
Multifunctional pseudohalide-based ionic liquid doping promotes efficient and stable perovskite solar cells†
Xing
Guo
a,
Zhenhua
Lin
 *ab,
Wenying
Cao
b,
Yumeng
Xu
b,
Qingrui
Wang
b,
Boyao
Zhang
b,
Yue
Hao
b and
Jingjing
Chang
*ab
*ab,
Wenying
Cao
b,
Yumeng
Xu
b,
Qingrui
Wang
b,
Boyao
Zhang
b,
Yue
Hao
b and
Jingjing
Chang
*ab
aAdvanced Interdisciplinary Research Center for Flexible Electronics, Academy of Advanced Interdisciplinary Research, Xidian University, 710071, Xi’an, China. E-mail: zhlin@xidian.edu.cn; jjingchang@xidian.edu.cn
bState Key Discipline Laboratory of Wide Band Gap Semiconductor Technology, School of Microelectronics, Xidian University, 710071, Xi’an, China
First published on 1st June 2023
Abstract
High-quality perovskite films are crucial to realizing high-efficiency stable perovskite solar cells. Doping is a direct and effective strategy to passivate defects and improve the crystal quality of perovskite films. Herein, we introduce pseudohalide-based ionic liquid TEAPF6 into perovskite films as an excellent additive. TEAPF6 doping has multiple effects: the PF6− anion could fill the halide vacancies, which helps to suppress ion migration through vacancies. The highly electronegative PF6− could bind with uncoordinated Pb2+ and organic cations to passivate defects. TEAPF6 doping improved the crystal quality and suppressed the charge recombination in perovskite films. More importantly, the TEA+ with long alkyl groups and PF6− could enhance the hydrophobicity of the perovskite films. As a result, the device with TEAPF6 achieved a PCE of 22.13%. Moreover, TEAPF6 doping enhanced the stability of PSCs. This work demonstrates the excellent effect of pseudohalide-based ionic liquid doping on improving the crystal quality of perovskite films.
 Xing Guo | Dr Xing Guo obtained his PhD degree in Microelectronics and Solid State Electronics from Xidian University in 2021. In 2021, he joined Academy of Advanced Interdisciplinary Research, Xidian University as a lecturer. His current research focuses on organic–inorganic hybrid perovskite solar cells, and energy conversion and storage device integration. |
 Zhenhua Lin | Prof. Zhenhua Lin received her PhD degree from the National University of Singapore in 2013. After graduation, she was a Research Fellow in Electrical and Computer Engineering, National University of Singapore from 2013 to 2014. She joined Xidian University, where she is currently a professor. Her research interests include perovskite and metal oxide materials, electronic devices (transistors and RRAM) and optoelectronic devices (solar cells, photodetectors, and X-ray detectors). |
1. Introduction
Over the past few years, metal halide perovskites have been extensively studied due to their tunable bandgap, strong light absorption, long carrier diffusion length, and low-temperature solution processing.1–5 Since 2009, the power conversion efficiency (PCE) of perovskite solar cells (PSCs) has rocketed from 3.8% to 25.7%.6–12 This efficiency has already met the requirements of commercial applications, but it is still far from reaching the theoretical limit of efficiency.13,14 It also continues to be challenging for PSCs to achieve the necessary stability for commercialization. High-quality perovskite films are the key to improving the efficiency and stability of PSCs. It is well known that perovskite films prepared by the solution process are polycrystalline, and contain a large number of defects. The defects as the recombination center will cause nonradiative recombination of the carrier. On the other hand, ion migration between defects under an electric field also causes perovskite decomposition. Therefore, defect passivation of perovskite films is an effective strategy to further improve the efficiency and stability of PSCs.15–19Ionic liquids (ILs) are a class of low-melting salts composed of anions and cations, and can achieve different properties by adjusting the combination of anions and cations. ILs have been widely used in perovskite defect passivation due to their excellent electrical conductivity and stability.20–26 Liu et al. doped CsPbI3 perovskites with 1-ethyl-3-methylimidazolium hydrogen sulfate (EMIMHSO4) ILs to manage the defects, energy alignment, and stability of perovskite films fabricated via blade-coating.27 The PSCs doped with EMIMHSO4 achieved a PCE of 20.01%, and the encapsulated PSCs retained 95% of their initial PCE after 1000 hours under ambient conditions. Akin et al. introduced 1-hexyl-3-methylimidazolium iodide (HMII) ILs as an additive into FAPbI3 perovskites.28 They found that HMII ILs could facilitate the grain coarsening of FAPbI3 crystals and reduce the grain-boundary migration activation energy. The grain size of HMII-doped perovskites reached the micrometer scale. The PSCs with HMII achieved a PCE of 20.6% with a notable increased open-circuit voltage (VOC) of about 80 mV. Moreover, the HMII-doped PSCs retained over 80% and about 90% of their initial PCE at 60 ± 10% relative humidity and 65 °C, respectively. Due to the high chemical activity, the I− in perovskite films is prone to form vacancies and other defects. The defect density, crystal quality, and stability of perovskite films can be effectively regulated by introducing other halide anions such as Br− and Cl−.29 Besides, some pseudohalide anions with similar properties to halide anions, such as BF4−, PF6−, SCN−, NO3−, and CH3COO−, are also introduced into perovskites.20,30–32 Among them, BF4− and PF6− not only have a similar ionic radius to I− but also have stronger electronegativity than single halide anions, which could substitute the I− and suppress the defects related to halides. ILs with pseudohalide anions can combine multiple advantages to improve the properties of perovskite films. Snaith et al. incorporated 1-butyl-3-methylimidazolium tetrafluoroborate (BMIMBF4) ILs into perovskites thereby increasing the efficiency and long-term stability of PSCs.33 The most stable packaged PSCs degraded by only about 5% over 1800 hours of continuous simulated full-spectrum sunlight at 70 to 75 °C, and they also estimated that it will take about 5200 hours for the PSCs to drop to 80% of their peak performance. Furthermore, they doped perovskite films with BMPBF4 ILs and found that BMPBF4 ILs could retard impurity phase inversion and pinhole formation during aging.34 The unencapsulated and encapsulated PSCs retained 80% and 95% of their peak PCE for 1010 and 1200 hours under full-spectrum simulated sunlight in an ambient atmosphere.
In this work, we introduced a pseudohalide-based IL tetraethylammonium hexafluorophosphate (TEAPF6) into Cs0.05MA0.68FA0.27PbI3−xClx perovskite films as a multifunctional additive. TEAPF6 contains a large structurally stable organic cation (TEA+) and a pseudohalide anion (PF6−). The PF6− could fill the halide vacancies and bond with Pb2+ and organic cations. The TEA+ with long alkyl groups and PF6− could enhance the hydrophobicity of the perovskite film. The TEAPF6 additive has an excellent effect on improving the crystal quality and effectively passivating the defects of perovskite films. As a result, the PSCs with TEAPF6 achieved the highest PCE of 22.13% with a significantly increased VOC from 1.10 V to 1.15 V. Moreover, TEAPF6 ILs increased the stability of the PSCs.
