
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest-controlled polymorphism and exceptionally marked bi-stability in a spin crossover 3D porous amino-functionalized coordination polymer†
Alejandro
Orellana-Silla
a,
Manuel
Meneses-Sánchez
a,
Rubén
Turo-Cortés
 a,
Víctor
Rubio-Giménez‡
b,
Giel
Arnauts‡
b,
M. Carmen
Muñoz
c,
Rob
Ameloot
b,
Carlos
Bartual-Murgui
*a and
José Antonio
Real
*a
a,
Víctor
Rubio-Giménez‡
b,
Giel
Arnauts‡
b,
M. Carmen
Muñoz
c,
Rob
Ameloot
b,
Carlos
Bartual-Murgui
*a and
José Antonio
Real
*a
aDepartamento de Química Inorgánica, Instituto de Ciencia Molecular (ICMol), Universitat de València, Catedrático José Beltrán Martínez, 2, 46980 Paterna, Spain. E-mail: carlos.bartual@uv.es; jose.a.real@uv.es
bCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy (cMACS), Katholieke Universiteit Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
cDepartamento de Física Aplicada, Universitat Politècnica de València, Camino de Vera s/n, 46022 Valencia, Spain
First published on 19th September 2023
Abstract
Functionalization of spin crossover (SCO) porous coordination polymers (PCPs) with hydrogen-bonding active groups is an efficient strategy for the study of multifunctional materials. Indeed, the interaction of guests with the host framework may improve the cooperativity of the SCO inducing eventual occurrence of bi-stability. However, comprehensive magneto-structural studies are still needed to understand the role of guests in the enhancement of the SCO properties. Here, we report the 3D Hofmann-type PCP {FeII(dpyan)[AuI(CN)2]2}·guest (dpyan = 2,5-di(pyridin-4-yl)aniline; guest = ethanol (1·EtOH), methanol (1·MeOH), nitrobenzene (1·NO2Bz), water/methanol (1·H2O·MeOH)) where the amino functionalized dpyan ligand has been selected to induce host–guest interactions. Our X-ray diffraction studies reveal that the packing mode of 1·guest is determined by the trapped guest yielding either orthorhombic (1·NO2Bz and 1·H2O·MeOH) or monoclinic (1·EtOH and 1·MeOH) phases differing by subtle distortions of the framework. The thermal dependence of the magnetic susceptibility shows that all derivatives present SCO properties modulated by the amount and nature of the encapsulated guest as well as by the crystal phase. The latter is clearly evidenced for the orthorhombic derivatives which, concomitantly with partial desorption of guest, transform into the monoclinic phase whose SCO behaviour is radically different to that of the original orthorhombic structure. As a relevant example, when 1·NO2Bz (orthorhombic) converts into 1·0.77NO2Bz (monoclinic), the SCO drastically changes from a non-hysteretic gradual spin transition to a cooperative SCO with a colossal hysteresis 105 K wide. These remarkable SCO properties have been interpreted relying on the obtained structural information.
Introduction
The spin crossover (SCO) phenomenon refers to the property exhibited by some octahedrally coordinated 3d4–3d7 transition metals of switching between two electronic configurations, as determined by the distribution of the valence electrons within the t2g and eg orbitals.1 A paradigmatic example of this molecular event involves the FeII ion (3d6) which, in an appropriate hexacoordinated coordination sphere (usually of the type FeIIN6), can be controllably, reversibly, and detectably interconverted between the low spin (LS, e6g, S = 0, diamagnetic) and the high spin (HS, e4gt22g, S = 2, paramagnetic) states through the application of external stimuli.2 Importantly, resulting from their different electronic configurations, the interconversion between both states involves changes in the structure, and magnetic, optical and dielectric properties of the complex. This coupling between external perturbation and physical property modification places the SCO materials as promising candidates for the fabrication of spintronic devices.3In the solid state, the change of the spin state is governed by the electron–phonon coupling between the switching metallic centres. Therefore, their degree of interaction will determine the efficiency with which the spin change is transmitted. Considering a thermally induced SCO, when the metallic centres are strongly bonded (e.g. by coordination and/or covalent bonds), the system will exhibit high cooperativity featuring an abrupt change of the physical properties. Indeed, this thermally-driven cooperative SCO can occasionally exhibit a hysteresis effect which confers bi-stability (i.e. memory effect)4 to the material, a very sought after property for applications in solid-state memories and actuators.5–9
A quite appealing archetypal structure for obtaining SCO compounds with highly cooperative and hysteretic profiles are the 3D Hofmann-type porous coordination polymers (PCPs) with general formula {FeII(L)[MI(CN)2]2}·guest (M = Au, Ag).10 In this structure, formed by doubly interpenetrated networks, the FeII ions are strongly connected, equatorially, through linear dicyanometallate units and, axially, by organic L molecules (typically bis-monodentate pyridine-type ligands). A very interesting feature presented by this type of compounds is that the SCO properties are affected by host–guest interactions which in turn may be tunned through the rational selection of specific guests and the functionalization of the L pillar ligands. Indeed, overall, the presence of a guest generally improves the cooperativity of the spin transitions inducing the occurrence of thermal hysteresis.11–17 However, although it is well stablished that host–guest interactions and guest-induced internal pressure are behind the emergence of cooperative and hysteretic effects,18–22 the lack of structural information prevents a deeper understanding of bi-stability. In this context, the occurrence of polymorphism can eventually provide insight about the magneto-structural correlations since it permits to directly compare the SCO properties of different structural phases of a given compound.23
Here, we present the 3D Hofmann-type PCP {FeII(dpyan)[AuI(CN)2]2}·guest (1·guest, dpyan = 2,5-di(pyridin-4-yl)aniline) whose amino functionalized pillaring dpyan ligand (Scheme 1) has been intentionally selected to promote the occurrence of host–guest interactions. Indeed, this compound crystallizes with different guest molecules depending on the solvents used during the synthesis (guest = ethanol (1·EtOH), methanol (1·MeOH), nitrobenzene (1·NO2Bz), water/methanol (1·H2O·MeOH)). Our structural data have revealed that the packing mode of 1·guest is determined by the nature of the trapped guest yielding either orthorhombic (1·NO2Bz and 1·H2O·MeOH) or monoclinic (1·EtOH and 1·MeOH) phases through subtle distortions of the framework. Although the thermal dependence of the magnetic susceptibility shows that all derivatives present spin transitions, it has been found that their characteristics are governed by both the network packing and the nature of the trapped guest. This is clearly showcased for the orthorhombic derivatives which, concomitantly to the partial desorption of guest, transform into the monoclinic phase whose SCO behaviour is radically different to that of the original orthorhombic structure. As a relevant example, when 1·NO2Bz (orthorhombic) converts into 1·0.77NO2Bz (monoclinic), the thermally induced switching properties drastically change from a gradual non-hysteretic spin transition to a cooperative SCO displaying a colossal hysteresis width of 105 K. These remarkable SCO properties have been interpreted by relying on the obtained structural information.
