
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning based models for high-throughput classification of human pregnane X receptor activators†
Yiyuan
Gou
ab,
Lilai
Shen
a,
Shixuan
Cui
 *ab,
Meiling
Huang
a,
Yiqu
Wu
a,
Penghan
Li
a and
Shulin
Zhuang
*ab
*ab,
Meiling
Huang
a,
Yiqu
Wu
a,
Penghan
Li
a and
Shulin
Zhuang
*ab
aKey Laboratory of Environment Remediation and Ecological Health, Ministry of Education, College of Environmental and Resource Sciences, Zhejiang University, Hangzhou 310058, China. E-mail: sxcui@zju.edu.cn; shulin@zju.edu.cn
bWomen's Reproductive Health Key Laboratory of Zhejiang Province, Women's Hospital, School of Medicine, Zhejiang University, Hangzhou 310006, China
First published on 23rd December 2022
Abstract
The pregnane X receptor (PXR) is a master receptor in regulating the metabolism and transport of structurally diverse endogenous compounds. Activation of PXR by xenobiotics potentially induces adverse effects and disrupts normal physiological states. Therefore, it is essential to filter out PXR activators despite challenges in the construction of PXR screening models. Herein, we developed a high-throughput model using machine learning to classify human PXR (hPXR) activators and non-activators. Molecular descriptors and eight fingerprints were calculated for a diverse dataset retrieved from the PubChem database. The dimension reduction procedure was adopted to define an optimal subset of fingerprints and 87 molecular descriptors before the model construction. Five machine learning methods coupled with molecular descriptors and fingerprints were compared and the XGBoost method combined with RDKit descriptors yielded the best performance with AUC values of 0.913 and 0.860 for the training set (4144 chemicals) and external test set (1037 chemicals). The model constructed with the XGBoost method has high prediction ability as revealed by the applicability domain analysis. Our built machine learning models are useful for identifying compounds of potential PXR activators and facilitating the prioritization of contaminants of emerging concern.
Environmental significancePregnane X Receptor (PXR) is a master receptor in regulating the metabolism and transport of structurally diverse endogenous compounds. Activation of PXR by xenobiotics potentially induce adverse effects and disrupt normal physiological states. Therefore, the identification of PXR activators is significant for the health risk assessment. In the present study, we developed machine learning based models to classify human PXR (hPXR) activators and non-activators based on a diverse dataset retrieved from PubChem database. Five machine learning methods coupled with molecular descriptors and fingerprints are compared to select optimal combinatorial model based on five-fold cross validation and external validation. Our model improved robustness and generalization capabilities, which can be served as a fast and reliable filter tool for early identification of PXR activators, facilitating the risk examination for potential PXR activators. |
1. Introduction
Pregnane xenobiotic receptor (PXR), also known as steroid and xenobiotic receptor (SXR), is a member of the nuclear receptor superfamily of ligand-activated transcription factors.1 It plays a critical role in the mediation of the metabolism and detoxification of exogenous compounds involving inflammatory response, cell proliferation, and migration, where dysregulation is associated with different disease states.2,3 PXR can be activated by a wide variety of chemicals, including bile acids, steroid hormones, dietary vitamins, prescription drugs, and environmental chemicals.4,5 The altered expression of PXR by xenobiotics may be involved in bone disorders, hepatic steatosis, inflammatory bowel disease, and cancers.6 Thus, the identification of PXR activators is significant for health risk assessment.Multiple in vitro or in vivo assays have been used to assess compounds for PXR activation.7 Considering the restrictions of 3R principles (reduction, replacement, and refinement) and cost of experimental testing, computational tools have emerged as an alternative way for rapid, efficient, and high-throughput screening of PXR activators. Ligand-based computational models employing pharmacophore mapping and quantitative structure–activity relationships (QSARs) have previously been developed to discriminate PXR activators from non-activators.5,8–15 To date, several QSAR models have been built using machine learning methods, including k-NN, Naïve Bayesian, probabilistic neural networks, artificial neural networks, and random forest.16–22 The lack of larger PXR datasets has restricted the application of PXR classification models to a large scale of compound screening. Therefore, a comprehensive and large dataset is required to develop machine learning driven models with a broader chemical space and higher generalization ability.
In this study, we developed predictive models with high prediction accuracy and stable generalization ability using diverse datasets retrieved from the PubChem database. Molecular descriptors and eight molecular fingerprints were adopted to represent chemical structures. Five machine learning approaches, including Bernoulli Naïve Bayes (BNB), random forest (RF), support vector machine (SVM), AdaBoost, and extreme gradient boosting (XGBoost) were evaluated by five-fold cross-validation and external validation for robustness and predictive performances. The applicability domain was further defined to elaborate on the regulatory acceptance of the established models. The model with the best performances can be used as a screening tool to assess PXR activation potential of chemicals and prioritize compounds for experimental validations.
