
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ (bio)remediation treatment options for U and Sr contaminated land: a comparison of radionuclide retention and remobilisation†
Gianni F.
Vettese‡
 *,
Katherine
Morris
,
Matthew
White-Pettigrew
,
Luke T.
Townsend§
,
Samuel
Shaw
,
Christopher
Boothman
and
Jonathan R.
Lloyd
*
*,
Katherine
Morris
,
Matthew
White-Pettigrew
,
Luke T.
Townsend§
,
Samuel
Shaw
,
Christopher
Boothman
and
Jonathan R.
Lloyd
*
Williamson Research Centre for Molecular Environmental Science, Research Centre for Radwaste Disposal, Department of Earth and Environmental Science, The University of Manchester, Manchester, England, UK. E-mail: Gianni.vettese@helsinki.fi; jon.lloyd@manchester.ac.uk
First published on 14th August 2023
Abstract
The past 60+ years of global nuclear activity has resulted in a significant legacy of radioactive contaminated lands which have high economic costs associated with their remediation. Developing clean-up technologies that are environmentally friendly, economically viable and effective in the long-term is key, with in situ remediation techniques as an important option. However, questions remain regarding the most favorable methods of remediation, and the long-term stability of any immobilised radionuclide(s). Here, we used sediment microcosms to assess the long-term (300 day) stability of immobilised U and Sr formed during anoxic microbial and chemical treatments, and assessed their stability during re-oxidation scenarios (with oxygen or nitrate additions, 100 days). We used six contrasting treatment approaches which resulted in 89 to >99%, and 65–95% removal efficiencies for U and Sr, respectively. These included two Zero Valent Iron (ZVI) based products (NANOFER 25S and Carbo-Iron); a slow-release electron donor (Metals Remediation Compound, MRC) to stimulate U(VI) bioreduction alongside a readily bioavailable electron donor control (lactate/acetate mix); electron donor (lactate/acetate) with elevated sulfate to stimulate metal and sulfate reduction; glycerol phosphate to promote both bioreduction of U(VI) and biomineralization of inorganic U/Sr phosphates; and finally a natural attenuation (no remediation agent added) control. X-ray Absorption Spectroscopy (XAS) revealed that whilst aqueous U was removed from solution via multiple mechanisms including sorption, reduction and incorporation, aqueous Sr was mostly removed via outer sphere complexation mechanisms. Re-oxidation with air led to increased U remobilisation (≤89%) compared to nitrate oxidation (≤73%), but neither oxygen or nitrate re-oxidation led to significant Sr remobilisation (≤38%), suggesting Sr speciation may be stable over extended timescales post remediation. Treatments amended with ZVI or glycerol phosphate not only removed the most U and Sr from solution (>99%) but they also retained the most U and Sr following re-oxidation (retaining ≥75% of the originally added U and Sr). XAS analyses suggests that enhanced immobilisation, as seen in the treatments amended with ZVI or glycerol phosphate, may be due to the U/Sr incorporation into mineral phases (i.e., iron oxyhydroxide and phosphate phases). This suggests that optimal (bio)remediation strategies should target both reduction and biomineralisation mechanisms to facilitate radionuclide-mineral incorporation, promoting longer-term stability.
Environmental significanceNuclear energy usage, both civilian and military, has resulted in a significant legacy of contaminated land worldwide. Protecting the environment demands a comprehensive understanding of the biogeochemical behaviour of radionuclides under environmental conditions. Uranium and strontium are key risk-driving radionuclides, that require remediation, and in situ (bio)remediation offers an effective, economic and robust approach that is applicable at industrial scale. Despite extensive research, the long term stability of the end-points formed during (bio)remediation hinders application for contaminated land clean-up. This study aims to improve our knowledge of leading (bio)remediation strategies for the removal of uranium and strontium from groundwater, quantifying their long term retention in contaminated land scenarios. Ultimately, this will help identify optimal treatments that offer the most stable end-points. |
Introduction
Elevated concentrations of redox active U and non-redox active 90Sr contaminate groundwaters at nuclear sites worldwide e.g., Sellafield, UK; Rifle and Hanford, USA; and Maya, Russia.1–5 In some circumstances, contaminated sites may require intervention to control U and 90Sr subsurface mobility. Here, remediation treatments would need to be amenable to in situ application, be robust for treatment of a range of co-contaminants (both radionuclides and non-active contaminants) and form end-points that remain stable over extended timescales. This study investigates the effectiveness of a range of commonly applied remediation approaches including bioremediation, biostimulation and chemical remediation to treat U and Sr before assessing the stability of these treatments during oxidative perturbations over 400 days.In both laboratory and field scale studies, U in groundwaters can be remediated by microbial reduction processes whereby soluble U(VI), as uranyl(VI) [U(VI)O22+], is enzymatically reduced to form a range of poorly soluble U(IV) species via addition of simple electron donors.6–11 These biogenic, poorly soluble U(IV) phases include nano-crystalline uraninite, and non-uraninite U(IV) reduction products; the latter are often bound to biomass via phosphoryl ligands and are referred to as non-crystalline U(IV). In both laboratory experiments and natural environments U(IV) is often found as a mixture of both nano-crystalline uraninite and non-crystalline(IV).7,9,12–19 Here some studies suggest nano-crystalline uraninite is more stable (e.g., less susceptible to oxidative remobilisation) than non-crystalline U(IV).20–22 Aqueous U(VI) immobilisation can also be achieved via sorption to geomedia,19,23–27 uranyl(VI) mineral precipitation (as e.g., carbonates/phosphates),28–30 and/or abiotic reduction to poorly soluble U(IV) on reaction with surface associated Fe(II) and or sulfide in bioreduced sediments.31–33 In addition, if U(V,VI) is present during the formation of the Fe(oxide)oxide phases, U can be incorporated into Fe minerals including Fe(III)- and Fe(II)-bearing phases during their formation.34–39 Other treatments that show promise for U remediation include nanoscale zero valent iron (nZVI),40–47 organo-phosphates (such as glycerol-phosphate),48,49 and also proprietary slow release electron donors.50 These treatments form different end-points including nanoscale uraninite, sorbed and incorporated U (onto/into minerals) and U(IV)-phosphate minerals which may offer control of U solubility over longer timescales than simple electron donor amendments and highlight the complexity of U-speciation in these systems.40–53
Under environmental conditions, 90Sr is present as the aqueous Sr(II) cation and its environmental mobility is largely governed by pH, sorption to negatively-charged mineral surfaces and/or mineral precipitation.54 At low ionic strengths and at circumneutral to basic conditions, Sr(II) can sorb to mineral surfaces including Fe-(oxy)hydroxides and clays due to their large cation exchange capacity and surface area.55–60 Here, Sr(II) typically forms outer-sphere complexes in competition with other cations (such as H+, Na+, K+, Mg2+),61–65 and Sr(II) bound via outer sphere sorption is considered susceptible to desorption and remobilisation with changes in pH and ionic strength.65–67 Simple electron donors (such as acetate, lactate or glycerol) which promote bioreduction processes typically do not impact Sr(II) removal from solution64,68,69 unless other biogeochemical processes impact on pH and lead to alkaline conditions.63,64 Higher pH not only increases the net negative charge on mineral surfaces (therefore increasing electrostatic attraction with Sr(II) cations), but eventually (at pH > 9) may promote formation of carbonate minerals which can incorporate Sr(II).70 Indeed, mineralisation of Sr(II) offers the potential for precipitation of recalcitrant phases. Here, Sr(II) incorporation into Ca-carbonate (e.g., calcite)71 and Ca-phosphate (e.g., hydroxy-apatite) phases67,68,72 has been noted. Sr(II) also has the potential to be remediated using ZVI, although to the best of our knowledge, there is only one study of Sr(II) removal using ZVI.73 Efecan et al. showed that although 40–77% Sr was removed from solution, almost two thirds of the immobilised Sr was remobilised during 24 hour desorption tests with DI water, suggesting that Sr(II) uptake was highly reversible and would therefore be an inefficient method of Sr(II) remediation over long periods.
In situ U and 90Sr remediation at contaminated sites has been trialled in field-scale studies.74–76 For example, at the U.S. Department of Energy's Rifle Field Research site, pilot-scale bioremediation trials have explored bioreduction of U(VI) in groundwaters mediated by in situ injections of electron donor.74,76–78 The trials successfully showed an initial drop in soluble U(VI) to below levels prescribed by the Environmental Protection Agency,76 however, the work found that prolonged U(VI) removal only occurred when electron donor amendment was ongoing.76,78 This suggests that for bioremediation or chemical remediation of soluble U(VI) to succeed over extended times, the U species formed during remediation need to be optimised so they are recalcitrant to re-oxidation and remobilisation even after treatment has ended. Similar field scale trials targeting 90Sr remediation have been conducted at the U.S. Department of Energy's Hanford nuclear legacy site.79 Here, after extensive lab and field scale research, an injectable apatite [Ca5(PO4)3(Cl,F,OH)] permeable reactive barrier down gradient of a 90Sr plume was installed to remediate 90Sr in groundwater via the in situ formation of Ca-phosphate phases, specifically targeting Sr(II) sorption and subsequent incorporation into apatite.80,81 In this case, the simultaneous in situ addition of Ca-citrate and Na-phosphate to the sub-surface led to the controlled breakdown of Ca-citrate, which in turn allowed Ca to react with inorganic phosphate to precipitate amorphous apatite, which adsorbed and incorporated 90Sr. This pilot-scale field trial successfully reduced 90Sr mobility away from the trial site into the adjacent Columbia River by 90% for at least 4 years.79
Overall, at many radioactively contaminated land sites, there is a clear need for a ‘toolkit’ of remediation methods that are applicable over extended timescales, to help guide the choice of optimal contaminant control strategies. Whilst the reduction of U(VI) to poorly soluble U(IV) is an attractive remediation route, the resultant reduced U(IV) phases may be susceptible to oxidative remobilisation after exposure to groundwater containing oxidants e.g., dissolved oxygen10,75,82–86 or nitrate.75,85,87–92 Dissolved oxygen can ingress into reduced sediments via oxygenated waters86 and nitrate is a ubiquitous groundwater contaminant originating from chemical and animal waste fertilisers.93 Nitrate is also present at many nuclear facilities at elevated concentrations due to the use of nitric acid in the nuclear fuel cycle.70,90,94–96 Indeed, the stability of the immobilised phases is key to the long term stewardship of radioactively contaminated land and there is a need to develop radionuclide immobilisation strategies, which are effective over long timescales.
Here, we have selected an untreated, ‘natural attenuation’, control and 6 targeted bioremediation and chemical treatments which have been shown through a number of past studies to offer promising avenues for exploration for U and Sr remediation.49,50,68,76,97,98 These comprised of four bioremediation treatments (electron donor only; electron donor plus excess sulfate; a slow release, commercially available, electron donor (MRC); glycerol phosphate), and two in situ chemical treatment approaches based on ZVI (NANOFER 25S; and Carbo-Iron). In these experiments, we explored the fate of U and Sr over 300 days of incubation, and then assessed the impact of oxidation with air and nitrate on U and Sr solubility and speciation in solids over a further 100 days. The study used aqueous geochemical analyses, 16S rRNA gene analysis and X-ray absorption spectroscopy (XAS) to understand the key biogeochemical processes controlling U and Sr behaviour. Our experiments provide clear insight into the optimal treatments for both U and Sr (glycerol phosphate, NANOFER 25S and Carbo-Iron) which all retained ≥80% of the initially added U and Sr in the solid phase for the duration of the re-oxidation perturbations.