 | ||
| Scheme 1 Structure of ligand dpyan (2,5-di(pyridin-4-yl)aniline) used in this work. | ||
Results
Synthesis and chemical characterization
All compounds were obtained as similar yellow plate-shaped single crystals through slow liquid–liquid diffusions of Fe(BF4)2·6H2O (or FeCl2·4H2O), dpyan and K[Au(CN)2] solutions giving rise, depending on the used solvents, to different solvates of compound {Fe(dpyan)[Au(CN)2]2}·guest (1·guest) (see Experimental Section in ESI† for the synthesis details). Whereas single crystals of 1·MeOH and 1·EtOH are produced in pure methanol and ethanol solutions, respectively, crystals of 1·H2O·MeOH are obtained in mixed water/methanol solutions. Besides, crystals of the compound 1·NO2Bz are achieved in a nitrobenzene/methanol diffusion medium.The rapid loss of crystallinity observed for the as-synthesized crystals once removed from the mother liquor suggests a spontaneous desorption of the guest solvents. However, the thermogravimetric analysis (TGA), carried out for each solvate, indicates that a certain amount of guest is retained after exposing the crystals to air. Indeed, the resulting TGAs are consistent with the presence of ca. 0.5, 1.5, and 1 molecule of methanol, ethanol and nitrobenzene per FeII ion which are released within the 310–370 K (1·MeOH), 345–410 (1·EtOH), and 390–550 K (1·NO2Bz) temperature range (Fig. S1, ESI†), respectively. For 1·H2O·MeOH, we performed mass spectrometry (MS) coupled with TGA to estimate if the remaining molecules of guest are either water or methanol. The results reveal a 2% of mass loss within the 300–400 K temperature range where the mass spectrum signals at 17 and 31 m/z confirm the retention of both water and methanol (Fig. S2, ESI†). This mass loss is far from the 5% expected for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (H2O:MeOH) stoichiometry inferred by crystallography (see below) indicating a clear partial solvent loss. When the same sample is measured after several weeks exposed to air, the recorded mass loss increases up to 3%. This fact, together with the observed larger relative amount of water with respect to methanol suggests a progressive adsorption of water from the humidity of air.
:1 (H2O:MeOH) stoichiometry inferred by crystallography (see below) indicating a clear partial solvent loss. When the same sample is measured after several weeks exposed to air, the recorded mass loss increases up to 3%. This fact, together with the observed larger relative amount of water with respect to methanol suggests a progressive adsorption of water from the humidity of air.
Given the lability of the guests in compounds 1·MeOH, 1·EtOH and 1·H2O·MeOH, the solvent free counterpart 1·ø can be easily obtained through a mild thermal treatment at 400 K for 10 minutes. In contrast, because of its higher temperature of desorption, the guest molecules of nitrobenzene are only partially removed at 400 K. Thus, as observed by TGA, different samples with varying nitrobenzene contents (1·0.77NO2Bz, 1·0.60NO2Bz and 1·0.34NO2Bz) were obtained after heating at 400 K during 20, 120 and 480 minutes, respectively, as verified by TGA (Fig. S3, ESI†). Treating the sample at a higher temperature (473 K) for 1 h was required to obtain the solvent free 1·ø compound from 1·NO2Bz.
Aiming at investigating the susceptibility to re-adsorb water from the air moisture, a TGA was carried out on two samples of 1·ø which were previously exposed to air overnight and for three days (Fig. S4, ESI†), respectively. The results indicate the loss of around 0.6 and 1.5% of mass, within the 300–400 K temperature range consistent with a gradual recovering of 0.3 and 0.7 molecules of water per FeII ion, respectively.
Single crystal X-ray diffraction
The structures of 1·NO2Bz, 1·H2O·MeOH and 1·EtOH were determined at different temperatures (see below) through single crystal X-ray diffraction (SCRXD). Tables S1–S4 (ESI†) contain the crystallographic data including selected distances and angles of each compound. 1·NO2Bz and 1·H2O·MeOH are isomorphous (orthorhombic phase) and their structures will be described together. In contrast, the ethanol solvate presents a monoclinic phase and its structure will be described in a separated section. To prevent the desorption of the guest molecules and the consequent deterioration of the crystallinity, the crystals were directly transferred from the mother liquor, protected with perfluoropolyether cryo-oil, rapidly mounted on a cryoloop and placed in the diffractometer chamber below room temperature (260 K). Despite many attempts, the structure of 1·MeOH, which is isostructural to 1·EtOH (see below) could not be solved due to the rapid loss of crystallinity even surrounded by the oil. Similarly, the structure of 1·ø, also isostructural to 1·EtOH, was not achieved due to the collapse of the single crystals after desorption of the guests. | ||
| Fig. 1 Fragment of the crystal structures of (a) 1·NO2Bz and (b) 1·H2O·MeOH showing the corresponding atom numbering of the asymmetric unit cells. | ||
Both equatorial [Au(CN)2]− and axial dpyan units act as bridging ditopic ligands, thereby forming an extended 3D network (Fig. 2a). The high porosity exhibited by this network permits the interpenetration of an identical network stabilized by strong aurophilic interactions [Au⋯Au = 3.122 Å (1·NO2Bz) and 3.154 Å (1·H2O·MeOH) at 250 and 260 K, respectively], in such a manner that the apical dpyan ligands of one of them pass through the centroid of the {Fe4[Au(CN)2]4} windows of the other (Fig. 2b and c). Despite the interpenetration, there is still enough void space to host guest molecules. In the case of 1·H2O·MeOH, the position of the methanol and water guests are clearly identified in the pores (Fig. 2d). The methanol molecule, located in the vicinity of the NH2 group, is stabilized through a strong H2N⋯HOCH3 hydrogen bond [(d(N⋯O) = 2.655 Å)]. Besides, the methanol molecule interacts with one of water (OMeOH⋯Owater = 3.143 Å), hence forming molecular chains stabilized through much weaker H-bonds that parallelly spread along the 1D channels of the network. In contrast, the nitrobenzene molecules in 1·NO2Bz are too disordered to identify their exact location. However, the electronic density found within the pores suggests that the position of the guests is not equivalent to that found for methanol and water in 1·H2O·MeOH. Instead, the electronic residue of the nitrobenzene molecules is homogenously distributed within the 1D pores rather than situated in the proximity of the NH2 groups as observed in 1·H2O·MeOH (Fig. S5, ESI†). This is likely the reason for the lack of NH2 groups pointing at the central part of the pores of 1·NO2Bz, as this space is required to accommodate the larger aromatic guest. Along the same lines, and as a consequence of the above mentioned Au1–N1–Fe1 angle differences, the acute Au⋯Fe⋯Au angle (ϕ) in the {Fe4[Au(CN)2]4} pseudo-square windows is slightly larger for 1·NO2Bz (78.8°, see Fig. 2c) than for 1·H2O·MeOH (71.6°). This effect is also related with the more regular arrangement of the network in 1·NO2Bz required to encapsulate the bulkier molecule of nitrobenzene. Similarly, the absence of disorder in the pyridine of 1·NO2Bz seems to be associated with the blocking action caused by the trapped nitrobenzene molecules. To estimate the amount of included nitrobenzene, an electron mask was applied in the structure of 1·NO2Bz determining the presence of 1 molecule of nitrobenzene per FeII ion, which is consistent with the TGA measurements (Fig. S1, ESI†).