2. Materials and methods
2.1 Data preparation
Chemicals with activity toward PXR were retrieved from the PubChem BioAssay database (AID 1347033), resulting in a dataset composed of 9667 chemicals.23 Based on the agonist potency and efficacy score, chemicals were divided into three categories: active, inactive, and inconclusive. The entries that were labeled as “inconclusive” in the activity outcome were deleted. Organometallics, inorganics, mixtures, and salts were removed and duplicated compounds were excluded. After the pretreatment, the final dataset contained 1367 active compounds and 3814 inactive compounds. To build classification models, active compounds were labeled as 1 while inactive compounds were labeled as 0.2.2 Molecule representation and feature extraction
Molecular descriptors were calculated by RDKit containing a total of 208 1D/2D molecular descriptors (https://www.rdkit.org).24 Eight molecular fingerprints, including MACCS keys (166 bits), PubChem fingerprint (881 bits), Klekota-Roth fingerprint (4860 bits), Extended fingerprint (1024 bits), Daylight (1024 bits), CDK GraphOnly (1024 bits), Morgan (1024 bits) and Morgan (2048 bits) were computed directly from the SDF files.25 Both molecular descriptors and eight molecular fingerprints were used to represent molecules. Molecular descriptors and fingerprint bits with all zero values or zero variance were deleted to avoid overfitting and to enhance the model generalization. The redundant descriptors or fingerprint bits with Pearson correlation coefficients higher than 0.95 in comparison to any descriptors or bits were also removed. Furthermore, the recursive feature elimination (RFE) method incorporated with the random forest was used to select molecular descriptors and fingerprints.26 The subset of features obtained by the best AUC scores was maintained for later modeling.2.3 Models building with machine learning approaches
Before constructing models, the dataset was randomly split into a training set (80%) and an external test set (20%). Equal proportions of the active to inactive class ratios in each split were maintained (stratified splitting). Five machine learning algorithms, including BNB,27 RF,28 SVM,29 AdaBoost,30 and XGBoost31 were used to build models. Tuning hyperparameters searches were conducted through five-fold stratified cross-validation on training data for better model performances (Table S3†).2.3.1 Bernoulli Naïve Bayes
The BernoulliNB class from the Naïve Bayes module of Scikit-learn was used to construct the BNB models. The CalibratedClassifierCV in Scikit-learn tuned our BNB models through five-fold stratified cross-validation based on isotonic regression. Strategy isotonic calibration could optimize the classifier by calibrating the probability scores, resulting in more reliable probability estimates.32 Based on these calibrated probabilities, balanced accuracy, the area under the receiver operating characteristic curve (AUC), and other metrics were computed.2.3.2 Support vector machine
SVM classification with the libsvm method from Scikit-learn was used. The grid search with five-fold stratified cross-validation using balanced classes was performed for C (1 × 10−3, 1 × 10−2, 1 × 10−1, 1, 10, 100), gamma values (1 × 10−1, 1 × 10−2, 1 × 10−3) and kernel (rbf, linear, sigmond). The model with optimal parameters was retrained with a training set and validated on an external test set.2.3.3 Random forest
The RandomForestClassifier method with balanced class weights was used to build the model. The five-fold stratified cross-validation grid search was performed using 5, 10, 25, 50, 75, 100, and 200 estimators with the AUC as a scoring function. The optimal number of estimators was kept for model validation.2.3.4 AdaBoost
The AdaBoostClassifier method of 200 estimators and 0.9 learning rate was used with a decision tree as a base classifier. Similar to BNB model construction, the five-fold stratified cross-validation based on isotonic regression was applied to tune AdaBoost models.2.3.5 XGBoost
The XGBClassifier method with balanced class weights was used to build the model. Five-fold stratified cross-validation was used in the grid search of learning_rate (0.01, 0.1, 1.0), n_estimators (10, 25, 50, 100), gamma (0, 0.05, 0.1, 0.3, 0.5, 0.7, 0.9, 1.0), max_depth (3, 5, 6, 7, 9, 12, 15, 17, 25) and min_child_weight (1, 3, 5, 7). The combination of parameters with the best performance was retained as the optimal values and saved for model comparison and prediction.2.4 Model evaluation
The robustness and predictivity of the developed models were assessed by internal and external validations. The internal validation was achieved by the five-fold stratified cross-validation for 10 iterations, while the external validation was testified by the predictions of the test set. All models were evaluated by balanced accuracy, precision, recall, F1 score, AUC, Cohen's Kappa (CK), and Matthews correlation coefficient (MCC) (Text S1†).2.5 Definition of the applicability domain
The applicability domain (AD) is essential for evaluating reliability regarding model predictions.33 Only predictions for external compounds that fall inside the applicability domains are considered valid. Thus, it is essential to develop models with defined applicability domains. In this study, a similarity-based applicability domain analysis was adopted with the evaluation of the distance between a query sample and its k-nearest neighbors in the training set.34 The Euclidean distance between any two molecules using MACCS keys was calculated. The AD threshold, DT, was obtained according to the following formula:| DT = ȳ + Zσ |
3. Results and discussion
3.1 Analysis of curated dataset
After data collection and curation, a total number of 5181 compounds were obtained and split into a training set (4144 chemicals) and an external test set (1037 chemicals) by stratified random sampling. The activators and non-activators in each data set are listed in detail in Table 1. The training set contained 1093 active compounds and 3051 inactive compounds, while the external test set contained 274 and 763 active and inactive compounds, respectively. Both datasets maintain equal proportions of active and inactive compounds to that of the original dataset.| Dataset | Activators | Non-activators | Total number |
|---|---|---|---|
| Training set | 1093 | 3051 | 4144 |
| External test set | 274 | 763 | 1037 |
| Total number | 1367 | 3814 | 5181 |
To explore the structural diversity of the training set intuitively, the Tanimoto similarity index for each pair of the training molecules was calculated using MACCS fingerprints. Fig. 1A shows the structural diversity among the training set. The overall similarity of the training set is low, indicating the significantly diverse chemical structures in the training set. Principal component analysis (PCA) was further used to compare the chemical space between the training set and the test set based on seven selected descriptors (molecular weight, Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, number of rotatable bonds, number of aromatic rings, number of rotatable bonds, number of hydrogen bond acceptors, and number of hydrogen bond donors).35 The top three principal components, accounting for 86.18% of the total descriptor variance, were used to visualize the chemical space of the training and test sets (Fig. 1B–D). Each molecule in the three-dimensional (3D) space was projected into the corresponding two-dimensional planes. A similar chemical space was observed between the training set (grey squares) and the test set (red circles), providing credibility to evaluate the generalization ability on the external test set. The diversity and complexity of our dataset facilitate the construction of machine learning based prediction models with high generalization ability.
P, number of rotatable bonds, number of aromatic rings, number of rotatable bonds, number of hydrogen bond acceptors, and number of hydrogen bond donors).35 The top three principal components, accounting for 86.18% of the total descriptor variance, were used to visualize the chemical space of the training and test sets (Fig. 1B–D). Each molecule in the three-dimensional (3D) space was projected into the corresponding two-dimensional planes. A similar chemical space was observed between the training set (grey squares) and the test set (red circles), providing credibility to evaluate the generalization ability on the external test set. The diversity and complexity of our dataset facilitate the construction of machine learning based prediction models with high generalization ability.