Experimental
Sediment characterisation
Subsurface sediments were sampled from a well characterised field site in west Cumbria representative of the Sellafield sub-surface.95,99 The sediments were sampled at approximately 10 cm depth into sealed, sterile HDPE bags, which were stored at 10 °C prior to use. For detailed analysis, see ESI Section SI1.†Batch microcosm experiments
Large batch microcosm experiments were set up with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 sediment to water ratio in sterile 2 L Schott bottles with 1.5 L of modified, sterile artificial groundwater and 150 ± 0.5 g of sediment under anaerobic conditions. The recipe for artificial groundwater was adapted from ref. 100 and contained (mM): KCl (0.09), MgSO4·7H2O (0.39), MgCl2·6H2O (0.39), CaCO3 (1.67), NaNO3 (0.32), NaCl (0.16), NaHCO3 (2.88) and SrCl2 (0.99). The six systems were set up, in singlet, as above with the following amendments: sodium acetate (5 mM) and sodium lactate (5 mM); sodium acetate (5 mM), sodium lactate (5 mM) and sodium sulfate (10 mM); Metals Remediation Compound (MRC) (5 g L−1);101 glycerol-3-phosphate (10 mM by mass); NANOFER 25S (5 g L−1), supplied as an aqueous suspension of nanoscale ZVI particles suspended in a bio-degradable stabiliser97 and; Carbo-Iron (5 g L−1), stabilised with carboxymethylcellulose (20 wt%).42,97 A seventh system was set up without amendment as a natural attenuation control. U, as U(VI)O22+ in 0.001 M HCl, was spiked into the artificial groundwater at 0.18 mM and stable Sr was added to the in the groundwater at 0.9 mM. These concentrations were modelled to be undersaturated with respect to U(VI) and Sr(II) in the groundwater using PHREEQC (version 3.0.0) thermodynamic modelling with the ANDRA SIT database102 (data not included). Experiments were then sealed with butyl rubber stoppers, incubated in the dark at room temperature and were swirled at regular intervals during the experiment. All chemicals used were of analytical grade.
:10 sediment to water ratio in sterile 2 L Schott bottles with 1.5 L of modified, sterile artificial groundwater and 150 ± 0.5 g of sediment under anaerobic conditions. The recipe for artificial groundwater was adapted from ref. 100 and contained (mM): KCl (0.09), MgSO4·7H2O (0.39), MgCl2·6H2O (0.39), CaCO3 (1.67), NaNO3 (0.32), NaCl (0.16), NaHCO3 (2.88) and SrCl2 (0.99). The six systems were set up, in singlet, as above with the following amendments: sodium acetate (5 mM) and sodium lactate (5 mM); sodium acetate (5 mM), sodium lactate (5 mM) and sodium sulfate (10 mM); Metals Remediation Compound (MRC) (5 g L−1);101 glycerol-3-phosphate (10 mM by mass); NANOFER 25S (5 g L−1), supplied as an aqueous suspension of nanoscale ZVI particles suspended in a bio-degradable stabiliser97 and; Carbo-Iron (5 g L−1), stabilised with carboxymethylcellulose (20 wt%).42,97 A seventh system was set up without amendment as a natural attenuation control. U, as U(VI)O22+ in 0.001 M HCl, was spiked into the artificial groundwater at 0.18 mM and stable Sr was added to the in the groundwater at 0.9 mM. These concentrations were modelled to be undersaturated with respect to U(VI) and Sr(II) in the groundwater using PHREEQC (version 3.0.0) thermodynamic modelling with the ANDRA SIT database102 (data not included). Experiments were then sealed with butyl rubber stoppers, incubated in the dark at room temperature and were swirled at regular intervals during the experiment. All chemicals used were of analytical grade.
Sediment/groundwater slurry was sampled aseptically at select time points using Ar flushed syringes. An aliquot of slurry was added immediately to 0.5 M HCl for analysis of % Fe(II)/Fetotal; calculated as the 0.5 N HCl extractable Fe(II) divided by the total 0.5 N HCl extractable Fe using the ferrozine assay.103 The samples were then centrifuged (14800g, 10 min), and the solid phases were used for 16S rRNA gene analysis, with the supernatant analysed for pH and Eh and a sub-aliquot acidified and analysed for total aqueous U, Sr and Fe by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Residual supernatant was frozen at −80 °C for analysis of acetate, lactate, sulfate, nitrate, nitrite, phosphate and glycerol phosphate by Ion Chromatography (IC).48 Here, key data are available in the ESI (Section 3†).
Oxidative remobilisation of U and Sr
After 300 days incubation in large-scale (1.5 L) microcosms with the different remediation treatments, the resultant slurries were distributed into smaller (25 mL) experiments for re-oxidation experiments. Here, re-oxidation experiments were set up in triplicate under anaerobic conditions. In air re-oxidation experiments, 25 mL of the different treatment slurries were added to 100 mL conical flasks with a sponge stopper, covered with Al foil and stored the dark at room temperature. Flasks undergoing re-oxidation were then gently agitated every 2–3 days to ensure air exchange, and sampled at selected time points over 100 days.97,104 For nitrate re-oxidation experiments, 25 mL of the sediment slurries were distributed into serum bottles under anoxic conditions, and then spiked with 10 mM Na-nitrate to provide significant excess nitrate at concentrations relevant to nuclear sites.70,94,96 At selected time points, slurries were centrifuged (14800g, 10 min) and the aqueous fraction analysed for U, Sr and Fe by ICPMS; sulfate, nitrate, nitrite, phosphate, glycerol phosphate by IC, and changes in the microbial community were monitored using 16S rRNA gene analysis. For the ZVI-based re-oxidation treatments, parallel re-oxidation samples were set up for bulk X-ray Absorption Spectroscopy (XAS) analysis after 60 days of re-oxidation.
Solid-phase analysis
Microbial analysis
In order to examine changes in the microbial community, DNA was extracted from the solids99,108 in each treatment at 0 and 300 days of treatment, and then after 100 days of re-oxidation with air and nitrate. Further details are provided in the ESI (Section 5†).Proprietary substrates
A range of commercially available treatments including: MRC, NANOFER 25S and Carbo-Iron were used in this study. MRC is a glycerol polylactate compound containing an organosulfur ester designed to be a slow-release substrate suitable for sustained stimulation of microbial activity in situ supplied by Regenesis. NANOFER 25S is an aqueous dispersion of zero-valent iron nano-particles with a biodegradable stabiliser supplied by NANO IRON.97 Carbo-Iron is a composite of activated carbon colloids and zero-valent iron clusters supplied by the Helmholtz Centre for Environmental Research.42 A stabilised suspension of Carbo-Iron was prepared by coating with carboxymethylcellulose to improve its transport properties.42 MRC, NANOFER 25S and Carbo-Iron have been used in past work relevant to the remediation of halogen, heavy metal, and radioactive contaminated soils and sediments.50,97,109–113Results and discussion
Here we discuss the changes in biogeochemistry for the treatments during the first 300 days; we also examine the U and Sr end-points at 300 days using XAS analysis, and report changes in microbial community composition by 16S rRNA gene sequencing. We then discuss the re-oxidation behaviour of U and Sr for a further 100 days with oxygen and nitrate.Batch microcosm experiments & XAS analyses
Sediment microcosms were set up using six different treatments to investigate their potential for removal of aqueous U (Fig. 1, left) and Sr (Fig. 1, middle) from contaminated sediments over a 300 day anoxic incubation experiment. Changes in the % Fe(II)/Fetotal (Fig. 1, right) and Eh (Fig. SI 2†) were used as indicators of the redox environment in the incubations.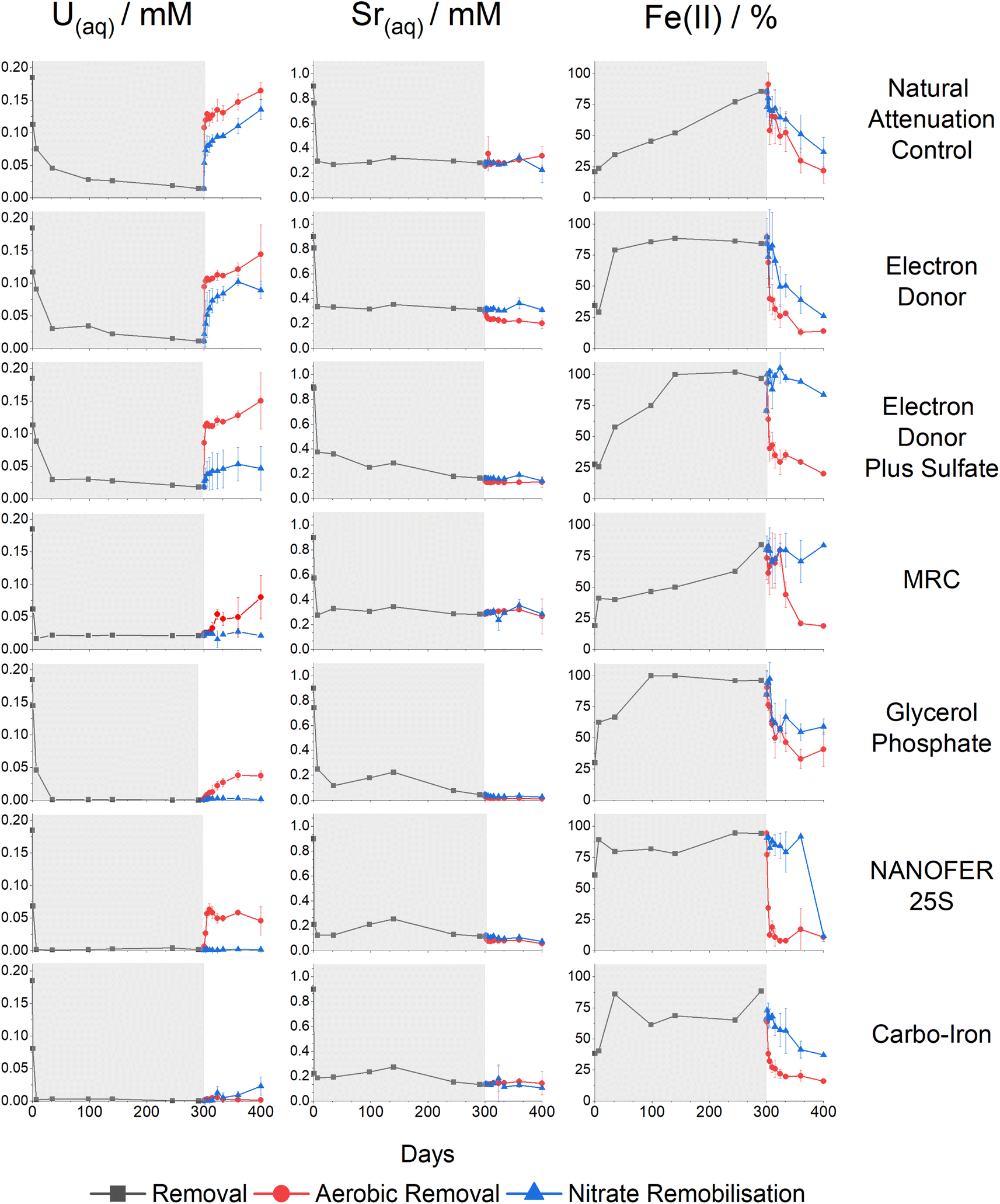 | ||
| Fig. 1 Aqueous U (left) and Sr (middle) concentrations in mM, and % Fe(II)/Fetotal (right). With data for the incubation period (black lines) and remobilisation data for each species after aeration (red lines) or when amended with nitrate (blue lines). Data for the incubation period are taken from singlet measurements, whilst data for the oxidative remobilisation were performed in triplicate with error bars representing 1 standard deviation (σ). Incubation periods are highlighted in grey and remobilisation phases are left blank. | ||