 | ||
| Fig. 2 (a) Perspective view of one of the two interpenetrated 3D networks of 1·H2O·MeOH. (b) Side and (c) top view showing the two interpenetrated 3D structures with the Au⋯Au (green-red dashed lines) internetwork interactions (the equivalent figures of 1·NO2Bz are at first glance indistinguible from those of 1·H2O·MeOH). (d) Perspective view of 1·H2O·MeOH showing the host–guest and guest–guest interactions between the water, methanol and the host framework. | ||
When cooling to 120 K, the crystals change their colour to red, but maintaining both the orthorhombic Cmma space group and the general structure described at 250/260 K. However, the observed 〈FeII–N〉av values are 1.955 Å (1·NO2Bz) and 1.966 Å (1·H2O·MeOH) which correspond well with that expected for the FeII in the LS state configuration and, therefore, indicate the occurrence complete HS-to-LS transitions. Furthermore, the aurophilic interactions become stronger [Au⋯Au = 3.064 (1·NO2Bz) Å and 3.109 Å (1·H2O·MeOH)] as well as the ϕ angles increase to 81.6 (1·NO2Bz) and 75.2° (1·H2O·MeOH).
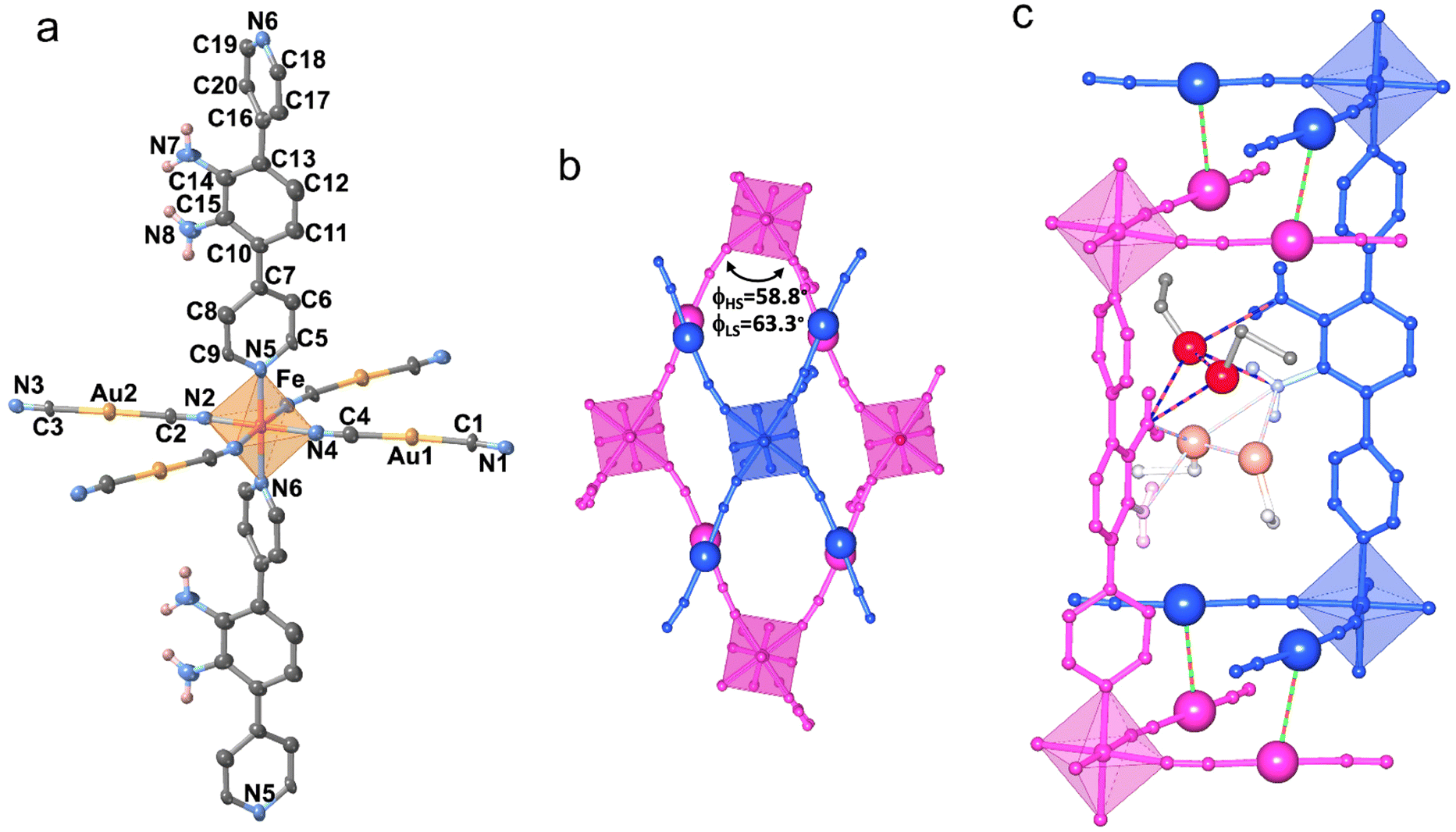 | ||
| Fig. 3 (a) Octahedral environment of compound 1·EtOH showing the atom labeling of the asymmetric unit. Corresponding (b) up and (c) side view of the interpenetration mode of 1·EtOH displaying the ethanol molecules accommodated througout the 1D cavities. Green-red and blue-red dashed lines represent the host–host and host–guest interactions, respectively. | ||
When cooling down to 120 K, the crystal of 1·EtOH, which changes its colour to red, shows the same monoclinic P2/c space group with a similar structure as that described at 200 K. However, the 〈Fe–N〉av (1.947 Å) is 0.220 Å shorter than at 200 K, reflecting a complete HS-to-LS transition upon cooling. Additionally, the aurophilic interactions become slightly shorter upon SCO (Au1⋯Au1 = 3.278 Å and Au2⋯Au2 = 3.234 Å) and the ϕ angle increases to 63.3°.