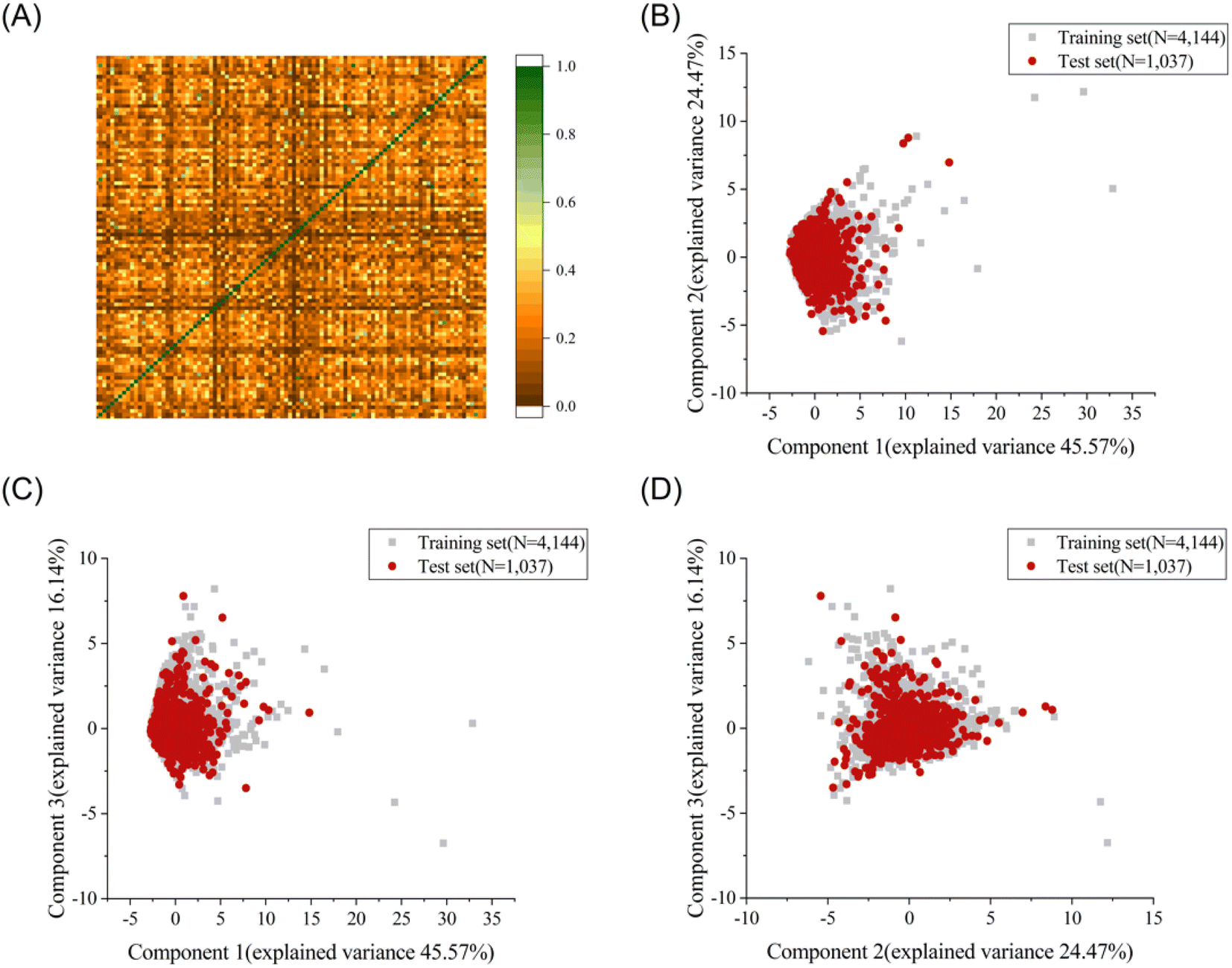 | ||
| Fig. 1 (A) Heat map of Tanimoto similarity for the training set characterized by MACCS fingerprint. Principal component analysis (PCA) of the dataset with indication of the training and test for chemical space comparison. The top three principal components account for 86.18% of the total descriptor variance. Plot of the first and second principal component (B), the first and third component (C) and the second and third component (D). | ||
3.2 Selection of molecular descriptors and fingerprints
Descriptors are symbolic representations that encode chemical information about the structures. By converting the molecules into numbers, chemical descriptors play a significant role in in silico QSAR modeling. However, applying a large number of descriptors for model building may increase the risk of model overfitting. Dimensionality reduction is necessary to develop models with fewer variables while maintaining the physical meanings of original features.36 After feature filtering for 208 molecular descriptors, in total 174 molecular descriptors with nonzero variances and low pairwise correlations were applied for feature selection and 87 descriptors were maintained (Table S1†). Eight fingerprints were filtered by deleting fingerprint bits with high correlation and zero variance. The sizes of the nine features, including eight fingerprints and molecular descriptors, and their selected ones are summarized in Table S2.†After the feature selection, the remaining 87 descriptors represented chemical properties related to topological, constitutional, and physicochemical aspects, consistent with the previous observations that descriptors associated with solubility, lipophilic properties, the balance between polarity and lipophilicity and electrostatic properties have a remarkable effect on the model performance.15 Descriptors such as MinAbsPartialCharge, Chiln, Chi2v and MolLogP were revealed as crucial factors for distinguishing hPXR molecules.21
3.3 Property distribution for PXR activators and non-activators