Natural attenuation control
During the 300 day anaerobic treatment period, the pH increased from 7.5 to 7.8 and significant U and Sr were removed from solution (Fig. 1 and SI 1†). An increase in the % Fe(II)/Fetotal in the natural attenuation control (from 21% to 86%) suggested that consumption of labile organic carbon in the sediment induced Fe(III) reduction, consistent with an elevated (4.1%wt) total organic carbon present in the sediments (Fig. 1 & ESI Section 1†).11 U(VI) removal was relatively fast during the first 5 days and continued steadily for the remaining incubation period with 94% (0.17 mM) of the added U removed after 300 days (Fig. 1). Sr sorption occurred over the first seven days (69% removal, 0.62 mM), after which there was no further change in Sr concentrations similar to past work.64,65,68 Approximately 25% (0.1 mM) of the initial 0.4 mM sulfate was consumed during incubation (Fig. SI 3A†). Overall these results confirm both Fe(III)- and sulfate-reducing conditions were stimulated in the organic matter rich sediment.Linear combination fitting (LCF) of the U XANES of the natural attenuation 300 day incubation endpoint identified U present as both U(VI) and U(IV) (approximately 70:30 U(VI):U(IV)) (Fig. SI 10†). Accordingly, the best fit for the EXAFS was obtained using these data to inform fitting (Fig. 2, Table SI 1†), with a 70% U(VI) contribution described by shells U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oax at 1.8 Å (CN 1.4) and U(VI)–Oeq at 2.4 Å (CN4.2) and a 30% U(IV) contribution U(IV)–O at 2.3 Å (CN 2.6), suggesting that a combination of sorption and reduction processes were controlling U solubility. Partial reduction to U(IV) was consistent with the development of Fe(III)- and sulfate-reducing conditions fuelled by natural organic matter in the sediment. The U(VI) fraction could be present as either: (i) sorbed, uranyl(VI) phases to the solid geomedia (e.g., ref. 19 and 23–27), (ii) uranyl(VI) carbonate/phosphate precipitates (e.g., ref. 28–30), or (iii) a combination of points (i) and (ii). The Sr K-edge EXAFS were best fit with a single shell of 9 O backscatterers at 2.6 Å consistent with outer sphere sorption to the sediments (Fig. 2, Table SI 1†).61,68
Oax at 1.8 Å (CN 1.4) and U(VI)–Oeq at 2.4 Å (CN4.2) and a 30% U(IV) contribution U(IV)–O at 2.3 Å (CN 2.6), suggesting that a combination of sorption and reduction processes were controlling U solubility. Partial reduction to U(IV) was consistent with the development of Fe(III)- and sulfate-reducing conditions fuelled by natural organic matter in the sediment. The U(VI) fraction could be present as either: (i) sorbed, uranyl(VI) phases to the solid geomedia (e.g., ref. 19 and 23–27), (ii) uranyl(VI) carbonate/phosphate precipitates (e.g., ref. 28–30), or (iii) a combination of points (i) and (ii). The Sr K-edge EXAFS were best fit with a single shell of 9 O backscatterers at 2.6 Å consistent with outer sphere sorption to the sediments (Fig. 2, Table SI 1†).61,68
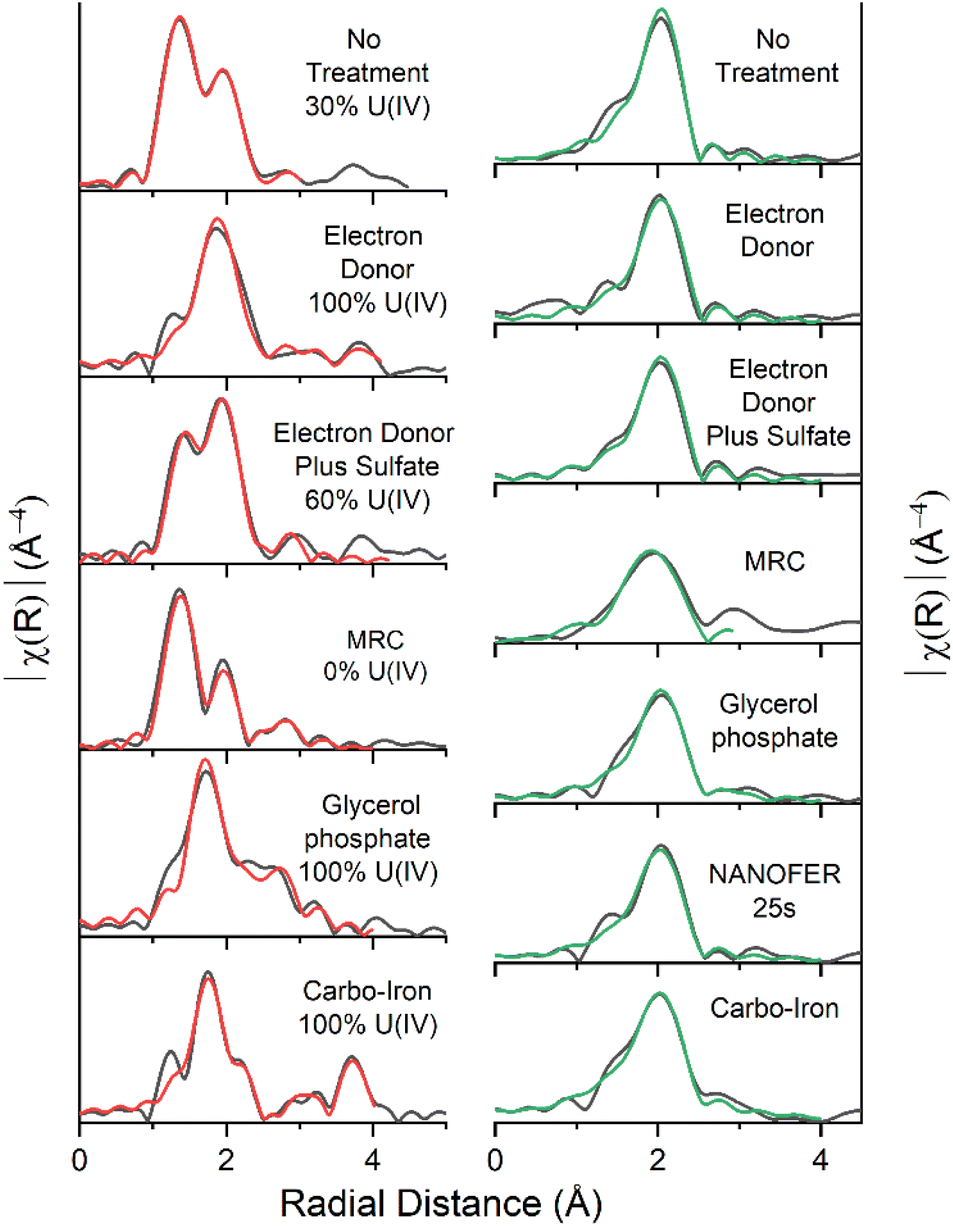 | ||
| Fig. 2 EXAFS Fourier transform data (black) and fits U L3-edge (left, red) and for Sr K-edge (right, green). % U(IV) contributions included. | ||
Electron donor addition
Microbial communities in the electron donor (5 mM Na-acetate & 5 mM Na-lactate) amended experiment consumed all of the acetate and lactate during the course of the 300 day incubation (Fig. SI 4A†). Consistent with the microbial consumption of electron donor, this system showed an increase in pH to 8.0 and an increase in the % Fe(II)/Fetotal, which stabilised at approximately 80% Fe(II) ingrowth within 35 days (Fig. 1 and SI 1†). These conditions were coupled to significant U removal (94%, 0.17 mM, similar to the natural attenuation control) over 300 days, and Sr also behaved similarly to the natural attenuation control experiment, with 65% (0.59 mM) Sr removed from solution (Fig. 1). This is consistent with previous studies which report that electron donor amendments have limited impact on Sr mobility unless bioreduction raises the pH > 9.64,68,69 Additionally, sulfate reduction also occurred with complete removal of the initial 0.4 mM sulfate occurring after 300 days, as further evidenced by a blackening of the sediments indicating insoluble Fe sulfide formation (e.g., FeS).In the electron donor amended experiment, LCF of the U XANES data suggested that the system was dominated by U(IV) (Fig. SI 10†), consistent with the formation of nano-crystalline uraninite and non-crystalline U(IV).9,16,17,21 Accordingly, the EXAFS data were best fitted as a mixture non-crystalline U(IV) bound to phosphate moieties on biomass, with a contribution from nano-crystalline uraninite indicated by a U–U shell at 3.9–4.0 Å (CN = 3) (Fig. 2 & Table SI 1†).9,14–16,18,19 Again, the Sr K-edge EXAFS were best fitted with a single shell of 9 O backscatterers at 2.6 Å consistent with outer sphere sorption to the sediments, indigenous P present in the sediment did not significantly affect Sr speciation (Fig. 2, Table SI 1†).61,68
Electron donor plus sulfate treatment
The third system contained acetate (5 mM), lactate (5 mM) electron donors and elevated concentrations of sulfate (10.4 mM compared to 0.4 mM in the electron donor only system). Here, we explored the impact of elevated sulfidation on U and Sr biogeochemistry as it may influence the rate and extent of U(VI) bioreduction, as well as the stability of the end-points.84,87,91,114,115 Additionally, elevated sulfate can lead to enhanced formation of Fe(II)-sulfides on bioreduction and these reportedly act as redox buffers for U(VI) during sediment oxidation.84,87,91,114,115 In this system, the pH increased to 8.9 presumably due to excess sulfate reduction, which was elevated compared to the natural attenuation and electron donor systems (pH 7.8 and 8.0 respectively) (Fig. SI 1†). Significant U(VI) was removed from solution (90%, 0.16 mM) concurrent with an increase in % Fe(II)/Fetotal (Fig. 1). Sr removal was enhanced compared to the natural attenuation control and the electron donor amended systems (82%, 0.74 mM, Fig. 1), presumably due to the increased pH (to 8.9) following sulfate reduction (Fig. SI 1†).116 Sulfidation was extensive in this experiment as evidenced by consumption of sulfate, a noticeable blackening of sediments and sulfide odour, and a pH increase and Eh decrease to below −400 mV (Fig. SI 2 and SI 5B†).Despite development of robust Fe(III)- and sulfate-reducing conditions, LCF of the U XANES suggested only approximately 60% of the added U was reduced to U(IV), suggesting that elevated sulfate hindered the extent of U(VI)-reduction (Fig. SI 10†).74–76 Here, the U EXAFS were best fit with a 40:60 U(VI):U(IV) ratio for the U–O shells, and the addition of modest U–P backscatterer contribution at 3.2 Å (F-test 98.8%) suggesting U was likely speciated as non-crystalline U(IV) associated with phosphate, similar to the electron donor only system (Fig. 2 & Table SI 1†). Non-crystalline U(IV) has previously been reported under sulfate-reducing conditions.8,9,17–19,22,51 Again, the Sr K-edge EXAFS data were best fitted with a single shell of 9 O backscatterers at 2.6 Å consistent with outer sphere sorption to the sediments (Fig. 2, Table SI 1†).61,68
MRC treatment
