
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tuneable gold nanostars: the role of adenosine monophosphate†
Carlos
Fernández-Lodeiro
 ab,
Javier
Fernández-Lodeiro
*cd,
Adrián
Fernández-Lodeiro
cd,
Silvia
Nuti
cd,
Carlos
Lodeiro
cd,
Alec
LaGrow
e,
Ignacio
Pérez-Juste
b,
Jorge
Pérez-Juste
*ab and
Isabel
Pastoriza-Santos
*ab
ab,
Javier
Fernández-Lodeiro
*cd,
Adrián
Fernández-Lodeiro
cd,
Silvia
Nuti
cd,
Carlos
Lodeiro
cd,
Alec
LaGrow
e,
Ignacio
Pérez-Juste
b,
Jorge
Pérez-Juste
*ab and
Isabel
Pastoriza-Santos
*ab
aCINBIO, Universidade de Vigo, Campus Universitario As Lagoas, Marcosende, 36310 Vigo, Spain
bDepartamento de Química Física, Universidade de Vigo, Campus Universitario As Lagoas, Marcosende, 36310 Vigo, Spain. E-mail: juste@uvigo.gal; pastoriza@uvigo.gal
cBIOSCOPE Group, LAQV@REQUIMTE, Chemistry Department, Faculty of Science and Technology, University NOVA of Lisbon, Caparica Campus, 2829-516 Caparica, Portugal
dPROTEOMASS Scientific Society, Rua dos Inventores, Madam Parque, Caparica Campus, 2829-516 Caparica, Portugal
eInternational Iberian Nanotechnology Laboratory, Braga, 4715-330, Portugal
First published on 2nd August 2023
Abstract
The seed-mediated growth of gold nanostructures is known to be strongly dependent not only on the gold seed nanocrystal structure but also on the presence of different additives that may influence the morphology, and therefore the crystalline structure of the final nanoparticle. Among the different additives or capping ligands, biomolecules are an interesting family due to their potential biomedical applications such as drug delivery, bioimaging, biosensing, phototherapy, and antimicrobial activities. Here, we develop a seed-mediated strategy for synthesizing uniform Au nanostars with tuneable optical properties which involves adenosine monophosphate (AMP) as a capping ligand. The experimental data reveal the key role of AMP not just providing colloidal stability and directing the reduction of the gold precursor via complexation but also mediating the anisotropic growth of the Au seeds via its selective adsorption on the different crystalline facets of Au nanoparticles. These observations agree with theoretical simulations carried out using molecular dynamics and density functional theory (DFT) calculations. Interestingly, the obtained Au nanostars showed high thermal stability as well as colloidal stability in polar organic solvents, which allowed their direct silica coating via the Stöber method. Importantly, we also explored the mimic enzymatic activity of the resulting gold nanostars and observed a superior catalytic activity compared with other gold nanoparticles reported in the literature.
Introduction
The morphology of Au nanoparticles is strongly conditioned by the presence of capping ligands during synthesis. In a seed-mediated growth process, the capping ligands may interact with specific crystalline facets of the seed or even complex with metal precursors. Therefore, apart from providing colloidal stability, they can strongly affect the reduction of the metal precursors or/and alter nanoparticle growth.1 Among the large list of capping ligands, biological molecules are rather appealing when we deal with biomedical applications such as drug delivery, bioimaging, biosensing, phototherapy, and antimicrobial activities. The use of different biomolecules, such as DNA, amino acids and peptides, as capping ligands to mediate the growth of Au nanoparticles has already been reported. DNA can absorb or covalently bind to metal nanoparticles and direct their growth into different morphologies.2,3 Interestingly, chiral amino acids and peptides can induce the dissymmetric growth of Au nanoparticles to obtain chiral nanostructures with unique optical responses.4,5 Nevertheless, a lack of systematic study and an understanding of the effect of a biomolecule on the NP growth are often observed.6Star-like gold nanoparticles (AuNSs) are perhaps the most studied anisotropic gold geometry at the nanometer scale after gold nanorods. Their tunable optical response in the visible-near infrared (Vis-NIR) region, dictated by the core size and number/length of branches, and the strong light confinement at their tips7 have fostered interest in these nanoparticles in a wide range of applications that include surface-enhanced spectroscopies,8,9 photonics,10 cancer therapy11 and (photo)catalysis.12 A myriad of protocols based on colloidal chemistry has been developed to obtain AuNSs, with seed-mediated growth approaches allowing for greater uniformity and better modulation of optical properties. In this regard, the features of the metal nanoparticles employed as seeds are a key factor in modulating the number of tips or overall dimensions of AuNSs without inducing noticeable changes in their optical response.13 For example, smaller seeds lead to AuNSs with fewer tips deriving into more narrow optical properties in comparison with larger seeds.13 Although multi-twinned seeds are most commonly used to promote the formation of nanostars, single crystalline seeds can also be employed by evolving them to NPs with twinned planes.
In the AuNS synthesis, polymers, surfactants, or discrete organic molecules were also reported as shape-directing agents. One of the first and widely used methods is based on the growth of tiny seeds using polyvinylpyrrolidone (PVP) as a reducing and capping agent and dimethylformamide (DMF) as a solvent.13,14 This strategy results in uniform nanostars with high yields and defined and tunable optical properties. Unfortunately, this method implies the use of DMF (toxic organic solvent) and PVP which may make the subsequent functionalization of the nanoparticles difficult due to the lack of control of the PVP absorbed on the nanoparticle surface.15 Water-soluble polymers, such as poly(allylamine hydrochloride) (PAH),16 pyromellitic dianhydride-p-phenylene diamine (PPDDs),17 and polystyrene sulfonate (PSS),18 have been recently reported for the synthesis of AuNSs in water using seed-mediated or seedless approaches, although in some cases the control in the optical properties is limited. Some surfactants can also direct the growth of AuNSs, such as quaternary ammonium surfactants,19,20 Triton X,21 sodium dodecyl sulfate (SDS),22 lauryl sulfobetaine (LSB)23 and dioctyl sodium sulfosuccinate (AOT). For instance, it is hypothesized that cetyltrimethylammonium bromide (CTAB) preferentially adsorbs on the {100} gold seed facets.24 However, the presence of twin defects in the seed will weaken the binding of the surfactant and result in branch growth instead of symmetrical elongation, inducing the anisotropic growth via the preferential deposition on the {111} facet.25 Nonetheless, surfactants are often highly cytotoxic or not environmentally friendly (especially in the case of CTAB or benzyldimethylhexadecylammonium chloride (BDAC)).
As an alternative to polymers or surfactants, discrete molecules such as hydroquinone (HQ), bis(p-sulfonatephenyl)phenylphosphine dihydrate dipotassium (BSPP) and the Good's buffers family (e.g. EPPS, MOPS, and HEPES) have also been employed for the synthesis of AuNSs.26–30 The latter is probably the most commonly used for the seedless synthesis of AuNSs which importantly does not require an additional surfactant; therefore, it is of interest due to its NP biocompatibility and ease to functionalize. The limitations of this strategy are that it requires the tight control of different synthetic factors, the colloidal stability, and the presence of other Au NPs as subproducts with some Good's buffers. Finally, biomolecules (albumin,31 gelatin protein,32L-DOPA,33 and tryptophan–glutaraldehyde (Trp–GA)34) have also been employed for AuNS synthesis although the reported results show broad plasmonic responses or limited plasmon tuning. Overall, different synthetic protocols can be classified into seedless or seed-mediated. Two main parameters could be identified as critical: (1) the reducing agent that allows the kinetically controlled growth and (2) the capping ligand that stabilizes the NP and may direct its anisotropic growth through the selective adsorption and/or blocking of certain crystalline facets. Thus, the development of new strategies for AuNS synthesis requires a more in-depth analysis of these key parameters. Table S1 (ESI†) summarizes different synthesis protocols of AuNSs and their main properties.
Therefore, the development of new synthetic strategies for AuNSs employing biomolecules that offer tuneability of plasmon response and with high colloidal stability remains a challenge. Nevertheless, it will facilitate the application of AuNSs in the medical area. In this scenario, nucleotides are interesting capping ligands for the synthesis of metallic NPs35–40 since they are composed of a nitrogenous base that provides affinity for the Au surface,41,42 a five-carbon sugar (ribose or deoxyribose), and at least one phosphate group which provides colloid stability.43 For instance, Wang et al. reported the use of a single-stranded DNA (ssDNA) consisting of homogenous oligonucleotides or mixed-base oligonucleotides to control the shape and surface roughness of Au NPs using Au plates as seeds.3 The resulting NPs had similar shapes regardless of the DNA length. Besides, the effect of the corresponding oligodeoxyribonucleotides was studied showing similar behavior. But, the use of mononucleotides led to particles with poor colloidal stability. Recently, Cho et al. showed that adenine-based ssDNA could be used as a chiral shape modifier to synthesize Au NPs with a chiral morphology.44 In all cases, the nucleobase-dependent morphology evolution can be correlated with the selective interaction at the Au–ssDNA surface. Nonetheless, there is a lack of an in-depth analysis of the effect of mononucleotides in the nanoparticle growth following seed-mediated approaches. Adenosine monophosphate (AMP) is a nucleotide consisting of an adenine base linked to a ribose sugar with a phosphate group. Interestingly, it has been reported that adenine could bind to Au(III) species through the N7 site of the purine ring forming Au(III)–AMP complexes. The high stability of such complexes has been ascribed to hydrogen bonding interactions and proton transfers.45 In addition, AMP has been reported to adsorb on noble metal surfaces through either a nitrogen ring and/or the external NH2 of the purine ring.41 Both the complexation ability of the adenine moiety towards gold salt and the adsorption capabilities to metal surfaces make AMP an interesting molecule with a dual role in the AuNP synthesis. That is, AMP as a complexing agent can affect the reduction kinetics of the metal precursor and as a capping agent can provide colloidal stability and mediate the growth of Au nanoparticles.