Powder X-ray diffraction (PXRD)
PXRD was carried out to verify the crystal phase purity of the bulk samples and to detect eventual crystallographic transformations accompanying the guest desorption processes. This technique has also been used to provide structural information for 1·MeOH and 1·ø whose structures could not be obtained through SCXRD.The diffraction patterns of all samples were recorded for the air-dried crystalline powders and for their fully desolvated counterparts (Fig. 4 and Scheme 2). Moreover, the diffraction patterns of solvated samples (1·MeOH, 1·EtOH and 1·H2O·MeOH) were also obtained whilst immersed in their mother liquor (ML) to avoid the loss of their volatile guests. Since the nitrobenzene molecules are not desorbed at room temperature, the compound 1·NO2Bz was directly analysed after being air-dried.
 | ||
| Fig. 4 PXRD patterns of solvates 1·guest [guest = MeOH and EtOH (a); NO2Bz and H2O·MeOH (b)] immersed in the mother liquor (ML), as air-dried samples and upon desolvation (or partial desolvation) after thermal treatment (1·ø). The simulated patterns (grey plots) of 1·EtOH (a) and 1·H2O·MeOH and 1·NO2Bz (b) in the HS are also depicted for comparison. Orthorhombic and monoclinic phases are highlighted in black and blue plots, respectively. The solvent in parentheses indicates the parent solvate. (c) Evolution with temperature of the PXRD pattern of 1·NO2Bz and d) the corresponding enlarged 2θ range between 15 and 22°. | ||
 | ||
| Scheme 2 Synthesis, desolvation processes and crystallographic transformations of all solvates reported in this work. *According to TGA, y and z values are lower than 1. | ||
For 1·EtOH, ML and air-dried diffractograms are very similar to that simulated from the SCXRD data. This observation reveals that both the as-synthesized and the air-dried crystals of 1·EtOH display exclusively the same monoclinic phase described above for 1·EtOH. Moreover, the diffraction pattern remains practically intact after heating the sample to fully desorb the ethanol molecules, indicating that the empty 1·ø retains the monoclinic structure of its parent solvate 1·EtOH (Fig. 4a). A similar situation is observed for 1·MeOH (Fig. 4a) whose ML and air-dried diffraction patterns are essentially the same and coincides with that of 1·EtOH, demonstrating that both solvates are isostructural. Indeed, the same pattern can be obtained after heating either 1·EtOH or 1·MeOH, the latter merely producing a slightly less crystalline material.
Contrarily to 1·EtOH and 1·MeOH, compounds 1·H2O·MeOH and 1·NO2Bz exhibit important structural changes induced by the loss of guests (Fig. 4b). As expected, the diffraction patterns of 1·H2O·MeOH (ML) and 1·NO2Bz (air-dried) are similar and consistent with those simulated from the orthorhombic structures obtained through SCXRD. However, in the case of air-dried 1·H2O·MeOH, the spontaneous partial loss of solvent is accompanied by a drastic modification of the diffraction pattern from the orthorhombic to the monoclinic phase. The subsequent heating of the sample does not significantly modify the diffraction pattern and retains the monoclinic phase to yield 1·ø.
Besides, when heating 1·NO2Bz at 400 K under air for 20 minutes, part of the nitrobenzene molecules desorbs yielding compound 1·0.77NO2Bz. The resulting diffraction pattern is that of the monoclinic phase showing that the slight loss of nitrobenzene provokes a crystal transformation from the orthorhombic to the monoclinic phase. Heating a fresh sample of 1·NO2Bz at 473 K under air for 1 h also yields the monoclinic compound 1·ø.
Aiming at studying in detail this heating-induced orthorhombic→monoclinic phase change, we performed temperature-dependent in situ PXRD of 1·NO2Bz. Measurements were carried out under dynamic vacuum (see experimental details in ESI†) to obtain equivalent conditions to SQUID magnetometry (see below). As visible in Fig. 4c and d (enlarged scale), the orthorhombic diffraction pattern is maintained in the 300–340 K temperature range. In contrast, when heating above 340 K, a mixture of phases appears, showing an evolution from the orthorhombic to the monoclinic pattern. This phase change is completed within 370–380 K when the diffraction pattern of the pure monoclinic form is obtained. At this point, we determined the amount of retained nitrobenzene in this sample to be 0.58 molecules per FeII (Fig. S8, ESI†) via ex situ TGA. This confirms that the crystallographic phase change is coupled to the loss of guest which in turn is accelerated by vacuum.
Furthermore, it is worth noting that the transition from the orthorhombic to the monoclinic form, upon 1·H2O·MeOH→1·ø and 1·NO2Bz→1·ø transformations, is fully irreversible. Attempts of re-establishing the orthorhombic phase by immersing 1·ø in pure nitrobenzene or water/methanol solutions for a week did not recover the original orthorhombic form as the monoclinic phase was retained instead (Fig. S9, ESI†).