The fourth system used a commercially available, slow release, organosulfur ester, known as “Metals Reducing Compound” (MRC) intended to target redox active metals including Cr(VI) and Tc(VII).50,101,117 The MRC treatment caused acidification of the sediment microcosm to pH 4, which is in contrast to past work where acidification to pH 6 occurred, suggesting that the sediment in the current work has a low buffering capacity (Fig. SI 1†).50 Here U removal was 89% (0.16 mM), and Sr removal was 69% (0.62 mM) (Fig. 1). By the experiment end-point, near complete reduction of Fe(III) was observed; this occurred at a much slower rate than the other experiments and was consistent with reported lower Fe(III) reduction rates (pH 4, Fig. SI† 1) in low pH sediments.95,118 Interestingly, there was no evidence for development of sulfate reduction over the 300 day incubation period (Fig. SI 6A†) and furthermore, all ion chromatography samples from the MRC treatment contained a fingerprint consistent with a complex mix of volatile fatty acids,50 presumably from biodegradation of the MRC compound.U XANES for the MRC system was essentially the same as the uranyl(VI) standard suggesting no significant reduction had occurred (Fig. SI 10†). Accordingly, the U EXAFS data were fitted with a UOax dioxygenyl oxygen at 1.8 Å (N = 2) and 6 equatorial U–Oeq backscatterers at 2.4 Å. Addition of a third S backscatterer shell at approximately 3.2 Å statistically improved the fit (F-test 96.5%) and was indicative of bidentate coordination to sulfate9,15,17,18,119,120 (present in the structure of MRC) (Fig. 2 & Table SI 1†). Sr K-edge EXAFS data were best fitted with a single shell of 9 O backscatterers at 2.6 Å consistent with outer sphere sorption to the sediments (Fig. 2, Table SI 1†).61,68
Glycerol phosphate treatment
Biostimulation via organophosphate amendments is a promising remediation method as it provides an electron donor capable of reducing contaminant species and the phosphate facilitates the precipitation of relatively insoluble phosphate biominerals.67,121–124 Metal-phosphate precipitation can be achieved in the subsurface via addition of inorganic phosphate, but this may cause localised clogging at injection wells if the injection rate is too high.76,125 To avoid these issues, slow release organophosphates, including glycerol phosphate, are designed to biodegrade after injection and dispersion have been explored in other experimental works and are included in this study.48,49 In the glycerol phosphate amended experiment, the pH remained stable at 7.5 (Fig. SI 1†)49,68 and glycerol phosphate biodegradation and phosphate release were confirmed in the solution analyses (Fig. SI 7A†). This was accompanied by U(VI)- and Fe(III)- bioreduction until sulfate-reducing conditions were observed (Fig. 1 and SI 7B†). At the end of the experimental incubation, significant inorganic phosphate remained in solution (2 mM, 20% of total added phosphate). Interestingly, U was removed rapidly from solution, with >75% (0.14 mM) removal within the first week and >99% (0.18 mM) by day 35 (Fig. 1) similar to previous relevant studies.48,49,126 Sr removal in this system was significantly enhanced (97%, 0.87 mM) compared to the natural attenuation control, with removal occurring over 300 days similar to past work (Fig. 1).68The U XANES data suggested that U(IV) dominated (Fig. SI 10†) in these treatments, and the EXAFS were successfully fitted as U(IV)-phosphate, consistent with literature49,126,127(Fig. 2; Table SI 1†). The Sr K-edge EXAFS data were initially fitted using a single shell of 9 oxygen atoms at a distance of 2.6 Å. This did not fully resolve several features in the Fourier transformed data, and (informed by relevant literature) a second Sr–P shell was added at 3.3 Å (CN 1.3, F-test 96.3%) consistent with a Sr-doped hydroxyapatite-like local coordination environment (Fig. 2 & Table SI 1†).66,68,128 Indeed, the fitted shell occupancy of 1.3 in the Sr–P shell suggests that up to a third of the Sr was inner sphere bound to phosphate. This is consistent with initial adsorption and then slower incorporation of a fraction of Sr into Ca-phosphate minerals (Fig. 2, Table SI 1†).68,79,80
Zero-valent iron treatments: NANOFER 25S and Carbo-Iron
Both U and Sr were removed rapidly from solution in the presence of NANOFER 25S and Carbo-Iron, within 1 week they both removed >99% (0.18 mM) U(VI), and 86% (0.77 mM) and 79% (0.71 mM) of the Sr, respectively (Fig. 1).44,98,113,129,130 The % Fe(II)/Fetotal remained >75% and 60% after 35 days for NANOFER 25S and Carbo-Iron, respectively (Fig. 1), suggesting that reducing conditions persisted in these incubations (Fig. 1). The pH for NANOFER 25S was 8.0 and for Carbo-Iron was 7.5 (Fig. SI 1†), and there was evidence of sulfate removal from solution confirming that sulfate reduction had occurred (Fig. SI 8A and SI 9A†). Here, the majority (>75%) of sulfate reduction occurred after U and Sr removal (Fig. 1, SI 8A and SI 9A†) suggesting sulfate reduction was not a major control on the initial removal mechanism for U or Sr.Linear combination fitting of the XANES data between uranyl(VI) and U(IV)-uraninite standards suggested U(IV) dominated in the 300 day NANOFER 25S incubated sample (approximately 80%) with a small contribution from U(VI) (Fig. SI 10†). In the Carbo-Iron system, XANES linear combination fitting suggested U(IV) dominated (>99%) (Fig. SI 10†). A fitting model based on UO2 with an additional single scattering U–Fe pathway (CN 1, 3.5 Å, F-test 98.0%) provided the best fit for the Carbo-Iron EXAFS (Table SI 1†). These data suggest that U is present as adsorbed U(IV) on the surface of an Fe-phase (e.g., magnetite) and was consistent with published fits for year-long U(VI) reduction by ZVI.41,131–134 In the Sr EXAFS both systems were successfully fitted with a single O shell at 2.6 Å (Table SI 1†) suggesting outer sphere sorption dominated.61,68
Microbial analysis after batch incubation
16S rRNA gene analysis was used to investigate the microbial community evolution in our experiments. Initially, sediments contained a relatively diverse range of soil bacteria and archaea (Fig. SI 12†).After 300 days of incubation under anaerobic conditions, the microbial community present in the natural attenuation control and electron donor systems were remarkably similar. Both saw an increase in Fe(III)-reducers from 0.5% to 2.4% and from 0.6% to 1.0% in the natural attenuation control and the electron donor addition, respectively (e.g., after 300 days Geobacter spp. was present in both (≤1.9%), but Geothrix spp. were only detected in the electron donor system (≤0.3%)). This is consistent with Fe(III) reduction, and the electron donor only system saw a significant increase in relative abundance of methane-oxidising Methanobacteria (from >0.1% to 7.5% of Operational Taxonomic Units (OTUs)) suggesting some evidence for methanogenesis (Fig. SI 12†). The electron donor with sulfate system, showed significant increases in the relative abundance of both Fe(III)- (e.g., Geobacter) and sulfate-reducing bacteria (e.g., Desulfobulbus, Desulfovibrio and Desulfosporosinus), but showed less evidence for active methane-oxidising communities (Methanobacteria represented only 2.2% of the microbial OTUs) suggesting sulfate-reducers outcompeted methane-oxidising bacteria in these experiments (Fig. SI 12†). In the system with MRC, which had a pH of 4, there was a relative decrease in the microbial diversity Shannon index (5.11), suggesting reduced microbial diversity, with the microbial community dominated by Bacilli (50.3%) (Fig. SI 12†). Following biostimulation with glycerol phosphate, a diverse microbial community was detected including sequences closely related to known Fe(III)- and sulfate-reducing bacteria (e.g., Geobacter, Geothrix and Desulfosporosinus) (Fig. SI 12†), consistent with past work.49,68 Again, Methanobacteria. (6.5% of the OTUs) were detected at the experimental end-point suggesting methanogenesis (and subsequent methane oxidation) had been stimulated. The NANOFER 25S incubation had a low Shannon index (5.91) suggesting reduced microbial diversity, potentially due to toxicity effects associated with ZVI (Fig. SI 12†).97,135 Conversely, the Carbo-Iron amended system showed a relatively similar Shannon index to the other systems (7.02). Here, similar Fe(III)- and sulfate-reducing genera were detected compared to the electron donor amended experiment (e.g., Geobacter, Desulfovibrio and Desulfobulbus) indicating increased microbial diversity and robust bioreduction after stimulation, which was potentially enhanced by hydrogen production (from ZVI corrosion) to the breakdown of the cellulose coating of these materials (Fig. SI 12†).