Therefore, herein we investigated the use of AMP in the synthesis of AuNSs using a seed-mediated method. The role of AMP as a complexing and capping ligand was deeply analyzed and the experimental data were supported by computational studies employing molecular dynamics and density functional theory (DFT) calculations. Besides, the effect of other parameters such as the concentration and size of Au seeds was studied. Next, we evaluated the behavior of AuNSs in polar organic solvents and under heating (100 °C) as well as the direct silica coating. The silica coating will enhance their colloidal, chemical, and thermal stabilities, facilitating their manipulation and integration into sensors, photovoltaic cells, catalysts, and other functional devices.46 Finally, the capabilities of the synthesized Au nanostars as peroxidase-mimicking nanozymes were studied. Nanozymes, defined as nanomaterials with enzyme-like characteristics, have emerged to overcome the intrinsic limitations of natural enzymes (such as high cost or low stability) and broaden their potential applications in sensing, imaging, and therapy among others.47,48 Particularly, the oxidation of uncolored 3,3′,5,5′-tetramethylbenzidine (TMB) into blue oxidized TMB (ox-TMB) in the presence of H2O2 has been employed as a model reaction.
Experimental
Materials
Adenosine 5′-monophosphate sodium salt (AMP, ≥99%), sodium citrate tribasic dihydrate (≥98%), sodium borohydride (NaBH4 ≥99), 3,3′,5,5′-tetramethylbenzidine (TMB, ≥99%), acetic acid (100.5%), L-ascorbic acid (≥99%), dimethyl sulfoxide (DMSO, ≥99.5), sodium hydroxide (NaOH, ≥97%), and tetraethyl orthosilicate (TEOS, ≥98%) were purchased from Sigma-Aldrich. Tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 99.99%) and hydrogen peroxide (H2O2, 33%) were supplied by Alfa Aesar. Absolute ethanol (EtOH, ≥99.9%) was purchased from Scharlau. Ammonia solution (NH3, 35%) was purchased from Fisher Chemical. All chemicals were used as received and ultra-pure water (type I) was used in all the preparations.Characterization
UV-Visible extinction spectra were recorded using a Cary 5000 or an Agilent 8453 spectrophotometer. Low-resolution transmission electron microscopy (TEM) images were obtained using a JEOL JEM 1010 transmission electron microscope operating at an acceleration voltage of 100 kV. Aberration-corrected transmission electron microscopy was carried out using an Image and Probe-Corrected FEI Titan Themis microscope at 60–300 kV, operating at 200 kV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and electron tomography series were acquired using a Probe-Corrected FEI Titan G2 80–200 kV ChemiSTEM operating at 200 kV, with a convergence angle of 8 mrad and a tilt range of ±70°. ζ Potential was determined using a Zetasizer Nano S (Malvern Instruments, Malvern, UK) at 22 °C in standard 1 mL polystyrene cuvettes.Computational methods
The initial gas phase conformational search of adenosine monophosphate (AMP) has been performed using the GMMX method included in the PCModel program.49 These conformations with relative energies below 10 kcal mol−1 have been further optimized employing DFT methods with the M062X functional and the 6-311+G* basis set and characterized as energy minima by the computation of their harmonic vibrational frequencies using Gaussian16.For studying the adsorption of AMP on different gold surfaces, molecular dynamic simulations were performed using GROMACS (version 2021.3)50 with the CHARMM36 force field parameters for AMP.51 The Lennard-Jones parameters employed for gold were those reported by Heinz et al.52 which are designed to provide good descriptions for adsorption on different facets of metal surfaces.53 We have employed three gold slabs representing the Au{100}, Au{110} and Au{111} surfaces with the following dimensions:
| Slab | Dimensions (Å3) | Number of gold atoms | Simulation box height (Å) |
|---|---|---|---|
| {100} | 32.6 × 32.6 × 12.2 | 768 | 30.8 |
| {110} | 32.6 × 31.7 × 11.5 | 704 | 31.5 |
| {111} | 31.7 × 30.0 × 14.1 | 792 | 34.6 |
As starting points for the MD simulations, we have chosen 5 different initial orientations of AMP at distances shorter than 5 Å from the surface and solvated the system with 1000 water molecules employing the TIP3P model. The size of the box was adjusted to maintain 1 g mL−1 density and avoid vertical interactions, which resulted in heights for the simulation box above the surface of around 30 Å. The energy of each system was minimized until the maximum force on any atom is lower than 1000 kJ mol−1 nm−1, followed by NVT equilibration during 500 ps with a time step of 2 fs. The temperature was kept constant at 300 K employing the V-rescale thermostat and the cut-off for van-der-Waals interactions was set at 1.2 nm. Production runs were performed for 20 ns and the results for the last 10 ns were used for analysis.
To obtain the adsorption energy of AMP on each slab, we employed the efficient approach of Heinz54 which requires separated NVT simulations and energy evaluations of the solute–solvent–surface (E(Au(slab)+AMP+H2O)), solute–solvent (E(AMP+H2O)), solvent–surface (E(Au(slab)+H2O)) and solvent (E(H2O)) systems maintaining the same number of solvent molecules and altering the vertical dimension of the simulation box to keep the density close to an ideal value of 1000 kg m−3. From these simulations, the adsorption energy is obtained as
| Eads = E(Au(slab)+AMP+H2O) − E(AMP+H2O) + E(H2O) − E(Au(slab)+H2O) |
The MD simulations for the solute–solvent system were also used for comparing the difference between isolated AMP in water and AMP adsorbed on different gold surfaces. Finally, it must also be noted that, although previous MD studies on nucleotides were performed using different unprotonated forms considering that the phosphate group is usually deprotonated in solution,55 our molecular dynamics simulations were performed employing the completely protonated AMP. This is due to the presence of ascorbic acid as a reducing agent which explains that the experimental pH of our media is around 6.5 and, according to the AMP pKa values,56 it is expected that most of the AMP should be present in its protonated form.
Synthesis of gold seeds (∼4 nm in diameter)
The synthesis of gold seeds was based on a previous report with minor modifications.57 20 mL of ultra-pure water with 0.125 mM HAuCl4 and 0.25 mM trisodium citrated were added into a 25 mL vial at RT. Under vigorous stirring, 250 μL of freshly prepared 10 mM NaBH4 was rapidly injected. Vigorous stirring was maintained for 15 more minutes. After this, the seed solution was heated for 60 minutes at 40 °C in an oven without agitation. Seeds can be stored at 4 °C, and they are used as it is.Synthesis of gold seeds (∼30 nm in diameter)
The synthesis of gold seeds was based on a previous method reported by Bastús et al.58 150 mL of an aqueous solution of 2.2 mM trisodium citrate was heated to boiling under vigorous stirring. After 15 min, 1 mL of 25 mM HAuCl4 in water was injected into the boiling solution. After 10 min, the reaction mixture was cooled down to 90 °C and subsequently, another injection of 1 mL of 25 mM HAuCl4 in water was performed. Another addition was repeated 30 min later. The mixture was allowed to react for 30 min and then 55 mL was extracted from the reaction mixture. Subsequently, 53 mL of water and 2 mL of 60 mM sodium citrate (in water) were added. The resulting solution was used as Au seeds. The process was repeated four times to yield 30 nm Au NPs. To remove the excess reagents, the colloids were centrifuged twice at 2000g for 30 min and redispersed in the same volume of water.Synthesis of gold nanostars with ∼4 nm gold seeds