Spin crossover properties
Thermally induced spin crossover (SCO) properties were analysed for representative crystalline powder samples of the corresponding solvates. The SCO was studied with a SQUID magnetometer by measuring the χMT product at variable temperature with a scan rate of 2 K min−1 (χM is the molar magnetic susceptibility and T is the temperature. See the Experimental Section in the ESI† for more details). Given the lability of the guests in compounds 1·MeOH, 1·EtOH and 1·H2O·MeOH and aiming to investigate the guest effect on the SCO behaviour, the samples were measured first, in the ML, then, after air-dried and, finally, after desorbing the samples at 400 K. Due to the stability of the nitrobenzene guests in 1·NO2Bz at room temperature, this sample was measured first in its air-dried state, later, partially desolvated at 400 K and finally fully desolvated at 473 K.When the crystals of 1·MeOH and 1·EtOH are removed from the ML and dried under ambient conditions they give rise to the χMT vs. T plots displayed in Fig. 5a and b (green dotted lines). 1·MeOH undergoes a noticeable decrease of the hysteresis width from 61 K (T↓1/2 = 169 K, T↑1/2 = 230 K) to 32 K (T↓1/2 = 200 K, T↑1/2 = 232 K). Besides, the SCO behaviour of 1·EtOH displays a switch from two-step to a single-step SCO accompanied by an enlargement of the hysteresis loop from 9 to 19 K (T↓1/2 = 168 K, T↑1/2 = 187 K) and a decrease of T1/2 by ca. 35 K. Interestingly, the measured thermal variation of the unit cell volume upon SCO of a single crystal of 1·EtOH, is parallel to the corresponding χMT vs. T plot for the air-dried sample (see blue and red circles in Fig. 5b). This indicates that the measured crystal has already experienced partial ethanol desorption upon SCXRD measurement.
 | ||
| Fig. 5 χ M T vs. T plots of compounds (a) 1·MeOH and (b) 1·EtOH measured in their ML (black plots), as air-dried samples (green plots) and after desolvation (dashed grey plots). The red (heating) and blue (cooling) plots in figure (b) represent thermal variation of the unit cell volume of a crystal of 1·EtOH. The solvent in parentheses indicates the parent solvate. | ||
Finally, the SCO properties resulting from the fully desolvation of 1·MeOH and 1·EtOH [1·ø(MeOH) and 1·ø(EtOH), respectively] are very similar. They exhibit complete spin transitions with T↓1/2/T↑1/2 equal to 180/189 K [1·ø (MeOH)] and 175/183 K [1·ø (EtOH)] featuring hysteresis widths of 9 and 8 K, respectively.
 | ||
| Fig. 6 (a) χMT vs. T plots of 1·H2O·MeOH measured in their ML (black plot) and as freshly (green plot)/aged (grey plot) air-dried samples. (b) χMT vs. T plots of 1·NO2Bz measured as air-dried sample (black plot) and upon removing partially (green plot) and completely (dashed grey plot) the nitrobenzene guest. (c) χMT vs. T behaviour of 1·xNO2Bz (monoclinic phase) with decreasing amounts of nitrobenzene. (d) Evolution of the χMT vs. T behaviour of a sample of 1·NO2Bz transforming from orthorhombic to monoclinic upon heating in situ within different thermal cycles. Note that in (a) and (b) compounds 1·H2O·MeOH and 1·NO2Bz change from the orthorhombic to the monoclinic phase when moving from the ML to the air-dried sample and from the air-dried sample to the desorbed sample, respectively. Figures (a) and (b) also depict the thermally induced unit cell volume change registered for the crystals of 1·H2O·MeOH and 1·NO2Bz, respectively (red and blue plots represent the cooling and heating modes). | ||
Once exposed to air, compound 1·H2O·MeOH experiences a partial solvent desorption concomitantly with an orthorhombic-to-monoclinic phase transformation as evidenced by TGA (Fig. S2, ESI†) and PRXD (Fig. 4b). As a result, the crystals of 1·H2O·MeOH undergo a marked increase of the hysteresis width up to 70 K (T↓1/2 = 175 K, T↑1/2 = 245 K, see grey dotted line curve in Fig. 6a). However, this large asymmetric hysteresis, recorded immediately after removing the crystals from the ML, is not stable over time and evolves to a less cooperative SCO behaviour at lower temperatures [i.e. T↓1/2 = 152 K/T↑1/2 = 172 K after 1 day (Fig. S10, ESI†) and T↓1/2 = 150 K/T↑1/2 = 161 K after 1 week (light grey dotted line in Fig. 6a)].
Ex situ thermal treatment at 400 K for 20 minutes of compound 1·NO2Bz triggers partial nitrobenzene desorption (1·NO2Bz → 1·0.77NO2Bz) and concomitant orthorhombic-to-monoclinic crystallographic phase change (Fig. S3, ESI† and Fig. 4). Analogously to 1·H2O·MeOH, the crystallographic phase change involves the aperture of an outstanding thermal hysteresis (105 K, T↓1/2 = 134 K, T↑1/2 = 240 K) (Fig. 6b). However, due to the stability of the nitrobenzene guest in the structure, in this case, it is stable over time if stored at room temperature (Fig. S11, ESI†). Additionally, the cycle remains unaltered when measuring at different temperature scan rates (Fig. S12, ESI†). More vigorous ex situ thermal treatments and subsequent higher release of nitrobenzene provokes the narrowing of the hysteresis loop width which is 72 K for 1·0.60NO2Bz (T↓1/2 = 152 K, T↑1/2 = 224 K, heating at 400 K for 120 min) and 8 K for 1·0.32NO2Bz (T↓1/2 = 155 K, T↑1/2 = 163 K, heating at 400 K for 20 min) (Fig. 6c).
Aiming at recording the intermediate SCO behaviours evolving from the orthorhombic (gradual without hysteresis) to the monoclinic (cooperative with large hysteresis) forms, a fresh sample of 1·NO2Bz was progressively heated in situ in the SQUID chamber at increasing temperatures (350, 370 and 380 K) and subsequent χMT vs. T cycles from 100 to 270 K were recorded after each heating treatment. The results (Fig. 6d) indicate that while the magnetic behaviour is practically not changed upon heating at 350 K (red dotted line), the SCO is clearly modified after heating at 370 K (orange dotted line) opening a hysteresis up to 25 K wide with T↓1/2 = 225 K and T↑1/2 = 250 K. Further heating treatments at 280 K provokes a progressive widening of the hysteresis loop (up to 91 K; T↓1/2 = 140 K; T↑1/2 = 231 K) displaying in the cooling mode a gradual evolution from a double to a single step. The TGA carried out on the resulting solvate after the thermal treatments reveals the presence of 0.72 molecules of nitrobenzene (Fig. S13, ESI†).
Finally, the complete desorption of solvent in 1·H2O·MeOH (20 min at 400 K) and 1·NO2Bz (1 h at 470 K) yields 1·ø (H2O·MeOH) and 1·ø (NO2Bz) which exhibit complete spin transitions with T↓1/2/T↑1/2 equal to 187/197 K (Fig. S12, ESI†) and 161/180 K (Fig. 6b) and hysteresis widths of 10 and 19 K, respectively.