Oxidative remobilisation of U and Sr
After 300 days incubation, remobilisation experiments mediated by oxidation with air or nitrate were conducted to determine the stability of U and Sr treatments. The red (air) and blue (nitrate) lines in Fig. 1 show aqueous U (left), Sr (middle) and the % Fe(II)/Fetotal (right). Changes in the percentage % Fe(II)/Fetotal and Eh (Fig. 1 and SI 2†) were used as indicators of the redox environment in the incubations. The percentage of remobilisation was calculated using the original concentrations of U, Sr.Natural attenuation control, electron donor and electron donor plus sulfate treatments
Oxic conditions were re-established over 100 days of aeration in the natural attenuation control, and the electron donor and the electron donor plus sulfate treatment systems, with % Fe(II)/Fetotal decreasing and Eh values increasing (Fig. 1 and SI 2†). Here, the U previously scavenged to sediments under reducing conditions showed significant remobilisation (>78%, 0.14 mM), suggesting these treatments are not resistant to re-oxidation via O2 ingress even in the case of the excess sulfate system, where sulfides have been reported to cause redox buffering in some systems (Fig. 1).84,87,91,114,115,136 EXAFS analysis on the incubated end-points for these samples suggested a mix of U(VI) and non-crystalline U(IV) which is consistent with re-oxidation to U(VI) and subsequent remobilisation (Fig. 1 and 2).9,20,21,137,138 When treated with 10 mM nitrate, the natural attenuation control and electron donor experiments showed essentially complete Fe(II)-re-oxidation, and the electron donor plus sulfate treatment showed only partial Fe(II)-re-oxidation, with the Eh values in all systems less oxidising than for the air re-oxidation equivalents (Fig. 1 and SI 2†). Here, U showed lower levels of remobilisation compared to the air re-oxidation experiments, with 73% (0.13 mM), 48% (0.09 mM) and 11% (0.02 mM) remobilisation of U from the natural attenuation control, electron donor, and electron donor plus sulfate systems respectively (Fig. 1). Presumably, the presence of carbonate which is at equilibrium with the atmospheric CO2 in the aerobic experiments, and restricted in the nitrate remobilisation studies, may enhance U oxidation/release to some extent. Indeed, higher remobilisation rates of bioreduced U are consistent with previous re-oxidation studies on bioreduced sediments containing U and Tc.85,139 In all of these experiments, Sr solubility remained largely unchanged confirming Sr sorption is largely unaffected by changes in redox chemistry caused by air or nitrate oxidation (Fig. 1).MRC treatment
For MRC, air oxidation showed an increase in Eh coupled to full re-oxidation of % Fe(II)/Fetotal; it also caused an increase in pH from 4 to 6, likely attributable to Fe(II)-oxidation or through dissolution of atmospheric CO2, which buffered the solution (Fig. 1 and SI 1†). Here, only 43% (0.08 mM) of the total added U was remobilised, suggesting that the immobilised phase was not highly susceptible to oxidative remobilisation (Fig. 1). This is consistent with XANES (Fig. SI 10†) and EXAFS (Fig. 2) fitting of U(VI) after 300 days incubation, as the U was already present as U(VI) re-oxidation is unlikely to significantly affect its solubility. Again, Sr levels did not change significantly following re-oxidation of the microcosms. Although nitrate amendment caused an increase in Eh (from +164 to +323 mV) it caused no change in pH (Fig. SI 1†) or % Fe(II)/Fetotal and this was reflected in no significant U or Sr remobilisation in this system, suggesting that pH is a key controlling factor in the biogeochemical behaviour of U and Sr (Fig. 1).Glycerol phosphate treatment
In past work, glycerol phosphate has shown promise as a treatment that affords long-term stability of U and Sr with significant removal and retention on air and nitrate re-oxidation.49,68 In the current study, air reoxidised sediments showed a decrease in % Fe(II)/Fetotal from 96% to 41% and an increase of Eh to +242 mV (Fig. 1 and SI 2†). This suggests oxidising conditions were re-established, but despite this, only 20% (0.04 mM) U was remobilised to solution (Fig. 1). With nitrate re-oxidation % Fe(II)/Fetotal decreased to 59% and Eh increased to +239 mV and similarly <1% U was remobilised to solution. Again, both air and nitrate treatments showed no significant (≤3%, <0.03 mM) remobilisation of Sr to solution (Fig. 1 and SI 2†). For both elements, the lack of remobilisation in air treated systems despite significant re-oxidation is reportedly due to the formation of stable, solid phosphate phases which have previously been shown as recalcitrant to oxidative remobilisation.49,68,79Zero-valent iron treatments: NANOFER 25S and Carbo-Iron
Following air re-oxidation of NANOFER 25S and Carbo-Iron systems, the pH did not change significantly (<0.1), the Eh increased to +360 and +330 mV and the % Fe(II)/Fetotal was 11% and 16% respectively (Fig. 1, SI 1 and SI 2†). Here, there was 25% (0.05 mM) U and 6% (0.05 mM) Sr remobilisation for NANOFER 25S and <1% U (<0.01 mM) and 16% (0.14 mM) Sr for Carbo-Iron. Both of the tailored ZVI treatments outperformed previous work with regular ZVI, where long-term U immobilisation was only maintained in the absence of carbonate, and where Sr treatment with ZVI was prone to Sr remobilisation on perturbation.73,98 Following nitrate re-oxidation of the NANOFER 25S and Carbo-Iron systems, the pH was 7.9 and 8.6, the Eh was +262 and +218 mV, and the % Fe(II)/Fetotal was 12% and 37% respectively (Fig. 1, SI 1 and SI 2†). Similarly, nitrate amendments only remobilised <1% (<0.01 mM) of U and 8% (0.07 mM) Sr for NANOFER 25S and 12% (0.02 mM) U and 12% (0.11 mM) Sr for Carbo-Iron respectively (Fig. 1). Overall, the Carbo-Iron and NANOFER 25S treatments show significant retardation of U and Sr on initial incubation, and low remobilisation on re-oxidation (Fig. 2). Interestingly, U re-oxidation after aeration was significantly lower and slower in the presence of Carbo-Iron compared to NANOFER 25S. Further analysis to explore the mechanism of retention of U in these phases was conducted using U EXAFS. Here, samples from the air re-oxidation of NANOFER 25S and Carbo-Iron were analysed after 60 days. LCF of the XANES for both samples suggested complete re-oxidation to U(VI) had occurred, despite the fact that oxidative remobilisation of U(VI) into solution was not significant (Fig. 1 and SI 10†). For both NANOFER 25S and Carbo-Iron, the re-oxidised EXAFS spectra were almost identical and were fitted with a full axial UOax shell, split equatorial U–Oeq shells and a single U–Fe backscatterer indicative of inner-sphere adsorption of uranyl to Fe(III) edge sites (Fig. 3 & Table SI 1†).24,29,140,141 In the case of NANOFER 25S the U EXAFS also contained a C shell at 2.92 Å which is consistent with bidentate, inner-sphere sorption of a U(VI)-carbonate complex binding to the Fe-oxide surface (Fig. 3 & Table SI 1†).24,29,140,141 In the Carbo-Iron sample a C shell could not be fitted to the spectra without constraining the U–C distance to 2.9 Å, suggesting that here uranyl(VI) was bound directly to the Fe-oxide surface in the Carbo-Iron sample (Fig. 3 & Table SI 1†). Interestingly, although the EXAFS models fit literature values for sorbed U(VI) to Fe-minerals,24,29,140,141 adsorbed U(VI) readily desorbs in air-equilibrated systems due to the formation of highly soluble uranyl carbonates between pH 7 and 8.142 The lack of remobilisation in these experiments suggests that U(VI) sorption to Fe is not likely the dominant retention mechanism. Rather, the recalcitrance of the U suggests some form of incorporation of U(VI) into an Fe oxide phase which has formed during the re-oxidation process.34–39 Indeed, due to the excellent removal and long-term retention capacities of the ZVI-amended systems, the mechanisms governing U(VI) reduction and remobilisation following treatment with ZVI merits further study.
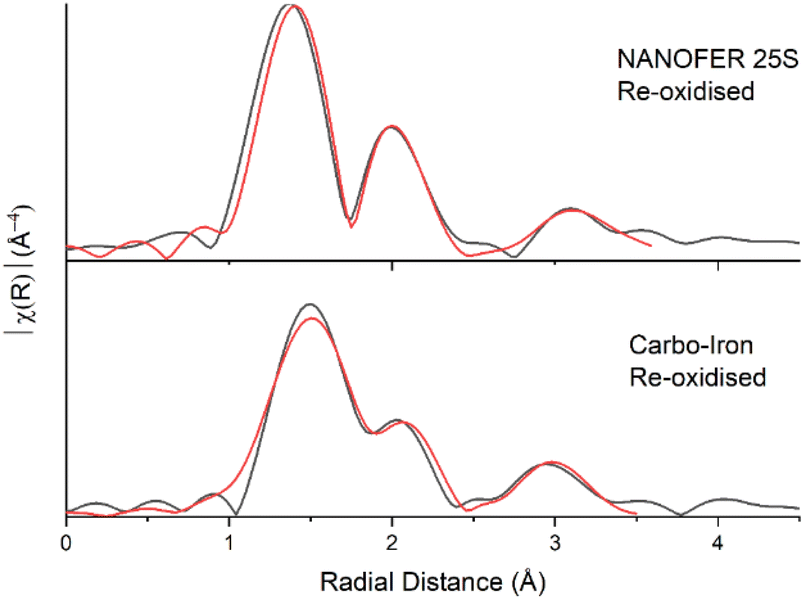 | ||
| Fig. 3 U L3-edge Fourier transform data (black) and fits (red) for the re-oxidised NanoFeR25s and Carbo-Iron samples. | ||
Microbial analysis of the remobilised samples
The microbial community that developed after oxidation with air was consistent with a response to oxic conditions. Close matches to Actinobacteria of the order Micrococcales and Micromonosporales, which are Fe(II)-oxidisers were observed across the oxic systems in significant quantities (Fig. SI 12†).143,144 Here, the genus Pseudoarthrobacter, strict aerobes144 represented 26% of the OTUs after air oxidation of the NANOFER 25S system. The large shifts in the microbial community in the day 400 aerobic samples are consistent with the re-establishment of oxic conditions (Fig. 1). Following nitrate additions, the microbial communities in the experiments were similar to the parallel day 300 bioreduction samples (Fig. SI 12†). This suggested that many of the organisms that proliferated during the initial bioreduction steps were also able to denitrify, consistent with the biogeochemical data (Fig. 1).Conclusions
Early radionuclide bioremediation concepts often involved biostimulation of radionuclide reduction (e.g., for U(VI) and Tc(VII)) using relatively simple electron donors (e.g., see ref. 145). However, there are concerns regarding the long term effectiveness of these approaches, due to the stabilities of the formed end-points, especially during re-oxidation. This study builds on more recent work that has led to the development of alternative processes that may deliver more recalcitrant end-points. We compared the long term effectiveness of several promising remediation techniques applied to redox active U and, non-redox active Sr contaminated sediments. Whilst many of these treatments have been investigated in separate studies, differences in experimental setups (e.g., methodologies, radionuclides, sediments used) hinder cross-comparisons of treatment performance.Generally U and Sr behaviour was largely similar across incubation experiments. Here, ≥89% and ≥65% of added U and Sr was removed from the aqueous phase in all experiments, respectively. During remobilisation experiments, aeration remobilised more U (presumably through the oxidation of U(IV) to U(VI)) than nitrate, and as expected Sr, which is not redox active, was largely unaffected by re-oxidation. Systems amended with glycerol phosphate or ZVI (NANOFER 25S and Carbo-Iron) were most efficient, achieving near complete removal of U and Sr and limited remobilisation (≤25% remobilisation of the initially added radionuclide compared to 43–89% remobilisation in other treatments). Whilst XAS identified a range of different U end-points, Sr speciation was dominated by weak-outer sphere sorption consistent with pH regimes below pH 11, with the exception of the glycerol phosphate amended system that yielded a highly recalcitrant, incorporated Sr-apatite phase. Bioremediation approaches (electron donor only, electron donor plus sulfate) yielded similar removal and remobilisation results to the ‘natural attenuation’ control, which had small quantities of organic carbon present. These are consistent with XAS analyses which demonstrated a combination of biogenic and nanocrystalline U(IV), and adsorbed U(VI) species in the natural attenuation, electron donor only and electron donor plus sulfate amended systems. Commercially available MRC treatments efficiently removed U and Sr from solution, although did not reduce U(VI). The fast rate of sorption and lack of reduction suggests that U removal was dictated by adsorption rather than reduction mechanisms in these treatments. Glycerol phosphate amendments proved to be one of the most effective treatments, removing >99% U and 97% Sr from solution, with ≤20% U and ≤3% Sr remobilised following re-oxidation. Interestingly, U EXAFS analyses yielded fits for noncrystalline/biogenic U(IV) (electron donor and electron donor plus sulfate amendments) and ningyoite-like U(IV)-phosphates (glycerol phosphate amendments), with the ningyoite-like U(IV) phosphates proving much more recalcitrant to remobilisation. The differences in these end-points have clear implications for establishing effective and long-lasting remediation strategies. ZVI based treatments (NANOFER 25S and Carbo-Iron) were equally as effective as glycerol phosphate amendments with >99% U and ≤86% Sr removal from solution and ≤25% U and ≤16% Sr remobilisation. Interestingly, EXAFS analysis of the aerobically reoxidised Carbo-Iron and NANOFER 25S yielded fits similar to adsorbed U(VI) species, however the lack of solubility of the U(VI) phases suggests that other mechanisms (e.g., incorporation) may govern U solubility. The mechanisms controlling this require further study in more controlled experiments.