2. Results and discussion
Fig. S1 (ESI†) shows the chemical structure of the TEAPF6 IL, and the TEAPF6 contains an organic cation (TEA+) and a pseudohalide anion (PF6−). The TEA+ cation has four alkyl groups, which are larger than MA+, FA+, and Cs+ cations in perovskites. The pseudohalide anion PF6− has similar properties and ionic radius to I− in the perovskite. The perovskite film acts as both a light absorption layer and a charge transport layer, and its surface morphology, crystalline quality, and other characteristics have a great impact on the efficiency and stability of PSCs. We first clarified the effect of TEAPF6 doping on the morphology of perovskite films, and we measured the top-view and cross-sectional scanning electron microscopy (SEM) images of perovskite films without and with TEAPF6. As shown in Fig. 1a–d and Fig. S2 (ESI†), the grain size of the perovskite films without and with TEAPF6 is similar. The pristine perovskite film shows wider grain boundaries, and the cross-section of the pristine perovskite film is unordered and has some holes, which may lead to charge recombination and device leakage, and it is also easier for water and oxygen to enter the interior and cause the decomposition of the perovskite film. While the perovskite films with TEAPF6 have denser grains and smaller grain boundaries, the cross-section exhibits more compact and vertically aligned crystal growth, which is beneficial to reducing the charge recombination at the grain boundaries and suppressing the decomposition of the perovskite film.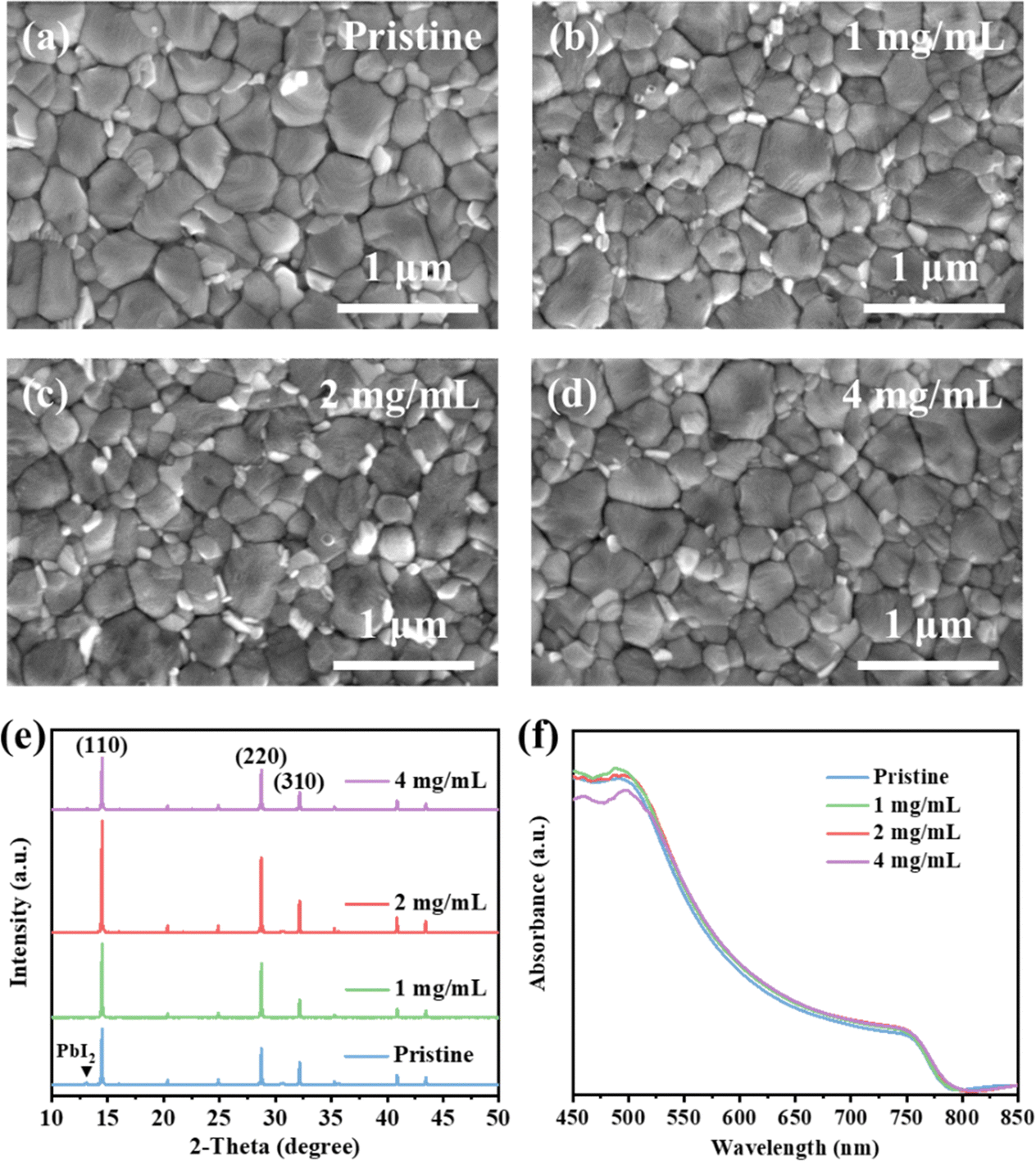 | ||
| Fig. 1 (a)–(d) SEM images of perovskite films with different concentrations of TEAPF6. (e) XRD pattern and (f) UV-vis spectra of perovskite films with different concentrations of TEAPF6. | ||
We further characterized the effect of the TEAPF6 additive on the crystal quality of perovskite films by X-ray diffraction (XRD). As shown in Fig. 1e, the perovskite films doped with different concentrations of TEAPF6 show three significant XRD diffraction peaks at 2θ of 14.49°, 28.73°, and 32.15°, corresponding to the (110), (220), and (310) crystal planes of the perovskite film, respectively.35–37 The perovskite films with TEAPF6 do not show any new peaks in the XRD patterns compared to the pristine film, indicating that the TEAPF6 additive did not change the crystal structure of the perovskite film, which is due to that the larger TEA+ cation cannot be embedded into the lattice of the perovskite. This phenomenon has also been demonstrated by Yang et al.38 For the perovskite films with 1 mg mL−1 and 2 mg mL−1 TEAPF6, the XRD diffraction peak intensity is enhanced compared to that of the pristine perovskite film, and the XRD peak intensity of the perovskite film with 2 mg mL−1 TEAPF6 is the highest. Meanwhile, the full width at half maximum (FHWM) of the (110) peak decreases slightly after TEAPF6 doping, as shown in Fig. S3 (ESI†). The increased XRD peak intensity and decreased FHWM indicate that TEAPF6 doping could improve the crystal quality of the perovskite film, which helps to enhance light absorption, reduce defect density, and reduce charge recombination. Notably, TEAPF6 doping can suppress the formation of PbI2 in perovskite films, corresponding to the disappearance of the XRD diffraction peak at a 2θ of 13.08°, which contributes to improving the stability of the perovskite film.9 When the doping concentration of TEAPF6 is increased to 4 mg mL−1, the XRD peak intensity of the perovskite film is decreased and the peak intensity of PbI2 is increased, indicating that the crystal quality of the perovskite film is decreased.
The crystal quality of the perovskite film affects its optical properties, and we further recorded the ultraviolet-visible (UV-vis) absorption spectra of perovskite films without and with TEAPF6. As shown in Fig. 1f, TEAPF6 doping enhances the light absorption of the perovskite films, which is associated with the improved morphology of the perovskite films after TEAPF6 doping. The enhanced light absorption is beneficial to improving the short-circuit current of the device. The bandgap of the perovskite can be calculated from UV-vis spectra, as shown in Fig. S4 (ESI†). The bandgap (Eg) of the perovskite slightly decreases from 1.58 eV to 1.57 eV after TEAPF6 doping, which is due to the decreased PbI2 in the perovskite film after TEAPF6 doping.
To investigate the charge recombination of the perovskite film without and with TEAPF6, we obtained the steady-state photoluminescence (PL) and time-resolved PL (TR-PL) spectra. As shown in Fig. 2a, the PL intensity of the perovskite film with 1 mg mL−1 and 2 mg mL−1 TEAPF6 is increased compared to that of the pristine perovskite film. The increased PL intensity indicates the decreased defects and suppressed charge recombination in perovskite films after TEAPF6 doping. As the doping concentration of TEAPF6 is further increased, the PL intensity of perovskite films is decreased, which is due to the deteriorated crystal quality of the perovskite film, corresponding to the XRD results. We also carried out the PL mapping measurements as shown in Fig. 2b and c and Fig. S5 (ESI†). The TEAPF6-doped perovskite film shows higher PL intensity and lifetime compared to the pristine perovskite film, indicating the improved film quality. Fig. 2d shows the TR-PL spectra of perovskite films with different concentrations of TEAPF6. The curves are fitted by a double exponential decay model as reported in our previous work,39,40 and the fitting results are listed in Table S1 (ESI†). The pristine perovskite film exhibits a carrier lifetime of 52.95 ns. After TEAPF6 doping, the carrier lifetime increases to 109.68 ns, 176.87 ns, and 145.41 ns for the perovskite film with 1 mg mL−1, 2 mg mL−1, and 4 mg mL−1 TEAPF6 doping, respectively. The TR-PL results of the perovskite films correspond to their PL results, indicating that TEAPF6 doping can suppress the charge recombination in perovskite films caused by defects.
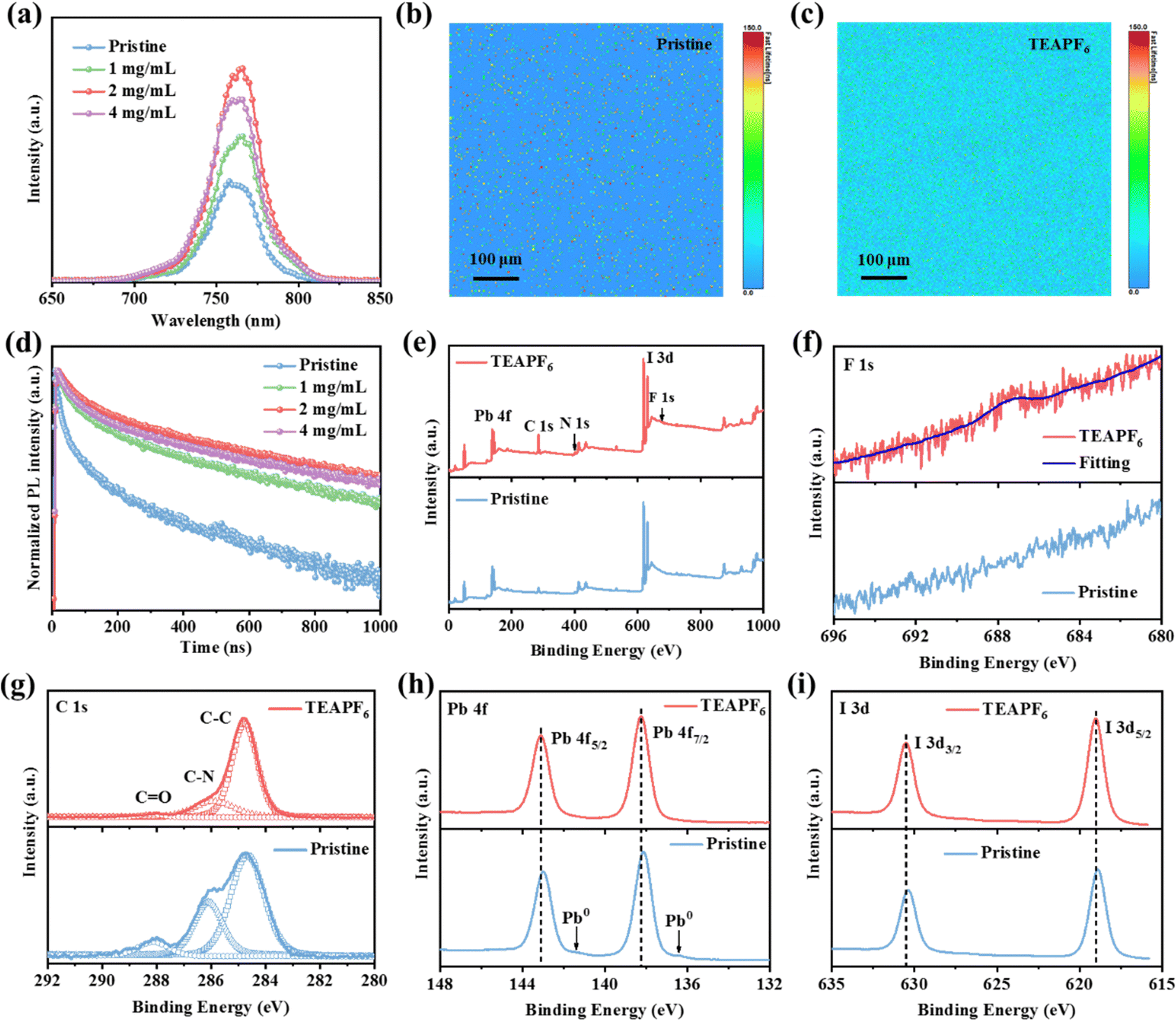 | ||
| Fig. 2 (a) Steady-state PL spectra of perovskite films. PL mapping of (b) pristine and (c) TEAPF6 doped perovskite films (2 mg mL−1). (d) TR-PL spectra of perovskite films. (e) The full XPS spectra of perovskite films without and with TEAPF6. The high-resolution XPS spectra of (f) F 1s, (g) C 1s, (h) Pb 4f, and (i) I 3d of perovskite films. | ||
To clarify the interaction between TEAPF6 and the perovskite, the X-ray photoelectron spectra (XPS) of perovskite films without and with TEAPF6 are obtained. Fig. 2e and f show the full XPS spectra and F1s spectra of perovskite films without and with TEAPF6, respectively. The presence of the F1s peak indicates the successful incorporation of TEAPF6 into the perovskite film. As shown in Fig. 2g, the C 1s spectra contain three peaks corresponding to the C–C, C–N, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks. Among them, the CO peak is considered to be related to the decomposition of organic cations in the perovskite film under the action of oxygen and moisture. The CO peak of the perovskite film is suppressed after TEAPF6 doping, indicating that TEAPF6 doping could improve the stability of the perovskite film.17,41Fig. 2h and i show the Pb 4f and I 3d XPS peaks. The Pb 4f peaks located at 138.13 eV and 143.00 eV shift to higher binding energies of 138.26 eV and 143.13 eV, respectively. The two peaks of I 3d shift from 618.87 eV and 630.38 eV to higher binding energies of 619.00 eV and 630.54 eV, respectively. The shift of Pb 4f and I 3d is because of the strong interaction between TEAPF6 and the perovskite film. The PF6− can form a covalent bond with Pb2+ and hydrogen bonds with organic cations due to its strong electronegativity. On the other hand, PF6− can fill halide vacancies in the perovskite film. The strong interaction can suppress the ion migration caused by defects in the perovskite film. It is worth noting that the Pb 4f spectra of the pristine perovskite film exhibit two peaks corresponding to Pb0, and the Pb0 in the perovskite film affect the optoelectronic characteristics and stability of the perovskite film.42 The Pb0 peak disappeared in the perovskite film with TEAPF6, indicating that TEAPF6 doping could suppress the formation of Pb0 defects. Furthermore, we recorded the Fourier-transform infrared spectra (FTIR) to confirm the interaction between the PF6− and perovskite, as shown in Fig. S6 (ESI†). The TEAPF6 film exhibits a peak at a wavenumber of 863.49 cm−1, which corresponds to the P–F peak;43 the TEAPF6-doped perovskite film also shows a P–F peak at 828.77 cm−1, which confirms the presence of PF6− in the perovskite film; the shift of P–F stretching vibration mode peaks indicates a strong interaction between the PF6− and perovskite. Moreover, the red-shift of the C–H bending vibration (from 1456.98 cm−1 to 1443.96 cm−1) is due to the formation of hydrogen bonds caused by the large electronegativity of PF6−.44
O peaks. Among them, the CO peak is considered to be related to the decomposition of organic cations in the perovskite film under the action of oxygen and moisture. The CO peak of the perovskite film is suppressed after TEAPF6 doping, indicating that TEAPF6 doping could improve the stability of the perovskite film.17,41Fig. 2h and i show the Pb 4f and I 3d XPS peaks. The Pb 4f peaks located at 138.13 eV and 143.00 eV shift to higher binding energies of 138.26 eV and 143.13 eV, respectively. The two peaks of I 3d shift from 618.87 eV and 630.38 eV to higher binding energies of 619.00 eV and 630.54 eV, respectively. The shift of Pb 4f and I 3d is because of the strong interaction between TEAPF6 and the perovskite film. The PF6− can form a covalent bond with Pb2+ and hydrogen bonds with organic cations due to its strong electronegativity. On the other hand, PF6− can fill halide vacancies in the perovskite film. The strong interaction can suppress the ion migration caused by defects in the perovskite film. It is worth noting that the Pb 4f spectra of the pristine perovskite film exhibit two peaks corresponding to Pb0, and the Pb0 in the perovskite film affect the optoelectronic characteristics and stability of the perovskite film.42 The Pb0 peak disappeared in the perovskite film with TEAPF6, indicating that TEAPF6 doping could suppress the formation of Pb0 defects. Furthermore, we recorded the Fourier-transform infrared spectra (FTIR) to confirm the interaction between the PF6− and perovskite, as shown in Fig. S6 (ESI†). The TEAPF6 film exhibits a peak at a wavenumber of 863.49 cm−1, which corresponds to the P–F peak;43 the TEAPF6-doped perovskite film also shows a P–F peak at 828.77 cm−1, which confirms the presence of PF6− in the perovskite film; the shift of P–F stretching vibration mode peaks indicates a strong interaction between the PF6− and perovskite. Moreover, the red-shift of the C–H bending vibration (from 1456.98 cm−1 to 1443.96 cm−1) is due to the formation of hydrogen bonds caused by the large electronegativity of PF6−.44
Next, we selected the pristine and 2 mg mL−1 TEAPF6 doped perovskite films to further study the effect of TEAPF6 doping on the defect density of perovskite films. We measured the defect density of the perovskite film without and with TEAPF6 by the space charge limited current (SCLC) method. The electron-only device with a structure of ITO/SnO2/MAFA (TEAPF6)/PCBM/Ag was fabricated. Fig. S7 (ESI†) shows the dark current–voltage (I–V) curves of devices without and with TEAPF6. The defect density is defined by the equation: Ndefect = (2εε0VTFL)/(eL2), where ε is the dielectric constant of the perovskite, ε0 is the vacuum dielectric constant, VTFL is the trap-filled limit voltage obtained from I–V curves, e is the electron charge, and L is the thickness of the perovskite film. The VTFL decreases from 0.93 V to 0.15 V after TEAPF6 doping, corresponding to the decreased Ndefect of the perovskite film from 2.68 × 1015 cm−3 to 4.32 × 1014 cm−3. The decreased defect density is due to that TEAPF6 doping improved the crystal quality and passivated the defects of the perovskite film, which will be conducive to suppressing the charge recombination.
To investigate the effect of TEAPF6 doping on the PSC performance, we fabricated planar PSCs with a structure of ITO/SnO2/perovskite (TEAPF6)/Spiro-OMeTAD/Ag. The details of the fabrication process are shown in the Experimental section in the ESI.†Fig. 3a shows the current density–voltage (J–V) curves of PSCs with different concentrations of TEAPF6 doping, and the corresponding parameters are listed in Table S2 (ESI†). The pristine PSCs exhibit a champion PCE of 20.23%, with a JSC of 23.89 mA cm−2, a VOC of 1.10 V, and an FF of 0.77. In contrast, the PSCs with 2 mg mL−1 TEAPF6 achieves the highest PCE of 22.13%, with a JSC of 24.26 mA cm−2, a VOC of 1.15 V, and an FF of 0.79. The PSCs with 1 mg mL−1 TEAPF6 exhibit a slightly increased average JSC and FF compared to the pristine PSCs due to the low doping concentration. When the doping concentration of TEAPF6 is further increased, the average PCE of PSCs is decreased, which is because the high concentration of TEAPF6 leads to the deterioration of the perovskite film quality and improvement of charge recombination, as proved by XRD and TR-PL. The increase in the PCE is mainly due to a significant increase in VOC and a slight increase in JSC and FF. The increased JSC and FF are due to the improved crystal quality and reduced grain boundary. The increased VOC is mainly related to passivated defects by TEAPF6 doping. Fig. 3b shows the forward and reverse J–V curves of the PSCs without and with 2 mg mL−1 TEAPF6. It is worth noting that hysteresis is suppressed after TEAPF6 doping. As listed in Table S3 (ESI†), the hysteresis index (HI) of PSCs, calculated using HI = (PCEreverse-PCEforward)/PCEreverse, decreases from 21.65% to 14.23% after TEAPF6 doping. Research shows that hysteresis is related to defects and ion migration,45 and the suppressed hysteresis indicates that TEAPF6 doping could effectively passivate defects in the perovskite film. To further investigate the effect of TEAPF6 doping on JSC, the external quantum efficiency (EQE) spectra are recorded. As shown in Fig. 3c, the integrated JSC of PSCs increases from 22.42 mA cm−2 to 23.41 mA cm−2 after TEAPF6 doping, which is consistent with the J–V measurements. The steady-state output of PSCs at maximum power point is also measured, as shown in Fig. 3d. The device with TEAPF6 exhibits a steady-state current density of 20.53 mA cm−2, which is higher than the 20.31 mA cm−2 of the pristine device. Moreover, the photocurrent of the device with TEAPF6 shows a shorter stabilization time compared to that of the pristine device, which is consistent with suppressed hysteresis in the TEAPF6-doped device.
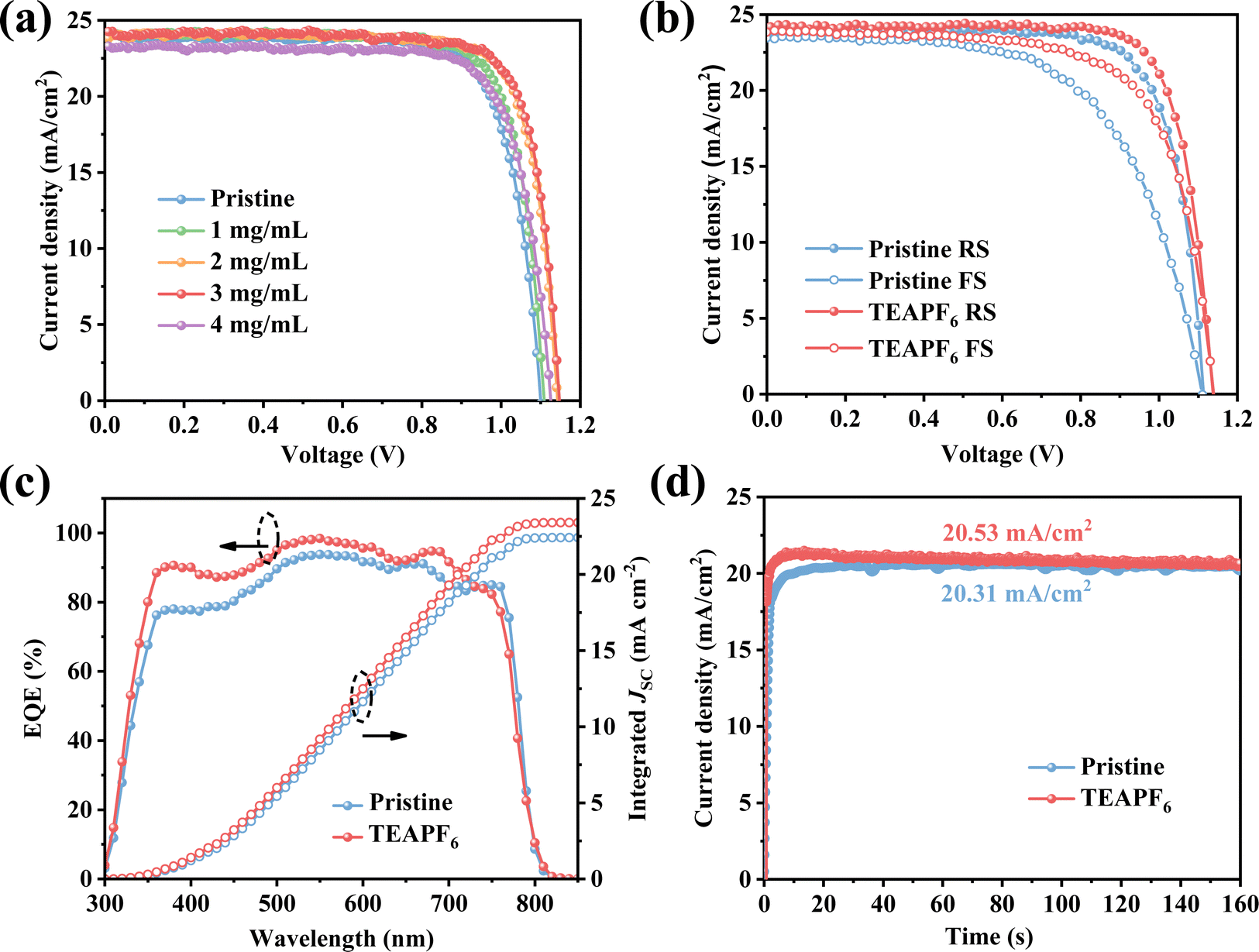 | ||
| Fig. 3 (a) J–V curves of PSCs with different concentrations of TEAPF6. (b) J–V curves in different scan directions. (c) EQE spectra and (d) steady-state output JSC of PSCs. | ||
To analyze the effect of TEAPF6 doping on charge transfer and recombination in the device, we carried out the transient photovoltage (TPV) and transient photocurrent (TPC) measurements. The photovoltage decay lifetime is associated with the charge recombination process in the device.46,47 As shown in Fig. 4a, the photovoltage decay time of the device increases from 18.39 μs to 81.05 μs after TEAPF6 doping, indicating that TEAPF6 doping can passivate defects and suppress the charge recombination in the device, which will contribute to improving the VOC of the device. The photocurrent decay lifetime is associated with the charge transfer process in the device. As shown in Fig. 4b, the photocurrent decay time of the device decreases from 1.45 μs to 1.11 μs after TEAPF6 doping, indicating that TEAPF6 can improve the charge extraction and transfer in the device, which helps to improve the JSC of the device. Fig. 4c shows the dark J–V curves of devices, and the dark current density of the device after TEAPF6 doping is 1 order of magnitude lower than that of the pristine devices. Dark current is related to charge recombination. The lower dark current further demonstrates the suppressed charge recombination by TEAPF6 doping. The relationship between VOC and light intensity (P) is also used to clarify the charge recombination process in devices. As shown in Fig. 4d, VOC and P exhibit a linear relationship that obeys the equation: VOC = nKT/q![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(P), where K is the Boltzmann constant, T is the absolute temperature, and q is the elementary charge, respectively. The slope of VOC–P curves (nKT/q) reflects the nonradiative recombination in the device.48 The slope of the device with TEAPF6 (1.27 KT/q) is much lower than that of the undoped device (1.45 KT/q), indicating that TEAPF6 doping can suppress the nonradiative recombination in the device. The electrical impedance spectra (EIS) are also recorded to investigate the charge transfer and recombination in the device. Fig. 4e shows the Nyquist plots of the device without and with TEAPF6; the curves are fitted by the equivalent circuit shown in Fig. S8 (ESI†), and the fitting parameters are listed in Table S4 (ESI†). The pristine device shows a series resistance (RS) of 60.93 Ω and a recombination resistance of (Rrec) of 2.08 × 106 Ω, and the device with TEAPF6 shows a RS of 41.48 Ω and a Rrec of 3.09 × 106 Ω. The increased Rrec indicates that TEAPF6 doping effectively suppresses charge recombination in PSCs. We further carried out the capacitance–voltage (C–V) measurement to clarify the effect of TEAPF6 doping on built-in potential (Vbi). The relationship between capacitance and Vbi obeys the Mott–Schottky formula.17,49 As shown in Fig. 4f, the Vbi of the device increases from 0.98 V to 1.04 V after TEAPF6 doping. The increased Vbi of the device after TEAPF6 doping is due to the suppressed charge recombination and helps to improve the charge extraction and transfer in the device.
ln(P), where K is the Boltzmann constant, T is the absolute temperature, and q is the elementary charge, respectively. The slope of VOC–P curves (nKT/q) reflects the nonradiative recombination in the device.48 The slope of the device with TEAPF6 (1.27 KT/q) is much lower than that of the undoped device (1.45 KT/q), indicating that TEAPF6 doping can suppress the nonradiative recombination in the device. The electrical impedance spectra (EIS) are also recorded to investigate the charge transfer and recombination in the device. Fig. 4e shows the Nyquist plots of the device without and with TEAPF6; the curves are fitted by the equivalent circuit shown in Fig. S8 (ESI†), and the fitting parameters are listed in Table S4 (ESI†). The pristine device shows a series resistance (RS) of 60.93 Ω and a recombination resistance of (Rrec) of 2.08 × 106 Ω, and the device with TEAPF6 shows a RS of 41.48 Ω and a Rrec of 3.09 × 106 Ω. The increased Rrec indicates that TEAPF6 doping effectively suppresses charge recombination in PSCs. We further carried out the capacitance–voltage (C–V) measurement to clarify the effect of TEAPF6 doping on built-in potential (Vbi). The relationship between capacitance and Vbi obeys the Mott–Schottky formula.17,49 As shown in Fig. 4f, the Vbi of the device increases from 0.98 V to 1.04 V after TEAPF6 doping. The increased Vbi of the device after TEAPF6 doping is due to the suppressed charge recombination and helps to improve the charge extraction and transfer in the device.
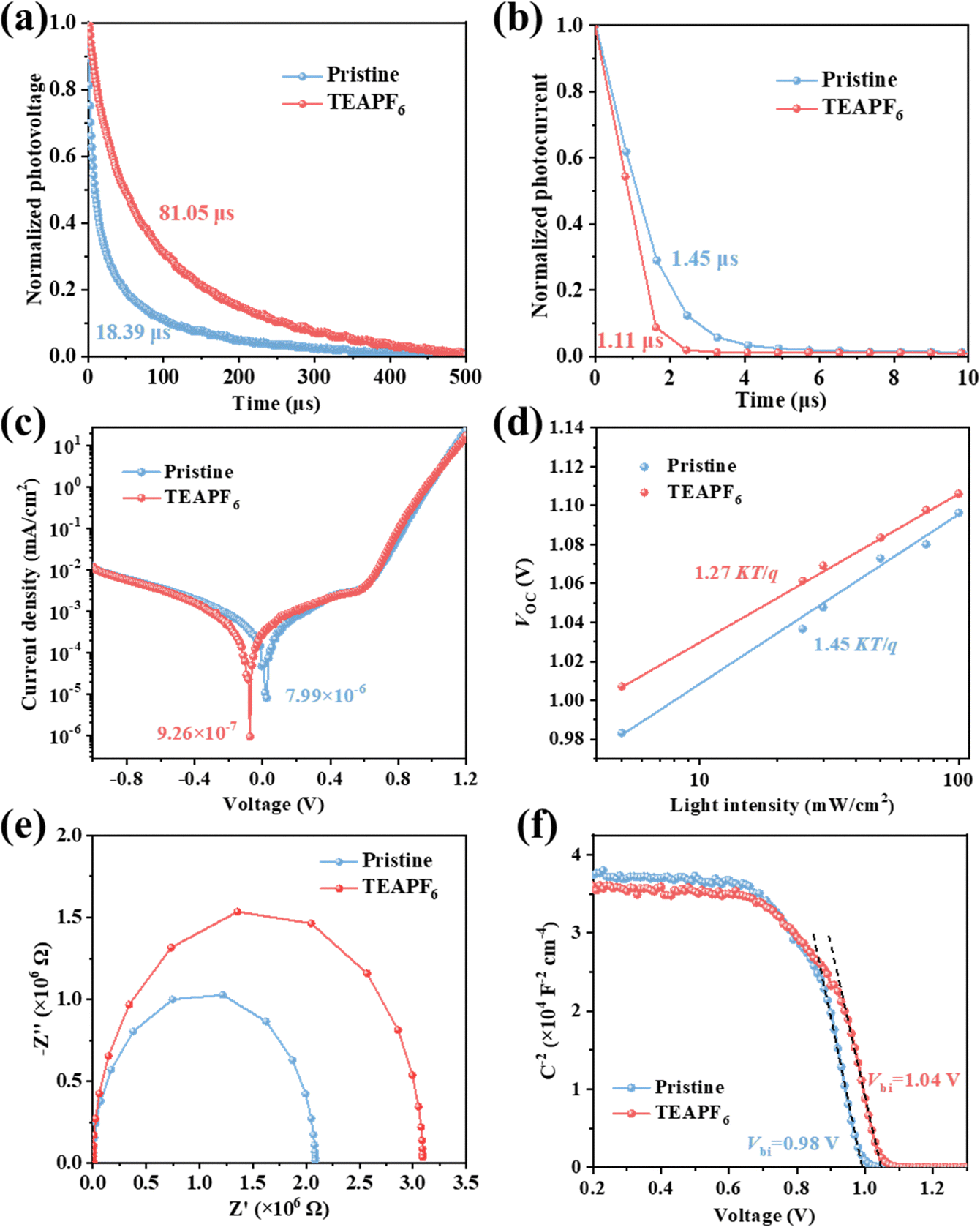 | ||
| Fig. 4 (a) TPV and (b) TPC measurements, (c) dark J–V measurements, (d) VOC as a function of light intensity, (e) EIS measurements, and (f) C–V measurements of the PSCs without and with TEAPF6. | ||
The stability of PSCs is critical for their commercial application. The stability of PSCs is not only affected by the properties of the perovskite itself but also related to external factors such as moisture, oxygen, and heat. We have demonstrated that TEAPF6 can improve the crystal quality of perovskite films and passivate defects. The pseudohalide PF6− can bond with Pb2+ and organic cations and fill the halide vacancies, which can enhance the crystal structure of perovskite films. Moreover, the larger TEA+ cation cannot be embedded into the lattice of the perovskite but can be absorbed onto the perovskite surface by a hydrogen bond, thus the hydrophobic TEA+ and PF6− help to improve the stability of the perovskite in moisture. First, we measured the water contact angle of perovskite films without and with TEAPF6. As shown in Fig. 5a and b, the water contact angle of the perovskite films is improved from 52.53° to 67.73° after TEAPF6 doping. The improved hydrophobicity prevents perovskite decomposition caused by water in the environment. Then, the PSCs without encapsulation are stored in an ambient environment with a relative humidity of 40% at room temperature in the dark. As shown in Fig. 5c, the pristine device retains 61% of its initial PCE after 750 h of storage, and the device with TEAPF6 retains a remarkable 86% of its initial PCE under the same conditions. The increased long-term stability of PSCs is due to the improved crystal quality and hydrophobicity of the perovskite film after TEAPF6 doping.
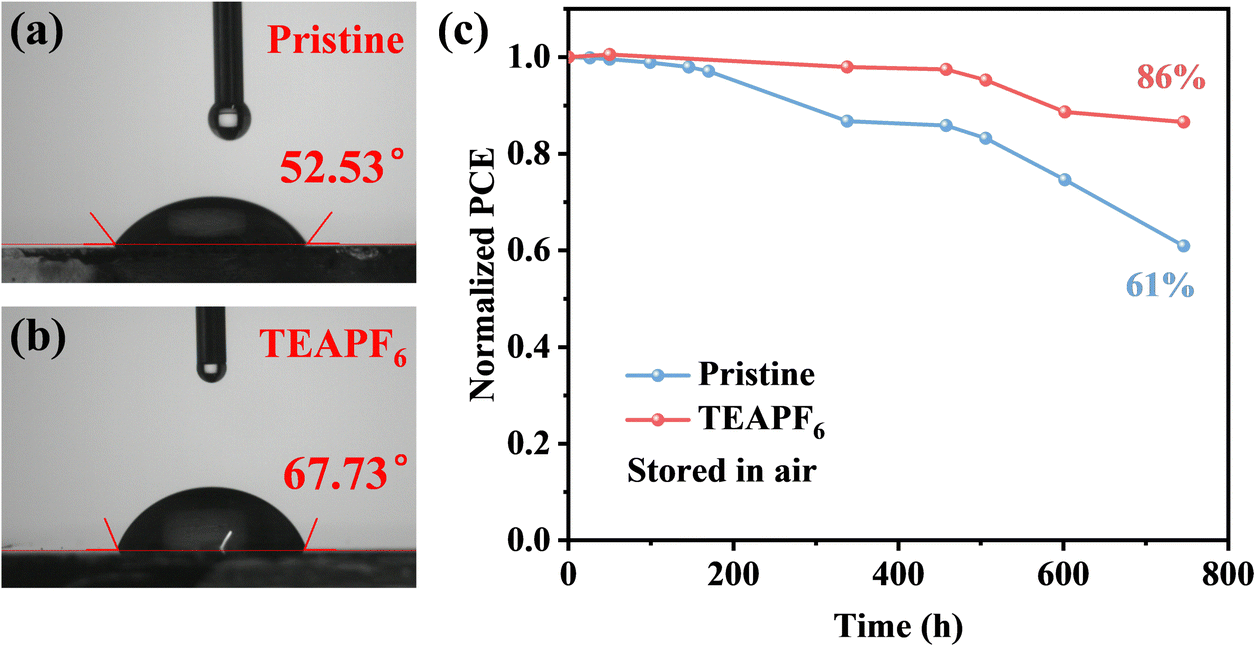 | ||
| Fig. 5 (a) and (b) The water contact angle of perovskite films without and with TEAPF6. (c) The long-term stability of PSCs. | ||
3. Conclusion
In conclusion, we have introduced the multifunctional IL TEAPF6 as an additive into the perovskite film. The pseudohalide PF6− can enhance the crystal structure and passivate the defects in the perovskite film by bonding with uncoordinated Pb2+ and the organic cation and filling the halide vacancies, which helps to suppress the ion migration and charge recombination in the perovskite film. Meanwhile, the TEAPF6 doping can also improve the hydrophobicity of the perovskite film. As a result, the PSCs with TEAPF6 achieve a PCE of 22.13%, with a significantly increased VOC from 1.10 V to 1.15 V. Remarkably, TEAPF6 doping enhances the stability of devices, and the unencapsulated device with TEAPF6 retains 86% of its initial PCE after aging in an ambient environment with a relative humidity of 40% for 750 h. This work provides an excellent strategy based on multifunctional pseudohalide-based ionic liquid doping to improve the efficiency and stability of PSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52192610, 62274127), the National Key Research and Development Program of China (2021YFA0715600, 2021YFA0717700, 2018YFB2202900), the Natural Science Basic Research Program of Shaanxi (2023-JC-QN-0681), Wuhu and Xidian University special fund for industry-university-research cooperation (XWYCXY-012021004), and Fundamental Research Funds for the Central Universities (XJS222214).References
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- L. Zhou, J. Chang, Z. Liu, X. Sun, Z. Lin, D. Chen, C. Zhang, J. Zhang and Y. Hao, Nanoscale, 2018, 10, 3053–3059 RSC.
- Z. Liu, J. Chang, Z. Lin, L. Zhou, Z. Yang, D. Chen, C. Zhang, S. (Frank) Liu and Y. Hao, Adv. Energy Mater., 2018, 8, 1703432 CrossRef.
- J. He, Q. Wang, Y. Xu, X. Guo, L. Zhou, J. Su, Z. Lin, J. Zhang, Y. Hao and J. Chang, Small, 2023, 19, 2205962 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- W. S. Yang, B. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. Il Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531–534 CrossRef CAS PubMed.
- M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J.-H. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 369, 1615–1620 CrossRef CAS PubMed.
- NREL, Best Research-Cell Efficiencies, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.pdf.
- J. Park, J. Kim, H.-S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2023, 1–3 Search PubMed.
- S. Rühle, Sol. Energy, 2016, 130, 139–147 CrossRef.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- L. Zhou, Z. Lin, Z. Ning, T. Li, X. Guo, J. Ma, J. Su, C. Zhang, J. Zhang, S. Liu, J. Chang and Y. Hao, Sol. RRL, 2019, 3, 1900293 CrossRef CAS.
- J. Ma, Z. Lin, X. Guo, L. Zhou, J. He, Z. Yang, J. Zhang, Y. Hao, S. Liu and J. Chang, J. Energy Chem., 2021, 63, 558–565 CrossRef CAS.
- X. Guo, X. Huang, J. Su, Z. Lin, J. Ma, J. Chang and Y. Hao, Chem. Eng. J., 2021, 417, 129184 CrossRef CAS.
- F. Gao, Y. Zhao, X. Zhang and J. You, Adv. Energy Mater., 2020, 10, 1902650 CrossRef CAS.
- R. Fan, W. Zhou, Z. Huang and H. Zhou, EnergyChem, 2020, 2, 100032 CrossRef.
- T. Niu, L. Chao, W. Gao, C. Ran, L. Song, Y. Chen, L. Fu and W. Huang, ACS Energy Lett., 2021, 6, 1453–1479 CrossRef CAS.
- M. Shahiduzzaman, E. Y. Muslih, A. K. M. Hasan, L. Wang, S. Fukaya, M. Nakano, M. Karakawa, K. Takahashi, M. Akhtaruzzaman, J.-M. Nunzi and T. Taima, Chem. Eng. J., 2021, 411, 128461 CrossRef CAS.
- L. Chao, T. Niu, Y. Xia, Y. Chen and W. Huang, Acc. Mater. Res., 2021, 2, 1059–1070 CrossRef CAS.
- M. Li, C. Zhao, Z. Wang, C. Zhang, H. K. H. Lee, A. Pockett, J. Barbé, W. C. Tsoi, Y. Yang, M. J. Carnie, X. Gao, W. Yang, J. R. Durrant, L. Liao and S. M. Jain, Adv. Energy Mater., 2018, 8, 1801509 CrossRef.
- D. He, T. Zhou, B. Liu, L. Bai, W. Wang, H. Yuan, C. Xu, Q. Song, D. Lee, Z. Zang, L. Ding and J. Chen, EcoMat, 2022, 4, 1–14 CrossRef.
- X. Zhu, M. Du, J. Feng, H. Wang, Z. Xu, L. Wang, S. Zuo, C. Wang, Z. Wang, C. Zhang, X. Ren, S. Priya, D. Yang and S. (Frank) Liu, Angew. Chem., Int. Ed., 2021, 60, 4238–4244 CrossRef CAS PubMed.
- Y. Wang, Y. Yang, N. Li, M. Hu, S. R. Raga, Y. Jiang, C. Wang, X. Zhang, M. Lira-Cantu, F. Huang, Y. Cheng and J. Lu, Adv. Funct. Mater., 2022, 32, 2204396 CrossRef CAS.
- Y. Du, Q. Tian, X. Chang, J. Fang, X. Gu, X. He, X. Ren, K. Zhao and S. (Frank) Liu, Adv. Mater., 2022, 34, 2106750 CrossRef CAS PubMed.
- S. Akin, E. Akman and S. Sonmezoglu, Adv. Funct. Mater., 2020, 30, 2002964 CrossRef CAS.
- N. Li, S. Tao, Y. Chen, X. Niu, C. K. Onwudinanti, C. Hu, Z. Qiu, Z. Xu, G. Zheng, L. Wang, Y. Zhang, L. Li, H. Liu, Y. Lun, J. Hong, X. Wang, Y. Liu, H. Xie, Y. Gao, Y. Bai, S. Yang, G. Brocks, Q. Chen and H. Zhou, Nat. Energy, 2019, 4, 408–415 CrossRef CAS.
- L. Chu, Matter, 2021, 4, 1762–1764 CrossRef CAS.
- J. Tao, X. Liu, J. Shen, S. Han, L. Guan, G. Fu, D. Kuang and S. Yang, ACS Nano, 2022, 16, 10798–10810 CrossRef CAS PubMed.
- H. Cheng, C. Liu, J. Zhuang, J. Cao, T. Wang, W. Wong and F. Yan, Adv. Funct. Mater., 2022, 32, 2204880 CrossRef CAS.
- S. Bai, P. Da, C. Li, Z. Wang, Z. Yuan, F. Fu, M. Kawecki, X. Liu, N. Sakai, J. T.-W. Wang, S. Huettner, S. Buecheler, M. Fahlman, F. Gao and H. J. Snaith, Nature, 2019, 571, 245–250 CrossRef CAS.
- Y. Lin, N. Sakai, P. Da, J. Wu, H. C. Sansom, A. J. Ramadan, S. Mahesh, J. Liu, R. D. J. Oliver, J. Lim, L. Aspitarte, K. Sharma, P. K. Madhu, A. B. Morales-Vilches, P. K. Nayak, S. Bai, F. Gao, C. R. M. Grovenor, M. B. Johnston, J. G. Labram, J. R. Durrant, J. M. Ball, B. Wenger, B. Stannowski and H. J. Snaith, Science, 2020, 369, 96–102 CrossRef CAS PubMed.
- B. Zhang, J. Su, X. Guo, L. Zhou, Z. Lin, L. Feng, J. Zhang, J. Chang and Y. Hao, Adv. Sci., 2020, 7, 1903044 CrossRef CAS PubMed.
- X. Guo, J. Du, Z. Lin, J. Su, L. Feng, J. Zhang, Y. Hao and J. Chang, Chem. Eng. J., 2021, 407, 127997 CrossRef CAS.
- X. Guo, X. Huang, J. Su, Z. Lin, J. Ma, J. Chang and Y. Hao, Chem. Eng. J., 2021, 417, 129184 CrossRef CAS.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef CAS.
- B. Zhang, J. Su, X. Guo, L. Zhou, Z. Lin, L. Feng, J. Zhang, J. Chang and Y. Hao, Adv. Sci., 2020, 7, 1903044 CrossRef CAS PubMed.
- L. Zhou, X. Guo, Z. Lin, J. Ma, J. Su, Z. Hu, C. Zhang, S. (Frank) Liu, J. Chang and Y. Hao, Nano Energy, 2019, 60, 583–590 CrossRef CAS.
- L. Zhou, J. Su, Z. Lin, X. Guo, J. Ma, T. Li, J. Zhang, J. Chang and Y. Hao, Research, 2021, 2021, 1–11 Search PubMed.
- L. Wang, H. Zhou, J. Hu, B. Huang, M. Sun, B. Dong, G. Zheng, Y. Huang, Y. Chen, L. Li, Z. Xu, N. Li, Z. Liu, Q. Chen, L.-D. Sun and C.-H. Yan, Science, 2019, 363, 265–270 CrossRef CAS PubMed.
- J. Tao, X. Liu, J. Shen, S. Han, L. Guan, G. Fu, D.-B. Kuang and S. Yang, ACS Nano, 2022, 16, 10798–10810 CrossRef CAS PubMed.
- Z. Li, Z. Li, G. Peng, C. Shi, H. Wang, S. Ding, Q. Wang, Z. Liu and Z. Jin, Adv. Mater., 2023, 2300480 CrossRef PubMed.
- C. Li, S. Tscheuschner, F. Paulus, P. E. Hopkinson, J. Kießling, A. Köhler, Y. Vaynzof and S. Huettner, Adv. Mater., 2016, 28, 2446–2454 CrossRef CAS PubMed.
- J. He, J. Su, J. Di, Z. Lin, S. Zhang, J. Ma, J. Zhang, S. Liu, J. Chang and Y. Hao, Nano Energy, 2022, 94, 106960 CrossRef CAS.
- X. Guo, J. Su, Z. Lin, X. Wang, Q. Wang, Z. Zeng, J. Chang and Y. Hao, iScience, 2021, 24, 102276 CrossRef CAS.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- L. Zhou, J. Su, Z. Lin, X. Guo, J. Ma, T. Li, J. Zhang, J. Chang and Y. Hao, Research, 2021, 2021, 1–11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc01028g |
| This journal is © The Royal Society of Chemistry 2023 |