To explore the relevance of chemical properties to hPXR activators, the distributions of six physicochemical properties, including molecular weight, log of the octanol/water partition coefficient (MolLogP), number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds and topological polar surface area (TPSA) between PXR activators and non-activators were investigated. Student's t-test was used to evaluate the significance of differences between the means of PXR activators and non-activators for each property.As shown in Fig. 2, the six physicochemical properties are significantly different between the two classes. The mean molecular weights for hPXR activators and non-activators are 314 and 214, respectively, suggesting that hPXR activators tend to obtain larger molecular weights than non-activators. MolLogP is related to the hydrophobicity of a molecule. The mean values of MolLogP were 3.69 and 1.91 for hPXR activators and non-activators, respectively. Considering the higher MolLogP values for hPXR activators, the increase in the hydrophobicity of the chemicals contributes to the stronger interaction with the hydrophobic binding sites of PXR, increasing the chance for activation.8,16,20
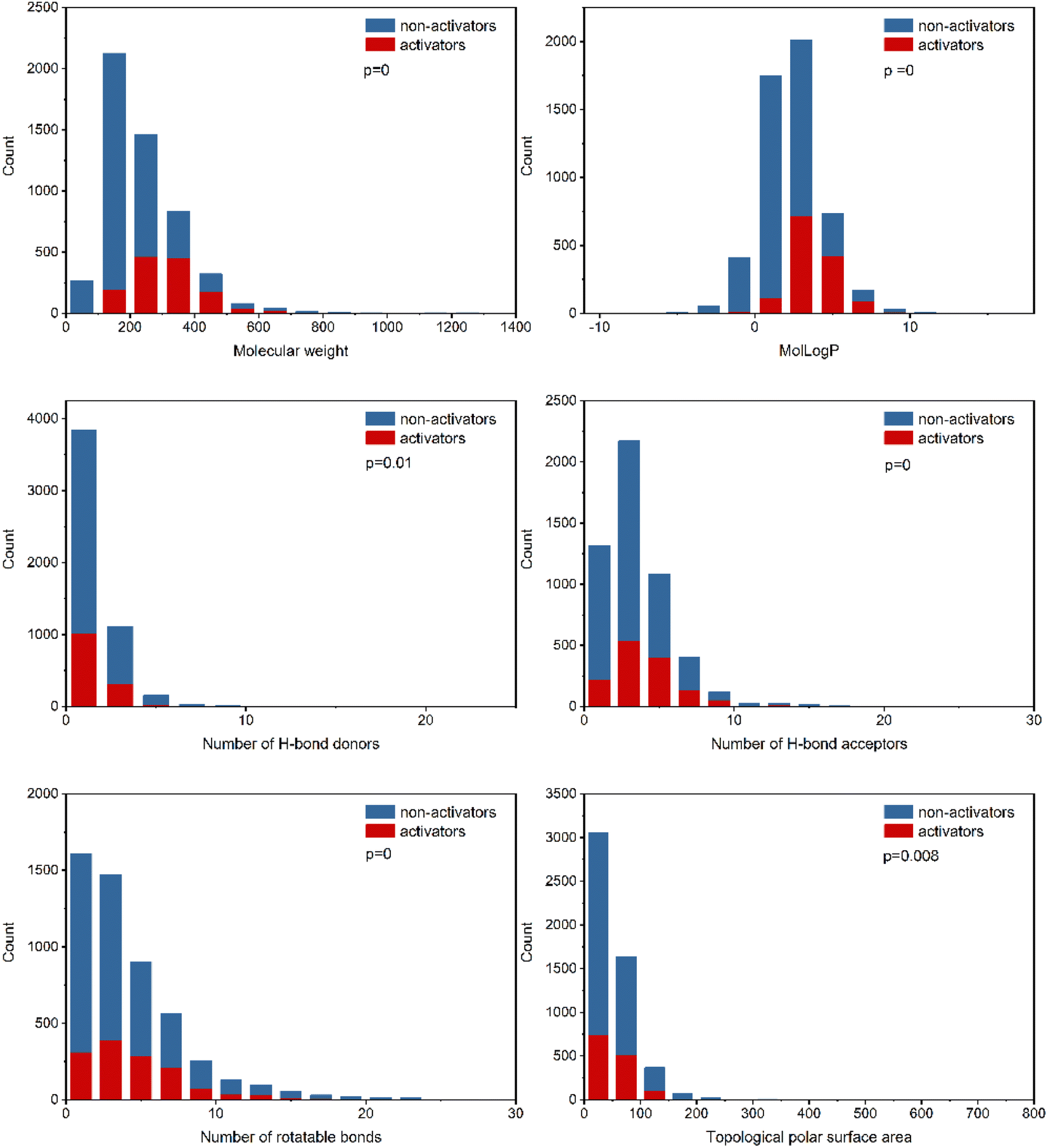 | ||
| Fig. 2 Distribution of six physicochemical descriptors (molecular weight, log of the octanol/water partition coefficient, number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds and topological polar surface area) for PXR activators and non-activators. Each bar indicates the number of positive activators (red) and non-activators (blue). | ||
The other four properties, the number of hydrogen bond acceptors, the number of hydrogen bond donors, the number of rotatable bonds, and the topological polar surface area (TPSA), represent the electrostatic or hydrogen bonding features of a molecule. It is observed that hPXR activators have a higher number of hydrogen bond acceptors than hydrogen bond donors, in line with the previously reported observations.8,16,19 Similar phenomena can be seen for TPSA and the number of rotatable bonds that hPXR activators have larger values than non-activators. The trends observed in these properties showed that hPXR activators tended to have relatively larger molecular weights and were more hydrophobic and structurally flexible. Compared with previous studies with the observation of no significant differences across all properties, despite the usage of the same physicochemical properties,19,20 it was indicated that the larger dataset contributes significantly to the difference in these six properties between hPXR activators and non-activators.