Overall, these data highlight the complexity of reductive remediation methods, which may form widely varying end-points and display variable rates of remobilisation when exposed to air or nitrate. Our work shows that for a remediation strategy to be effective it must include a removal (e.g., reduction, adsorption, precipitation) and a retention process (e.g. formation of-, or incorporation into- an environmentally stable mineral phase) which is largely unaffected by environmental perturbations (e.g., oxidation and salinity increases). Here, the best performing methods used glycerol phosphate or ZVI based treatments (NANOFER 25S or Carbo-Iron), which all have the potential to reduce redox active radionuclides (e.g., U(VI) and Tc(VII))68,97 and also (likely) mineral-incorporation (e.g., U/Sr into ningyoite/apatite respectively). These are good candidates for long-term site remediation strategies and, they therefore warrant further analysis in long-term column experiments and eventually field-scale studies to check their stability under in situ conditions.
Author contributions
Conceptualisation: G. F. V., J. R. L., K. M. & S. S.; funding acquisition: J. R. L., K. M. & S. S.; investigation: all; supervision: J. R. L., K. M. & S. S.; writing, reviewing and editing: all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a PhD bursary to Gianni F. Vettese from the Nuclear Decommissioning Authority, (R120085), managed through the National Nuclear Laboratory and was undertaken in the EPSRC funded NNUF RADER Facility at the University of Manchester (EP/T011300/1). Beamtime at the beamline B18 was funded by the grant SP13559-2 from the Diamond Light Source. We thank Alastair Bewsher and Paul Lythgoe (The University of Manchester) for IC and ICP-MS analytical support respectively. We also thank Katrin Mackenzie and Steffen Bleyl (UFZ Leipzig) for providing Carbo-Iron, Petra Skácelová (NANOIRON) for providing NANOFER 25S and Jack Shore (Regenesis) for providing MRC.Notes and references
- NDA, 2019 UK Radioactive Waste and Material Inventory, 2019 Search PubMed.
- W. J. F. Standring, D. H. Oughton and B. Salbu, Environ. Sci. Technol., 2002, 36, 2330–2337 CrossRef CAS PubMed.
- J. P. Mckinley, C. J. Zeissler, J. M. Zachara, R. Jeffrey Serne, R. M. Lindstrom, H. T. Schaef and R. D. Orr, Environ. Sci. Technol., 2001, 35, 3433–3441 CrossRef CAS PubMed.
- D. B. Watson, J. E. Kostka, M. W. Fields and P. M. Jardine, The Oak Ridge Field Research Center Conceptual Model, 2004 Search PubMed.
- J. M. Zachara, J. Serne, M. Freshley, F. Mann, F. Anderson, M. Wood, T. Jones and D. Myers, Vadose Zone J., 2007, 6, 985 CrossRef CAS.
- H. A. Vrionis, R. T. Anderson, I. Ortiz-Bernad, K. R. O'Neill, C. T. Resch, A. D. Peacock, R. Dayvault, D. C. White, P. E. Long and D. R. Lovley, Appl. Environ. Microbiol., 2005, 71, 6308–6318 CrossRef CAS PubMed.
- G. F. Vettese, K. Morris, L. S. Natrajan, S. Shaw, T. Vitova, J. Galanzew, D. L. Jones and J. R. Lloyd, Environ. Sci. Technol., 2020, 54, 2268–2276 CrossRef CAS PubMed.
- K. M. Campbell, R. K. Kukkadapu, N. P. Qafoku, A. D. Peacock, E. Lesher, K. H. Williams, J. R. Bargar, M. J. Wilkins, L. Figueroa, J. Ranville, J. A. Davis and P. E. Long, Appl. Geochem., 2012, 27, 1499–1511 CrossRef CAS.
- R. Bernier-Latmani, H. Veeramani, E. D. Vecchia, P. Junier, J. S. Lezama-Pacheco, E. I. Suvorova, J. O. Sharp, N. S. Wigginton and J. R. Bargar, Environ. Sci. Technol., 2010, 44, 9456–9462 CrossRef CAS PubMed.
- L. Newsome, K. Morris, S. Shaw, D. Trivedi and J. R. Lloyd, Chem. Geol., 2015, 409, 125–135 CrossRef CAS.
- L. Newsome, K. Morris, D. Trivedi, N. Atherton and J. R. Lloyd, Appl. Geochem., 2014, 51, 55–64 CrossRef CAS.
- Y. Suzuki, S. D. Kelly, K. M. Kemner and J. F. Banfield, Nature, 2002, 419, 134 CrossRef CAS PubMed.
- W. D. Burgos, J. T. McDonough, J. M. Senko, G. Zhang, A. C. Dohnalkova, S. D. Kelly, Y. Gorby and K. M. Kemner, Geochim. Cosmochim. Acta, 2008, 72, 4901–4915 CrossRef CAS.
- M. Stylo, D. S. Alessi, P. P. Shao, J. S. Lezama-Pacheco, J. R. Bargar and R. Bernier-Latmani, Environ. Sci. Technol., 2013, 47, 12351–12358 CrossRef CAS PubMed.
- D. S. Alessi, J. S. Lezama-Pacheco, J. E. Stubbs, M. Janousch, J. R. Bargar, P. Persson and R. Bernier-Latmani, Geochim. Cosmochim. Acta, 2014, 131, 115–127 CrossRef CAS.
- A. Bhattacharyya, K. M. Campbell, S. D. Kelly, Y. Roebbert, S. Weyer, R. Bernier-Latmani and T. Borch, Nat. Commun., 2017, 8, 15538 CrossRef CAS PubMed.
- M. I. Boyanov, K. E. Fletcher, M. J. Kwon, X. Rui, E. J. O'Loughlin, F. E. Löffler, K. M. Kemner, E. J. O'Loughlin, F. E. Löffler and K. M. Kemner, Environ. Sci. Technol., 2011, 45, 8336–8344 CrossRef CAS PubMed.
- J. R. Bargar, K. H. Williams, K. M. Campbell, P. E. Long, J. E. Stubbs, E. I. Suvorova, J. S. Lezama-Pacheco, D. S. Alessi, M. Stylo, S. M. Webb, J. A. Davis, D. E. Giammar, L. Y. Blue and R. Bernier-Latmani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4506–4511 CrossRef CAS.
- A. J. Fuller, P. Leary, N. D. Gray, H. S. Davies, J. F. W. Mosselmans, F. Cox, C. H. Robinson, J. K. Pittman, C. M. McCann, M. Muir, M. C. Graham, S. Utsunomiya, W. R. Bower, K. Morris, S. Shaw, P. Bots, F. R. Livens and G. T. W. Law, Chemosphere, 2020, 254, 126859 CrossRef CAS PubMed.
- D. S. Alessi, B. Uster, H. Veeramani, E. I. Suvorova, J. S. Lezama-Pacheco, J. E. Stubbs, J. R. Bargar and R. Bernier-Latmani, Environ. Sci. Technol., 2012, 46, 6150–6157 CrossRef CAS PubMed.
- J. M. Cerrato, M. N. Ashner, D. S. Alessi, J. S. Lezama-Pacheco, R. Bernier-Latmani, J. R. Bargar and D. E. Giammar, Environ. Sci. Technol., 2013, 47, 9756–9763 CrossRef CAS PubMed.
- J. O. Sharp, J. S. Lezama-Pacheco, E. J. Schofield, P. Junier, K. U. Ulrich, S. Chinni, H. Veeramani, C. Margot-Roquier, S. M. Webb, B. M. Tebo, D. E. Giammar, J. R. Bargar and R. Bernier-Latmani, Geochim. Cosmochim. Acta, 2011, 75, 6497–6510 CrossRef CAS.
- D. R. Brookshaw, R. A. D. Pattrick, P. Bots, G. T. W. Law, J. R. Lloyd, J. F. W. Mosselmans, D. J. Vaughan, K. Dardenne and K. Morris, Environ. Sci. Technol., 2015, 49, 13139–13148 CrossRef CAS PubMed.
- E. H. Winstanley, K. Morris, L. G. Abrahamsen-Mills, R. Blackham and S. Shaw, J. Hazard. Mater., 2019, 366, 98–104 CrossRef CAS PubMed.
- S. Nair, L. Karimzadeh and B. J. Merkel, Environ. Earth Sci., 2014, 71, 1737–1745 CrossRef CAS.
- Z. Guo, Y. Li and W. Wu, Appl. Radiat. Isot., 2009, 67, 996–1000 CrossRef CAS PubMed.
- Y. Gao, Z. Shao and Z. Xiao, J. Radioanal. Nucl. Chem., 2015, 303, 867–876 CrossRef CAS.
- R. J. Reeder, M. Nugent, G. M. Lamble, C. D. Tait and D. E. Morris, Environ. Sci. Technol., 2000, 34, 638–644 CrossRef CAS.
- J. G. Catalano and G. E. Brown, Geochim. Cosmochim. Acta, 2005, 69, 2995–3005 CrossRef CAS.
- D. Gorman-Lewis, P. C. Burns and J. B. Fein, J. Chem. Thermodyn., 2008, 40, 335–352 CrossRef CAS.
- S. Regenspurg, D. Schild, T. Schäfer, F. Huber and M. E. Malmström, Appl. Geochem., 2009, 24, 1617–1625 CrossRef CAS.
- D. E. Latta, M. I. Boyanov, K. M. Kemner, E. J. O'Loughlin and M. M. Scherer, Appl. Geochem., 2012, 27, 1512–1524 CrossRef CAS.
- S. P. Hyun, J. A. Davis and K. F. Hayes, Appl. Geochem., 2014, 50, 7–15 CrossRef CAS.
- H. E. Roberts, K. Morris, G. T. W. Law, J. F. W. Mosselmans, P. Bots, K. Kvashnina and S. Shaw, Environ. Sci. Technol. Lett., 2017, 4, 421–426 CrossRef CAS.
- T. A. Marshall, K. Morris, G. T. W. Law, F. R. Livens, J. F. W. Mosselmans, P. Bots and S. Shaw, Environ. Sci. Technol., 2014, 48, 3724–3731 CrossRef CAS PubMed.
- T. A. Marshall, K. Morris, G. T. W. Law, J. F. W. Mosselmans, P. Bots, H. Roberts and S. Shaw, Mineral. Mag., 2015, 79, 1265–1274 CrossRef.
- L. T. Townsend, S. Shaw, N. E. R. Ofili, N. Kaltsoyannis, A. S. Walton, J. F. W. Mosselmans, T. S. Neill, J. R. Lloyd, S. Heath, R. Hibberd and K. Morris, Environ. Sci. Technol., 2019, 54, 129–136 CrossRef PubMed.
