (a) Recycled wastewater. (b) Reusability of photocatalyst LGT. | |
Due to ERG-CH3 BBG has responded exceptionally to overcome a QEB of holes of the GO and 4f8e of Tb3+. The conduction band has 4f6e unpaired electron (UNPE) which are robustly excited to reach a conduction band of 5d and then an electron partially comes back and a phonon is emitted. That approaches the excited electron of a dye, especially its reducing site, and it gets reduced. The LGT of the LSNRs followed the same trends, which have proven the 4f6e UNPE of Tb to robustly interact with FE to catalyse the generation of holes. These holes reach the dyes in a larger amount in a shorter time, because 4f6e could have gained similar energy from the hν to get them excited with a similar φ to PCR the dyes in a shorter time than other LGT. Ho3+ has 4f4e UNPE which could not induce an impact on the hν vis-à-vis dye to generate holes, and so took a bit longer to reduce the same dye compared to the LGT of Tb with 4f6e UNPE. A similar action mechanism is followed for Ce2S3:GO and La2S3:GO as for the UNPE generating phonons. The @4f#e UNPE directly interact with the electron which could get excited to interact with the holes. Hence, the higher DOS of increasing 4f#e UNPE interacts with the intensified DOS of the dyes to get reduced as BBR > BBG > MB > MO and with increasing time as MO > MB > BBG > BBR. Thereby, the BBR and BBG with adequate ERG-CH3 as well as maximum π-conjugation have taken a shorter time with each LSNR and LGT (Fig. 28). This process is denoted as @4f#e, but only the DOS of the dye develops an interacting interfacial energy to overcome the QEB in a shorter time. The MO dye has azo and SO3− groups, which generate their own intensified and localized ψ(SO3−) and ψ(NN) that could not synchronize with the π-conjugation of the two benzene rings of MO dye. The EEC and EER have both neutralized the energy of the dyes on receiving the hν that has taken a longer time, despite the presence of LSNRs and LGT. Therefore, the electronic constituents of the dyes matter a lot for reduction as the MO have DOS of ψ(SO3−) and ψ(NN) unlike the DOS of the ψBenzene ring expressed as ψ(SO3−) ≠ ψ(NN) ≠ ψBenzene. Thus, the DOS of BBR and BBG could have been similar except for the ERG with BBG which take a shorter time. Additionally, the MB due to two different ethyl groups with an almost nil effect of ERGs could not develop an intensified DOS and has taken more time for reduction than BBR or BBG (Fig. 22).
7. Sonocatalytic reduction (SCR) of MB, MO, BBR and BBG
LSNRs, GO, and LGT were not homogeneously dissolved in water and phased out without electrostatically engaging the dipoles of water to transfer a cohesive force as PE to oscillatory energy. The hν was not captured by photocatalysts which were sonicated for monodispersion to SCR the dyes by generating functional sites. The sonication generates localized energy centres denoted as charge cavitation. The charge cavitation gains a higher surface area and available electronic oscillatory or resonating activity to develop ψ that cross over a QEB of electronic charge sites of dyes, and the species remained monodispersed. The sonication was further extended from monodispersion to generate photonic waves with a certain hν, equally capable of reducing dyes. Therefore, in both PCR and SCR, a monodispersion of photocatalyst and dyes is a prerequisite. Therefore, the PCPs as a database predict thermodynamic and kinetically stable states for receiving hν to generate ROC. SCR was conducted to equate PCR efficiency with monodispersion and then the hν was supplied and sonication continued to SCR. Sonication weakened the PE of shared electrons and FGs with both oscillating as charged clouds on a cavitation that aligns the electronic cloud shifting to a higher energy state to generate e− and h+ holes. The sonication induces mechanical energy to generate an electron-rich domain e− or electronic-charge-deficient h+ holes. LGT, LSNRs and GO solutions with dyes dissociated their bonds to move the electrons towards the respective holes of dyes for SCR.48 Sonication could also break the bonds and release energy due to oscillations.39 ΔS < 0 and ΔS > 0 indicate ordered and disordered configurations, and oscillations of LGT might be mildly disordered on aligning dyes with 40–90% Φ (Tables S7.7–S8.0, ESI†). The SCR route is effective for dipolar LSNRs and LGT by sonicating at 28 KHz for 5, 10, 20, 30, and 60 min, consequently raising the temperature to 298.15, 301.15, 305.15, 308.15, and 313.15 K, respectively. SCR structurally reduces a dye as a sono-frequency attacks the π-conjugation to enhance the electronic charge density. The structurally damaged dye does not respond to environmental and biological processes. A highly oxidised structure with ΔS < 0 is phased out (Tables S4.2–S5.8, ESI†). ΔG > 0 indicates interaction between dyes and LGT on sonication. Ea is not needed for SCR, as adequate energy already exists. ΔG > 0 values indicate that the sonicating frequency drastically disrupts a shared electron pair of covalent bonds (Fig. S2.4–S2.7, ESI†). The ΔG indicates simultaneous oscillation by a molecule to gain ΔS < 0 with ΔH < 0 values. ΔH < 0 and ΔS < 0 while the ΔG > 0 for all the LSNR systems with BBR, BBG and MB, except MO, have values ΔS > 0 (Fig. 29, and Fig. S2.4, S3.1, S3.2, ESI†). ΔS > 0 of MO is strongly disordered due to an azo group with the least @SCR. The excess π-conjugation increases the ability of @SCR and @PCR due to collisions and ROC (Fig. 23b). ΔS ≪ 0 with the least disorder has a higher @SCR. The least disordered and maximum @SCR as BBR > BBG > MB > MO indicate alignment (Tables S4.2–S5.8, ESI†). SCR in the order BBR > MB > BBG > MO with binding constants (kb) 0.0211 < 0.0255 < 0.0658 < 0.0800, respectively, indicate restricted MO activities as the binding causes a PCR and SCR. The oxidized ≈ reduced on binding dye with LSNRs or LGT as| | 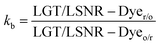 | (19) |
Eqn (19) indicates LGT/LSNR–Dyeo/r ≠ LGT/LSNR–Dyer/o with an equilibrium state. The higher kb for MO indicates a lower @PCR and @SCR as an azo group causes many collisions. The @PCR and @SCR for BBR, BBG and MB are maxima with a lower kb as a monodispersion. BBR, BBG and MB with a maximum π-conjugation is sensitive, unlike MO with weaker @SCR activity (Fig. 30).