In a 25 mL vial containing a certain volume of water (ca. 18 mL), 0.3 mL of HAuCl4 (20 mM) and 0.8 mL of AMP (20 mM) were added under vigorous magnetic stirring. After 5 minutes of stirring, 0.5 mL of AA (20 mM) and 100 μL of the as-prepared seed solution were added. The addition of seeds must be performed immediately after the addition of AA. The stirring is kept for 5 minutes and then, the vial is introduced into an oven for 24 hours at 40 °C without stirring. The volume of seeds was varied from 50 μL to 600 μL, and the AMP concentration was varied from 0.5 mM to 0.9 mM by adding an appropriate amount of AMP solution. To maintain the final volume equal in all the experiments, the total final volume was adjusted with the volume of ultra-pure water. After synthesis, the colloid is centrifuged twice at 3500 rpm for 30 min (5 mL). The pellet of the first centrifugation was redispersed in 5 mL of 10 mM NaOH and the pellet of the second centrifugation in 5 mL of ultra-pure water.Synthesis of gold nanostars with ∼30 nm gold seeds
In a 25 mL vial containing 17.4 mL of water, 0.3 mL of HAuCl4 (20 mM) and 0.8 mL of AMP (20 mM) were added under vigorous magnetic stirring. After 5 minutes of stirring, 0.5 mL of AA (20 mM) and 1000 μL of the as-prepared seed solution were added. The addition of seeds must be immediately after the addition of AA. The stirring is kept for 5 minutes and then, the vial is introduced into an oven for 24 hours at 40 °C without stirring. After synthesis, the colloid is centrifuged twice at 2000 rpm for 30 min (5 mL). The pellet of the first centrifugation was redispersed in 5 mL of 10 mM NaOH and the pellet of the second centrifugation in 5 mL of ultra-pure water.Silica coating of gold nanostars
In a 25 mL vial containing 3.715 mL of EtOH, 0.5 mL of nanostars (0.5 mM Au0) and 0.1 mL of NH3 were added. Under vigorous stirring, 10 μL of an ethanolic solution of 0.4 M TEOS was added. The stirring was kept for 3 hours and, finally, the colloid was centrifuged twice at 4000 rpm for 15 min redispersing the pellet in the same volume of EtOH.Temperature stability
In a 20 mL round bottom flask with a reflux system, 10 mL of gold nanostars (0.06 mM Au0) were added. The round bottom reaction flask was then introduced into an oil bath at 100 °C or 75 °C and stirred. After two hours, the colloid was brought to room temperature and UV-Vis absorption spectra were recorded.Catalytic activity of nanostar nanozymes
2 mL of acetate buffer solution (1.0 mM, pH = 4.0), 100 μL of nanostars (0.5 mM of Au0) and 50 μL of TMB (32 mM in DMSO) were added into a 1 cm cuvette. After the homogenization of the sample, 90.5 μL of H2O2 (33%) was quickly added. The mixture was homogenized again and UV-Vis spectra were recorded for 600 s. All the experiments were performed at 25 °C. To calculate the steady-state enzymatic kinetic parameters of the nanostars, various concentrations of either TMB or H2O2 were prepared in buffer solution always using 10 μg of nanozymes in each experiment (100 μL, 0.5 mM Au0 of gold nanostars). For TMB studies, TMB stock solutions of 60 mM, 48 mM, 32 mM, 20 mM, 12 mM, and 4 mM were prepared in DMSO and 50 μL of these solutions and 90.5 μL of H2O2 were used. In the case of study H2O2, 50 μL of 32 mM TMB was employed and 25, 50, 100, 200, 300, and 600 μL of H2O2 were added. All the experiments were performed at 25 °C.Results and discussion
The complexation ability of the adenine moiety from AMP towards the gold salt can strongly affect the formation kinetics of AuNPs. Therefore, we studied the influence of the complexation of AMP with the gold salt precursor (AuCl4−) on its redox properties. It is known that the addition of ascorbic acid to a AuCl4− solution in the absence of any complexation agent leads to fast and uncontrollable nucleation and growth processes and the eventual nanoparticle aggregation due to the absence of stabilizing agents.59 In the present case, the addition of ascorbic acid to an aqueous solution containing AuCl4− and AMP at molar ratios of 1:2 and 1:4 just led to the partial reduction of AuCl4− to AuCl2−, as indicated by the disappearance of the Au(III) CTT band located at ca. 305 nm (Fig. S1 in the ESI†). This suggests that the complexation of AuCl4− with AMP shifts its redox potential to more negative values. To give further insight into this experimental observation, different energy profiles for the binding mechanism of AMP to AuCl4− were studied by means of DFT calculation (see details in the ESI† and Fig. S2). Fig. 1 shows the most favorable energy profile suggesting that AMP interacts initially with the Au(III) salt to form a complex, [AuCl4⋯AMP]−, mainly stabilized by a parallel interaction between the square planar AuCl4− and the adenine unit in AMP. Upon reduction, the [AuCl4⋯AMP]− complex is transformed into [AuCl2⋯AMP]−, which presents a strong direct interaction between the Au(I) atom and the N7 site in the adenine moiety. Interestingly, the additional reduction of the [AuCl2⋯AMP]− complex to the final [Au⋯AMP] species is energetically unfavorable. These theoretical data predicted that additional Au seeds were needed to achieve the [AuCl2⋯AMP]− reduction to Au(0). Fig. S3 and S4 in the ESI† show alternative complexation mechanisms predicting, in all cases, less favored energy paths.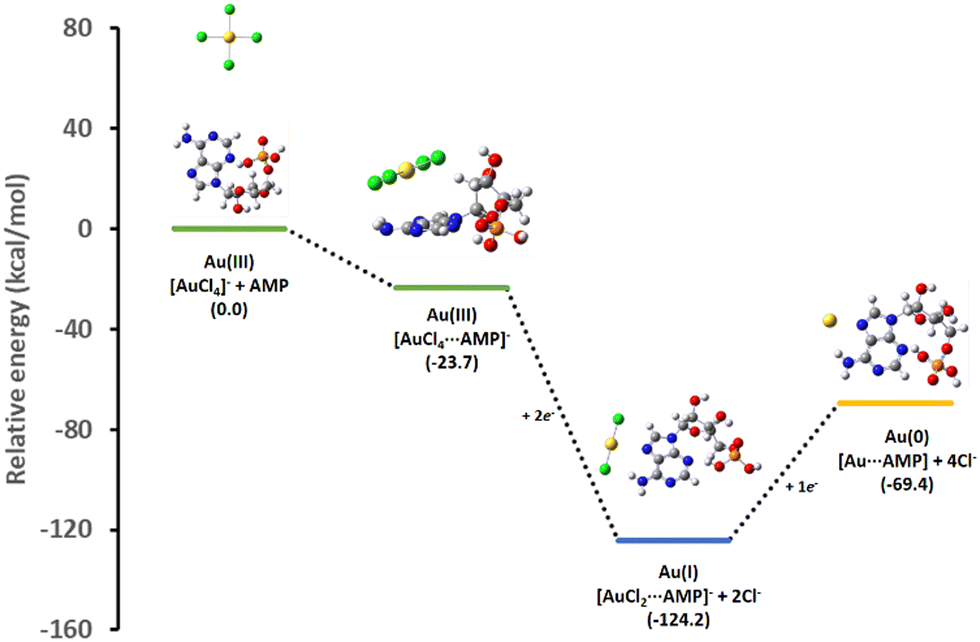 | ||
| Fig. 1 Theoretical relative energy profile for the interaction of AuCl4− with AMP and its subsequent reduction intermediates. | ||
To prove the autocatalytic process, tiny Au seeds stabilized by citrate were prepared using a strong reducing agent (NaBH4). Firstly, the influence of the AMP concentration on the growth of Au seeds was studied while keeping constant the concentrations of the gold salt (0.5 mM), ascorbic acid (0.3 mM) and Au seed (320 pM). Fig. 2 shows the influence of the AMP concentration on the optical properties as well as on the morphology of the overgrown Au nanoparticles. Thus, in the presence of 0.5 mM AMP (an AMP:Au(0) molar ratio of 800), the resulting Au nanoparticles are characterized by a broad LSPR band located at ca. 650 nm with a shoulder at ca. 550 nm.60 Besides, the TEM characterization revealed a star-shaped morphology with an overall size of 38.7 ± 5.0 nm and small tips of 5–10 nm in length (see Fig. 2(B) and Fig. S5 in the ESI†). With this information, we could assign the main LSPR to the tip localized plasmon mode and the shoulder at 550 nm to the core mode. Interestingly, the gradual increase in the AMP concentration from 0.5 mM to 0.9 mM led to a gradual red-shift of the main LSPR from 650 nm to 720 nm, 800 nm and 850 nm, corresponding to an increase in the AMP:Au(0) molar ratio from 960 to 1120, 1280 and 1440, respectively. The careful TEM analysis of the morphology of the Au nanostars (see Fig. 2 and Fig. S5 in the ESI†) revealed that as the AMP:Au(0) molar ratio increased the number of tips per particle decreased but their length notably increased. Thus, Au nanostars synthesized at the highest AMP:Au(0) molar ratio (1440) presented tips with lengths up to 25–30 nm. Overall, the experimental results seem to indicate that the preferential adsorption of AMP on certain crystalline facets of Au seeds may be responsible for the anisotropic growth.
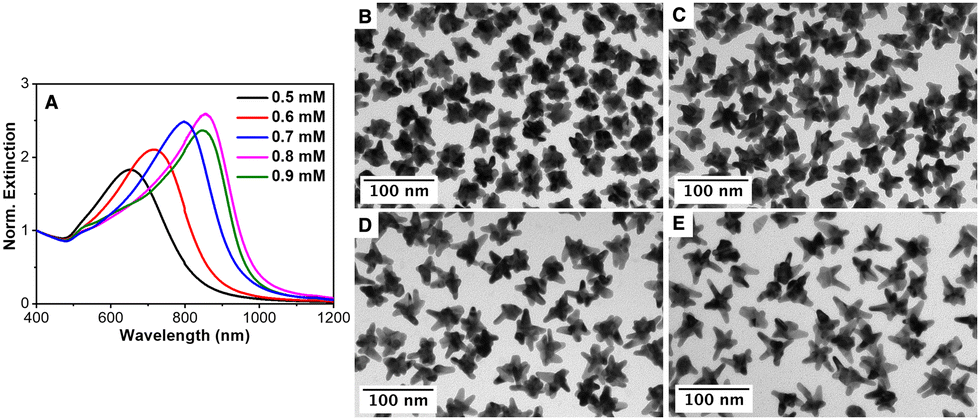 | ||
| Fig. 2 (A) Normalized extinction spectra of AuNSs synthesized using different AMP concentrations, as indicated. (B)–(E) Representative TEM images: (B) 0.5 mM AMP, (C) 0.6 mM AMP, (D) 0.7 mM AMP and (E) 0.8 mM AMP. [Au]seed = 320 pM, [AuCl4−] = 0.3 mM and [AA] = 0.5 mM, kept constant in all cases. | ||
Next, the influence of the Au seed concentration was explored while keeping the AMP concentration at 0.8 mM. Fig. 3(A) shows the optical properties of the resulting Au nanostars upon decreasing the concentration of Au seeds from 1264 pM to 632 pM, 474 pM, 316 pM and 158 pM, corresponding to an increase in the AMP:Au(0) molar ratio from 320 to 640, 853, 1280 and 2560, respectively. It should be noted that the LSPR band red-shifted with the decreasing seed concentration from ca. 680 nm ([Au]seed = 1264 pM) to ca. 950 nm ([Au]seed 158 pM). The TEM characterization revealed that the red shift is associated with i) an increase in the overall dimension of the nanostars from 29.7 ± 8.4 nm to 64.4 ± 4.1 nm and ii) an increase in the average length of the spikes (see Fig. 3(B)–(E) and Fig. S6 in the ESI†). These observations agree with the previously reported data.13,16 Besides, an increase in the number of spikes per particle was also observed. It should be noted that in the absence of Au seeds, the formation of Au nanoparticles is marginal even after 24 hours of reaction (see Fig. S7 and S8, ESI†).
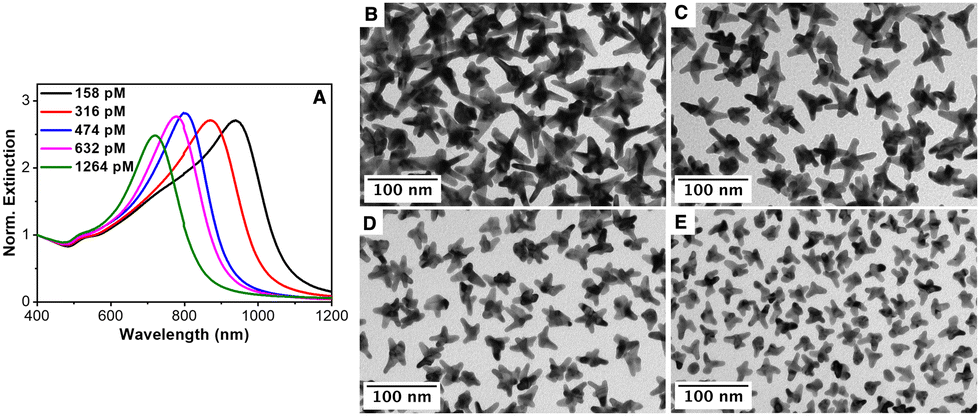 | ||
| Fig. 3 (A) Normalized extinction spectra of different AuNSs synthesized using different concentrations of Au seeds, as indicated. (B)–(E) Corresponding TEM images: (B) 158 pM, (C) 316 pM, (D) 632 pM and (E) 1264 pM. (E) Au seed concentration. [AuCl4−] = 0.3 mM, [AA] = 0.5 mM and [AMP] = 0.8 mM, kept constant in all cases. | ||
To perform a more comprehensive study of the growth mechanism, a time-resolved analysis of the synthesis of AuNSs was carried out by Vis-NIR spectroscopy and TEM. As can be seen in Fig. 4(A), at the initial stages (30 minutes), the nanoparticles present the main LSPR located at ca 730 nm that gradually red-shifts with time until reaching a constant value at ca. 880 nm. The TEM characterization of the particles at different stages, Fig. 4(B)–(D), showed that the tips/spikes are formed at the early stages of the reaction (30 minutes), and they grow in length throughout the reaction, being responsible for the observed red-shift in the LSPR.60 Finally, the impact of the reaction temperature and the concentration of AA was studied. Fig. S9 (ESI†) shows the influence of the AA, from 0.3 mM to 0.6 mM, in a standard AuNS synthesis. At a low AA concentration (0.3 mM), the gold salt reduction is rather low (ca. 10%). Nevertheless, at 0.4 mM, the reduction is ca. 90% with a LSPR band at 780 nm and at 0.5 mM and 0.6 mM the reduction is complete, and the optical properties remain unchanged (LSPR band at ca. 800 nm). Therefore, a AA concentration of 0.5 mM was chosen to assure the complete gold salt reduction. Fig. S10 (ESI†) shows the optical properties of the AuNSs synthesized at room temperature, 40 °C and 70 °C. At room temperature, only ca. 50% of the gold salt is reduced, while at 40 °C and 70 °C the reduction is complete and the optical properties in both cases are quite similar with an intense LSPR band located at ca. 800 nm. Taking this into account, a temperature of 40 °C was selected to guarantee the complete gold salt reduction with 0.5 mM AA.
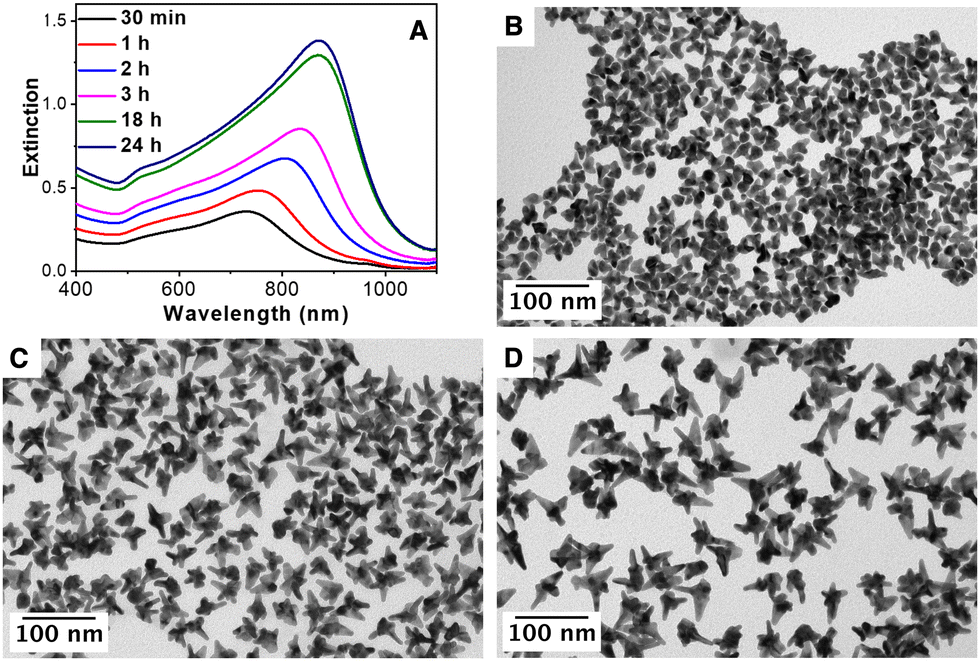 | ||
| Fig. 4 (A) Time evolution extinction spectra recorded during the formation of AuNSs. (B)–(D) TEM images of nanoparticles after (B) 30 minutes, (C) 3 hours and (D) 24 hours of reaction. [Au]seed = 316 pM, [AuCl4−] = 0.3 mM, [AA] = 0.5 mM and [AMP] = 0.8 mM. | ||
The crystalline structure and morphology of AuNSs were carefully investigated by aberration-corrected high-resolution TEM (HRTEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). Fig. 5 and Fig. S11 in the ESI† show representative HRTEM analyses of two different AuNSs synthesized using the same concentration of Au seeds (320 pM) but different AMP concentrations (0.8 mM and 0.6 mM). As it is shown in Fig. 5(A)–(C), most of the spikes present crystalline planes (111) and (200) and seem to grow perpendicular to the (111) direction where the defects are located and grow along the defect planes with a (211) preferential direction. Furthermore, most of the tips usually showed a twinning plane running down the branch to the core of the particle, which is evident in the tilted HAADF images at a range of angles depending on the arm (Fig. 5(G)–(H)). Interestingly, variations in the AMP concentration did not give rise to changes in the crystalline structure of the AuNSs and just affected their morphology. Fig. 5(I) and Fig. S11I in the ESI† show a 3D tomographic reconstruction of AuNSs obtained with 0.8 mM and 0.6 mM of AMP, respectively, where it is demonstrated that the larger tips for AuNSs are obtained with 0.8 mM AMP. Interestingly, and regardless of the AMP concentration, HRTEM images showed that the center of the gold nanostars exhibited a multi-twinned structure resembling that of an icosahedron (Fig. S12 in ESI†). Hence, concerning the twins, it is highly plausible that the branches originate from the twins inside the seeds, resembling the characteristics observed in icosahedral or decahedral particles.13 Notably, the branches exhibit distinct (111) surface facets, accompanied by some higher-order features towards both the tips and the base of the branches. This is evident in the high-resolution image, where the surface facet running along the edge of the branch is the (111) facet. Additionally, twinning can be observed along the (111) direction, contributing to the growth of the branch. This phenomenon is further supported by the presence of twins in the other arms as observed for other particle orientations (Fig. 5(D)–(H)).
 | ||
| Fig. 5 (A) Aberration corrected HRTEM of a gold nanostar synthesized using 0.8 mM AMP and 320 pM of Au seeds, and the white arrows point towards twins in the tips. (B) Zoomed image of the tip from the white box in (A). A defect plane running down the center of the tip is observed. (C) The FFT of (B) shows two zone axes both viewed down the [110] axis twinned along the (111) spacing. (D)–(H) HAADF-STEM images taken from the tomography tilt series. White arrows indicate when the defect plane extends along the tips. (I) 3D tomographic reconstruction of the nanostar. | ||
Importantly, this AMP-mediated synthesis of gold nanostars may also be extended to larger Au seeds. As a proof of concept, citrate-stabilized spherical Au NPs with a mean diameter of ca. 30 nm were employed as seeds (see the experimental section for details). Fig. 6 shows the optical properties together with the representative TEM images of the Au seed and the nanostars obtained upon increasing the AMP concentration from 0.4 mM to 0.9 mM (AMP:Au(0) molar ratios of 10 to 17.5 and 22.9) while keeping constant the Au seed concentration ([Au]seed = 480 pM). For the smaller Au seeds (see Fig. 2), the increase in the AMP concentration gave rise to a redshift and a broadening of the main LSPR. It is in agreement with the TEM results. Thus, the TEM analysis revealed that the overall diameter ranged between 70 and 90 nm regardless of the AMP concentration (Fig. S13 in the ESI†). In addition, the mean length of the spikes increased from 5 to 10 nm for 0.4 mM AMP up to 30 nm in the presence of 0.9 mM AMP. On the other hand, the broadening in the optical response (see Fig. 3(A)) could be ascribed to a larger core size and an increase in the number of spikes per particle. The morphological features and the optical response are in line with those previously reported PVP-stabilized AuNSs.61
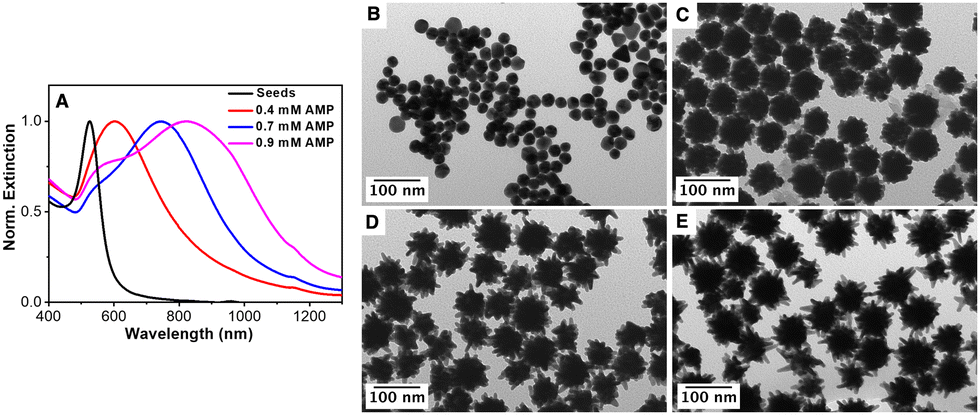 | ||
| Fig. 6 (A) Normalized extinction spectra of Au seeds and AuNSs synthesized using different AMP concentrations. (B) TEM image of 30 nm citrate-stabilized Au seeds. (C)–(E) TEM images of AuNSs synthesized using different AMP concentrations: (C) 0.4 mM, (D) 0.7 mM, and (E) 0.9 mM. [Au]seed = 480 pM, [AuCl4−] = 0.3 mM, [AA] = 0.5 mM. | ||
To propose a plausible mechanism for the synthesis of Au nanostars, we have to take into consideration the following: (i) the higher stability of the [AuCl2⋯AMP]− complex in comparison with the [AuCl4⋯AMP]− and [Au⋯AMP] complexes, as demonstrated by DFT calculations (Fig. 1), (ii) both types of citrate-stabilized Au seeds present the multiple-twinned crystalline structure,16 (iii) the presence of twinning planes running along most of the tips (Fig. 5), and (iv) the increase in the length of the tips upon increasing the AMP concentration (Fig. 2). Therefore, the experimental data demonstrate that AMP plays a fundamental role in the morphological transformation from the spherical shape of the seed to the star shape of the overgrown particles. Such morphological transformation may be initially ascribed to the preferential adsorption of the AMP molecules towards certain crystal facets stabilizing them and promoting the deposition of gold adatoms towards less stabilized facets leading to the formation of tips/spikes through the Stranski–Krastanov growth mode.62 Therefore, as previously reported for other Au NS synthesis such as Good's buffers,30,63 the preferential adsorption to certain crystallographic facets has been ascribed as the key parameter to promote the AuNS growth.
Molecular dynamic calculations were employed to study the adsorption of AMP on different gold surfaces. To this purpose, we built up three model slabs for the Au{111}, Au{110} and Au{100} facets and the adsorption energy for a single AMP molecule on each slab was evaluated as previously proposed by Heinz54 (see the computational methods section in the experimental section).
| Eads = E(Au{slab}+AMP+H2O) − E(AMP+H2O) + E(H2O) − E(Au{slab}+H2O) | (1) |
According to the results obtained, adsorption of AMP on the Au{111} surface (Eads = −137.1 kcal mol−1) was energetically more favorable than that on Au{100} and Au{110} surfaces (Eads = −103.9 kcal mol−1 and Eads = −97.6 kcal mol−1, respectively). A representative configuration for the adsorption of AMP on the Au{111} surface is depicted in Fig. 7(A), where it can be seen that AMP adopts a rather elongated disposition with the adenine moiety almost parallel to the metal surface (the average angle between the adenine plane and the top atomic layer of the gold slab is 6.3°). This disposition tends to maximize the number of contacts between the gold and nitrogen atoms in the adenine ring. Besides, the oxygen atom of the ribose ring is directed toward the surface at an average distance of 2.7 Å and the two OH bonds of the phosphate ring are also in close interaction with the metal. Interestingly, due to adsorption, there is a considerable reduction of the AMP conformational flexibility compared to its behavior in an aqueous solution (see the detailed description in Fig. S14–S16 in the ESI†). In particular, the adenine ring in the isolated AMP adopts syn and anti orientations with respect to the ribose unit due to rotation around the central C–N bond between both rings. However, this rotation is hindered due to adsorption and the adenine and ribose units are always syn-oriented (Fig. 7).
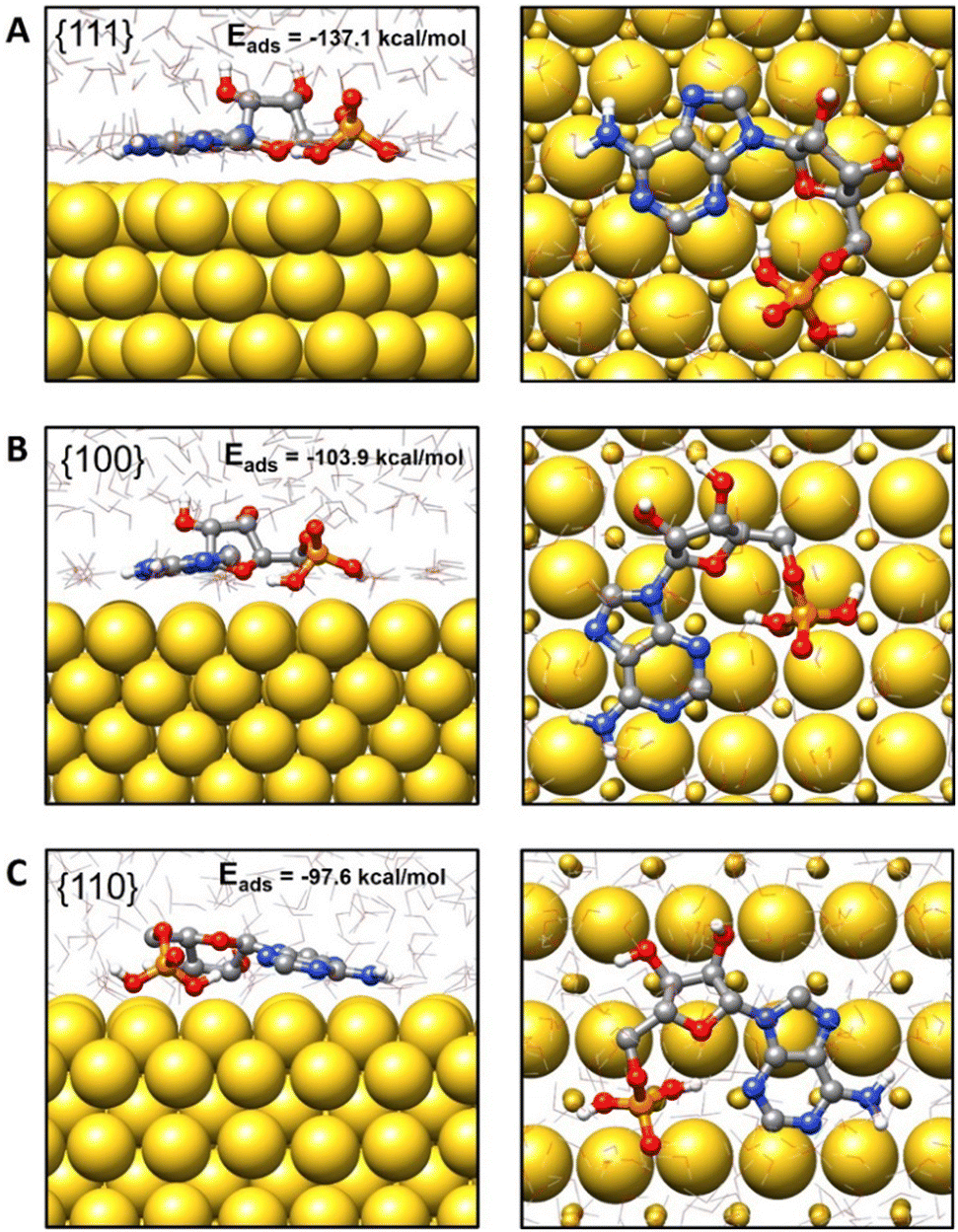 | ||
| Fig. 7 Side and top views of representative configurations for AMP adsorbed on the (A) {111}, (B) {100} and (C) {110} Au surfaces. The adsorption energies have been included. | ||
AMP also adopts extended or stretched configurations when adsorbed on the Au{100} and Au{110} surfaces (see Fig. 7(B) and (C)). Nevertheless, the average angles between the adenine ring and the metal surface are slightly larger than for Au{111} (8.6° and 11.0° for {100} and {110}, respectively), and the average distances from the ribose oxygen atom to the first metal layer are also larger (2.8 Å and 3.8 Å, respectively, see Fig. S17, ESI†). These trends correlate with the smaller preference for the adsorption of AMP on the Au{100} and Au{110} surfaces and seem to be related to the gold atom density on the slab top layer. Thus, due to the high gold atom density on the Au{111} layer, there is a larger number of direct contacts between the nitrogen and oxygen atoms of AMP and the gold atoms in Au{111} in agreement with the stronger interaction predicted between AMP and the Au{111} facet. However, the number of direct atom interactions reduces in Au{100} and Au{110} with respect to Au{111} (see Fig. 7) and this reduction explains the smaller adsorption energies in the former cases.
Finally, it must be remarked here that, according to our theoretical results, during the nanoparticle growth in the presence of AMP and due to its larger affinity for the Au{111} layer, it can be expected that AMP would preferentially cover this {111} facet, deviating the deposition of gold adatoms towards less stabilized facets. This would cause the final shape of the nanoparticle to present a larger proportion of Au{111} facets, as observed in the experimental results. It should be pointed out that other nucleotides, such as guanosine monophosphate (GMP), were tested, but the fine-tuning of the morphology of the final particles with the concentration was poor (see Fig. S18, ESI†). The interaction of different nucleotides with the Au seed has already been reported as the key factor that controls the final morphology when employing several homogenous oligonucleotides.3
In summary, it seems reasonable to propose that the growth of the twin boundaries of the multi-twinned Au seeds, and therefore the formation of tips, is stimulated by the preferential adsorption of AMP molecules in [111] and [200] planes.
To further study the versatility of AMP as a capping agent, the stability of the particles in different water-miscible organic solvents was tested. Fig. 8(A) shows the optical properties of the same particles when redispersed in water, ethanol, DMF and DMSO. The spectra reveal the high colloidal stability of the particles in polar organic solvents. In addition, the position of the LSPR band red-shifts upon increasing the refractive index from 1.33 to 1.36, 1.41 and 1.48, respectively. With these results, the refractive index sensitivity (RIS), defined as the shift in the LSPR band with the refractive index of the medium (Δλ/Δn), was analyzed64 observing a good linear dependence of the LSPR with the refractive index (Fig. S19 in the ESI†) and a RIS of 359 nm/RIU. This value aligns with other reported gold nanoparticles.16,65
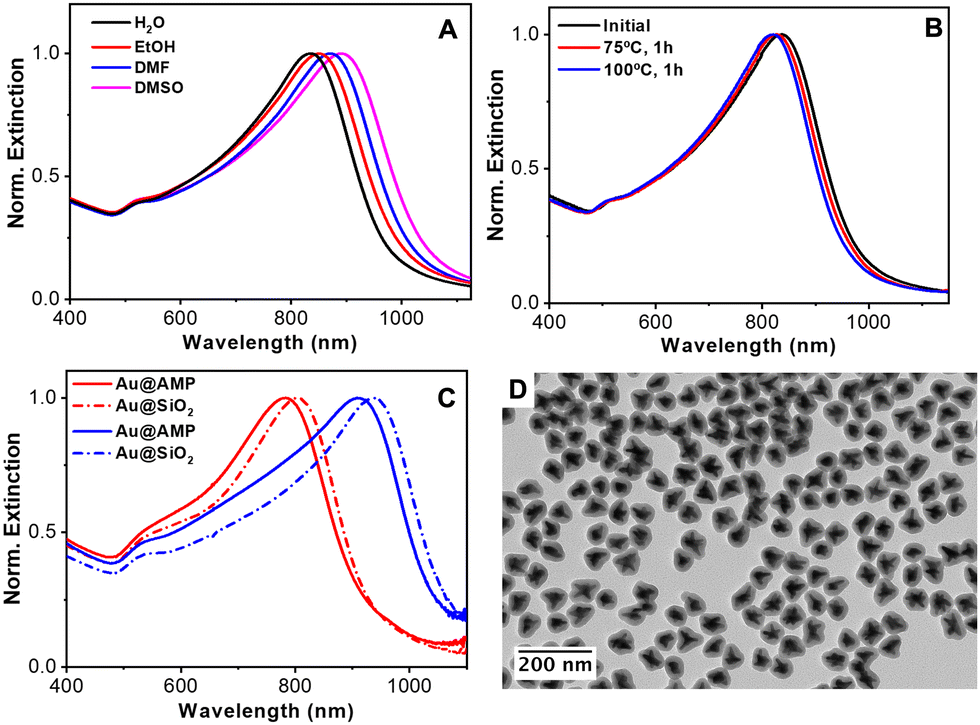 | ||
| Fig. 8 (A) Normalized extinction spectra of AMP-stabilized AuNSs redispersed in different solvents as indicated. (B) Normalized extinction spectra of nanostars before and after thermal treatment at 75 and 100 °C. (C) Normalized extinction spectra of AuNSs before (solid lines) and after (dashed lines) the silica coating. (D) TEM images of silica-coated AuNSs with LSPR at 750 nm. | ||
Next, the thermal stability of nanostars in water was also examined by heating the colloidal NP dispersion at 75 °C or 100 °C for one hour. As shown in Fig. 8(B), the nanostars suffer only a small blue shift in their main LSPR (ca. 11 and 17 nm at 75 and 100 °C, respectively), demonstrating their good thermal stability. Note that gold nanostars have high surface energy located at the tips, where gold atoms tend to migrate towards more favorable regions, inevitably degrading their optical properties.66
Finally, taking into account the high stability of the particles in ethanol and the negative surface ζ potential due to the adsorption of AMP, the silica coating was tested using the well-known Stöber method.67 The silica coating of metal nanoparticles improves their colloidal stability, facilitates their bioconjugation, etc.68,69 The coating process typically requires colloids dispersed in an ethanolic solution and also the presence of a capping ligand that promotes the nucleation and growth of silica on the metal surface. A range of polymers and surfactants such as polyvinylpyrrolidone (PVP)69 and thiolated poly(ethylene glycol) (PEG)70 have been reported for the controlled silica deposition on metal nanoparticles. Nonetheless, these approaches required a post-synthetic nanoparticles functionalization step. In the present case, we found that AMP-stabilized Au nanoparticles can be directly coated with silica without any further functionalization by applying the Stöber method67 (see the Experimental section for details). Fig. 8(C) shows the UV-vis-NIR spectra of two different Au nanostars before and after the silica coating. In both cases, the LSPR band red shifts upon silica coating due to the increase in the local refractive index surrounding the particles.71Fig. 8(D) and Fig. S20 in the ESI† show the representative TEM images of the nanostars after the silica coating. All the particles are individually coated with a uniform silica shell. Both, the optical and morphological characterization studies demonstrate that the spikes/tips retain their integrity, confirming the remarked ability of AMP as the stabilizing agent of Au nanostars.
The catalytic activity of natural peroxidases and their artificial mimics stem essentially from their unique capabilities to mediate the electron transfer from organic molecular substrates to H2O2. It catalyzes the homolytic split of H2O2 to generate surface-adsorbed hydroxyls that are highly reactive toward the oxidation of the substrates. Particularly, it has been reported that gold nanoparticles can mimic the peroxidase-like activity of enzymes.72,73 Furthermore, Au nanoparticles combined with certain nucleotides can improve the catalytic activity. Specifically, adenosine triphosphate (ATP) and adenosine diphosphate (ADP) were found to improve their peroxidase-like activity.74,75 With this idea in mind, the catalytic performance of the AuNSs was tested employing as a model reaction the oxidation of the uncolored TMB into a blue ox-TMB in the presence of H2O2. AuNSs with the main LSPR at 850 nm (AuNS850, Fig. 9(A) and (B)) and 700 nm (AuNS700, Fig. S21 in the ESI†) were employed for this study. The oxidation of TMB mediated by the AuNSs was studied by UV-Vis absorption spectroscopy. As shown in Fig. 9(C), in a few seconds, three absorption bands corresponding to the ox-TMB are evidenced which gradually increased in intensity with time. In contrast, in the absence of H2O2 and/or gold NPs, no ox-TMB formation is detected (Fig. S22 in the ESI†). To determine the peroxidase-like performance of the AuNSs, experiments at different concentrations of TMB were carried out. Fig. 9(D) shows the time evolution curves of the ox-TMB band at 652 nm observing a gradual intensity increase till reaching a plateau. Next, the initial reaction rates (V0) were determined by taking the initial linear segment in different curves and using a molar absorption coefficient for ox-TMB of 39.000 M−1 cm−1.76
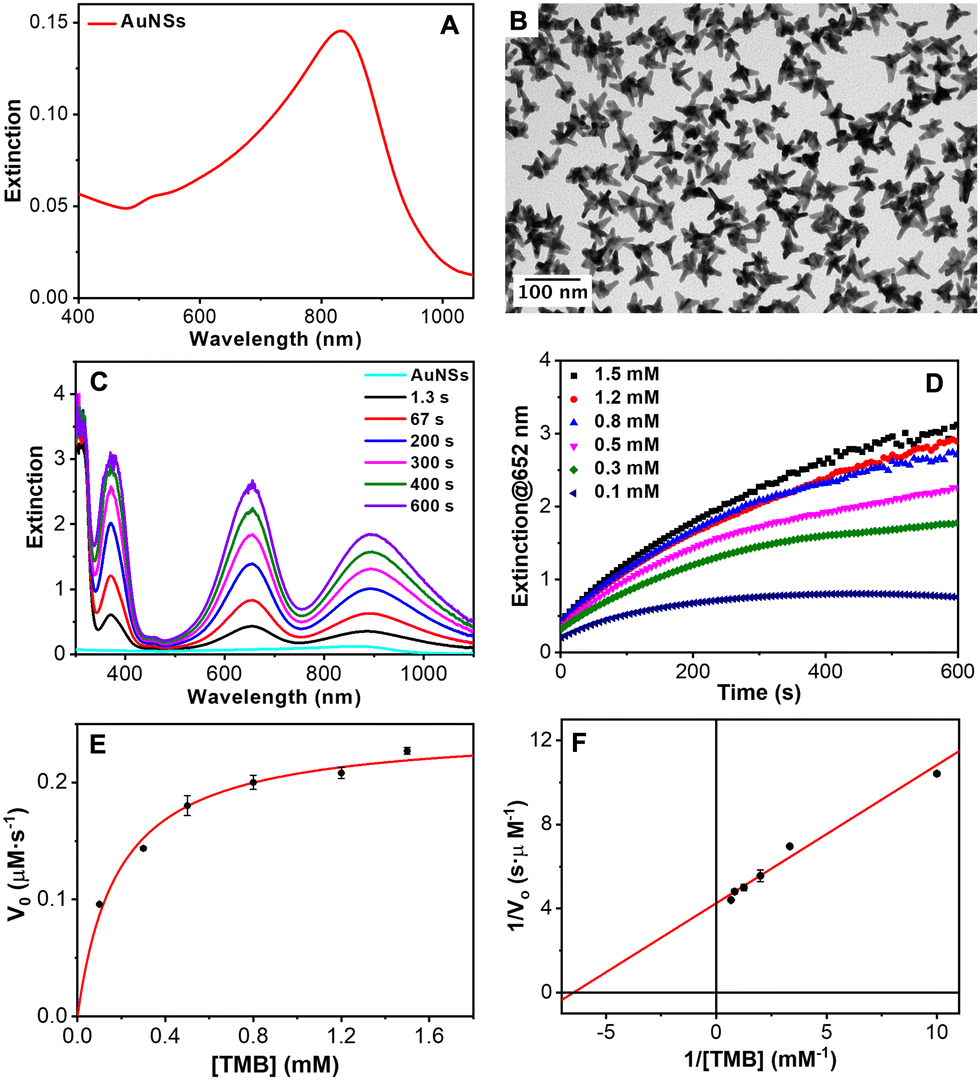 | ||
| Fig. 9 (A) Vis-NIR extinction spectrum and (B) TEM image of AuNSs used as catalysts. (C) Time evolution absorption spectra of the TMB oxidation in the presence of AuNSs. (D) Time evolution of the absorbance at 652 nm for different TMB concentrations, as indicated. (E) Michaelis–Menten curve of AuNS nanozymes. (F) Lineweaver–Burk plot derived from the Michaelis–Menten curve. Error bars represent the standard deviation derived from three independent experiments. | ||
To fully characterize the nanoparticles as catalysts, V0 was plotted versus TMB concentration (Fig. 9(E)). The data fitted very well with the Michaelis–Menten model. Finally, we estimated the constant (Km) and maximum reaction velocity (Vmax) using the Lineweaver–Burk equation (eqn (1) and Fig. 9(F)).
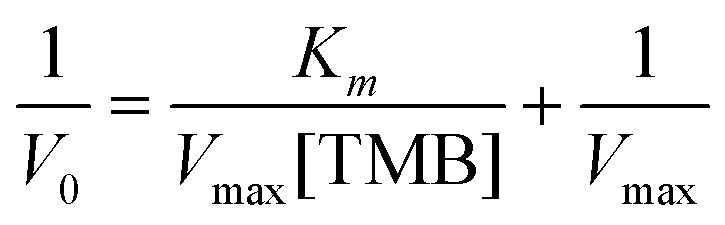 | (2) |
Besides, V0 and Vmax were estimated by performing experiments with different concentrations of H2O2 (Fig. S23 and S24 in the ESI†).
For AuNS850, the Km and Vmax values of TMB were 0.154 mM and 23.5 × 10−8 M s−1, respectively, being very similar for AuNS700 (0.155 mM and 23.7 × 10−8 M s−1, respectively). Regarding H2O2, the Km and Vmax values were 692.5 mM and 47.8 × 10−8 M s−1, respectively, for AuNS850 and 721.0 mM and 49.0 × 10−8 M s−1, respectively, for AuNS700. Therefore, no significant differences were observed between both types of AuNSs. Besides, the obtained Km values (TMB and H2O2) are similar to those reported in the literature for other gold nanostructures. The values of the Km for TMB are smaller than the ones of the H2O2, meaning that the affinity of the nanozyme for TMB is higher.77 Regarding the second catalytic parameter, Vmax, the values obtained for the AuNSs were found one of the highest values reported for gold nanozymes only composed of this metal. Similar or even better results can be found in the literature but using Au alloys with other metals or hybrids with organic structures such as metal–organic frameworks (MOFs) as it is summarized in Table S2 in the ESI.†
Conclusions
We developed a versatile seed-mediated approach to synthesize gold nanostars in a wide size range (between 30 and 90 nm). The presence of AMP was a key point to obtain well-defined gold nanostars with tunable size and optical properties (between 580 and 950 nm). The number and the length of the tip, which rule out the optical properties, can be controlled through the AMP:Au molar ratio. Advanced microscopy analysis showed that the gold nanostar presents a polycrystalline nature with several tips branching out the surface. Interestingly, most of the tips analyzed usually showed a twinning plane running down the tip to the core of the particle, which may indicate a preferential growth of the multi-twinned crystallographic structure of the seeds used. DFT and molecular dynamics calculations support the experimental data suggesting the role of AMP to modulate the reduction of the gold precursor via complexation and as a shape-directing agent via preferential adsorption on certain Au crystalline facets. Furthermore, AMP was found to be a robust capping agent since nanoparticles are colloidally stable in polar organic solvents, and also offer considerable resistance to reshaping up to 100 °C with a minimal variation in their optical response. Interestingly, we noted that the AMP allows the direct silica coating, expanding the future applications of this nanomaterial. Finally, the nanostars capped with AMP displayed a very good capability to mimic enzyme activity for TMB oxidation since the obtained Vmax was one of the best values reported in the literature until today for gold nanozymes (see Table S2 in the ESI† for a comparison with other Au nanozymes).Data availability
The data that support the findings of this study are openly available in zenodo.org at https://doi.org/10.5281/zenodo.8206693.Author contributions
The manuscript was written through the contributions of all authors. All authors gave approval for the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge financial support from the European Innovation Council (Horizon 2020 Project: 965018—BIOCELLPHE), the MCIN/AEI/10.13039/501100011033 (grant PID2019-108954RB-I00), the FSE (“El FSE invierte en tu futuro”), the Xunta de Galicia/FEDER (grant GRC ED431C 2020/09), the European Regional Development Fund (ERDF), and the Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior, FCT-MCTES (grants UIDB/50006/2020, UIDP/50006/2020 and Met4cat, EXPL/QUI-COL/0263/2021). J. F.-L. thanks FCT-UNL for the research contract through the Program DL 57/2016−Norma Transitória. S. N. thanks the FCT-MCTES Portugal for her doctoral grant associated with the Chemistry PhD program (SFRH/BD/144618/2019). C. F.-L. acknowledges Xunta de Galicia for a predoctoral scholarship (Programa de axudas á etapa predoutoral). C. L., A. F. L., S. N and J. F. L thank the financial support of the PROTEOMASS Scientific Society (Portugal) (General Funding Grants 2022-2023) and the Associate Laboratory Research Unit for Green Chemistry-Clean Processes and Technologies - LAQV/REQUIMTE. This work was carried out in part through the use of the INL Advanced Electron Microscopy, Imaging and Spectroscopy Facility and Microscopy Facility at CACTI (Universidade de Vigo). The authors thank Dr. Jamila Djafari for her assistance with the graphical abstract.Notes and references
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783 RSC.
- C. Lu, S. Zhou, F. Gao, J. Lin, J. Liu and J. Zheng, TrAC Trends Anal. Chem., 2022, 148, 116533 CrossRef CAS.
- Z. Wang, L. Tang, L. H. Tan, J. Li and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 9078–9082 CrossRef CAS PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- B. Ni, M. Mychinko, S. Gómez-Graña, J. Morales-Vidal, M. Obelleiro-Liz, W. Heyvaert, D. Vila-Liarte, X. Zhuo, W. Albrecht, G. Zheng, G. González-Rubio, J. M. Taboada, F. Obelleiro, N. López, J. Pérez-Juste, I. Pastoriza-Santos, H. Cölfen, S. Bals and L. M. Liz-Marzán, Adv. Mater., 2023, 35, 2208299 CrossRef CAS.
- M.-Q. He, Y.-L. Yu and J.-H. Wang, Nano Today, 2020, 35, 101005 CrossRef CAS.
- J. Reguera, J. Langer, D. Jiménez de Aberasturi and L. M. Liz-Marzán, Chem. Soc. Rev., 2017, 46, 3866–3885 RSC.
- I. B. Becerril-Castro, I. Calderon, N. Pazos-Perez, L. Guerrini, F. Schulz, N. Feliu, I. Chakraborty, V. Giannini, W. J. Parak and R. A. Alvarez-Puebla, Anal. Sens., 2022, 2, e202200005, DOI:10.1002/anse.202200005.
- M. Tavakkoli Yaraki, M. Wu, E. Middha, W. Wu, S. Daqiqeh Rezaei, B. Liu and Y. N. Tan, Nano-Micro Lett., 2021, 13, 58 CrossRef PubMed.
- Z. Kang, M. Liu, Z. Li, S. Li, Z. Jia, C. Liu, W. Qin and G. Qin, Photonics Res., 2018, 6, 549 CrossRef CAS.
- A. Espinosa, A. K. A. Silva, A. Sánchez-Iglesias, M. Grzelczak, C. Péchoux, K. Desboeufs, L. M. Liz-Marzán and C. Wilhelm, Adv. Healthcare Mater., 2016, 5, 1040–1048 CrossRef CAS PubMed.
- P. Priecel, H. Adekunle Salami, R. H. Padilla, Z. Zhong and J. A. Lopez-Sanchez, Chin. J. Catal., 2016, 37, 1619–1650 CrossRef CAS.