- L. T. Townsend, K. Morris, R. Harrison, B. Schacherl, T. Vitova, L. Kovarik, C. I. Pearce, J. F. W. Mosselmans and S. Shaw, Chemosphere, 2021, 276, 130117 CrossRef CAS PubMed.
- O. Stagg, K. Morris, A. Lam, A. Navrotsky, J. M. Velázquez, B. Schacherl, T. Vitova, J. Rothe, J. Galanzew, A. Neumann, P. Lythgoe, L. Abrahamsen-Mills and S. Shaw, Environ. Sci. Technol., 2021, 55, 16445–16454 CrossRef CAS PubMed.
- O. Riba, T. B. Scott, K. Vala Ragnarsdottir and G. C. Allen, Geochim. Cosmochim. Acta, 2008, 72, 4047–4057 CrossRef CAS.
- S. Tsarev, R. N. Collins, A. Fahy and T. D. Waite, Environ. Sci. Technol., 2016, 50, 2595–2601 CrossRef CAS PubMed.
- K. Mackenzie, S. Bleyl, A. Georgi and F. D. Kopinke, Water Res., 2012, 46, 3817–3826 CrossRef CAS PubMed.
- R. A. Crane and T. B. Scott, J. Hazard. Mater., 2011, 211–212, 112–125 Search PubMed.
- S. Tsarev, R. N. Collins, E. S. Ilton, A. Fahy and T. D. Waite, Environ. Sci.: Nano, 2017, 4, 1304–1313 RSC.
- Z. Dong, Z. Zhang, R. Zhou, Y. Dong, Y. Wei, Z. Zheng, Y. Wang, Y. Dai, X. Cao and Y. Liu, RSC Adv., 2020, 10, 34859–34868 RSC.
- A. Chen, C. Shang, J. Shao, J. Zhang and H. Huang, Sci. Total Environ., 2017, 575, 1291–1306 CrossRef CAS PubMed.
- D. Q. Liu, Z. R. Liu, C. F. Wang, Y. Lai and X. Huang, Rengong Jingti Xuebao, 2016, 45, 1328–1334 CAS.
- L. Newsome, K. Morris and J. R. Lloyd, PLoS One, 2015, 10, 1–14 CrossRef CAS PubMed.
- L. Newsome, K. Morris, D. Trivedi, A. Bewsher and J. R. Lloyd, Environ. Sci. Technol., 2015, 49, 11070–11078 CrossRef CAS PubMed.
- L. Newsome, A. Cleary, K. Morris and J. R. Lloyd, Environ. Sci. Technol., 2017, 51, 1595–1604 CrossRef CAS PubMed.
- K. E. Fletcher, M. I. Boyanov, S. H. Thomas, Q. Wu, K. M. Kemner and F. E. Löffler, Environ. Sci. Technol., 2010, 44, 4705–4709 CrossRef CAS PubMed.
- C. M. Smeaton, C. G. Weisener, P. C. Burns, B. J. Fryer and D. A. Fowle, Am. Mineral., 2008, 93, 1858–1864 CrossRef CAS.
- H. Wen, Z. Pan, D. Giammar and L. Li, Environ. Sci. Technol., 2018, 52, 5841–5850 CrossRef CAS PubMed.
- S. H. Wallace, S. Shaw, K. Morris, J. S. Small and I. T. Burke, Environ. Sci. Technol., 2013, 47, 3694–3700 CrossRef CAS PubMed.
- A. Dyer, J. Chow and I. M. Umar, J. Mater. Chem., 2000, 10, 2734–2740 RSC.
- S. A. Carroll, S. K. Roberts, L. J. Criscenti and P. A. O’day, Geochem. Trans., 2008, 9, 2 CrossRef PubMed.
- V. Balek, Z. Málek, J. Šubrt and A. Ždimera, J. Radioanal. Nucl. Chem., 1996, 212, 321–331 CrossRef CAS.
- D. García, J. Lützenkirchen, M. Huguenel, L. Calmels, V. Petrov, N. Finck and D. Schild, Minerals, 2021, 11, 1093 CrossRef.
- J. C. Mendez and T. Hiemstra, Geochim. Cosmochim. Acta, 2020, 286, 289–305 CrossRef CAS.
- M. S. Ho, G. F. Vettese, P. H. Keto, S. P. Lamminmäki, M. Vikman, E. Myllykylä, K. Dardenne and G. T. W. Law, Minerals, 2023, 13, 436 CrossRef CAS.
- A. J. Fuller, S. Shaw, C. L. Peacock, D. Trivedi and I. T. Burke, Langmuir, 2016, 32, 2937–2946 CrossRef CAS PubMed.
- P. A. O'day, M. Newville, P. S. Neuhoff, N. Sahai and S. A. Carroll, J. Colloid Interface Sci., 2000, 222, 184–197 CrossRef PubMed.
- C. L. Thorpe, C. Boothman, J. R. Lloyd, G. T. W. Law, N. D. Bryan, N. Atherton, F. R. Livens and K. Morris, Appl. Geochem., 2014, 40, 135–143 CrossRef CAS.
- C. L. Thorpe, J. R. Lloyd, G. T. W. Law, I. T. Burke, S. Shaw, N. D. Bryan and K. Morris, Chem. Geol., 2012, 306–307, 114–122 CrossRef CAS.
- S. H. Wallace, S. Shaw, K. Morris, J. S. Small, A. J. Fuller and I. T. Burke, Appl. Geochem., 2012, 27, 1482–1491 CrossRef CAS.
- S. Handley-Sidhu, J. A. Hriljac, M. O. Cuthbert, J. C. Renshaw, R. A. D. Pattrick, J. M. Charnock, B. Stolpe, J. R. Lead, S. Baker and L. E. Macaskie, Environ. Sci. Technol., 2014, 48, 6891–6898 CrossRef CAS PubMed.
- S. Handley-Sidhu, J. C. Renshaw, S. Moriyama, B. Stolpe, C. Mennan, S. Bagheriasl, P. Yong, A. Stamboulis, M. Paterson-Beedle, K. Sasaki, R. A. D. Pattrick, J. R. Lead and L. E. Macaskie, Environ. Sci. Technol., 2011, 45, 6985–6990 CrossRef CAS PubMed.
- A. Cleary, J. R. Lloyd, L. Newsome, S. Shaw, C. Boothman, G. Boshoff, N. Atherton and K. Morris, Chem. Geol., 2019, 509, 213–222 CrossRef CAS.
- D. R. Brookshaw, J. R. Lloyd, D. J. Vaughan and R. A. D. Pattrick, Geomicrobiol. J., 2016, 33, 206–215 CrossRef CAS.
- C. L. Thorpe, G. T. W. Law, C. Boothman, J. R. Lloyd, I. T. Burke and K. Morris, Geomicrobiol. J., 2012, 29, 484–493 CrossRef CAS.
- Y. Fujita, G. D. Redden, J. C. Ingram, M. M. Cortez, F. G. Ferris and R. W. Smith, Geochim. Cosmochim. Acta, 2004, 68, 3261–3270 CrossRef CAS.
- L. E. Macaskie and A. C. R. Dean, Biotechnol. Lett., 1985, 7, 627–630 CrossRef CAS.
- N. Efecan, T. Shahwan, A. E. Eroǧlu and I. Lieberwirth, Desalination, 2009, 249, 1048–1054 CrossRef CAS.
- W. M. Wu, J. Carley, T. Gentry, M. A. Ginder-Vogel, M. Fienen, T. Mehlhorn, H. Yan, S. Caroll, M. N. Pace, J. Nyman, J. Luo, M. E. Gentile, M. W. Fields, R. F. Hickey, B. Gu, D. Watson, O. A. Cirpka, J. Zhou, S. Fendorf, P. K. Kitanidis, P. M. Jardine and C. S. Criddle, Environ. Sci. Technol., 2006, 40, 3986–3995 CrossRef CAS PubMed.
- J. M. Senko, J. D. Istok, J. M. Suflita and L. R. Krumholz, Environ. Sci. Technol., 2002, 36, 1491–1496 CrossRef CAS PubMed.
- R. T. Anderson, H. A. Vrionis, I. Ortiz-Bernad, C. T. Resch, P. E. Long, R. Dayvault, K. Karp, S. Marutzky, D. R. Metzler, A. Peacock, D. C. White, M. Lowe and D. R. Lovley, Appl. Environ. Microbiol., 2003, 69, 5884–5891 CrossRef CAS PubMed.
- J. R. Lloyd and J. C. Renshaw, Curr. Opin. Biotechnol., 2005, 16, 254–260 CrossRef CAS PubMed.
- K. H. Williams, P. E. Long, J. a. Davis, M. J. Wilkins, a. L. N'Guessan, C. I. Steefel, L. Yang, D. Newcomer, F. a. Spane, L. J. Kerkhof, L. McGuinness, R. Dayvault and D. R. Lovley, Geomicrobiol. J., 2011, 28, 519–539 CrossRef CAS.
- V. R. Vermeul, J. E. Szecsody, B. G. Fritz, M. D. Williams, R. C. Moore and J. S. Fruchter, Groundwater Monit. Rem., 2014, 34, 28–41 CrossRef CAS.
- J. E. Szecsody, J. S. Fruchter, M. L. Rockhold, J. P. Mckinley, M. Oostrom, V. R. Vermeul, R. C. Moore, M. T. Covert, C. Burns, T. W. Wietsma, M. D. Williams, A. T. Breshears, L. Zhong and B. J. Garcia, Sequestration of Sr-90 Subsurface Contamination in the Hanford 100-N Area by Surface Infiltration of a Ca-Citrate-Phosphate Solution, 2009 Search PubMed.
- J. E. Szecsody, J. S. Fruchter, C. A. Burns, M. L. Rockhold, M. Oostrom, M. D. Williams and V. R. Vermeul, in WM2008 Conference, February 24–28, 2008, Phoenix, AZ, 2008 Search PubMed.
- S. H. Moon, J. Komlos and P. R. Jaffé, Environ. Sci. Technol., 2007, 41, 4587–4592 CrossRef PubMed.
- J. Komlos, A. Peacock, R. K. Kukkadapu and P. R. Jaffé, Geochim. Cosmochim. Acta, 2008, 72, 3603–3615 CrossRef CAS.
- Y. Bi, S. P. Hyun, R. Kukkadapu and K. F. Hayes, Geochim. Cosmochim. Acta, 2013, 102, 175–190 CrossRef CAS.
- G. T. W. Law, A. Geissler, I. T. Burke, F. R. Livens, J. R. Lloyd, J. M. McBeth and K. Morris, Geomicrobiol. J., 2011, 28, 497–506 CrossRef CAS.
- D. Pan, K. H. Williams, M. J. Robbins and K. A. Weber, Environ. Sci. Technol., 2018, 52, 8133–8145 CrossRef CAS PubMed.
- H. S. Moon, J. Komlos and P. R. Jaffé, J. Contam. Hydrol., 2009, 105, 18–27 CrossRef CAS PubMed.
- K. T. Finneran, M. E. Housewright and D. R. Lovley, Environ. Microbiol., 2002, 4, 510–516 CrossRef CAS PubMed.
- H. R. Beller, Appl. Environ. Microbiol., 2005, 71, 2170–2174 CrossRef CAS PubMed.
- J. D. Istok, J. M. Senko, L. R. Krumholz, D. Watson, M. A. Bogle, A. Peacock, Y. J. Chang and D. C. White, Environ. Sci. Technol., 2004, 38, 468–475 CrossRef CAS PubMed.
- J. D. Istok, M. Park, L. D. McKay, D. B. Watson, T. C. Hazen, S. Jagadamma and C. J. Paradis, J. Contam. Hydrol., 2016, 187, 55–64 CrossRef PubMed.
- J. Nolan and K. A. Weber, Environ. Sci. Technol. Lett., 2015, 2, 215–220 CrossRef CAS.
- K. R. Burow, B. T. Nolan, M. G. Rupert and N. M. Dubrovsky, Environ. Sci. Technol., 2010, 44, 4988–4997 CrossRef CAS PubMed.
- J. M. Senko, T. A. Dewers and L. R. Krumholz, Appl. Environ. Microbiol., 2005, 71, 7172–7177 CrossRef CAS PubMed.
- G. T. W. Law, A. Geissler, C. Boothman, I. T. Burke, F. R. Livens, J. R. Lloyd and K. Morris, Environ. Sci. Technol., 2010, 44, 150–155 CrossRef CAS PubMed.
- J. M. McBeth, G. Lear, J. R. Lloyd, F. R. Livens, K. Morris and I. T. Burke, Geomicrobiol. J., 2007, 24, 189–197 CrossRef CAS.
- L. Newsome, K. Morris, A. Cleary, N. K. Masters-Waage, C. Boothman, N. Joshi, N. Atherton and J. R. Lloyd, J. Hazard. Mater., 2019, 364, 134–142 CrossRef CAS PubMed.
- R. A. Crane, M. Dickinson, I. C. C. Popescu and T. B. B. Scott, Water Res., 2011, 45, 2931–2942 CrossRef CAS PubMed.
- M. S. Ho, G. F. Vettese, K. Morris, J. R. Lloyd, C. Boothman, W. R. Bower, S. Shaw and G. T. W. Law, Sci. Total Environ., 2022, 834, 155332 CrossRef CAS PubMed.
- M. J. Wilkins, F. R. Livens, D. J. Vaughan, I. Beadle and J. R. Lloyd, Geobiology, 2007, 5, 293–301 CrossRef CAS.
- Regenesis, Metals Remediation Compound (MRC®), https://regenesis.com/eur/remediation-products/metals-remediation-compound-mrc/, accessed 9 May 2019 Search PubMed.
- Andra, Thermo-Chimie (version applicative 4.3.0), https://www.thermochimie-tdb.com/, accessed 14 June 2017 Search PubMed.
- D. R. Lovley and E. J. Phillips, Appl. Environ. Microbiol., 1987, 53, 1536–1540 CrossRef CAS PubMed.
- N. K. Masters-Waage, K. Morris, J. R. Lloyd, S. Shaw, J. F. W. Mosselmans, C. Boothman, P. Bots, A. Rizoulis, F. R. Livens and G. T. W. W. Law, Environ. Sci. Technol., 2017, 51, 14301–14310 CrossRef CAS PubMed.
- A. J. Dent, G. Cibin, S. Ramos, A. D. Smith, S. M. Scott, L. Varandas, M. R. Pearson, N. A. Krumpa, C. P. Jones and P. E. Robbins, J. Phys.: Conf. Ser., 2009, 190, 012039 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- L. Downward, C. H. Booth, W. W. Lukens, F. Bridges, C. S. Division and L. Berkeley, X-Ray Absorpt. Fine Struct. - XAFS13 13th Int. Conf., 2007, vol. 882, pp. 2–4 Search PubMed.
- B. M. Macaulay, C. Boothman, B. E. van Dongen and J. R. Lloyd, Front. Microbiol., 2020, 11, 330 CrossRef PubMed.
- S. Bleyl, F. D. Kopinke and K. Mackenzie, Chem. Eng. J., 2012, 191, 588–595 CrossRef CAS.
- J. Němeček, O. Lhotský and T. Cajthaml, Sci. Total Environ., 2014, 485–486, 739–747 CrossRef PubMed.
- M. Vogel, I. Nijenhuis, J. Lloyd, C. Boothman, M. Pöritz and K. Mackenzie, Sci. Total Environ., 2018, 628–629, 161–169 Search PubMed.
- J. Kašlík, J. Kolařík, J. Filip, I. Medřík, O. Tomanec, M. Petr, O. Malina, R. Zbořil and P. G. Tratnyek, Chem. Eng. J., 2018, 354, 335–345 CrossRef.
- S. Klimkova, M. Cernik, L. Lacinova, J. Filip, D. Jancik and R. Zboril, Chemosphere, 2011, 82, 1178–1184 CrossRef CAS PubMed.
- J. Carpenter, Y. Bi and K. F. Hayes, Environ. Sci. Technol., 2015, 49, 1078–1085 CrossRef CAS PubMed.
- Y. Bi and K. F. Hayes, Environ. Sci. Technol., 2014, 48, 632–640 CrossRef CAS PubMed.
- C. L. Thorpe, G. T. W. W. Law, J. R. Lloyd, H. A. Williams, N. Atherton and K. Morris, Environ. Sci. Technol., 2017, 51, 12104–12113 CrossRef CAS PubMed.
- E. L. Brodie, D. C. Joyner, B. Faybishenko, M. E. Conrad, C. Rios-Velazquez, J. Malave, R. Martinez, B. Mork, A. Willett, S. Koenigsberg, D. J. Herman, M. K. Firestone and T. C. Hazen, Chemosphere, 2011, 85, 660–665 CrossRef CAS PubMed.
- L. Petrie, N. N. North, S. L. Dollhopf, D. L. Balkwill and J. E. Kostka, Appl. Environ. Microbiol., 2003, 69, 7467–7479 CrossRef CAS PubMed.
- C. Hennig, K. Schmeide, V. Brendler, H. Moll, S. Tsushima and A. C. Scheinost, Inorg. Chem., 2007, 46, 5882–5892 CrossRef CAS PubMed.
- M. Walter, T. Arnold, T. Reich and G. Bernhard, Environ. Sci. Technol., 2003, 37, 2898–2904 CrossRef CAS PubMed.
- C. C. Fuller, J. R. Bargar, J. A. Davis and M. J. Piana, Environ. Sci. Technol., 2002, 36, 158–165 CrossRef CAS PubMed.
- R. Finch and T. Murakami, in Reviews in Mineralogy, ed. P. C. Burns and R. J. Finch, Golden, 1999, vol. 38, pp. 91–179 Search PubMed.
- A. J. Pinto, M. A. Gonçalves, C. Prazeres, J. M. Astilleros and M. J. Batista, Chem. Geol., 2012, 312–313, 18–26 CrossRef CAS.
- J. L. Jerden and A. K. Sinha, Appl. Geochem., 2003, 18, 823–843 CrossRef CAS.
- D. M. Wellman, J. P. Icenhower and A. T. Owen, Environ. Chem., 2006, 3, 219–224 CrossRef CAS.
- G. Kuippers, K. Morris, L. T. Townsend, P. Bots, K. Kvashnina, N. D. Bryan and J. R. Lloyd, Environ. Sci. Technol., 2021, 55(8), 4597–4606 CrossRef CAS PubMed.
- X. Rui, M. J. Kwon, E. J. O'Loughlin, S. Dunham-Cheatham, J. B. Fein, B. Bunker, K. M. Kemner and M. I. Boyanov, Environ. Sci. Technol., 2013, 47, 5668–5678 CrossRef CAS PubMed.
- L. Foster, K. Morris, A. Cleary, H. Bagshaw, D. Sigee, J. K. Pittman, K. Zhang, G. Vettese, K. F. Smith and J. R. Lloyd, Front. Earth Sci., 2020, 8, 556244 CrossRef.
- J. Farrell, W. D. Bostick, R. J. Jarabek and J. N. Fiedor, Ground Water, 1999, 37, 618–624 CrossRef CAS.
- G. Sheng, P. Yang, Y. Tang, Q. Hu, H. Li, X. Ren, B. Hu, X. Wang and Y. Huang, Appl. Catal., B, 2016, 193, 189–197 CrossRef CAS.
- M. I. Boyanov, E. J. O'Loughlin, E. E. Roden, J. B. Fein and K. M. Kemner, Geochim. Cosmochim. Acta, 2007, 71, 1898–1912 CrossRef CAS.
- S. Chakraborty, F. Favre, D. Banerjee, A. C. Scheinost, M. Mullet, J. J. Ehrhardt, J. Brendle, L. Vidal and L. Charlet, Environ. Sci. Technol., 2010, 44, 3779–3785 CrossRef CAS PubMed.
- Z. Wang, K. U. Ulrich, C. Pan and D. E. Giammar, Environ. Sci. Technol. Lett., 2015, 2, 227–232 CrossRef CAS.
- A. Satpathy, J. G. Catalano and D. E. Giammar, Environ. Sci. Technol., 2022, 56, 4111–4120 CrossRef PubMed.
- L. Newsome, et al. , J. Hazard. Mater., 2019, 364, 134–142 CrossRef CAS PubMed.
- R. K. Sani, B. M. Peyton, J. E. Amonette and G. G. Geesey, Geochim. Cosmochim. Acta, 2004, 68, 2639–2648 CrossRef CAS.
- J. Komlos, H. S. Moon and P. R. Jaffé, J. Environ. Qual., 2008, 37, 2058–2062 CrossRef CAS PubMed.
- J. Komlos, B. Mishra, A. Lanzirotti, S. C. B. Myneni and P. R. Jaffé, J. Environ. Eng., 2008, 134, 78–86 CrossRef CAS.
- A. Geissler, G. T. W. W. Law, C. Boothman, K. Morris, I. T. Burke, F. R. Livens and J. R. Lloyd, Geomicrobiol. J., 2011, 28, 507–518 CrossRef CAS.
- T. D. Waite, J. A. Davis, T. E. Payne, G. A. Waychunas and N. Xu, Geochim. Cosmochim. Acta, 1994, 58, 5465–5478 CrossRef CAS.
- J. R. Bargar, R. Reitmeyer, J. J. Lenhart and J. A. Davis, Geochim. Cosmochim. Acta, 2000, 64, 2737–2749 CrossRef CAS.
- T. E. Payne and T. D. Waite, Radiochim. Acta, 2022, 110, 549–559 CrossRef CAS.
- C. Bryce, N. Blackwell, C. Schmidt, J. Otte, Y. M. Huang, S. Kleindienst, E. Tomaszewski, M. Schad, V. Warter, C. Peng, J. M. Byrne and A. Kappler, Environ. Microbiol., 2018, 20, 3462–3483 CrossRef CAS PubMed.
- H. J. Busse, Int. J. Syst. Evol. Microbiol., 2016, 66, 9–37 CrossRef CAS PubMed.
- K. H. Williams, J. R. Bargar, J. R. Lloyd and D. R. Lovley, Curr. Opin. Biotechnol., 2013, 24, 489–497 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00104k |
| ‡ Present address: Radiochemistry Unit, Department of Chemistry, The University of Helsinki, Helsinki, Finland. |
| § Present address: Immobilisation Science Laboratory, Department of Materials Science and Engineering, University of Sheffield, Sheffield S1 3JD, UK. |
| This journal is © The Royal Society of Chemistry 2023 |