In order to prove the reversibility of the SCO properties throughout the 1·guest(as synthesized) → 1ø → 1·guest(re-adsorbed) sequence, a sample of 1·ø(MeOH) was immersed in methanol, ethanol or nitrobenzene solutions and measured in suspension. Successive thermal cycles were carried out until stabilization of the SCO properties upon guest adsorption. In all cases, the resulting SCO behaviours are quite different to those of the as synthesized compounds (Fig. S15, ESI†). The χMT vs. T plots of the resolvated compounds 1·NO2Bz and 1·EtOH display non-hysteretic gradual and incomplete spin transitions with T1/2 values below that of the desolvated compounds [155 K (1·NO2Bz); 172 K (1·EtOH)]. In contrast, in the case of 1·MeOH, the SCO shifts towards higher temperatures showing T↓1/2/T↑1/2 of 219 and 226 K and 5 K of hysteresis.
Discussion
In this work, we have explored the use of an amino functionalized organic ligand (dpyan) to assemble the 3D porous Hofmann-type FeII-based coordination polymer {Fe(dpyan)[Au(CN)2]2}·guest (1·guest), in order to combine synergistic host–guest and spin crossover properties. Depending on the solvent used during the synthesis, two types of polymorphic forms are predictably adopted by 1·guest. When the guest is nitrobenzene (1·NO2Bz) or water/methanol (1·H2O·MeOH) we obtain an orthorhombic phase whereas when it is methanol (1·MeOH) or ethanol (1·EtOH) we observe a monoclinic structure. Although both polymorphs are characterized by a comparable double interpenetrated network, they present differences in the flexion degrees of the rhombic {Fe(Au(CN2)2} grids which are more (monoclinic) or less (orthorhombic) distorted as a function of the nature of the trapped guest.From a structural point of view, it seems that the stability of one polymorph versus the other is determined by the competition between the host–guest and host–host interactions. Thus, in the orthorhombic 1·NO2Bz and 1·H2O·MeOH species, the interactions stablished between the disordered guests and the main network controls the stabilization of an orthorhombic high symmetry structure even if this conformation disables the establishment of significant host–host interactions other than the aurophilic ones. In contrast, in the monoclinic 1·MeOH and 1·EtOH compounds, the alcohol guests are highly oriented and occupy specific positions. These guest locations are compatible with the deformation and loss of symmetry of the framework which allows the attainment of concomitant host–guest and host–host π⋯π interactions together with the Au⋯Au contacts. Although, the latter become significantly weaker. In the absence of guest (1·ø), the driving force of the structural conformation relays essentially in the host–host π⋯π interactions which lead to the monoclinic form. Importantly, the stabilization of the monoclinic phase is irreversible since the transformation back to the orthorhombic phase induced by the re-adsorption of nitrobenzene or water/methanol is not produced. It turns out that the energy required to revert the π⋯π interactions stablished between the interpenetrated networks in the monoclinic phase cannot be compensated by the entry of guest. The higher stability of the monoclinic versus the orthorhombic phase is also evidenced by the orthorhombic-to-monoclinic transformation observed for the as-synthesised 1·NO2Bz and 1·H2O·MeOH upon the partial desorption (even a small amount) of guest.
The SCO properties exhibited by the solvates 1·guest reflect a marked dependence on the nature of the adsorbed guest which is explained by the sensitivity that the -NH2 confers to the host framework. However, while a clear guest-dependence of the SCO is observed for the monoclinic polymorph, the magneto-structural data reveals a poor SCO guest dependence for the orthorhombic one. Indeed, the orthorhombic forms of 1·NO2Bz and 1·H2O·MeOH present very similar gradual SCO behaviours without meaningful modifications when passing from nitrobenzene to H2O·MeOH (Fig. 6a and b, black curves). It appears that, in this case, the resulting SCO profiles are rather determined by the polymorph packing which lacks π⋯π interactions affording a bad communication between the FeII centres. Besides, the difference of size between nitrobenzene and water/methanol guests does not result in a substantial T1/2 modification indicating the absence of significant steric effects.
In contrast, the guest induced wide range modulation of the SCO in the monoclinic polymorph is remarkable. The most notable example is indeed observed for 1·NO2Bz. When 1·NO2Bz is gently heated, a small fraction of nitrobenzene is released (1·NO2Bz → 1·0.77NO2Bz) and, concomitantly, the system undergoes a crystallographic change from the orthorhombic to the monoclinic phase. As a result, the SCO properties change drastically moving from a very gradual 2 K wide hysteresis to a cooperative 105 K wide hysteresis. This value is only 5 K narrower than that of [FeII(1,2,3-triazolate)2]24 which exhibits, for the best of our knowledge, the widest hysteresis for a 3D SCO polymeric complex.
The suggested mechanism explaining the marked effect of the crystallographic phase change on the SCO properties is based on the intrinsic structural characteristics of the involved polymorphs and their respective interactions with the nitrobenzene guest. The exceptionally low T↓1/2 value observed for the monoclinic 1·0.77NO2Bz compound can be attributable to steric factors which would gain importance with respect to that observed for the orthorhombic form. This, in turn, can be explained by the considerable decrease of the calculated accessible void space (in the HS state) when moving from the orthorhombic (868.0 Å3) to the monoclinic (627.9 Å3) phase. However, once the HS → LS transition has occurred, the LS state may be stabilized through electronic factors induced by directional H-bond interactions between the nitrobenzene guests and the NH2 groups. This suggested mechanism for the occurrence of hysteresis is indeed consistent with the SCO behaviours observed after successive losses of nitrobenzene. Thus, when decreasing the amount of trapped nitrobenzene, both steric and electronic effects diminish yielding, as a result, hysteresis loops with increasing T↓1/2 and decreasing T↑1/2 and thereby with decreasing hysteresis widths. Similar conclusions of the source of bi-stability were reported by Konar et al. describing the increase of hysteresis width displayed by a 3D compound when substituting the guest molecules of acetonitrile by stilbene.25
Indeed, the same mechanism may explain the SCO behaviour observed for compound 1·H2O·MeOH and 1·MeOH. The former exhibits a 70 K hysteresis wide appearing upon the orthorhombic-to-monoclinic crystallographic change. However, in this case, the hysteresis width decreases with time due to the progressive spontaneous loss of guest. The latter shows, in the mother liquor, a hysteresis 61 K wide which narrows to 32 K upon exposing to air, also prompted by a methanol loss. Thus, although 1·H2O·MeOH and 1·MeOH exhibit SCO behaviours with analogue characteristics to that of 1·0.77NO2Bz, the higher volume and capacity to generate host–guest interactions of nitrobenzene versus methanol or water account for the more pronounced bi-stability of the latter.