3.4 Comparison of the classification models through internal validation
A total of 45 models were built by five machine-learning methods combined with molecular descriptors and eight molecular fingerprints. 10 iterations of five-fold stratified cross-validation were conducted with the training set to assess the model robustness (Table S4†). Fig. S1† shows the performances of different algorithms and features. The BNB models executed the poorest results with any features across all the metrics compared with SVM, RF, XGBoost, and AdaBoost. The top 10 combinatorial classification models for the training set are listed in Table 2.| Methods | BA | Precision | Recall | F1 | AUC | CK | MCC | |
|---|---|---|---|---|---|---|---|---|
| a XGBoost: extreme gradient boosting. RF: random forest. AdaBoost: adaptive boosting. SVM: support vector machine. RDKitMD: molecular descriptors calculated by RDKit. Pub: PubChem fingerprints. Ext: extended fingerprints. MAC: MACCS keys. Day: daylight fingerprints. σ: standard error. BA: balanced accuracy. AUC: the area under receiver operating characteristic curve. CK: Cohen's Kappa. MCC: Matthews correlation coefficient. | ||||||||
| RDKitMD-XGBoost | Mean | 0.841 | 0.726 | 0.788 | 0.756 | 0.913 | 0.663 | 0.665 |
| σ | 0.01 | 0.02 | 0.02 | 0.02 | 0.01 | 0.03 | 0.02 | |
| RDKitMD-RF | Mean | 0.829 | 0.702 | 0.773 | 0.735 | 0.907 | 0.634 | 0.636 |
| σ | 0.01 | 0.03 | 0.03 | 0.02 | 0.01 | 0.03 | 0.03 | |
| RDKitMD-AdaBoost | Mean | 0.797 | 0.736 | 0.682 | 0.708 | 0.895 | 0.608 | 0.610 |
| σ | 0.02 | 0.03 | 0.03 | 0.02 | 0.01 | 0.03 | 0.03 | |
| Pub-XGBoost | Mean | 0.812 | 0.663 | 0.763 | 0.709 | 0.891 | 0.595 | 0.598 |
| σ | 0.01 | 0.02 | 0.03 | 0.02 | 0.01 | 0.03 | 0.03 | |
| Ext-XGBoost | Mean | 0.808 | 0.652 | 0.762 | 0.702 | 0.884 | 0.584 | 0.588 |
| σ | 0.01 | 0.02 | 0.02 | 0.02 | 0.01 | 0.03 | 0.03 | |
| Pub-SVM | Mean | 0.806 | 0.607 | 0.797 | 0.689 | 0.883 | 0.556 | 0.567 |
| σ | 0.01 | 0.02 | 0.02 | 0.02 | 0.01 | 0.03 | 0.03 | |
| Pub-RF | Mean | 0.795 | 0.656 | 0.727 | 0.689 | 0.882 | 0.570 | 0.572 |
| σ | 0.01 | 0.02 | 0.03 | 0.02 | 0.01 | 0.03 | 0.03 | |
| Ext-SVM | Mean | 0.811 | 0.612 | 0.804 | 0.694 | 0.881 | 0.564 | 0.575 |
| σ | 0.01 | 0.02 | 0.02 | 0.02 | 0.01 | 0.02 | 0.02 | |
| Day-XGBoost | Mean | 0.799 | 0.642 | 0.747 | 0.690 | 0.877 | 0.568 | 0.571 |
| σ | 0.01 | 0.02 | 0.03 | 0.02 | 0.01 | 0.02 | 0.02 | |
| MAC-XGBoost | Mean | 0.800 | 0.649 | 0.744 | 0.693 | 0.875 | 0.572 | 0.575 |
| σ | 0.01 | 0.02 | 0.03 | 0.02 | 0.01 | 0.02 | 0.02 | |
The comparison of the overall performances of various classification models showed that the XGBoost algorithms with molecular descriptors (RDKitMD-XGBoost) are the best combinatorial classification model in the internal validations. It has the highest AUC value of 0.913 and the highest BA of 0.841, suggesting its good capability for discriminating hPXR activators. The random forest algorithm with molecular descriptors has similar performance with the second highest AUC values of 0.907 and the second highest balanced accuracy of 0.829. The robustness of the model performance was assessed by calculating the standard deviations. As shown in Table 2, the standard deviations of the RDKitMD-XGBoost model are relatively small (0.01, 0.01 for AUC and BA, respectively), indicating the statistical robustness of the model to the training set.
3.5 Comparison of the classification models through external validation
To evaluate the generalization ability, the models constructed by the training set were validated using the external test set (Table S5†). Most models achieved good performances in the five-fold stratified cross-validation and have good predictive capabilities validated by external validation (Fig. 3). The top 10 combinatorial classification models for the external test set are listed in Table 3. The RDKitMD-XGBoost model yielded the best predictive performance, achieving the BA, AUC, precision, and recall values of 0.860, 0.860, 0.728, and 0.832, respectively. The F1 score, Cohen's Kappa and Matthews correlation coefficient are also highest in the external validation, indicating that the RDKitMD-XGBoost model has a relatively very high generalization ability.| Methods | BA | Precision | Recall | F1 | AUC | CK | MCC |
|---|---|---|---|---|---|---|---|
| a XGBoost: extreme gradient boosting. RF: random forest. AdaBoost: adaptive boosting. SVM: support vector machine. RDKitMD: molecular descriptors calculated by RDKit. Pub: PubChem fingerprints. Ext: extended fingerprints. MAC: MACCS keys. Day: daylight fingerprints. BA: balanced accuracy. AUC: the area under receiver operating characteristic curve. CK: Cohen's Kappa. MCC: Matthews correlation coefficient. | |||||||
| RDKitMD-XGBoost | 0.860 | 0.728 | 0.832 | 0.777 | 0.860 | 0.689 | 0.692 |
| RDKitMD-RF | 0.849 | 0.691 | 0.832 | 0.755 | 0.849 | 0.656 | 0.661 |
| Pub-XGBoost | 0.845 | 0.674 | 0.836 | 0.746 | 0.845 | 0.641 | 0.648 |
| Ext-XGBoost | 0.829 | 0.643 | 0.821 | 0.721 | 0.829 | 0.604 | 0.613 |
| Day-SVM | 0.829 | 0.611 | 0.854 | 0.712 | 0.829 | 0.584 | 0.602 |
| Ext-SVM | 0.827 | 0.606 | 0.854 | 0.709 | 0.827 | 0.578 | 0.587 |
| MAC-RF | 0.825 | 0.682 | 0.781 | 0.728 | 0.825 | 0.621 | 0.624 |
| Pub-SVM | 0.822 | 0.596 | 0.850 | 0.701 | 0.822 | 0.582 | 0.588 |
| Ext-RF | 0.814 | 0.628 | 0.799 | 0.703 | 0.814 | 0.578 | 0.587 |
| RDKitMD – AdaBoost | 0.813 | 0.728 | 0.723 | 0.725 | 0.813 | 0.627 | 0.627 |
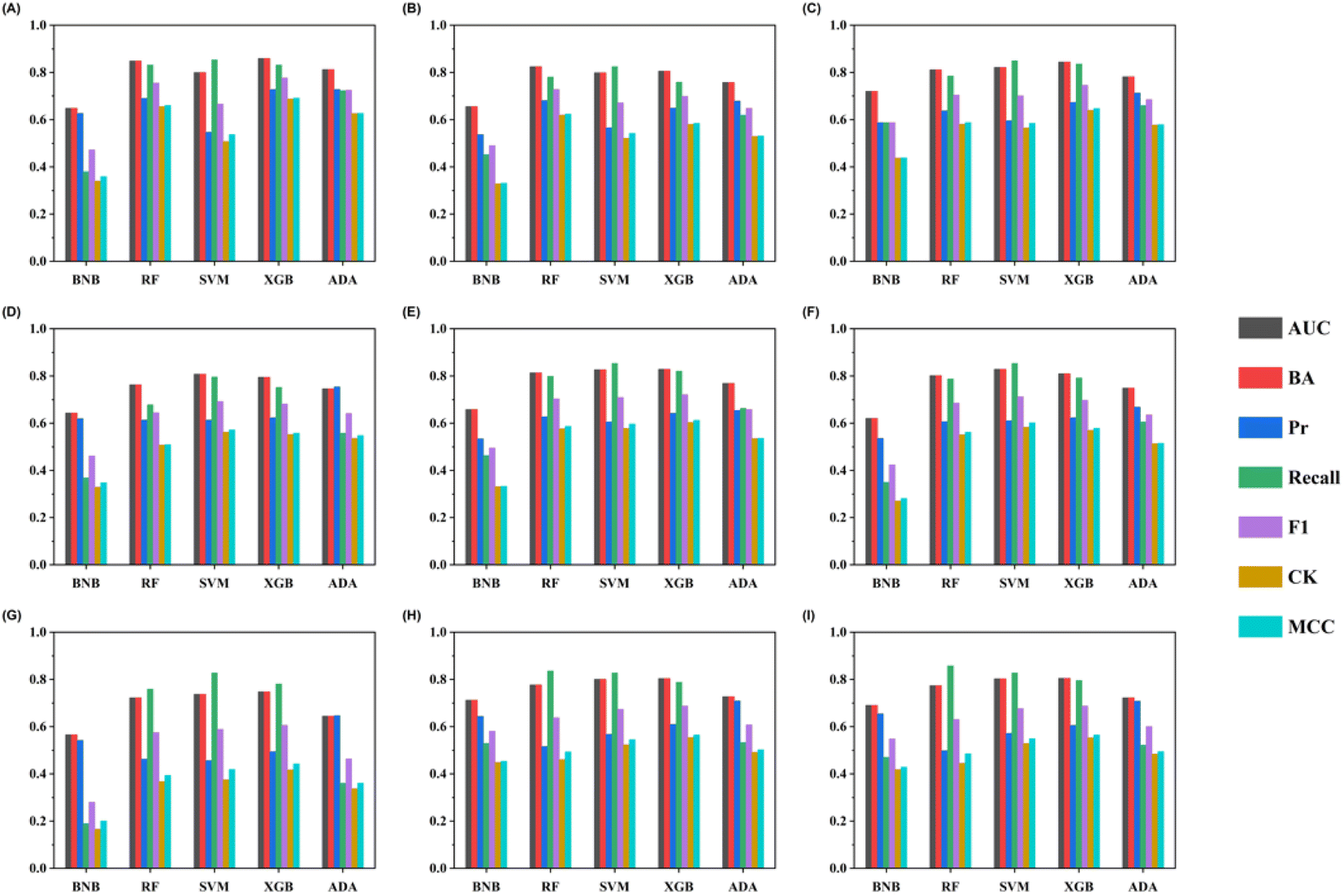 | ||
| Fig. 3 External validation performances of individual models constructed by five machine learning algorithms and nine molecular features. The sub-figures show the results using nine molecular features. The y-axis gives the performance values and different metrics are depicted by colors. Five machine learning algorithms are grouped and labeled at the x-axis. (A) RDKit molecular descriptors (B) MACCS fingerprint (C) PubChem fingerprint (D) KlekotaRoth fingerprint (E) CDK extended fingerprint (F) Daylight fingerprint (G) CDK graphonly fingerprint (H) Morgan (1024) fingerprint (I) Morgan (2048) fingerprint. | ||
The performances of the machine learning models are affected by both input features and chosen algorithms. RDKitMD-XGBoost performed the best in both the five-fold stratified cross-validations and external validation. The XGBoost algorithm was developed by minimizing the loss using a gradient descent algorithm. It adopts a sparse-aware splitting-finding approach to train more efficiently on sparse data, which is beneficial when input features are chemical descriptors (most entries are zero).37 Regularization options were further utilized to enhance the generalization ability of the model, contributing to the best performance of XGBoost-related models. A similar result was observed when comparing XGBoost, RF, and deep neural networks for toxicity prediction.38 The choice of molecular representation is also crucial to model development. In this study, RDKit descriptors outperform other fingerprints in most cases, which is potentially attributed to their low dimensionality and sparsity.
3.6 AD analysis
The Euclidean distance-based method was employed to analyze the applicability domain (Table S6†). According to this definition, the calculated threshold of AD was 0.22. Chemicals with a Euclidean distance exceeding 0.22 are considered outside the domain. The performances of the ten best combinatorial classification models for compounds within and outside AD are presented in Table 4. By applying AD, the predictive results for chemicals within AD were improved in comparison with those outside AD. For example, the metric AUC values for compounds within the domain were higher than those outside AD in the corresponding models, indicating that the AD can effectively isolate poor predictions. It should be noted that the recall for out-of-domain chemicals are higher than those within the domain. This can be attributed to the small number of active compounds in the out-of-domain chemicals. This observation confirmed that the use of AD succeeded in ruling out prediction errors, and thus enhanced the predictive performance of models.39| Models | ID | OD | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BA | Precision | Recall | F1 | AUC | CK | MCC | BA | Precision | Recall | F1 | AUC | CK | MCC | |
| RDKitMD-XGBoost | 0.869 | 0.758 | 0.802 | 0.779 | 0.869 | 0.722 | 0.722 | 0.832 | 0.708 | 0.856 | 0.775 | 0.832 | 0.634 | 0.642 |
| RDKitMD-RF | 0.852 | 0.729 | 0.777 | 0.752 | 0.852 | 0.687 | 0.688 | 0.819 | 0.667 | 0.876 | 0.757 | 0.819 | 0.595 | 0.611 |
| Pub-XGBoost | 0.862 | 0.688 | 0.818 | 0.747 | 0.862 | 0.677 | 0.681 | 0.808 | 0.663 | 0.850 | 0.745 | 0.808 | 0.578 | 0.591 |
| Ext-XGBoost | 0.864 | 0.680 | 0.826 | 0.746 | 0.864 | 0.674 | 0.680 | 0.770 | 0.616 | 0.817 | 0.702 | 0.770 | 0.503 | 0.517 |
| Day-SVM | 0.852 | 0.643 | 0.818 | 0.720 | 0.852 | 0.639 | 0.646 | 0.775 | 0.590 | 0.882 | 0.707 | 0.775 | 0.493 | 0.525 |
| Ext-SVM | 0.864 | 0.650 | 0.843 | 0.734 | 0.864 | 0.656 | 0.665 | 0.759 | 0.576 | 0.863 | 0.691 | 0.759 | 0.466 | 0.496 |
| MAC-RF | 0.864 | 0.697 | 0.818 | 0.753 | 0.864 | 0.684 | 0.688 | 0.775 | 0.669 | 0.752 | 0.708 | 0.775 | 0.534 | 0.537 |
| Pub-SVM | 0.845 | 0.628 | 0.810 | 0.708 | 0.845 | 0.622 | 0.630 | 0.764 | 0.574 | 0.882 | 0.696 | 0.764 | 0.470 | 0.506 |
| Ext-RF | 0.838 | 0.674 | 0.769 | 0.718 | 0.838 | 0.641 | 0.644 | 0.761 | 0.597 | 0.824 | 0.692 | 0.761 | 0.480 | 0.499 |
| RDKitMD – AdaBoost | 0.801 | 0.741 | 0.661 | 0.699 | 0.801 | 0.628 | 0.630 | 0.804 | 0.720 | 0.771 | 0.744 | 0.804 | 0.598 | 0.599 |
4. Conclusion
We built high throughput screening models for PXR activators using five machine learning algorithms with a combination of chemical descriptors or fingerprints as the training features. The classifier based on the XGBoost algorithm and RDKit descriptors showed the best robustness and prediction ability, achieving AUC values of 0.913 for the training set and 0.860 for the external test set. Our model showed improved robustness and generalization capabilities based on a large dataset, which can be used as a fast and reliable filter tool for the preliminary identification of PXR activators. Efforts are still needed to optimize the performances and promote the application through prospective screenings, further facilitating the risk assessment for potential PXR activators.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (No. 21876153, 22136001) and the Zhejiang Lab Open Research Project (K2022MF0AB01).References
- S. A. Kliewer, B. Goodwin and T. M. Willson, The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism, Endocr. Rev., 2002, 23(5), 687–702 CrossRef CAS PubMed.
- J. Yan and W. Xie, A brief history of the discovery of PXR and CAR as xenobiotic receptors, Acta Pharm. Sin. B, 2016, 6(5), 450–452 CrossRef.
- P. O. Oladimeji and T. Chen, PXR: more than just a master xenobiotic receptor, Mol. Pharmacol., 2018, 93(2), 119–127 CrossRef CAS PubMed.
- Y. Chen and D. Nie, Pregnane X receptor and its potential role in drug resistance in cancer treatment, Recent Pat. Anti-Canc., 2009, 4(1), 19–27 CAS.
- M. Dybdahl, N. G. Nikolov, E. B. Wedebye, S. Ó. Jónsdóttir and J. R. Niemelä, QSAR model for human pregnane X receptor (PXR) binding: screening of environmental chemicals and correlations with genotoxicity, endocrine disruption and teratogenicity, Toxicol. Appl. Pharmacol., 2012, 262(3), 301–309 CrossRef CAS.
- M. Banerjee, D. Robbins and T. Chen, Targeting xenobiotic receptors PXR and CAR in human diseases, Drug Discov. Today, 2015, 20(5), 618–628 CrossRef CAS.
- A. Hall, H. Chanteux, K. Ménochet, M. Ledecq and M. E. D. Schulze, Designing out PXR activity on drug discovery projects: a review of structure-based methods, empirical and computational approaches, J. Med. Chem., 2021, 64(10), 6413–6522 CrossRef CAS PubMed.
- S. Ekins and J. A. Erickson, A pharmacophore for human pregnane X receptor ligands, Drug Metab. Dispos., 2002, 30(1), 96–99 CrossRef CAS PubMed.
- D. Schuster and T. Langer, The identification of ligand features essential for PXR activation by pharmacophore modeling, J. Chem. Inf. Model., 2005, 45(2), 431–439 CrossRef CAS PubMed.
- G. Lemaire, C. Benod, V. Nahoum, A. Pillon, A. M. Boussioux, J. F. Guichou, G. Subra, J. M. Pascussi, W. Bourguet, A. Chavanieu and P. Balaguer, Discovery of a highly active ligand of human pregnane X receptor: a case study from pharmacophore modeling and virtual screening to “in vivo” biological activity, Mol. Pharmacol., 2007, 72(3), 572–581 CrossRef CAS.
- C. N. Chen, Y. H. Shih, Y. L. Ding and M. K. Leong, Predicting activation of the promiscuous human pregnane X receptor by pharmacophore ensemble/support vector machine approach, Chem. Res. Toxicol., 2011, 24(10), 1765–1778 Search PubMed.
- N. Torimoto-Katori, R. Huang, H. Kato, R. Ohashi and M. Xia, In silico prediction of hPXR activators using structure-based pharmacophore modeling, J. Pharm. Sci., 2017, 106(7), 1752–1759 CrossRef CAS.
- C. Yin, X. Yang, M. Wei and H. Liu, Predictive models for identifying the binding activity of structurally diverse chemicals to human pregnane X receptor, Environ. Sci. Pollut. Res. Int., 2017, 24(24), 20063–20071 CrossRef CAS.
- S. A. Rosenberg, M. Xia, R. Huang, N. G. Nikolov, E. B. Wedebye and M. Dybdahl, QSAR development and profiling of 72524 REACH substances for PXR activation and CYP3A4 induction, Comput. Toxicol., 2017, 1, 39–48 CrossRef.
- H. Matter, L. T. Anger, C. Giegerich, S. Güssregen, G. Hessler and K. H. Baringhaus, Development of in silico filters to predict activation of the pregnane X receptor (PXR) by structurally diverse drug-like molecules, Bioorg. Med. Chem., 2012, 20(18), 5352–5365 CrossRef CAS PubMed.
- C. Y. Ung, H. Li, C. W. Yap and Y. Z. Chen, In silico prediction of pregnane X receptor activators by machine learning approaches, Mol. Pharmacol., 2007, 71(1), 158–168 CrossRef CAS.
- A. Khandelwal, M. D. Krasowski, E. J. Reschly, M. W. Sinz, P. W. Swaan and S. Ekins, Machine learning methods and docking for predicting human pregnane X receptor activation, Chem. Res. Toxicol., 2008, 21(7), 1457–1467 Search PubMed.
- H. Rao, Y. Wang, X. Zeng, X. Wang, Y. Liu, J. Yin, H. He, F. Zhu and Z. Li, In silico identification of human pregnane X receptor activators from molecular descriptors by machine learning approaches, Chem. Res. Toxicol., 2012, 118, 271–279 Search PubMed.
- M. D. AbdulHameed, D. L. Ippolito and A. Wallqvist, Predicting rat and human pregnane X receptor activators using Bayesian classification models, Chem. Res. Toxicol., 2016, 29(10), 1729–1740 Search PubMed.
- H. Shi, S. Tian, Y. Li, D. Li, H. Yu, X. Zhen and T. Hou, Absorption, distribution, metabolism, excretion, and toxicity evaluation in drug discovery. 14. Prediction of human pregnane X receptor activators by using naive Bayesian classification technique, Chem. Res. Toxicol., 2015, 28(1), 116–125 Search PubMed.
- V. Rathod, V. Belekar, P. Garg and A. T. Sangamwar, Classification of human pregnane X receptor (hPXR) activators and non-activators by machine learning techniques: a multifaceted approach, Comb. Chem. High T. Scr., 2016, 19(4), 307–318 Search PubMed.
- S. Hirte, O. Burk, A. Tahir, M. Schwab, B. Windshügel and J. Kirchmair, Development and experimental validation of regularized machine learning models detecting new, structurally distinct activators of PXR, Cells, 2022, 11(8), 1253 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem in 2021: new data content and improved web interfaces, Nucleic Acids Res., 2021, 49(D1), 1388–1395 CrossRef.
- M. Lovrić, J. M. Molero and R. Kern, PySpark and RDKit: moving towards big data in cheminformatics, Mol. Inf., 2019, 38(6), e1800082 CrossRef.
- H. Ji, H. Deng, H. Lu and Z. Zhang, Predicting a molecular fingerprint from an electron ionization mass spectrum with deep neural networks, Anal. Chem., 2020, 92(13), 8649–8653 CrossRef CAS PubMed.
- P. M. Granitto, C. Furlanello, F. Biasioli and F. Gasperi, Recursive feature elimination with random forest for PTR-MS analysis of agroindustrial products, Chemom. Intell. Lab. Syst., 2006, 83(2), 83–90 CrossRef CAS.
- X. Xia, E. G. Maliski, P. Gallant and D. Rogers, Classification of kinase inhibitors using a Bayesian model, J. Med. Chem., 2004, 47(18), 4463–4470 CrossRef CAS PubMed.
- V. Svetnik, A. Liaw, C. Tong, J. C. Culberson, R. P. Sheridan and B. P. Feuston, Random forest: a classification and regression tool for compound classification and QSAR modeling, J. Chem. Inf. Comput. Sci., 2003, 43(6), 1947–1958 CrossRef CAS.
- C. Cortes and V. Vapnik, Support-vector networks, Mach. Learn., 1995, 20(3), 273–297 Search PubMed.
- Y. Freund and R. E. Schapire, A decision-theoretic generalization of on-line learning and an application to boosting, J. Comput. Syst. Sci., 1997, 55(1), 119–139 CrossRef.
- T. Chen and C. Guestrin, XGBoost: a scalable tree boosting system, 22nd ACM SIGKDD Int Conf, 2016, 785–794 Search PubMed.
- B. Zadrozny and C. Elkan, Obtaining calibrated probability estimates from decision trees and naive Bayesian classifiers, ICML, 2001, 609–616 Search PubMed.
- H. Dragos, M. Gilles and V. Alexandre, Predicting the predictability: a unified approach to the applicability domain problem of QSAR models, J. Chem. Inf. Model., 2009, 49(7), 1762–1776 CrossRef CAS PubMed.
- M. Shen, A. LeTiran, Y. Xiao, A. Golbraikh, H. Kohn and A. Tropsha, Quantitative structure–activity relationship analysis of functionalized amino acid anticonvulsant agents using k nearest neighbor and simulated annealing PLS methods, J. Med. Chem., 2002, 45(13), 2811–2823 CrossRef CAS PubMed.
- V. O. Gawriljuk, P. P. K. Zin, A. C. Puhl, K. M. Zorn, D. H. Foil, T. R. Lane, B. Hurst, T. A. Tavella, F. T. M. Costa, P. Lakshmanane, J. Bernatchez, A. S. Godoy, G. Oliva, J. L. Siqueira-Neto, P. B. Madrid and S. Ekins, Machine learning models identify inhibitors of SARS-CoV-2, J. Chem. Inf. Model., 2021, 61(9), 4224–4235 CrossRef CAS.
- Danishuddin and A. U. Khan, Descriptors and their selection methods in QSAR analysis: paradigm for drug design, Drug Discov. Today, 2016, 21(8), 1291–1302 CrossRef CAS PubMed.
- Z. Wu, M. Zhu, Y. Kang, E. L. Leung, T. Lei, C. Shen, D. Jiang, Z. Wang, D. Cao and T. Hou, Do we need different machine learning algorithms for QSAR modeling? A comprehensive assessment of 16 machine learning algorithms on 14 QSAR data sets, Brief. Bioinform., 2021, 22(4), bbaa321 CrossRef.
- R. P. Sheridan, W. M. Wang, A. Liaw, J. Ma and E. M. Gifford, Extreme gradient boosting as a method for quantitative structure–activity relationships, J. Chem. Inf. Model., 2016, 56(12), 2353–2360 CrossRef CAS PubMed.
- Z. Wang, J. Chen and H. Hong, Developing QSAR models with defined applicability domains on PPARγ binding affinity using large data sets and machine learning algorithms, Environ. Sci. Technol., 2021, 55(10), 6857–6866 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details of model evaluation metrics, tables regarding the brief definitions of 87 selected descriptors, the features employed for model development, the optimal hyperparameters of the five algorithms, five-fold stratified cross-validation performances of combinatorial models, external validation performances of combinatorial models, the performance in the domain and out of domain chemicals in the external test set for the top ten combinatorial classification models, figure regarding five-fold stratified cross-validation performances of individual models (PDF). See DOI: https://doi.org/10.1039/d2va00182a |
| This journal is © The Royal Society of Chemistry 2023 |