 |
| | Fig. 29 The number of unpaired electrons directly enhances the @PCR and @SCR. | |
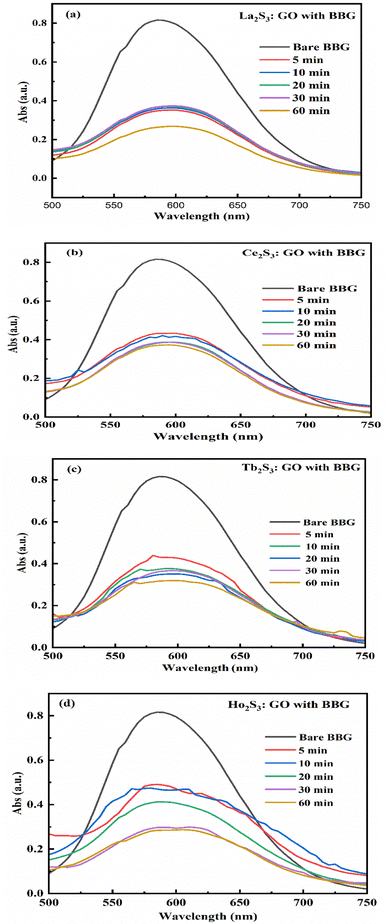 |
| | Fig. 30 SCR of BBG with LGT and other dyes given (Fig. S8–S11, ESI†). | |
The dye alone absorbed maximum UV light at λmax at 550, 585, 665, and 465 nm for BBR, BBG, MB, and MO, respectively, in both PCR and SCR, due to the structural stability of LSNRs and LGT predicted by the PCPs. The reduction of dyes at the same λmax indicates no degradation of dyes into fragments and also no specific complex formation between the dyes and photocatalysts. However, the broadening of the λmax peaks indicates the distribution of similar species forming or yet to be reduced. Thus, the PCPs predict a stronger monodispersion of LSNRs, GO, and LGT. Absorbance of dyes decreases with time and 4f#e during an SCR. The time and 4f#e as vital parameters analyse the ability of a photocatalyst to reduce a dye. The active sites of the dye were trying to capture the opposite holes with substantial sonication energy and get reduced. The sonocatalyzing performance is observed by plotting (C4fe/C0) vs. 4f#e (Fig. 31). The increasing 4f#e induced SCR activities to catalyse wastewater effluents. The time-dependent thermodynamic parameters for LSNRs, LGT with BBR, BBG and MO under SCR indicate reduction activities (Fig. S3.1 and S3.2, ESI†). The increasing rate of sonication energy has electronically disordered a structure with ψ of LGT oscillation. Compared to PCR, the SCR streamlined the dipoles and reduced dyes with 40–90% Φ. The ΔS with dyes from La2S3 to La2S3:GO decreases as La2S3:GO is highly ordered and partially oscillates due to valence electrons (Fig. S3.1, ESI†). The ΔS with La2S3 to Ce2S3:GO increases as 4f1e oscillates, but Tb2S3:GO to Ho2S3:GO are ordered with a least ΔS, so BBR has interacted with 4f#e and FE via π-conjugation (Fig. S3.1, ESI†). Tb2S3:GO to Ho2S3:GO with ΔS < 0 due to stable dipoles SCR dyes in shorter time. LSNRs with higher ΔS than LGT but could not SCR MO due to an azo group compared to LGT. LSNRs have disordered an alignment of dipoles causing recombination of holes. Sonication to induce undesired collisions with @SCR as the dipoles attract the holes of GO. BBG with –CH3 could not establish patterns of ΔS values causing higher @SCR (Fig. S3.1, ESI†). ΔS values are scattered with MO unlike BBG due to –CH3 with different electronic junctions causing SCR (Tables S4.2–S5.8, ESI†). ΔG values for MB, BBR, and MO with increasing time are increased except the BBG as it has an extra ER group that disrupts a bond on a sonication (Fig. S2.4–S2.7, ESI†). Insoluble LSNRs and LGT could not SCR itself with ΔG > 0, but MB with LSNRs having ΔG < 0 indicates an LSNR ∼ MB solubilizing combination with H2O while LGT having ΔG > indicates weaker spontaneity (Tables S4.2–S5.8, ESI†). MO with ΔG < 0 indicates a stronger kb of azo dye, and a milder sonication effect occurring than for other dyes (Fig. S2.7d, ESI†). The ΔG > 0 and ΔH < 0 of LSNRs, GO, and LGT indicate a stronger binding with BBG due to –CH3 (Tables S4.2–S5.8, ESI†). Sonication breaks the NHS and splits H2O, releasing ΔH, and fragments become vibrant (Fig. 27). BBG and BBR have released a higher ΔH than MB and MO to align LSNRs, GO and LGT and causing the higher SCR activities (Tables S4.2–S5.8, ESI†).
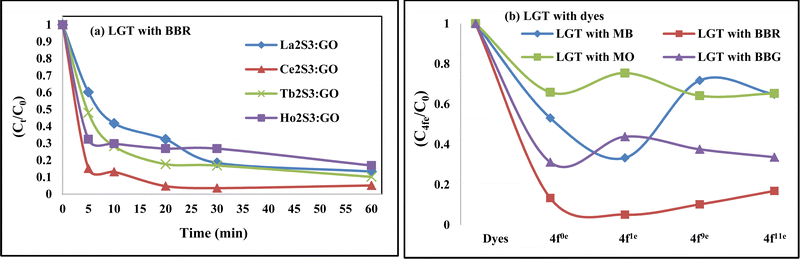 |
| | Fig. 31 (a) Sonocatalytic activity as a function of time and (b) as a function of 4f electrons. | |
8. Tables
Table 5 Dynamic light scattering analysis of GO, LSNRs and LGT aqueous solution
| Parameter |
GO |
La2S3 |
La2S3:GO |
Ce2S3 |
Ce2S3:GO |
Tb2S3 |
Tb2S3:GO |
Ho2S3 |
Ho2S3:GO |
| PDI |
— |
0.425 |
1.591 |
1.002 |
0.175 |
0.619 |
1.023 |
5.35 |
1.369 |
| Diameter (nm) |
6.10 |
457.0 |
307 |
17.66 |
263.7 |
486 |
212.9 |
16.57 |
325 |
| — |
230.3 |
— |
— |
109.3 |
— |
97.60 |
— |
157.7 |
| Volume (%) |
100 |
74.8 |
100 |
100 |
47.2 |
100 |
52.1 |
100 |
20.9 |
| — |
25.2 |
— |
— |
52.8 |
— |
47.9 |
— |
79.1 |
| Width |
2.32 |
344.00 |
158.70 |
9.28 |
161.10 |
169.30 |
126.80 |
6.18 |
98.00 |
| — |
54.90 |
— |
— |
44.40 |
— |
36.50 |
— |
104.30 |
| Mobility (μ s−1) |
0.81 |
1.74 |
−0.04 |
−0.04 |
1.22 |
0.30 |
−1.40 |
0.62 |
0.87 |
| Zeta pot. (mV) |
10.41 |
22.24 |
−0.49 |
−0.51 |
15.64 |
3.88 |
−17.60 |
7.97 |
11.08 |
| Charge (fC) |
0.00052 |
0.327 |
−0.004 |
−0.0064 |
0.033 |
0.036 |
−0.057 |
0.059 |
0.032 |
| Polarity |
+ve |
+ve |
−ve |
−ve |
+ve |
+ve |
−ve |
+ve |
+ve |
| Conductivity (μs cm−1) |
50 |
43 |
18 |
27 |
17 |
24 |
20 |
38 |
22 |
Table 6 Viscosities (mPa S) of LSNRs and LGT
| Temp. (K) |
La2S3 |
Ce2S3 |
Tb2S3 |
Ho2S3 |
La2S3:GO |
Ce2S3:GO |
Tb2S3:GO |
Ho2S3:GO |
| 283.15 |
1.0152 |
0.9035 |
0.8705 |
0.8514 |
0.8944 |
0.8680 |
0.8596 |
0.9281 |
| 288.15 |
1.0475 |
0.9736 |
0.9565 |
0.9497 |
0.9704 |
0.9565 |
0.9668 |
0.9668 |
| 298.15 |
0.8746 |
0.8787 |
0.8827 |
0.8945 |
0.8825 |
0.8827 |
0.9142 |
0.8275 |
| 310.15 |
0.7095 |
0.6986 |
0.6986 |
0.6950 |
0.7058 |
0.6913 |
0.6986 |
0.6950 |
| 315.15 |
0.6379 |
0.5930 |
0.5833 |
0.5650 |
0.5990 |
0.5748 |
0.5586 |
0.6301 |
Table 7 Viscosities (mPa S) of LSNRs and LGT with BSA
| Temp. (K) |
BSA |
La2S3 BSA |
Ce2S3 BSA |
Tb2S3 BSA |
Ho2S3 BSA |
La2S3:GO BSA |
Ce2S3GO BSA |
Tb2S3:GO BSA |
Ho2S3:GO BSA |
| 283.15 |
0.8699 |
0.8668 |
0.8837 |
0.8727 |
0.8617 |
0.8638 |
0.8699 |
0.8973 |
0.8669 |
| 288.15 |
0.9634 |
0.9630 |
0.9702 |
0.9668 |
0.9531 |
0.9599 |
0.9600 |
0.9770 |
0.9599 |
| 298.15 |
0.8827 |
0.8824 |
0.8788 |
0.8866 |
0.8748 |
0.8866 |
0.8788 |
0.8747 |
0.8826 |
| 310.15 |
0.6367 |
0.6364 |
0.6403 |
0.6402 |
0.6439 |
0.6465 |
0.6474 |
0.6437 |
0.6543 |
| 315.15 |
0.4945 |
0.4937 |
0.5067 |
0.4977 |
0.5099 |
0.5054 |
0.5128 |
0.5157 |
0.5209 |
Table 8 Surface tension (mN m−1) of LSNRs and LGT
| Temp. (K) |
La2S3 |
Ce2S3 |
Tb2S3 |
Ho2S3 |
La2S3:GO |
Ce2S3:GO |
Tb2S3:GO |
Ho2S3:GO |
| 283.15 |
65.97 |
63.82 |
66.01 |
65.56 |
66.01 |
64.25 |
65.11 |
67.41 |
| 288.15 |
70.25 |
68.19 |
69.21 |
69.21 |
68.72 |
67.70 |
68.19 |
69.21 |
| 298.15 |
71.06 |
71.64 |
71.64 |
72.22 |
71.63 |
71.64 |
71.64 |
71.64 |
| 310.15 |
58.481 |
63.725 |
65.07 |
66.48 |
68.95 |
67.44 |
68.95 |
70.00 |
| 315.15 |
51.98 |
58.38 |
60.56 |
62.15 |
66.52 |
63.47 |
66.01 |
67.99 |
Table 9 Surface tension (mN m−1) of LSNRs and LGT with BSA
| Temp. (K) |
BSA |
La2S3 BSA |
Ce2S3 BSA |
Tb2S3 BSA |
Ho2S3 BSA |
La2S3:GO BSA |
Ce2S3:GO BSA |
Tb2S3:GO BSA |
Ho2S3:GO BSA |
| 283.15 |
74.21 |
66.91 |
71.97 |
72.52 |
74.20 |
73.58 |
73.07 |
73.06 |
70.88 |
| 288.15 |
70.799 |
67.67 |
69.21 |
69.73 |
70.26 |
70.26 |
70.26 |
69.21 |
68.19 |
| 298.15 |
66.868 |
66.84 |
65.89 |
65.89 |
65.41 |
66.37 |
66.87 |
65.41 |
65.40 |
| 310.15 |
65.995 |
61.98 |
64.16 |
63.71 |
63.71 |
65.89 |
65.97 |
65.50 |
65.47 |
| 315.15 |
67.00 |
59.36 |
64.27 |
63.86 |
64.27 |
67.28 |
66.51 |
66.95 |
66.83 |
Table 10 Friccohesites (cm S−1) of LSNRs and LGT
| Temp. (K) |
La2S3 (10−4) |
Ce2S3 (10−4) |
Tb2S3 (10−4) |
Ho2S3 (10−4) |
La2S3:GO (10−4) |
Ce2S3:GO (10−4) |
Tb2S3:GO (10−4) |
Ho2S3:GO (10−4) |
| 283.15 |
154 |
140 |
132 |
136 |
135 |
135 |
132 |
155 |
| 288.15 |
149 |
143 |
138 |
137 |
141 |
141 |
142 |
140 |
| 298.15 |
119 |
114 |
123 |
124 |
123 |
123 |
128 |
116 |
| 310.15 |
121 |
110 |
107 |
105 |
102 |
099 |
100 |
099 |
| 315.15 |
122 |
101 |
096 |
091 |
090 |
090 |
084 |
049 |
Table 11 Friccohesity (cm S−1) of LSNRs and LGT LGT with BSA
| Temp. (K) |
BSA (10−4) |
La2S3 (10−4) |
Ce2S3 (10−4) |
Tb2S3 (10−4) |
Ho2S3 (10−4) |
La2S3:GO (10−4) |
Ce2S3:GO (10−4) |
Tb2S3:GO (10−4) |
Ho2S3:GO (10−4) |
| 283.15 |
117 |
130 |
122 |
120 |
116 |
117 |
119 |
123 |
122 |
| 288.15 |
136 |
142 |
140 |
139 |
136 |
137 |
137 |
141 |
141 |
| 298.15 |
132 |
132 |
133 |
135 |
134 |
134 |
131 |
134 |
135 |
| 310.15 |
096 |
103 |
100 |
100 |
101 |
092 |
092 |
092 |
093 |
| 315.15 |
074 |
083 |
079 |
078 |
079 |
075 |
077 |
077 |
078 |
9. Conclusion
Multifunctional LSNR and LGT photocatalysts and adsorbents were prepared and had enhanced solubility of BSA by initiating interactions with the peptide bonds and electrostatic dipoles of aq-BSA with solubilizing activities. Physicochemical properties and thermodynamic functions of aq-LSNR, and aq-LGT alone and with 0.010 g% BSA at 283.15–315.15 K and within 0.002–0.01 g% @0.004 g% at 298.15 K indicating structural stability by doing PCR and SCR activities. The physicochemical properties and thermodynamic functions of BSA with LSNRs and LGT indicates a stronger 4f#e ∼ FE interface with electrostatic dipoles, peptide bonds, and salt bridges as a robust approach in the area of lanthanide chemistry with biomolecules. The stronger temperature and concentration effect with structural stability, functionality, and thermal stability of LSNRs and LGT enabling their reusability for PCR and SCR activities. LSNRs with a stronger hν trapping ability to photocatalyze the fluorescent dyes in a longer time while LGT has shortened reduction time due to 4f#e ∼ FE interfaces. LGT recycled wastewater containing fluorescent effluents in ∼60 min compared to LSNRs and GO with ∼24 h and 120 min, respectively. LGT has reduced BBG, BBR, MB, and MO as 90–100, 87–97, 90–96, and 93–95% @PCR with 94–100, 94–99, 93–97, and 93–95% quantum yields, respectively. Each dye was sonicated @28 kHz with LSNRs and LGT separately for 5, 10, 20, 30, and 60 min were consequently raised to 298.15, 301.15, 305.15, 308.15, and 313.15 K, respectively, as an exothermic activity of LSNRs and an LGT support with thermodynamic functions. SCR has reduced the dyes with 40 to 90% Φ. PCR and SCR activities indicated that LGT is a good photocatalyst and a solubilizing agent with excellent reusability.
Abbreviations
| GO | Graphene oxide |
| LSNRs | Lanthanide sulphide nanorods |
| LGT | Lanthanide GO template |
| MB | Methylene blue |
| BBR | Brilliant blue red |
| BBG | Brilliant blue green |
| MO | Methylene orange |
| BSA | Bovine serum albumin |
| PCPs | Physicochemical properties |
| FT-IR | Fourier transform infrared spectroscopy |
| GT | Graphite |
| GtO | Graphite oxide |
| RB | Round bottom flask |
| RPM | Revolutions per minute |
| aq | Aqueous |
| PDN | Pendant drop number |
| VFT | Viscous flow time |
| wt | Weight |
| SCR | Sonocatalytic reduction |
| FE | Functional edge |
| RE | Resonating energy |
| HHI | Hydrophilic–hydrophilic interactions |
| HB | Hydrogen bond |
| PB | Peptide bond |
| CAT | Critical aggregation temperature |
| SL | Sunlight |
| PCR | Photocatalytic reduction |
| ERG | Electron releasing group |
| TGA | Thermogravimetric |
| DTG | Differential thermal gravimetry |
Author contributions
Krishan Kumar conducted the bench work, calculations, plotting figures and other designs for effective discussion and conclusive. Bhargab Sahoo initiated formulating the molecular solution preparation for accurate stoichiometry to measure the primary data of binary systems. Tara Chand Meghwal initiated formulating the molecular solution preparation for accurate stoichiometry to measure the primary data of ternary systems. Prof. Man Singh was in charge of overall supervision for conducting the research work, frequent discussions, exploring new models outer experiment observations and matching with the explored scientific exhalations to substantially create the new science out of the research. Funding Sources There was no funding source.
Conflicts of interest
The authors confirm that there is no conflict of interest.
Acknowledgements
The authors thankful to Central University of Gujarat, India for facilities. Mr Krishan Kumar is thankful to UGC-DAE CSR Indore, was acknowledged for financial support (ref. CRS/2021-22/379) and Dr Mukul Gupta as a Principal Collaborator, and Dr Vasant Sathe, UGC-DAE CSR Indore, was acknowledged for providing Raman facility. The authors are thankful to CSIR-CMCRI Bhavnagar, Gujarat for FESEM + EDS analysis.
References
- X. Zeng, Y. Liu, T. Zhang, J. C. Jin, J. L. Li, Q. Sun, Y. J. Ai, M. L. Feng and X. Y. Huang, Ultrafast and selective uptake of Eu3+ from aqueous solutions by two layered sulfides, Chem. Eng. J., 2021, 420, 127613 CrossRef CAS.
- R. Kumar, S. Sahoo, E. Joanni, R. K. Singh, K. Maegawa, W. K. Tan, G. Kawamura, K. K. Kar and A. Matsuda, Heteroatom doped graphene engineering for energy storage and conversion, Mater. Today, 2020, 39, 47–65 CrossRef CAS.
- S. Dev and M. Singh, Metallic sulfide nanoparticles anchored graphene oxide: Synthesis, characterization and reduction of methylene blue to leuco methylene blue in aqueous mixtures, J. Phys. Chem. Solids, 2020, 139, 109335 CrossRef CAS.
- A. T. Smith, A. M. LaChance, S. Zeng, B. Liu and L. Sun, Synthesis, properties, and applications of graphene oxide/reduced graphene oxide and their nanocomposites, Nano Mater. Sci., 2019, 1, 31–47 CrossRef.
- K. Kumar, R. P. Dave, S. Dev and M. Singh, Study of molar properties of GO after doping with transition metals for photodegradation of fluorescent dyes, RSC Adv., 2022, 29734–29756 RSC.
- J. Dadashi, M. A. Ghasemzadeh and M. Salavati-Niasari, Recent developments in hydrogels containing copper and palladium for the catalytic reduction/degradation of organic pollutants, RSC Adv., 2022, 12, 23481–23502 RSC.
- S. Roméro, A. Mosset, J. C. Trombe and P. Macaudière, Low-temperature process of the cubic lanthanide sesquisulfides: Remarkable stabilization of the γ-Ce2S3 phase, J. Mater. Chem., 1997, 7, 1541–1547 RSC.
- İ. A. Kariper, Synthesis and characterization of cerium sulfide thin film, Prog. Nat. Sci.: Mater. Int., 2014, 24(6), 663–670 CrossRef.
- İ. A. Kariper, The production of UV absorber amorphous cerium sulfide thin film, Mater. Res., 2017, 20, 1345–1349 CrossRef.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Graphene-based supercapacitor with an ultrahigh energy density, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- G. Darabdhara, M. R. Das, S. P. Singh, A. K. Rengan, S. Szunerits and R. Boukherroub, Ag and Au nanoparticles/reduced graphene oxide composite materials: Synthesis and application in diagnostics and therapeutics, Adv. Colloid Interface Sci., 2019, 271, 101991 CrossRef CAS PubMed.
- M. Ubaidullah, A. M. Al-Enizi, T. Ahamad, S. F. Shaikh, M. A. Al-Abdrabalnabi, M. S. Samdani, D. Kumar, M. A. Alam and M. Khan, Fabrication of highly porous N-doped mesoporous carbon using waste polyethylene terephthalate bottle-based MOF-5 for high performance supercapacitor, J. Energy Storage, 2021, 33, 102125 CrossRef.
- A. Roca-Sabio, M. Regueiro-Figueroa, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas and C. Platas-Iglesias, Density functional dependence of molecular geometries in lanthanide(III) complexes relevant to bioanalytical and biomedical applications, Comput. Theor. Chem., 2012, 999, 93–104 CrossRef CAS.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. van Grondelle, Govindjee and G. D. Scholes, Light absorption and energy transfer in the antenna complexes of photosynthetic organisms, Chem. Rev., 2017, 117(2), 249–293 CrossRef CAS PubMed.
- X. Wei, H. Feng, L. Li, J. Gong, K. Jiang, S. Xue and P. K. Chu, Synthesis of tetragonal prismatic γ-In2Se3 nanostructures with predominantly {110} facets and photocatalytic degradation of tetracycline, Appl. Catal., B, 2020, 260, 118218 CrossRef CAS.
- R. Tondwal and M. Singh, Chitosan functionalization with a series of sulfur-containing α-amino acids for the development of drug-binding abilities, J. Appl. Polym. Sci., 2018, 135, 1–14 CrossRef.
- S. Mahapatra, G. Madras and T. N. G. Row, Ind. Eng. Chem. Res., 2007, 46, 1013–1017 CrossRef CAS.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C. K. Tsung, Aperture-Opening Encapsulation of a Transition Metal Catalyst in a Metal-Organic Framework for CO2 Hydrogenation, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed.
- P. Nuengmatcha, S. Chanthai, R. Mahachai and W. C. Oh, Sonocatalytic performance of ZnO/graphene/TiO2 nanocomposite for degradation of dye pollutants (methylene blue, texbrite BAC-L, texbrite BBU-L and texbrite NFW-L) under ultrasonic irradiation, Dyes Pigm., 2016, 134, 487–497 CrossRef CAS.
- M. Sun, J. Ma, M. Zhang, Y. Xiao, Y. Zhu and S. Zhang, Thiourea-modified Fe3O4/graphene oxide nanocomposite as an efficient adsorbent for recycling Coomassie brilliant blue from aqueous solutions, Mater. Chem. Phys., 2020, 241, 122450 CrossRef CAS.
- C. M. Marin, L. Wang, J. R. Brewer, W. N. Mei and C. L. Cheung, Crystalline α-Sm2S3 nanowires: Structure and optical properties of an unusual intrinsically degenerate semiconductor, J. Alloys Compd., 2013, 563, 293–299 CrossRef CAS.
- J. Choi, J. C. Kim, Y. B. Lee, I. S. Kim, Y. K. Park and N. H. Hur, Fabrication of silica-coated magnetic nanoparticles with highly photoluminescent lanthanide probes, Chem. Commun., 2007, 1644–1646 RSC.
- D. G. Papageorgiou, I. A. Kinloch and R. J. Young, Mechanical properties of graphene and graphene-based nanocomposites, Prog. Mater. Sci., 2017, 90, 75–127 CrossRef CAS.
- O. P. Kumar, M. N. Ashiq, M. Ahmad, S. Anjum and A. Ur Rehman, Silver and yttrium-doped bismuth vanadate for photoluminescent activity and boosted visible light-induced photodegradation, J. Mater. Sci. Mater. Electron., 2020, 31, 21082–21096 CrossRef CAS.
- S. Aime, S. G. Crich, E. Gianolio, G. B. Giovenzana, L. Tei and E. Terreno, High sensitivity lanthanide(III) based probes for MR-medical imaging, Coord. Chem. Rev., 2006, 250, 1562–1579 CrossRef CAS.
- P. Macaudie, Low-temperature process of the cubic lanthanide sesquisulfides: remarkable stabilization of the c-Ce S phase, J. Mater. Chem., 1997, 7, 1541–1547 RSC.
- V. J. Mane, S. B. Kale, S. B. Ubale, V. C. Lokhande, U. M. Patil and C. D. Lokhande, Lanthanum sulfide-manganese sulfide/graphene oxide (La2S3-MnS/GO) composite thin film as an electrocatalyst for oxygen evolution reactions, J. Solid State Electrochem., 2021, 25, 1775–1788 CrossRef CAS.
- J. Wu, K. Fujii, M. Yashima, A. Staykov, T. Akbay, T. Ishihara and J. A. Kilner, A systematic evaluation of the role of lanthanide elements in functional complex oxides; implications for energy conversion devices, J. Mater. Chem. A, 2018, 6, 11819–11829 RSC.
- R. Chetty, S. R. Pandya and M. Singh, Physicochemical interaction of cerium oxide nanoparticles with simulated biofluids, hemoglobin, insulin, and ds-DNA at 310.15 K, New J. Chem., 2020, 44, 1825–1845 RSC.
- J. Yin, Y. Hu and J. Yoon, Fluorescent probes and bioimaging: Alkali metals, alkaline earth metals and pH, Chem. Soc. Rev., 2015, 44, 4619–4644 RSC.
- D. Kumar, A. Chandra and M. Singh, Influence of Urea on Shifting Hydrophilic to Hydrophobic Interactions of Pr(NO3)3, Sm(NO3)3, and Gd(NO3)3 with BSA in Aqueous Citric Acid: A Volumetric, Viscometric, and Surface Tension Study, J. Chem. Eng. Data, 2014, 59(11), 3643–3651 CrossRef CAS.
- K. A. Gschneidner, Preparation and Processing of Rare Earth Chalcogenides, J. Mater. Eng. Perform., 2000, 7, 656–660 CrossRef.
- Y. Han, Preparation of Calcium lanthanum Sulfide Powders via Alkoxide Su, J. Am. Ceram. Soc., 1991, 19, 2815–2819 CrossRef.
- A. B. Ghosh, N. Saha, A. Sarkar, D. N. Srivastava, P. Paul and B. Adhikary, RSC Advances Solvent assisted and solvent free orientation of growth of nanoscaled lanthanide sulfides: tuning of morphology and manifestation of photocatalytic, RSC Adv., 2015, 3, 102818–102827 RSC.
- X. Luo, M. Zhang, L. Ma and Y. Peng, Preparation and stabilization of γ-La2S3 at low temperature, J. Rare Earths, 2011, 29, 313–316 CrossRef CAS.
- S. Rezania, A. Mojiri, J. Park, N. Nawrot, E. Wojciechowska, N. Marraiki and N. S. S. Zaghloul, Removal of lead ions from wastewater using lanthanum sulfide nanoparticle decorated over magnetic graphene oxide, Environ. Res., 2022, 204, 111959 CrossRef CAS PubMed.
- L. Li, S. Hirai and Y. Tasaki, Synthesis and sintering of samarium rich SmSx and its electrical property, J. Rare Earths, 2016, 34, 1042–1047 CrossRef CAS.
- K. R. Kolte, K. Shah and B. S. Chakrabarty, Exploring lanthanum sulphide characteristics for its physical properties, Int. J. Sci. Technol. Res., 2019, 8, 957–961 Search PubMed.
- G. Avashthi and M. Singh, Ultrasound accelerated near-edge functionalized heterogeneous graphene oxide sonocatalyst for surface optical bandwidth efficacy and in situ sonothermocatalysis, New J. Chem., 2021, 45, 5463–5483 RSC.
- S. Dev and M. Singh, Lanthanide sulfide decorated graphene oxide nanorods as an effective photocatalysts for reducing dyes, free radicals, and water split to produce H2 in sunlight, Solid State Sci., 2022, 134, 107045 CrossRef CAS.
- N. Vashistha, A. Chandra and M. Singh, HSA functionalized Gd2O3:Eu3+ nanoparticles as an MRI contrast agent and a potential luminescent probe for Fe3+, Cr3+, and Cu2+ detection in water, New J. Chem., 2020, 44, 14211–14227 RSC.
- S. S. Maktedar, S. S. Mehetre, G. Avashthi and M. Singh, In situ sonochemical reduction and direct functionalization of graphene oxide: A robust approach with thermal and biomedical applications, Ultrason. Sonochem., 2017, 34, 67–77 CrossRef CAS PubMed.
- P. Aryanrad, H. R. Naderi, E. Kohan, M. R. Ganjali, M. Baghernejad and A. Shiralizadeh Dezfuli, Europium oxide nanorod-reduced graphene oxide nanocomposites towards supercapacitors, RSC Adv., 2020, 10, 17543–17551 RSC.
- B. Hou, X. Wang, J. Wang, J. Yao, H. Zhang, W. Yu, G. Liu, X. Dong and L. Wang, In situ synthesis of homogeneous Ce2S3/MoS2 composites and their electrochemical performance for lithium ion batteries, RSC Adv., 2017, 7, 6309–6314 RSC.
-
M. Singh and S. Singh, Survismeter: Fundamentals, Concepts, and Applications, Jenny Stanford Publishing, 1st edn, 2019, DOI:10.1201/9780429027611.
- M. Singh, Survismeter-Type I and II for surface tension, viscosity measurements of liquids for academic, and research and development studies, J. Biochem. Biophys. Methods, 2006, 67, 151–161 CrossRef CAS PubMed.
- M. Singh, Upper critical solution temperatures for immiscible solvent systems with halide salts, carboxylic acids, surfactants and polynuclear aromatic compounds and benzene derivatives, J. Chem. Thermodyn., 2007, 39, 240–246 CrossRef CAS.
- R. Mirzajani and S. Karimi, Ultrasonic assisted synthesis of magnetic Ni-Ag bimetallic nanoparticles supported on reduced graphene oxide for sonochemical simultaneous removal of sunset yellow and tartrazine dyes by response surface optimization: Application of derivative spectrophot, Ultrason. Sonochem., 2019, 50, 239–250 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*
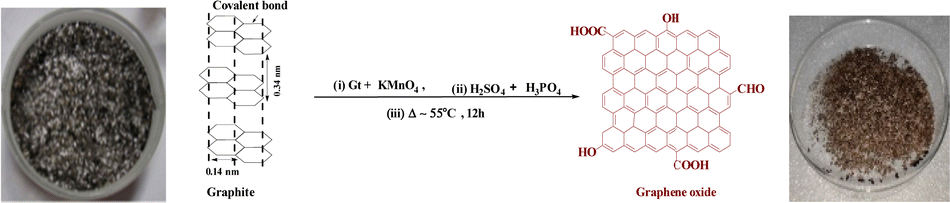
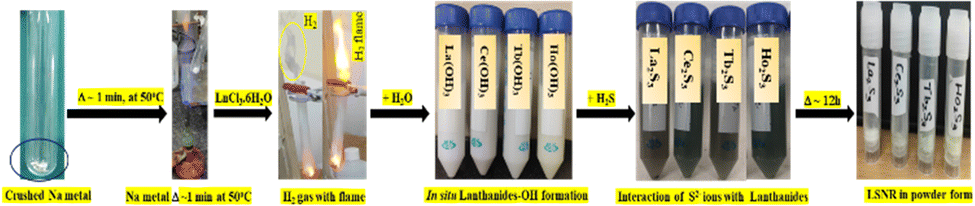


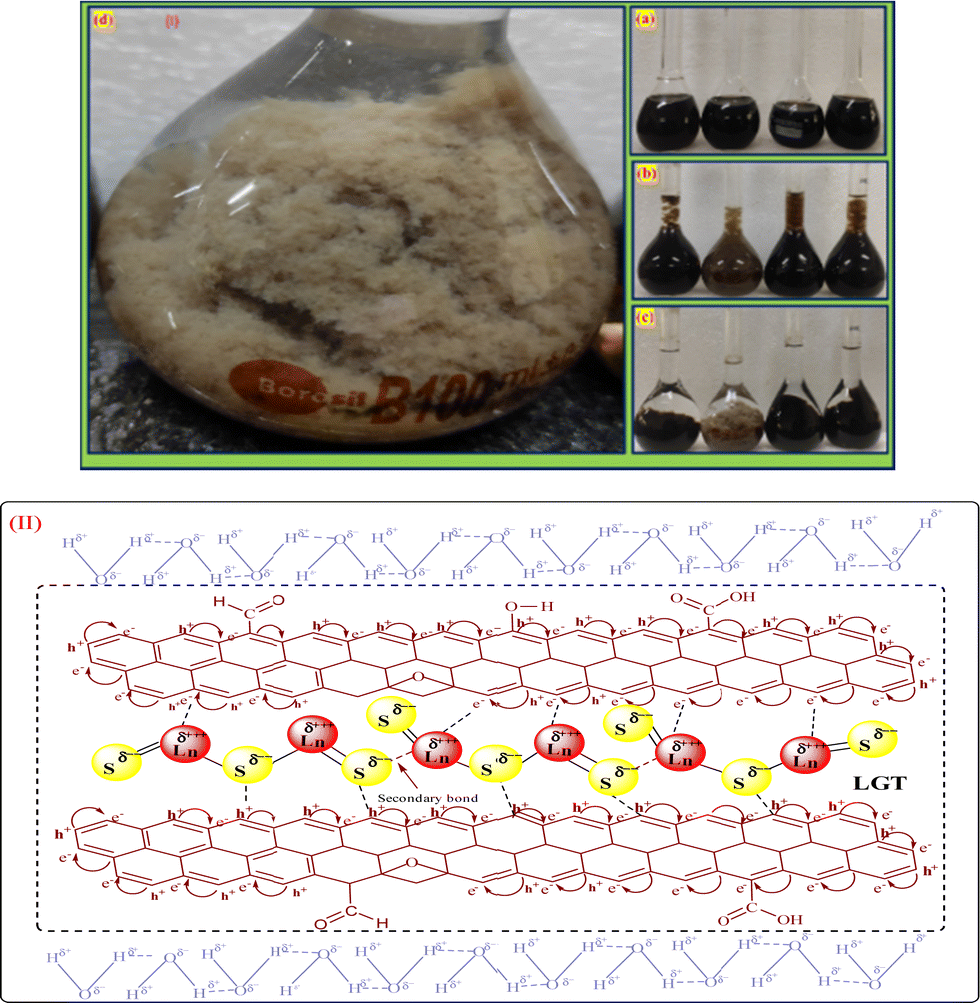


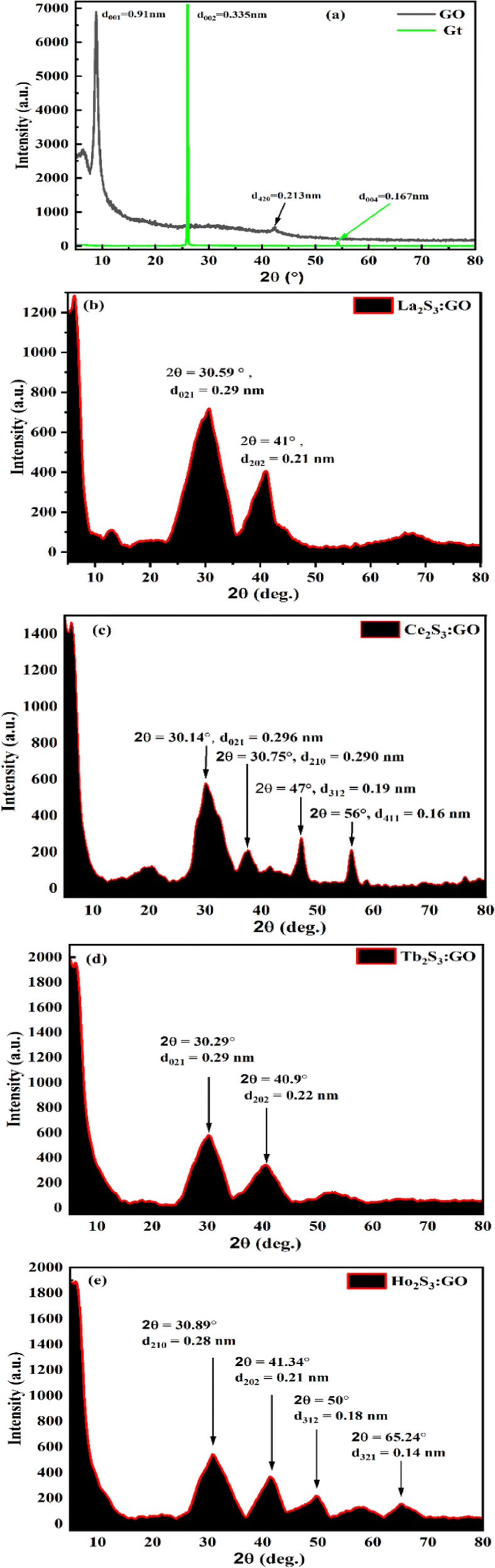


 , where the ψ could have π/2, π, and 2π coordinates of the wavevector. Hence, these hardcore wave mechanics laid down a function of interfacial alignment of the LSNRs and 2D GO sheets (Fig. 10). Therefore, the LGT attained a matrix with the most QDs, highly integrated with the uniformly distributed lattice of the LSNR ≈ GO energy plane without disrupting the basic structural framework of the LSNRs and GO. In fact, at 100 nm scale highly uniform energetic ODs are seen without energy spreading. Hence, the ψLGT ≈ [∂p2/2m ± V(x, t)]ψ integrate with the most synchronized arrangement depicted with the sharpest single light order.
, where the ψ could have π/2, π, and 2π coordinates of the wavevector. Hence, these hardcore wave mechanics laid down a function of interfacial alignment of the LSNRs and 2D GO sheets (Fig. 10). Therefore, the LGT attained a matrix with the most QDs, highly integrated with the uniformly distributed lattice of the LSNR ≈ GO energy plane without disrupting the basic structural framework of the LSNRs and GO. In fact, at 100 nm scale highly uniform energetic ODs are seen without energy spreading. Hence, the ψLGT ≈ [∂p2/2m ± V(x, t)]ψ integrate with the most synchronized arrangement depicted with the sharpest single light order.

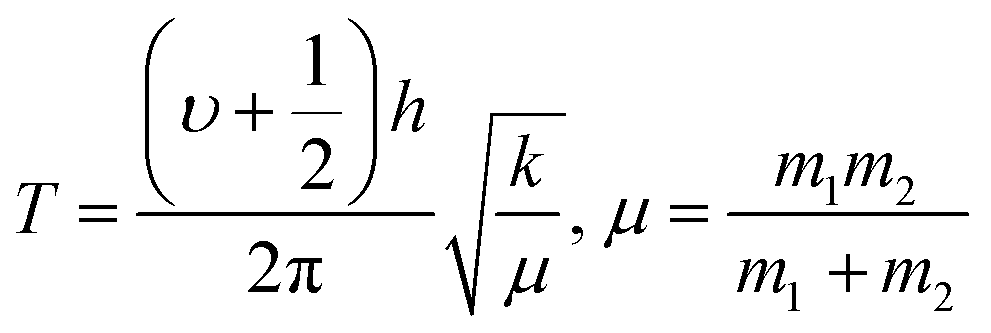
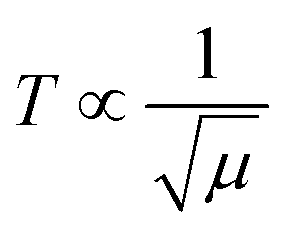 and
and 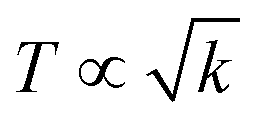 , and frequency depends on temperature and oscillations as per Hooke's law, and for the bond constant k,
, and frequency depends on temperature and oscillations as per Hooke's law, and for the bond constant k, 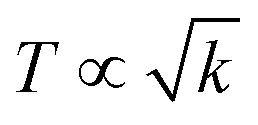 .
.


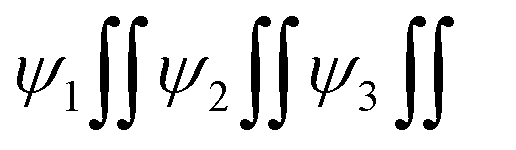 unpaired electronic states with [ψ1 ≈ ψ2 ≈ ψ3]2 probability. Subscripts 1, 2, 3 on ψ denote electron transitions from 1 → 2 → 3 energy states of the 4f#e suborbital. Critical analysis of the micrographs of GO, La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO indicate topographical differences at 4f#e (Fig. 14). The changes are caused by the splitting energy of Ln2S3:GO with GO due to 4f#e. LSNR dopants have influenced the GO morphology due to 4f#e ∼ FE on aligning a GO matrix vis-à-vis LGT. La2S3:GO, Tb2S3:GO, and Ho2S3:GO produced almost equally distributed energy patterns, unlike Ce2S3:GO. Equal distribution of similar lacings of GO sheets are split into equal distributions as each GO sheet participates in LGT formation. The unpaired electron of Tb3+ symmetrises a GO topography, accepting more hν for PCR and SCR with Tb2S3:GO (Fig. 14d). Ho2S3 with 4f11e develops ψ4f11 with the least QEB, inducing a symmetric topography RE of FE. The ψ4f11 and ψFE in the same φ have developed a single crystal with a sharp peak, as depicted with TDA/DTG (Fig. 11e).
unpaired electronic states with [ψ1 ≈ ψ2 ≈ ψ3]2 probability. Subscripts 1, 2, 3 on ψ denote electron transitions from 1 → 2 → 3 energy states of the 4f#e suborbital. Critical analysis of the micrographs of GO, La2S3:GO, Ce2S3:GO, Tb2S3:GO and Ho2S3:GO indicate topographical differences at 4f#e (Fig. 14). The changes are caused by the splitting energy of Ln2S3:GO with GO due to 4f#e. LSNR dopants have influenced the GO morphology due to 4f#e ∼ FE on aligning a GO matrix vis-à-vis LGT. La2S3:GO, Tb2S3:GO, and Ho2S3:GO produced almost equally distributed energy patterns, unlike Ce2S3:GO. Equal distribution of similar lacings of GO sheets are split into equal distributions as each GO sheet participates in LGT formation. The unpaired electron of Tb3+ symmetrises a GO topography, accepting more hν for PCR and SCR with Tb2S3:GO (Fig. 14d). Ho2S3 with 4f11e develops ψ4f11 with the least QEB, inducing a symmetric topography RE of FE. The ψ4f11 and ψFE in the same φ have developed a single crystal with a sharp peak, as depicted with TDA/DTG (Fig. 11e).