The ability of the desolvated compounds 1·ø to readsorb guest molecules is manisfested in the modification of the SCO properties upon guest re-uptake (Fig. S15, ESI†). These guest re-adsorptions, performed by preparing a suspension of 1·ø in the different solvents, do not culminate in the reestablishment of the SCO properties of the original as-synthesized samples. For example, the re-adsorption of nitrobenzene and ethanol leads to very gradual and incomplete spin transitions without hysteresis which contrast with the hysteretic and cooperative plots of the original 1·EtOH and 1·0.77NO2Bz monoclinic compounds. This observation can be attributed to the mechanism of diffusion of the ethanol and nitrobenzene molecules into the host framework channels (Scheme 3). Due to the bulky volume of these guests, the diffusion may be slow filling and saturating the structural channels from the surface as the guest is being adsorbed. This guest saturation, which may hinder the SCO, is not produced in the genuine synthesis of the solvates which naturally crystallize with a homogeneous quantity of guest in the pores with a closed structure that avoids the entry of extra solvent.
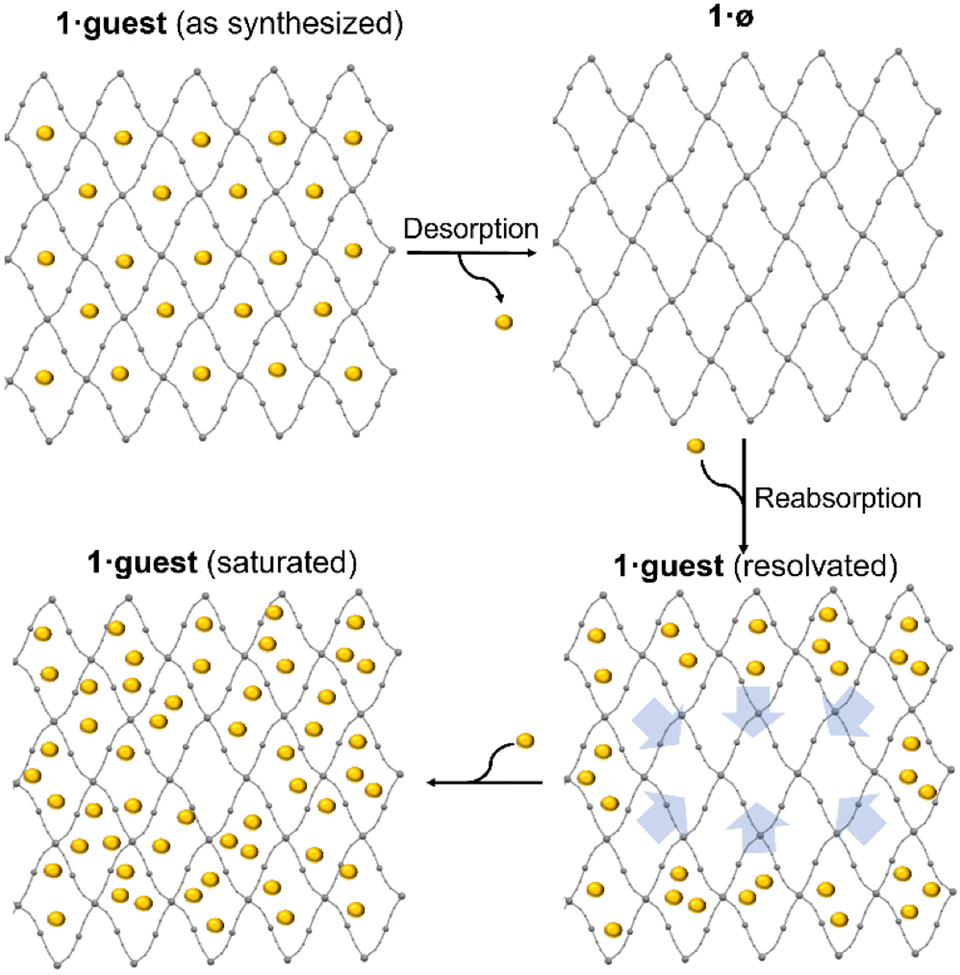 | ||
| Scheme 3 Suggested mechanism for the desorption and re-adsorption of guest leading to saturated pores. | ||
Besides, whereas the re-adsorption of methanol increases notably the T1/2 with respect to 1·ø, it slightly decreases the cooperativity of the SCO while maintaining a hysteresis of 8 K. Similarly, the adsorption of water also decreases the cooperativity conserving a hysteresis of 8 K although at lower temperatures (Fig. S14, ESI†). Similar loss of cooperativity has been observed in related 2D Hofmann networks and explained by elastic frustration within the crystal lattice upon guest loading.26,27 It turns out that, likely due to the lower volume of water and methanol, the steric effects are less marked than with ethanol and nitrobenzene and indeed, they are combined with electronic effects affecting notably to the spin crossover temperature. Further studies of the re-adsorption process with solvents of different nature, functional groups and size would be desirable to explore in detail the guest effect of the title compound.