- S. Barbosa, A. Agrawal, L. Rodríguez-Lorenzo, I. Pastoriza-Santos, R. A. Alvarez-Puebla, A. Kornowski, H. Weller and L. M. Liz-Marzán, Langmuir, 2010, 26, 14943–14950 CrossRef CAS PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CrossRef CAS PubMed.
- D. Jana, C. Matti, J. He and L. Sagle, Anal. Chem., 2015, 87, 3964–3972 CrossRef CAS PubMed.
- S. Nuti, C. Fernández-Lodeiro, J. Fernández-Lodeiro, A. Fernández-Lodeiro, J. Pérez-Juste, I. Pastoriza-Santos, A. P. LaGrow, O. Schraidt, J. Luis Capelo-Martínez and C. Lodeiro, J. Colloid Interface Sci., 2022, 611, 695–705 CrossRef CAS PubMed.
- V. M. Kariuki, J. C. Hoffmeier, I. Yazgan and O. A. Sadik, Nanoscale, 2017, 9, 8330–8340 RSC.
- J. Djafari, A. Fernández-Lodeiro, D. García-Lojo, J. Fernández-Lodeiro, B. Rodríguez-González, I. Pastoriza-Santos, J. Pérez-Juste, J. L. Capelo and C. Lodeiro, ACS Sustainable Chem. Eng., 2019, 7, 8295–8302 CrossRef CAS.
- F. Liebig, R. Henning, R. M. Sarhan, C. Prietzel, C. N. Z. Schmitt, M. Bargheer and J. Koetz, RSC Adv., 2019, 9, 23633–23641 RSC.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- A. J. Blanch, M. Döblinger and J. Rodríguez-Fernández, Small, 2015, 11, 4549 CrossRef.
- C.-H. Kuo and M. H. Huang, Langmuir, 2005, 21, 2012–2016 CrossRef CAS PubMed.
- P. Pallavicini, G. Chirico, M. Collini, G. Dacarro, A. Donà, L. D’Alfonso, A. Falqui, Y. Diaz-Fernandez, S. Freddi, B. Garofalo, A. Genovese, L. Sironi and A. Taglietti, Chem. Commun., 2011, 47, 1315–1317 RSC.
- N. Almora-Barrios, G. Novell-Leruth, P. Whiting, L. M. Liz-Marzán and N. López, Nano Lett., 2014, 14, 871–875 CrossRef CAS PubMed.
- M. S. Verma, P. Z. Chen, L. Jones and F. X. Gu, RSC Adv., 2014, 4, 10660–10668 RSC.
- J. A. Webb, W. R. Erwin, H. F. Zarick, J. Aufrecht, H. W. Manning, M. J. Lang, C. L. Pint and R. Bardhan, J. Phys. Chem. C, 2014, 118, 3696–3707 CrossRef CAS.
- G. Maiorano, L. Rizzello, M. A. Malvindi, S. S. Shankar, L. Martiradonna, A. Falqui, R. Cingolani and P. P. Pompa, Nanoscale, 2011, 3, 2227 RSC.
- R. M. Pallares, T. Stilson, P. Choo, J. Hu and T. W. Odom, ACS Appl. Nano Mater., 2019, 2, 5266–5271 CrossRef.
- K. Chandra, K. S. B. Culver, S. E. Werner, R. C. Lee and T. W. Odom, Chem. Mater., 2016, 28, 6763–6769 CrossRef CAS.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- J. Li, R. Cai, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2015, 3, 5806–5814 RSC.
- L. Lu, K. Ai and Y. Ozaki, Langmuir, 2008, 24, 1058–1063 CrossRef CAS PubMed.
- M. Sajitha, A. Vindhyasarumi, A. Gopi and K. Yoosaf, RSC Adv., 2015, 5, 98318–98324 RSC.
- P. Bian, J. Yuan, H. Han and Z. Ma, J. Nanosci. Nanotechnol., 2016, 16, 7503–7508 CrossRef CAS.
- M. Green and D. Smyth-Boyle, J. Mater. Chem., 2007, 17, 3588 RSC.
- D. Ungor, E. Csapó, B. Kismárton, Á. Juhász and I. Dékány, Colloids Surf., B, 2017, 155, 135–141 CrossRef CAS PubMed.
- W. Zhao, F. Gonzaga, Y. Li and M. A. Brook, Adv. Mater., 2007, 19, 1766–1771 CrossRef CAS.
- P. Huang, O. Pandoli, X. Wang, Z. Wang, Z. Li, C. Zhang, F. Chen, J. Lin, D. Cui and X. Chen, Nano Res., 2012, 5, 630–639 CrossRef CAS.
- F. Pu, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 1285–1306 RSC.
- Z. Wang, J. Zhang, J. M. Ekman, P. J. A. Kenis and Y. Lu, Nano Lett., 2010, 10, 1886–1891 CrossRef CAS.
- J. Kundu, O. Neumann, B. G. Janesko, D. Zhang, S. Lal, A. Barhoumi, G. E. Scuseria and N. J. Halas, J. Phys. Chem. C, 2009, 113, 14390–14397 CrossRef CAS.
- H. Kimura-Suda, D. Y. Petrovykh, M. J. Tarlov and L. J. Whitman, J. Am. Chem. Soc., 2003, 125, 9014–9015 CrossRef CAS PubMed.
- L. Berti and G. A. Burley, Nat. Nanotechnol., 2008, 3, 81–87 CrossRef CAS PubMed.
- N. H. Cho, Y. B. Kim, Y. Y. Lee, S. W. Im, R. M. Kim, J. W. Kim, S. D. Namgung, H.-E. Lee, H. Kim, J. H. Han, H. W. Chung, Y. H. Lee, J. W. Han and K. T. Nam, Nat. Commun., 2022, 13, 3831 CrossRef CAS PubMed.
- Y. He and L. Zhou, Comput. Theor. Chem., 2016, 1093, 20–28 CrossRef CAS.
- C. Hanske, M. N. Sanz-Ortiz and L. M. Liz-Marzán, Adv. Mater., 2018, 30, 1707003 CrossRef PubMed.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- M. Liang and X. Yan, Acc. Chem. Res., 2019, 52, 2190–2200 CrossRef CAS PubMed.
- A. L. Gille, B. C. Dutmer and T. M. Gilbert, J. Am. Chem. Soc., 2009, 131, 5714 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CAS.
- H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, J. Phys. Chem. C, 2008, 112, 17281–17290 CrossRef CAS.
- H. Ramezani-Dakhel, L. Ruan, Y. Huang and H. Heinz, Adv. Funct. Mater., 2015, 25, 1374–1384 CrossRef CAS.
- H. Heinz, J. Comput. Chem., 2010, 31, 1564–1568 CAS.
- P. Gruszczyński, K. Smalara, M. Obuchowski and R. Kaźmierkiewicz, J. Mol. Model., 2011, 17, 1081–1090 CrossRef PubMed.
- R. Alberty, R. Smith and R. Bock, J. Biol. Chem., 1951, 193, 425–434 CrossRef CAS PubMed.
- J. Pérez-Juste, L. M. Liz-Marzán, S. Carnie, D. Y. C. Chan and P. Mulvaney, Adv. Funct. Mater., 2004, 14, 571–579 CrossRef.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. Javier García de Abajo and L. M. Liz-Marzán, Nanotechnology, 2008, 19, 015606 CrossRef PubMed.
- S. Barbosa, A. Agrawal, L. Rodríguez-Lorenzo, I. Pastoriza-Santos, R. A. Alvarez-Puebla, A. Kornowski, H. Weller and L. M. Liz-Marzán, Langmuir, 2010, 26, 14943–14950 CrossRef CAS PubMed.
- I. V. Markov, Crystal Growth for Beginners, World Scientific, 2003 Search PubMed.
- W. Xi and A. J. Haes, J. Am. Chem. Soc., 2019, 141, 4034–4042 CrossRef CAS PubMed.
- B. Sepúlveda, P. C. Angelomé, L. M. Lechuga and L. M. Liz-Marzán, Nano Today, 2009, 4, 244–251 CrossRef.
- S. Rodal-Cedeira, V. Montes-García, L. Polavarapu, D. M. Solís, H. Heidari, A. La Porta, M. Angiola, A. Martucci, J. M. Taboada, F. Obelleiro, S. Bals, J. Pérez-Juste and I. Pastoriza-Santos, Chem. Mater., 2016, 28, 9169–9180 CrossRef CAS.
- L. Fabris, J. Phys. Chem. C, 2020, 124, 26540–26553 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS PubMed.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef.
- A. M. Fales, H. Yuan and T. Vo-Dinh, Langmuir, 2011, 27, 12186–12190 CrossRef CAS PubMed.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329–4335 CrossRef.
- X. Ji, Q. Li, R. Su, Y. Wang and W. Qi, Langmuir, 2023, 39, 3216–3224 CrossRef CAS PubMed.
- S. Nuti, A. Fernández-Lodeiro, L. E. Chinchilla, A. B. Hungría, J.-L. Capelo-Martinez, C. Lodeiro and J. Fernández-Lodeiro, J. Phys. Chem. Lett., 2023, 14, 6315–6320 CrossRef CAS PubMed.
- J. Shah and S. Singh, 3 Biotech, 2018, 8, 67 CrossRef PubMed.
- J. Shah, R. Purohit, R. Singh, A. S. Karakoti and S. Singh, J. Colloid Interface Sci., 2015, 456, 100–107 CrossRef CAS PubMed.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CrossRef CAS.
- M. Ma, J. Cao, A. Fang, Z. Xu, T. Zhang and F. Shi, Front. Chem., 2022, 9, 812083, DOI:10.3389/fchem.2021.812083.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc01567j |
| This journal is © The Royal Society of Chemistry 2023 |