Conclusions
In summary, we have investigated the dynamic host–guest interplay in a doubly interpenetrated porous 3D Hofmann-type coordination polymer functionalized with amino groups. The compound features a primitive cubic topology stemming from self-assembling the [Au(CN)2]− and the 2,5-di(pyridin-4-yl)aniline (dpyan) bis-monodentate rod-like ligands around the FeII node. During the crystal growth, two different polymorphs have been isolated depending on the nature of the guests included in the pores: one orthorhombic and another monoclinic. The former, displaying a reasonably regular framework, is metastable and strongly distorts like a flexible wine rack structural motif, to give a denser structure in the monoclinic form. This irreversible transformation is essentially triggered by a threshold of spontaneous or induced removal of guests from the pores. Interestingly, this structural change involves a drastic modification of the SCO properties moving from gradual without hysteresis to cooperative with very large hysteresis spin transitions. We associate these dramatic SCO variations upon phase change to the increase of the guest induced steric effects and to an enhancement of the host–guest interactions delaying, respectively, the HS → LS and the LS → HS transitions, giving rise therefore to wide regions of bi-stability. Besides, the irreversibility of the orthorhombic to monoclinic transition nicely illustrates that, during the crystal growth, the guests act as templates to afford the metastable form. However, once in the monoclinic desolvated form, the distortion of the framework, stabilized by closer interactions between the interpenetrated frameworks, does not allow the solvents reverting the system to the orthorhombic form. Indeed, due to the inhomogeneous guest distribution, the SCO properties of the resulting re-solvated monoclinic are clearly different to the as-synthesized compounds. The results reported in this work give insight about reaching the optimal pathways for the design of SCO bi-stable PCPs.Author contributions
Alejandro Orellana-Silla: investigation, methodology, writing – review & editing; Manuel Meneses-Sánchez: investigation, writing – review & editing; Rubén Turo-Cortés: investigation, methodology, writing review & editing; Víctor Rubio-Giménez: investigation, writing-review & editing; Giel Arnauts: investigation, writing – review & editing; M. Carmen Muñoz: supervision, writing – review & editing, funding acquisition; Rob Ameloot: supervision, writing – review & editing, funding acquisition Carlos Bartual-Murgui: supervision, writing, original draft, investigation; J. Antonio Real: funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by: Grant PID2019-106147GB-I00 funded by MCIN/AEI/10.13039/501100011033) and Unidad de Excelencia María de Maeztu (CEX2019-000919-M). A. O. S. thanks MCIN for the predoctoral (FPI) grant PRE2020-092798. R. T.-C. and M. M.-S. thank MCIN and FSE invierte en tu futuro for the predoctoral (FPI) grants PRE2018-084918 and BES2017-081393, respectively. V. R.-G. and G. A. thank the Research Foundation Flanders (FWO Vlaanderen) for a Junior Postdoctoral Fellowship (1263622N) a PhD fellowship (1SA7320N), respectively.References
- Spin crossover in transition metal compounds I-III, Top. Curr. Chem., ed. P. Gütlich and G. Goodwin, 2004, vol. 233–235 Search PubMed.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Eng., 1994, 33, 2024–2054 CrossRef.
- C. Lefter, V. Davesne, L. Salmon, G. Molnár, P. Demont, A. Rotaru, A. Bousseksou, C. Lefter, V. Davesne, L. Salmon, G. Molnár, P. Demont, A. Rotaru and A. Bousseksou, Magnetochemistry, 2016, 2, 18 CrossRef.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- O. Kahn and J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- T. K. Ekanayaka, G. Hao, A. Mosey, A. S. Dale, X. Jiang, A. J. Yost, K. R. Sapkota, G. T. Wang, J. Zhang, A. T. N’Diaye, A. Marshall, R. Cheng, A. Naeemi, X. Xu and P. A. Dowben, Magnetochemistry, 2021, 7, 3, DOI:10.3390/magnetochemistry7030037.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8(3), 194–197 CrossRef CAS PubMed.
- T. Miyamachi, M. Gruber, V. Davesne, M. Bowen, S. Boukari, L. Joly, F. Scheurer, G. Rogez, T. K. Yamada, P. Ohresser, E. Beaurepaire and W. Wulfhekel, Nat. Commun., 2012, 3, 938 CrossRef PubMed.
- M. Piedrahita-Bello, L. Salmon, G. Molnar, P. Demont, B. Martin and A. Bousseksou, J. Mater. Chem. C, 2020, 8, 6042–6051 RSC.
- M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068–2093 CrossRef.
- W. Liu, Y.-Y. Peng, S.-G. Wu, Y.-C. Chen, M. N. Hoque, Z.-P. Ni, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2017, 56, 14982–14986 CrossRef CAS PubMed.
- K. Yoshida, D. Akahoshi, T. Kawasaki, T. Saito and T. Kitazawa, Polyhedron, 2013, 66, 252–256 CrossRef CAS.
- J. E. Clements, J. R. Price, S. M. Neville and C. J. Kepert, Angew. Chem., Int. Ed., 2016, 55, 15105–15109 CrossRef CAS PubMed.
- J.-Y. Li, Z. Yan, Z.-P. Ni, Z.-M. Zhang, Y.-C. Chen, W. Liu and M.-L. Tong, Inorg. Chem., 2014, 53, 4039–4046 CrossRef CAS PubMed.
- J.-Y. Li, C.-T. He, Y.-C. Chen, Z.-M. Zhang, W. Liu, Z.-P. Ni and M.-L. Tong, J. Mater. Chem. C, 2015, 3, 7830–7835 RSC.
- J. Y. Li, Y. C. Chen, Z. M. Zhang, W. Liu, Z. P. Ni and M. L. Tong, Chem. – Eur. J., 2015, 21, 1645–1651 CrossRef CAS PubMed.
- A. Martinez-Martinez, E. Resines-Urien, N. S. Settineri, S. J. Teat, E. C. Sañudo, O. Fabelo, J. A. Rodriguez-Velamazan, L. Piñeiro-López and J. S. Costa, Cryst. Growth Des., 2023, 23(6), 3952–3957 CrossRef CAS.
- Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- R. Ohtani and S. Hayami, Chem. – Eur. J., 2017, 23(10), 2236–2248 CrossRef CAS PubMed.
- M. Nakaya, R. Ohtani and S. Hayami, Eur. J. Inorg. Chem., 2020,(39), 3709–3719 CrossRef CAS.
- Y. Xie, R.-B. Lin and B. Chen, Adv. Sci., 2022, 9(1), 2104234 CrossRef CAS PubMed.
- E. Resines-Urien, E. Fernandez-Bartolome, A. Martinez-Martinez, A. Gamonal, L. Piñeiro-López and J. S. Costa, Chem. Soc. Rev., 2023, 52(2), 705–727 RSC.
- J. Tao, R. J. Wei, R. Bin Huang and L. S. Zheng, Chem. Soc. Rev., 2012, 41, 703–737 RSC.
- M. Grzywa, R. Röb-Ohlenroth, C. Muschielok, H. Oberhofer, A. Błachowski, J. Żukrowski, D. Vieweg, H.-A. K. von Nidda and D. Volkmer, Inorg. Chem., 2020, 59, 10501–10511 CrossRef CAS PubMed.
- D. J. Mondal, B. Kumar, A. Paul and S. Konar, J. Mater. Chem. C, 2023, 11, 6750–6759 RSC.
- K. A. Zenere, S. G. Duyker, E. Trzop, E. Collet, B. Chan, P. W. Doheny, C. J. Kepert and S. M. Neville, Chem. Sci., 2018, 9, 5623–5629 RSC.
- D. J. Mondal, A. Mondal, A. Paul and S. Konar, Inorg. Chem., 2022, 61, 4572–4580 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of samples, experimental and instrumental details. CCDC 2277962–2277967. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc02363j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |