
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New shades of photochromism – yellow sodalites for the detection of blue light†
Hannah
Byron
 ab,
Teppo
Kreivilä
a,
Pauline
Colinet
c,
Tangui
Le Bahers
cd and
Mika
Lastusaari
*a
ab,
Teppo
Kreivilä
a,
Pauline
Colinet
c,
Tangui
Le Bahers
cd and
Mika
Lastusaari
*a
aDepartment of Chemistry, University of Turku, FI-20014 Turku, Finland. E-mail: miklas@utu.fi
bUniversity of Turku Graduate School (UTUGS), Doctoral Programme in Exact Sciences (EXACTUS), FI-20014 Turku, Finland
cENSL, CNRS, UCBL, Laboratoire de Chimie, UMR 5182, 46 allée d’Italie, 69634, Lyon, France
dInstitut Universitaire de France, 5 rue Descartes, 75005 Paris, France
First published on 15th February 2023
Abstract
The absorption energy of the F-center in photochromic sodalite can be lowered by expanding the unit cell with larger ions, changing the color from pink to blue. Blue-shifting this absorption to produce other colors is not so straightforward. In this work, sodalites of the formula Na2−2xCax(Na,K)6(AlSiO4)6(Cl,S)2 displaying white-to-yellow photochromism have been prepared and thoroughly characterized. Combining spectroscopic experiments and quantum chemical calculations, the formation of a Na2Ca entity inside the sodalite cage surrounding the trapped electron responsible for the yellow color is postulated. Optimal yellow photochromism occurs for 0.13 ≤ x ≤ 0.18, while at x ≥ 0.27 the formation of the by-product davyne begins to affect the structure and optical properties. Finally, the sensitivity of these materials to blue light is shown to make them well-suited as sensors for blue light over-exposure e.g. from computer screens or smart phones.
1. Introduction
Photochromism describes the ability of a material to change color upon exposure to light. This phenomenon is perhaps most familiar in the context of photochromic eyeglasses, which darken in sunlight and fade again when the wearer steps indoors.1 The first photochromic materials to be discovered were organic molecules,2 and due to the versatility of organic chemistry and the low density of organic molecules, these have long since replaced their silver chloride counterparts in commercially available photochromic lenses.3 However, despite the high tunability of organic photochromic molecules, they are often sensitive to humidity, pH and extreme temperatures. Inorganic photochromic materials, on the other hand, are generally superior in terms of robustness. Perhaps the most famous of these is the aforementioned borosilicate glass doped with silver salts from Corning Inc.4 The color in these glasses arises from the formation of silver nanoparticles, similar to the Ag-doped TiO2 nanoparticles of Ohko et al.5,6 Tungsten and molybdenum oxides have also been popular inorganic photochromic materials showing a yellow-to-blue color change considered for smart windows.3,7–9 Other matrices and activators have been developed to bring inorganic photochromic materials up to par with their organic counterparts in terms of reversibility10,11 and diversity of color range,12–17 though the majority of these contain at least one so-called “endangered” element, whose availability will become uncertain in the near future:18 for example P, V, Cu, Zn, Sr, Y, Nb, Mo, Ag and W.Photochromic sodalites, sometimes called hackmanites, have been a topic of research for almost a century. Recent developments into their synthetic analogues, some of which also display attractive luminescence properties, have shown that they have potential to be developed for a huge range of possible applications. The unique combination of white-to-pink reversible photochromism sensitive to UV,19 X-rays20 and gamma radiation,21 along with orange and blue-white photoluminescence as well as persistent luminescence, means these materials could find future use as safety lighting,22 X-ray imaging solutions,20 radiation detectors,20,21 dosimeters,20,21,23 in diagnostics22 and in energy storage,22 to name just a few. These materials are also highly robust and do not require any expensive, toxic or endangered elements.
The mechanism of photochromism (tenebrescence) in these materials is well established under UV excitation: the interaction between a disulfide S22− impurity in the structure, which replaces a Cl− anion inside the sodalite cage, and a nearby chloride vacancy (VCl) caused by the need for charge neutrality. When excited with UV radiation, an electron transfers from S22− to VCl, where it forms an F-center that absorbs visible light.24 In chlorosodalite this F-center absorbs green light of around 500–540 nm, giving the colored form a pink shade. The effect of the unit cell size and thus the size of the chloride vacancy on the absorption spectrum of the F-center has been known since the 1970s, where Phillips substituted Cl for Br and I, and noticed as the unit cell expanded that the F-center absorbed longer wavelengths of light, shifting the colored form from pink to purple to blue.25 In 2010 Williams et al. utilized gallium and germanium to expand the unit cell, and observed the same trends.16 It is indeed straightforward to expand the unit cell with larger ions and decrease the absorption energy of the F-center, however to go in the other direction is more challenging. Fluoride sodalites are tricky to prepare due to the high melting points and stabilities of alkali metal fluorides, and smaller cations like lithium do not have enough of an effect on the unit cell to change the photochromism color dramatically.16,22 A more radical approach is required to blue-shift the F-center's absorption significantly.
Some members of the sodalite family are known to contain calcium: haüyne ((Na,K)3(Ca,Na)(Al3Si3O12)(SO4,S,Cl)) and lazurite (Na7Ca(Al6Si6O24)(SO4)(S3)·H2O) are calcium- and sulfur-containing members of the sodalite family which crystallize in the same P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3n space group as sodalite. Haüyne and lazurite are brightly colored by polysulfide anions, often giving an ultramarine blue color, as in lapis lazuli.26,27 There is no record of these minerals being photochromic, however this may be due to their intense body colors rendering any color change all but invisible. Also related to these are the minerals nepheline (Na3K(Al4Si4O16)), cancrinite ((Na,Ca,□)8(Al6Si6O24)(CO3,SO4)2·2H2O) and davyne ((Na,K)6Ca2(Al6Si6O24)(Cl2,SO4)2). These minerals crystallize in hexagonal space groups P63 (nepheline, cancrinite) and P63/m (davyne), and both nepheline and cancrinite-like by-products have been observed during the synthesis of sodalites and their analogues.28,29 It is thus expected that the introduction of calcium to the sodalite system could result in the formation of one or more of these phases.
3n space group as sodalite. Haüyne and lazurite are brightly colored by polysulfide anions, often giving an ultramarine blue color, as in lapis lazuli.26,27 There is no record of these minerals being photochromic, however this may be due to their intense body colors rendering any color change all but invisible. Also related to these are the minerals nepheline (Na3K(Al4Si4O16)), cancrinite ((Na,Ca,□)8(Al6Si6O24)(CO3,SO4)2·2H2O) and davyne ((Na,K)6Ca2(Al6Si6O24)(Cl2,SO4)2). These minerals crystallize in hexagonal space groups P63 (nepheline, cancrinite) and P63/m (davyne), and both nepheline and cancrinite-like by-products have been observed during the synthesis of sodalites and their analogues.28,29 It is thus expected that the introduction of calcium to the sodalite system could result in the formation of one or more of these phases.
In this work, we investigated the effect of introducing calcium to photochromic sodalites, and present for the first time their remarkable yellow tenebrescence when synthesized with the correct Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ca ratio. We go on to explain the mechanism of the yellow photochromism through the assistance of computational simulations, we determine the optimal calcium concentrations for yellow photochromism and discuss the detrimental effect too much calcium has on the structure and photochromic properties. Finally, we show that these sodalites can be used as blue light sensors to help people detect over-exposure to blue light, which has been reported to have adverse effects on e.g. eye health and the human circadian system.30,31
:Ca ratio. We go on to explain the mechanism of the yellow photochromism through the assistance of computational simulations, we determine the optimal calcium concentrations for yellow photochromism and discuss the detrimental effect too much calcium has on the structure and photochromic properties. Finally, we show that these sodalites can be used as blue light sensors to help people detect over-exposure to blue light, which has been reported to have adverse effects on e.g. eye health and the human circadian system.30,31
2. Experimental
2.1 Synthesis
Samples of nominal formula Na2−2xCax(Na,K)6(AlSiO4)6(Cl,S)2 were synthesized using 0.7 g of dry zeolite 4A (Sigma Aldrich) or dry zeolite 3A (Zeochem), 0.06 g dry Na2SO4 (E. Merck, 99%), and stoichiometric amounts of NaCl (Sigma Aldrich, 99.5%) and CaCl2·6H2O (Riedel De Haën, 99%) corresponding to a total of 0.0040 mol. In a sample with x = 0.00, 0.0040 mol of NaCl was used; when x = 1.00, 0.0040 mol CaCl2·6H2O was used. For 0.00 < x < 1.00, the appropriate stoichiometric amounts of NaCl and CaCl2·6H2O were used. The starting materials were ground together by hand in an agate mortar and then placed in an oven, where they were heated in air to 850 °C for 5 h. After cooling, samples were ground again and placed in an oven, where they were heated to 850 °C in a reducing atmosphere of 88%N2/12%H2 for 2 h. Once cool, the samples were collected and analyzed. In some cases, NaBr (J. T. Baker), NaI (E. Merck), CaBr2 (British Drug Houses) and CaI2 (Alfa Aesar, 99.5%) were used in stoichiometric amounts instead of NaCl and CaCl2·6H2O.2.2 Analysis
The materials’ structure and purity were analyzed using room temperature X-ray powder diffraction (XRPD). XRPD was performed using a Panalytical Aeris benchtop instrument, with copper Kα1,2 radiation (λ = 1.5406 Å (Kα1) and 1.5444 Å (Kα2)) with a step size of 2θ = 0.011° and an integration time of 0.25 s per step. Some samples were measured multiple times and their diffractograms added to produce a good enough signal-to-noise ratio for Rietveld refinement with Panalytical HighScore Plus software connected to the ICDD PDF4+ 2021 database. Sample compositions were investigated with X-ray fluorescence spectroscopy (XRF) using a Panalytical Epsilon 1 device with internal Omnian calibration and a 50 kV Ag-anode X-ray tube. Results were compiled from four scans using a silver filter, a copper filter, an aluminum filter and no filter, respectively.The change of color of the materials after UV irradiation was investigated using reflectance measurements under illumination of a 40 W incandescent lamp, with an incident irradiance of 1.34 W m−2 in all measurements. The reflectance of the sample before excitation was measured and then used as the white background color. Samples were excited with 254 nm radiation from a UVP model UVLS-24 EL, 4 W 254/365 nm handheld lamp (UVC irradiance approx. 3.0 mW cm−2) and their reflectance measured again 5 s after the lamp was switched off to show the change in reflectance. Spectra were collected using an Avantes FC-IR600-1-ME-HTX optical fiber connected to an Avantes HS-TEC spectrometer with a data collection time of 1.6 s (160 ms integration time, 10 averages). The materials’ colors were quantified as L*a*b* coordinates using a Konica Minolta CM-2300d handheld spectrometer with its own white calibration disc.
Cumulative tenebrescence excitation spectra were measured by irradiating the sample with 340–220 nm radiation produced by a SB522 150 W Xe-arc lamp coupled to a LOT MSH 300 monochromator, for 7.5 minutes per wavelength. 10 nm intervals were used between irradiation wavelengths. The reflectance of the sample was measured using the same HS-TEC spectrometer and optical fiber as previously mentioned, but under the illumination of an OceanOptics LS-1-CAL incandescent torch. Reflectance was measured before irradiation and set as the white background; after irradiation reflectance was measured again. The reflectance wells were integrated to give the color intensity. Optical bleaching of tenebrescence was measured by first measuring the sample's reflectance and setting this as the white reference. The sample was then excited with 260 nm radiation from the SB522 150 W Xe-arc lamp coupled to a LOT MSH 300 monochromator for 5 minutes and its reflectance measured. The sample was allowed to sit in darkness for a further 5 min, after which its reflectance was measured to obtain the spontaneous fading in darkness of the sample. The sample was subsequently re-coloured using 260 nm radiation for 5 min, then bleached with light ranging from 300–680 nm. The cycle was repeated using 20 nm steps in the bleaching wavelength. The reflectance wells were integrated to give colour intensity, and the decrease in colour intensity caused by each bleaching wavelength relative to the previous intensity after 260 nm excitation was calculated accounting for spontaneous fading in darkness. These results produced the optical bleaching spectra. Reflectance was measured using the same HS-TEC spectrometer and optical fibre as previously mentioned under the illumination of a LS-1-CAL OceanOptics incandescent torch. The data collection time was 1.6 s (160 ms integration time, 10 averages).
Thermotenebrescence23,32 was measured using the same optical fiber and HS-TEC spectrometer as before, with a data collection time of 1 s (50 ms integration time, 20 averages). The sample's reflectance was measured before irradiation and used as the background. Similarly to before, the sample's reflectance was measured before irradiation as the white background, then measured again after 2 min of using a 254 nm handheld lamp as mentioned before (UVC irradiance approx. 2.2 mW cm−2). After coloration, the sample was then heated from 20–250 °C at a rate of 3 °C s−1 using a MikroLab Thermoluminescent Materials Laboratory Reader RA’04. Using the data collection time of 1 s, a reflectance spectrum was acquired every second during the heating. The reflectance signal was corrected to account for the effects of heating on an uncolored sample, and then the curves were integrated to calculate the color intensity at each temperature. The color intensity values were also corrected for spontaneous fading under white light. Color intensity with respect to temperature was plotted to obtain the thermotenebrescence curve. Thermotenebrescence data was handled in a similar way to thermoluminescence data to calculate the depth of the trap involved in thermal bleaching. The curves were analyzed and the trap depths calculated using the initial rise method.33
The rise of tenebrescence coloration were measured by constantly exciting samples with 254 nm radiation from a UVP model UVLS-24 EL, 4 W 254/365 nm handheld lamp (UVC irradiance approx. 1.3 mW cm−2) or with 302 nm radiation from a UVP model UVM-57, 6 W lamp (UVB irradiance approx. 1.3 mW cm−2) while measuring their reflectance under a 40 W incandescent lamp giving constant incident irradiance. In this case, MgO was used as the white reference. Reflectance spectra were measured using the same optical fiber coupled to the same HS-TEC spectrometer as before, with a 200 ms integration time and 10 averages. In this way, samples were irradiated for 60 minutes with data collected every two seconds. The integrals of the reflectance curves were used to give the color intensity.
The fade of tenebrescence was measured under incandescent lighting immediately after the rise of coloration by turning off the UV lamp and measuring the reflectance every two seconds for two hours. The fade of tenebrescence under an Airam 2700 K 9 W warm LED light, cool white LED ambient lighting, a blue LED from a North GLGID-02 RGB lamp, and a HP Compaq LA2306x WLED-backlit LCD computer monitor, were measured after 15 min of coloration under 254 nm UV in the same way. For fading curves, the white reference was the sample in its uncolored form. Irradiances of the light sources from 400–1050 nm were measured with a Delta Ohm HD 2102.1 photo/radiometer coupled to a LP 471 RAD irradiance meter. Dose correction was carried out for the pink and yellow F-center absorptions under incandescent light using the relative intensities of the incandescent lamp in those wavelength ranges; for the sodalite's entire absorption spectrum dose correction was done using the measured irradiances.
2.3 Computational simulations of F-center's absorption
During the computational study, only calcium substitutions of sodium atoms in the pristine sodalite of formula Na8(Al6Si6O24)Cl2 were considered.The methodology includes 3 main steps:
1. Optimization of the geometry of pristine sodalite's bulk in PBC, followed by the generation and optimization of the corresponding 2 × 2 × 2 supercell.
2. From this supercell, addition of the defect (F-center/trapped electron) at a Cl− site, as well as substitution of 1 to 4 of the sodium atoms surrounding this site by calcium ones, and optimization of the corresponding 2 × 2 × 2 supercells’ geometries. Three possibilities were considered in order to go from the unmodified [Na4VCl]3+ composition to: [Na3CaVCl]4+, [Na2CaVCl]3+ and [Ca2VCl]3+. They correspond to systems that will be referred to as Na3Ca, Na2Ca and Ca2 in opposition to the unmodified Na4 structure. In the case of Na3Ca, the addition of one positive charge was offset by the substitution of one Si4+ by one Al3+ in the surrounding β-cage.
3. Extraction of a cluster containing the β-cage and the corresponding modified “tetrahedron” structures for the simulation of the F-center's absorption with an embedded cluster approach. Atom positions are fixed to the ones of the geometry optimized in periodic boundary conditions.
Computational details are given in the following paragraphs:
All optimized structures obtained with the ab initio CRYSTAL1734 code were conducted at the DFT/PBE035 level of theory using localized basis sets. HF and KS equations were solved self-consistently with convergence criterion for the SCF cycles fixed at 10−7 Ha per unit cell. The reciprocal space was sampled according to a sublattice with a 12 × 12 × 12 k-points mesh for the geometry optimization of the bulk system, while a single k point (the Γ point) was used for the geometry optimization of the supercells. When considering the F-center in the structure (i.e. a trapped electron), calculations were performed in an unrestricted formalism to account for the singly-occupied orbital. A numerical frequency calculation was performed to assess the nature of the stable geometry found. This calculation was performed only on the sodium and calcium atoms since the β-cage is known to be very rigid and not affected by chemical modifications in the sodium tetrahedron.16,32
Localized basis set used in CRYSTAL PBC calculations was then kept for the F-center's absorption simulations. The basis set for the electron in the vacancy has been previously optimized and is of the form 111G(d).36 All-electron double-ζ basis sets with polarization functions were used for Si ([4s3p1d]/(20s12p1d)),37 Al ([4s3p1d]/(17s9p1d)),38 O ([3s2p1d]/(10s4p1d)),39 and Cl ([4s3p1d]/(16s10p1d)).40 All-electron triple-ζ basis sets with polarization functions were used for Na ([4s3p1d)]/(15s7p1d),41 and Ca ([6s4p1d]/(17s11p1d)).42
Spectroscopic investigations led on the extracted clusters (embedded in a sphere of pseudopotentials and an array of point charges),36 were done at the TD-DFT/B3LYP level with the same basis set as the one used for periodic boundary condition calculations with the Gaussian 16 package based on benchmark calculations.36,43
3. Results and discussion
3.1 Notes on the composition of the materials
The precise compositions and formulae of these materials are difficult to determine, due to the presence of defects in the structure. Nonetheless, the following general formula can be used to describe these materials: Na2−2xCax(Na,K)6(AlSiO4)6(Cl,S)2. The exact ratio of Na:K is uncertain, as the main starting material used was zeolite 3A powder, which is obtained from zeolite 4A that has undergone some degree of ion exchange to substitute Na with K. The extent of ion exchange is difficult to quantify, and many manufacturers do not state precise Na:K ratios in their material specifications sheets, however the amount of K is higher than that of Na in 3A zeolites. Potassium chloride is also a common impurity in the NaCl starting material, so it can be assumed that some potassium is present in all the materials, regardless of which zeolite was used. The same batch of zeolite 3A from the same company was used in all syntheses done with zeolite 3A, and thus it can be assumed that the ratio of Na:K in the zeolite 3A starting material was always the same. The formulae of the starting materials zeolite 4A and zeolite 3A can be expressed as the following: zeolite 4A, NaAlSiO4; zeolite 3A, (Na,K)AlSiO4. XRF measurements confirm the presence of potassium in all materials, and experimentally it was shown that the photochromic properties of calcium-containing sodalities were marginally improved by the presence of extra potassium from the zeolite 3A used (ESI,† Section S1). Thus, we used zeolite 3A as a starting material for most materials in this work. The effect of small amounts of potassium on the optical properties of these materials are visible. However, for simplicity, when discussing the mechanism of photochromism, we only consider sodium and calcium ions inside the sodalite cage.
NaCl is typically used in excess when synthesizing photochromic sodalite, to prevent the formation of halogen-free phases like nepheline.28 When preparing these materials, the same molar ratios of salt (i.e., NaCl and/or CaCl2) and sodium sulfate were used in all syntheses, i.e. 0.00042 mol sodium sulfate and 0.0040 mol of salt. NaCl was substituted with various mol% CaCl2 to produce a range of different compositions. Due to the salt always being used in excess, it is uncertain exactly how much sodium and calcium pass into the structures of these materials, however it is assumed that the Na:Ca ratio of the starting mixture and that of the product are roughly constant. X-ray fluorescence analyses have shown that an increased amount of CaCl2 in the starting mixture results in a greater proportion of calcium in the product (ESI,† Section S1), and according to the XRD diffractograms only very small amounts of Ca-containing by-products, such as CaCl2, are present, suggesting Ca does indeed pass into the sodalite structure. In lieu of exact formulae, the general formula with nominal values of x based on the molar ratios of NaCl:CaCl2 used in synthesis are given to describe these materials. The differences in valence and therefore stoichiometry of Na and Ca have been taken into account.
3.2 Change from pink to yellow photochromism
The ionic radii of Na+ and Ca2+ are rather similar (1.02 and 1.00 Å in coordination number 6),44 but their charges are different. Thus one can expect some difficulties in the solubility of Ca2+ into the sodalite structure. This is why calcium was added gradually with small steps. Here, we show the results for five samples with compositions around those found critical for the color of tenebrescence.We observed that tenebrescence is pink, as in calcium-free sodalites, with one absorption band at 540 nm, until the mole fraction x of calcium reaches 0.11. Above this threshold, the tenebrescence color is clearly yellow (Fig. 1a and b), with an asymmetric absorbance band with a maximum at 435 nm. As the calcium content is increased above 0.11, the color becomes more intense up to x = 0.18, then weakens again. From this we can determine that the optimum compositions for yellow photochromism are 0.13 ≤ x ≤ 0.18 (Fig. 1c). The pink absorption band is also least significant in the colored form of materials with these compositions. There is therefore an optimal distribution of defects able to form blue-absorbing F-centers in these materials. Photochromism is very weak close to the changeover, but appears to be a pale pinkish-orange color, with both absorption bands visible in the reflectance spectrum. L*a*b* color coordinate data also shows the transition of the color from pinker to yellower (Fig. 1d). All five samples had similar L* values, so their a* and b* values are comparable in terms of saturation.
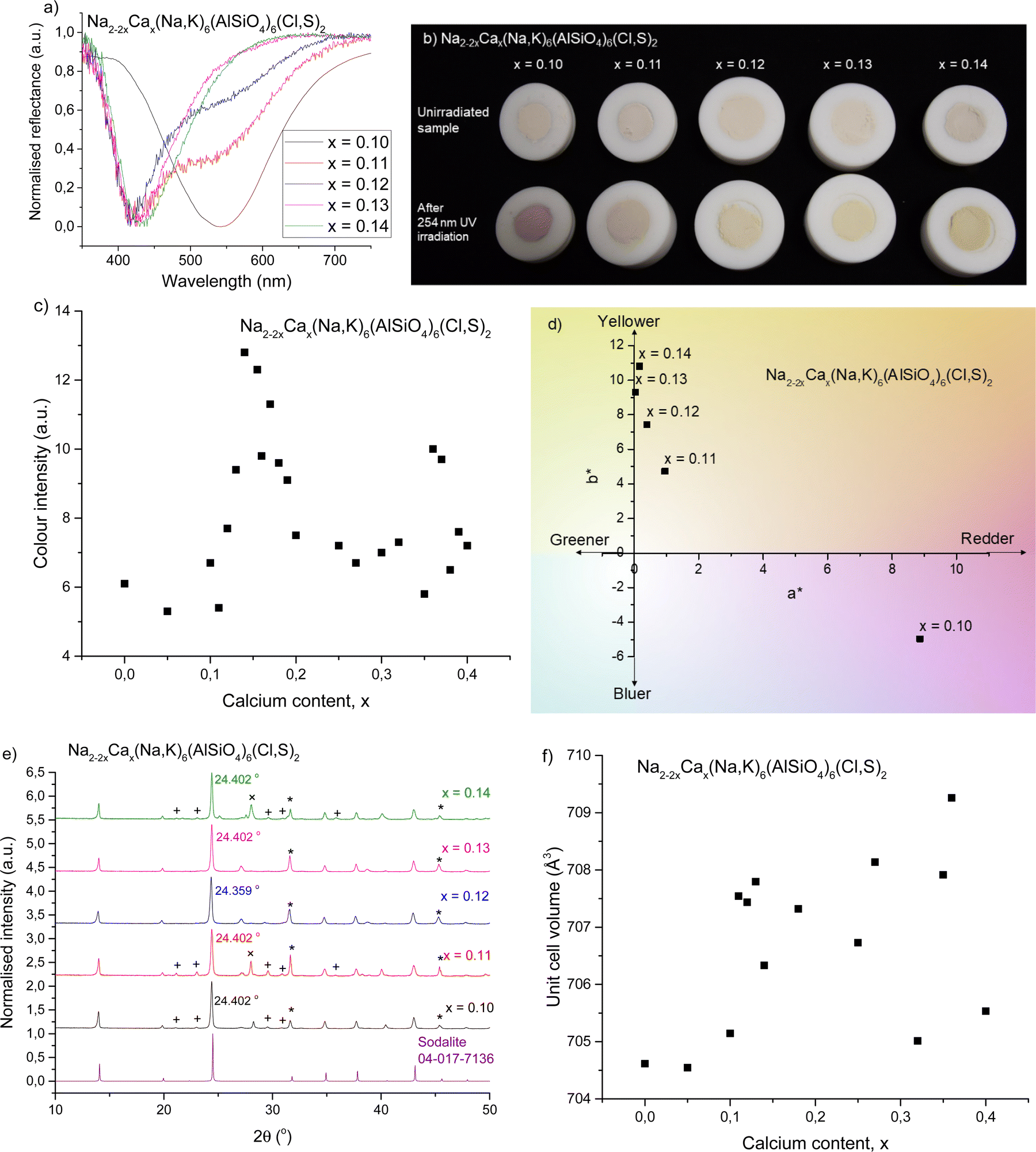 | ||
| Fig. 1 (a) Normalized reflectance curves of colored forms with 0.10 ≤ x ≤ 0.14 showing the changeover from pink to yellow photochromism. (b) Photographs of the same samples before and after irradiation with 254 nm UV showing their uncolored and colored forms, respectively. (c) Reflectance well depth (absorption intensity) at 420–435 nm for 0.00 ≤ x ≤ 0.40, showing variation in yellow color intensity with respect to calcium content. (d) a* and b* color coordinates of the colored forms of the samples, showing the shift from pink to yellow. (e) XRPD patterns compared to a sodalite ref. 16, with the main impurities indicated as follows: + = nepheline (PDF 04-015-8159),45 * = NaCl (PDF 04-013-1689),46 and × = KCl (PDF 04-007-3595).47 (f) Unit cell volumes obtained by Rietveld refinement with respect to calcium content. | ||
Fig. 1e presents the XRPD diffractograms, showing sodalite as the main phase, along with NaCl (marked with *), KCl (+) and nepheline (×) (ESI,† Section S2) as the most significant impurities. In order to study the structural effects surrounding this change, Rietveld refinements were performed to determine quantities such as the unit cell parameter and unit cell volume. The ratios of sodalite to the main impurities were also calculated during refinement (Table 1). Fig. 1f presents the unit cell volume with respect to calcium content. The variation in unit cell size is very small, with all values within 5 Å3 of each other, and there isn’t any clear trend in the unit cell volume's evolution as a function of Ca content. The slight variations may be due to distortion inside the cage caused by calcium's 2+ charge, since Na+ and Ca2+ are otherwise similar in size.
| x | a axis (Å) | V (Å3) | Nepheline (%) | NaCl (%) | KCl (%) | CaCl2 (%) |
|---|---|---|---|---|---|---|
| 0.10 | 8.901 | 705.15 | 6.6 | 5.0 | 6.4 | 0.0 |
| 0.11 | 8.911 | 707.54 | 21.7 | 9.9 | 9.0 | 0.0 |
| 0.12 | 8.910 | 707.44 | 0.0 | 15.6 | 0.0 | 1.4 |
| 0.13 | 8.912 | 707.80 | 0.0 | 16.2 | 0.0 | 0.0 |
| 0.14 | 8.906 | 706.33 | 9.0 | 7.8 | 11.9 | 0.0 |
When considering the mechanism of this phenomenon, the first thing to consider was the role of sulfur, which has been established to be essential in the photochromism of sodalites.19,48–50 Several low oxidation state polysulfide species are present in natural sodalite, usually giving a permanent body color such as yellow (S2−) or blue (S3−).26,27,51 We were able to detect some permanent coloration from S2− (ESI,† Section S3), but despite its low formation energy, the yellow-absorbing S3− was not present in these materials. However, Norrbo et al. proved that the reversible white to pink photochromism in typical photochromic sodalites arises from electron transfer from S22− to VCl upon UV irradiation to form an F-center capable of absorbing green light.24 Therefore, the possible presence of other polysufides than S22− does not affect the photochromism. We used this mechanism as a starting point to investigate the source of white-to-yellow photochromism in calcium-containing sodalites. Computational studies led by some of us provided a very strong agreement with experiments in the simulation of F-center's absorption in sodalite-type minerals.36 Here, the same protocol was used on calcium sodalites. Since only the close environment would impact the F-center's absorption, only modification of the surrounding tetrahedron was considered when adding calcium in the supercell. Three possibilities referred to as Na3Ca, Na2Ca and Ca2 were considered and compared to the unmodified Na4 structure (cf. Section 2.3, ESI,† Section S4). When two Na+ surrounding VCl were replaced with Ca2+, the absorbance profile of the F-center matched the experimental data remarkably well (Fig. 2a). From the agreement between simulated and experimental data we concluded that the Na2Ca species surrounding a chloride vacancy is the system responsible for yellow photochromism. The presence of calcium in the sodalite cage has two effects: first it modifies the crystal field felt by the trapped electron, inducing a blue shift of the absorption compared to the unmodified (Na4) structure. Secondly, it reduces the symmetry of the trapped electron embedding, resulting in the loss of orbital degeneracy, in turn leading to the splitting of the single absorption of the trapped electron in several absorptions.
 | ||
| Fig. 2 (a) Reflectance curves of photochromic sodalites showing pink (x = 0.00) and yellow (x = 0.14) photochromism, along with simulated reflectance curves for the F-center in Na4 and Na2Ca systems (TD-DFT/cam-B3LYP absorption energies convoluted by a Gaussian, using 0.3 eV as FWHM). The Na2Ca system's simulated reflectance spectrum matches well with the experimental result for x = 0.14. (b) Mechanism of F-center formation followed by energetic diagram of the three first electronic transitions for the Na4 and Na2Ca systems computed by TD-DFT. The red arrow represents excitation of an electron from S22−(π*) to VCl and the green arrows spin relaxation from a singlet to triplet state.32 Grey boxes represent the distribution of energies of ground state 1[S22−(π*),VCl(D0)] and excited state 1[S2−(π*),VCl−(D0)] due to variations in local structure. The transitions are dominated by the HOMO to the three lowest LUMOs characters (presented as inserts). Excited states are of doublet spin-multiplicity explaining the notation D0, D1, D2 and D3 for ground and excited states. (c–f) Structures of the pristine system called Na4 (c and e) and the defective Na2Ca (d and f) system from DFT optimized geometries showing the full beta cage (c and d) or focusing on the inside of the beta cage (e and f). Dark and light blue polyhedra surround the Si and Al atoms respectively. Red, yellow, pink and green spheres represent the oxygen, sodium and calcium atoms and the center of the cavity respectively. | ||
The switch from white → pink to white → yellow photochromism can be explained by considering the statistical distribution of the vacancies in the material. For x = 0.00, the only chloride vacancies present are surrounded by four Na+ (Na4, Fig. 2b, c and e), i.e., typical of pink photochromic sodalite, and this has been proven to give an absorption around 500–540 nm upon excitation (Fig. 2a and b). The broader band from experimental data may be the result of small amounts of potassium from the zeolite 3A starting material.52 As the amount of calcium in the material is increased, the cages increasingly contain the Na2Ca structure (Fig. 2d and f), which simulations predict would give a yellow F-center if a chloride vacancy was present inside this particular cage. However, due to the greater amount of Na4 cages in materials with a low calcium concentration (x = 0.00–0.10), the observed color is pink, as the likelihood of the vacancy occurring at the Na4 cage is much higher. As we approach the switchover point there are now enough vacancies in Na2Ca-type cages that the yellow band begins to appear, and for x ≥ 0.13, the majority of vacancies are in Na2Ca cages and only the yellow color is observed. The absorption spectrum of the colored form for x ≥ 0.13 is, however, asymmetrical, suggesting that some Na4 cages still contain vacancies, and upon irradiation a small amount of green-absorbing F-centers are still formed. This is discussed further in Section 3.4.
3.3 Return to pink and the loss of photochromism
The best yellow photochromism was observed in samples with 0.13 ≤ x ≤ 0.25. At these values of x, the 435 nm absorption dominates in the colored form, and the 540 nm absorption is very weak. However, as soon as x ≥ 0.27 the reappearance of the pink band was observed (Fig. 3a), and for x ≥ 0.50 the photochromism disappears completely (Fig. 3c). This can be correlated with a structural change – as calcium content increases, the XRD patterns show that a second phase, davyne ((Na,K)6Ca2(Al6Si6O24)(Cl2,SO4)2), begins to appear (Fig. 3b). | ||
| Fig. 3 (a) Reappearance of the pink absorption band as calcium content increases past x = 0.25. (b) Appearance of the davyne phase at x ≥ 0.27.16,53 The other main impurities are indicated as * = NaCl (PDF 04-013-1689)46 and × = KCl (PDF 04-007-3595).47 (c) Strongest yellow photochromic response compared to high calcium content reflectance curves, showing very weak or absent of photochromism for x ≥ 0.50. (d) Diffractograms of the samples in c, showing davyne has become the dominant phase at x ≥ 0.50. Increased noise levels indicate reduced crystallinity. | ||
The presence of davyne explains the changes in the photochromic properties at higher calcium contents: at first yellow photochromism starts to weaken (x > 0.18), and then the pink band begins to appear. This is likely due to calcium preferentially locating in the davyne phase as it begins to form, resulting in a reduced number of sodalite cages containing Na2Ca and thus weaker yellow photochromism. The presence of chloride and sulfate in davyne may also affect the number of S22−–VCl pairs in the material, and thus the intensity of photochromism. At x > 0.18 davyne may already be forming, but it is not yet crystalline enough to show in the XRPD diffractograms. At x = 0.27 enough davyne has formed to be visible by XRPD, and at the same time the pink band has returned, suggesting a greater number of remaining Cl vacancies are located in Na4 cages due to less Ca being present in the sodalite structure. Eventually davyne becomes the dominant phase (Table 2), and the materials also become less crystalline (Fig. 3d). This results in the gradual decrease in photochromic sodalite content, and at x ≥ 0.5 we observe complete absence of the sodalite phase and therefore only very weak photochromism.
| x | Sodalite (%) | Davyne (%) | Sodalite:Davyne ratio |
NaCl (%) | KCl (%) |
|---|---|---|---|---|---|
| 0.25 | 71.9 | 0.0 | 1:0 |
7.4 | 8.5 |
| 0.27 | 52.7 | 24.6 | 1:0.47 |
14.3 | 6.1 |
| 0.32 | 32.9 | 47.7 | 1:1.45 |
15.8 | 3.6 |
| 0.35 | 31.8 | 47.6 | 1:1.50 |
15.7 | 5.0 |
| 0.40 | 16.5 | 62.9 | 1:3.81 |
17.4 | 3.2 |
| 0.50 | 0.0 | 79.2 | 0:1 |
18.5 | 0.0 |
| 0.75 | 0.0 | 74.1 | 0:1 |
18.7 | 0.0 |
| 1.00 | 0.0 | 78.0 | 0:1 |
8.1 | 0.0 |
3.4 Further characterizations of yellow photochromism
The yellow photochromism was further characterized and compared to a typical Na2(Na,K)6(AlSiO4)6(Cl,S)2 photochromic sodalite. The excitation spectra of tenebrescence (Fig. 4a) indicate that all the materials begin to color under 340 nm UV, though significant coloration only begins under 320 nm or shorter wavelength radiation. The yellow materials are best excited by 290–300 nm or higher energy UV; on the other hand, the pink material is not fully colored until excitation with 260 nm. The slight decrease in color intensity at short wavelengths is likely due to harmonic wavelengths of light capable of bleaching the materials escaping the monochromator. | ||
| Fig. 4 (a) Tenebrescence excitation spectra for a yellow photochromic sodalite compared to a pink one. (b) Tenebrescence excitation spectra for the pink and yellow F-centers within a calcium-containing sodalite. (c) Reflectance spectra of the colored forms of a pink and a yellow photochromic sodalite and their corresponding optical bleaching spectra. (d) Reflectance spectra of a material clearly showing both yellow and pink absorption bands, and optical bleaching spectra of each of the two bands. | ||
When we look more closely at the excitation profiles of the pink and yellow F-centers in this material (Fig. 4b), we observe that full coloration of the pink component is achieved by 300 nm or higher energy UV, whereas the yellow component is fully colored by 280 nm and higher energy radiation. This is likely due to different, broader distributions in the energy levels of S22− and VCl surrounding the blue-absorbing Na2Ca F-centers compared to their green-absorbing Na4 counterparts (see Fig. 2b).
The optical bleaching spectrum of the yellow photochromism is also quite predictable – the material bleaches best under similar wavelengths of light to the F-center's absorption maximum (Fig. 4c). In this way it behaves similarly to typical photochromic sodalite, further supporting the proposed mechanism. For samples containing two absorption bands, we also obtained two different optical bleaching spectra, one for each band (Fig. 4d). This confirms that these two bands do indeed originate from separate color centers, some appearing pink and some yellow, and they function independently of one another.
Thermotenebrescence measurements were carried out to investigate the thermal bleaching energy of the yellow photochromism. The yellow photochromic material with x = 0.18 was found to have a thermal bleaching energy of 0.39 eV, which is close to that of the calcium-free reference, x = 0.00 (Table 3). When we examine the fading of the reflectance curves under thermal tenebrescence, we see that the pink side fades faster and a small yellow component remains at the maximum temperature (ESI,† Section S6). The calculated trap depth would therefore correspond to the fading of the pink component. In fact, the calculated trap depth values for the pink component of both x = 0.18 and x = 0.00 lie between previously presented thermal bleaching energies of sodium-only (0.48 eV) and sodium–potassium (0.28 eV) photochromic sodalites,23 likely due to the presence of some potassium in the materials from the starting material zeolite 3A.23,52 The F-centers responsible for the yellow color require more thermal energy to be emptied than was possible to obtain with our measurement setup.
| x | Absorption maximum (nm) | Optical bleaching max wavelength (nm) | Optical bleaching energy (eV) | Thermal trap depth (eV) |
|---|---|---|---|---|
| 0.00 | 540 | 480 | 1.8–3.6 | 0.44 ± 0.02 |
| 0.18 | 438 | 380 | 2.5–3.6 | 0.39 ± 0.03 |
| 0.11 (yellow) | 422 | 420 | 2.4–3.4 | — |
| 0.11 (pink) | 535 | 560 | 1.8–3.0 | — |
The rise and fade of color intensity in calcium-free and calcium-containing sodalites were measured under both 254 nm and 302 nm excitation and compared. Calcium-free sodalite colors rapidly under 254 nm UV, reaching 90% of the saturation intensity after only two minutes (Table 4). It colors more slowly under 302 nm and does not become as intense (Fig. 5a). It is expected that, due to the slight variation of local structure around the disulfide ions, there is a distribution of energies needed to create the color center. Therefore, 302 nm can activate only some of the color centers. The fact that the rise rate is slower with 302 nm may be because the help of lattice vibrations is needed to induce the coloration, i.e. because 302 nm falls a little short of the lowest energy required.
| x | Excitation source (nm) | Integrated region (nm) | Max intensity (a.u.) | Time to reach 90% max intensity (min) |
|---|---|---|---|---|
| 0.00 | 254 | 545–565 | 17804 |
1.9 |
| 302 | 9799 | 9.0 | ||
| 0.18 | 254 | 417–437 | 6961 | 24.8 |
| 302 | 566 | 7.3 | ||
| 254 | 515–535 | 361 | 28.2 | |
| 302 | 208 | 27.0 | ||
 | ||
| Fig. 5 (a) Coloration rise curves showing the difference in color rise functions for the pink and yellow color centers compared to a calcium-free sodalite. (b) Dose-corrected coloration fade curves showing the difference in fading rate in white incandescent light for the pink and yellow components, including a calcium-free reference. | ||
When we consider the rise curves of the calcium-containing sodalites, we observe under both 254 nm and 302 nm the pink F-centers located in Na4-containing cages color equally rapidly, reaching 90% saturation in approximately half an hour (Table 4 and Fig. 5a). The pink component is more intense under 254 nm, implying the presence of a range of energies needed to induce the color center, but all color centers form rapidly under higher energy radiation. The blue-absorbing Na2Ca F-centers behave more unusually. The yellow color is significantly more intense when excited with 254 nm radiation, but colors much more slowly. Under 302 nm radiation we reach 90% of the maximum saturation under that wavelength in less than half the time it takes under 254 nm, suggesting a much broader range of energies able to form the color center, the majority of which are only created after long exposure to high energy radiation.
The tenebrescence fades under white incandescent light in a bi-exponential fashion (Fig. 5b). The yellow color center fades very slowly under incandescent lighting, because an incandescent bulb produces comparatively little blue light, which is the optimum color for bleaching the yellow F-center (ESI,† Section S7). However, when the relative doses of blue and green light from the incandescent source are taken in to account, we observe that the yellow F-center fades rapidly (Table 5). In fact, the yellow F-centers would bleach almost an order of magnitude more quickly than the pink ones if exposed to a sufficient dose of blue light. This sensitivity to blue light is desirable when considering possible applications of the material, as discussed in Section 3.6. The pink F-centers in x = 0.18 fade slightly faster than the calcium-free reference, which is likely a result of the lower thermal bleaching activation energy of the x = 0.18 material (Table 3).
| x | Integrated region (nm) | τ 1 (h) | A(τ1) (%) | τ 2 (h) | A(τ2) (%) | Average τ (h) |
|---|---|---|---|---|---|---|
| 0.00 | 545–565 | 0.103 ± 0.002 | 25 | 1.11 ± 0.01 | 75 | 0.86 |
| 0.18 | 417–437 | 0.0124 ± 0.0004 | 32 | 0.202 ± 0.009 | 68 | 0.14 |
| 0.18 | 515–535 | 0.10 ± 0.01 | 23 | 0.47 ± 0.02 | 77 | 0.38 |
3.5 Effects of halogen substitution
We have established that calcium-containing sodalites give us access to a yellow F-center that absorbs in the 420–435 nm region; meanwhile, calcium-free chlorosodalites’ pink F-centers usually absorb between 520–550 nm.16 However, an orange F-center is missing from the series, and could be achieved by tuning the F-center to absorb light of between 450–480 nm. In the work of Phillips et al.25 as well as Williams et al.16 we see that by introducing larger ions into the crystal structure, the unit cell expands and thus the absorbance of the colored form shifts to longer wavelengths. It was therefore theorized that replacing some chlorine with bromine or iodine would expand the structure and shift the 430 nm absorbance towards longer wavelengths, potentially giving rise to this desired white-to-orange photochromism.In order to investigate the effect of introducing other halogens to the structure in the hope of achieving orange photochromism, we took a specific calcium ratio known to reliably produce yellow photochromism with no davyne by-product, namely x = 0.25, and replaced chloride partially or completely with bromide or iodide. Surprisingly, gradual introduction of a larger halogen did not produce the expected effect of shifting the F-center's absorbance minimum to longer wavelengths. Instead, the presence of larger halide ions caused the material to behave completely differently: the yellow color was lost and the usual pink photochromism of calcium-free sodalite was observed instead. We found that for bromide the loss of yellow photochromism occurs somewhere between 50–75 mol% Br (50–25 mol% Cl); for the iodide series this change happens between 0–25 mol% I (100–75 mol% Cl) (Fig. 6a). Rietveld refinements were performed on the XRPD data (ESI,† Section S8) and there is a noticeable expansion in the unit cell with increasing Br−/I− content, as expected (Fig. 6b).
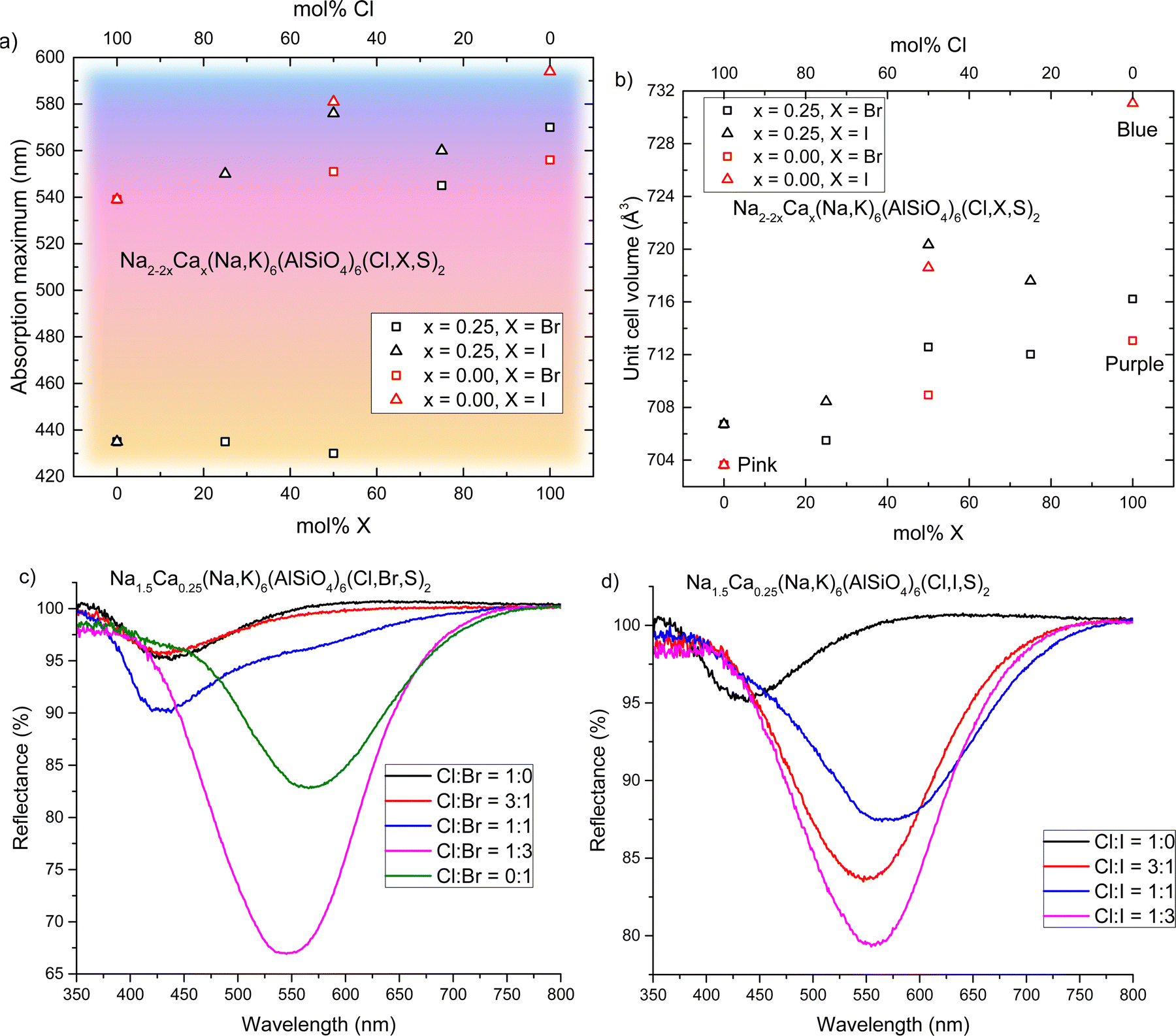 | ||
| Fig. 6 (a) Absorption maxima of the colored forms of Na2−2xCax(Na,K)6(AlSiO4)6(Cl,X,S)2 sodalites, where x = 0.25 or 0.00 and X = Br or I. The background represents the observed color of the colored form with respect to absorption maximum. (b) Unit cell volumes of the same sodalites with respect to Cl:X ratio, where X = Br or I. (c) Reflectance curves of x = 0.25 sodalites containing a mix of Cl and Br. (d) Reflectance curves of x = 0.25 sodalites containing a mix of Cl and I. | ||
For the samples containing bromine, we see that as the Cl:Br ratio reaches 1:1, the pink band begins to reappear (Fig. 6c). This suggests that as the amount of Br in the material increases, the ratio of vacancies in the Na4 and Na2Ca sites begins to change, increasingly favoring pink F-centers. At Cl:Br = 1:3, the pink centers dominate, and we see a similar absorbance maximum as in Ca-free bromide sodalite, close to 560 nm. In the case of iodide, the switch from yellow to pink is immediate (Fig. 6d). We propose that this is due to the size of the anion and the relative amounts of space in Na4versus Na2Ca cages: the larger Br− and I− anions fit more easily into the Na2Ca cages, which in turn increases the likelihood of any vacancies occurring inside Na4 cages. The larger size of iodide means this occurs even at Cl:I = 3:1, whereas smaller bromide still facilitates the formation of some F-centers in Na2Ca cages at higher Cl:Br ratios. Absorbance maxima of pink F-centers are again typical of Ca-free sodalites of the same Cl:X ratio. This is also reflected in the calculated unit cells, where regardless of calcium content the unit cell volumes are similar for samples of the same Cl:X ratio. Thus, we conclude that the pink/purple absorption comes from classic Na4-VX and S22− interactions, as in the calcium-free sodalites.
Whilst halide ion substitution was an unsuccessful in producing orange photochromism in this instance, this study did further confirm our conclusions that the color of these materials depends on the distribution of vacancies between Na4 and Na2Ca cages in these materials. It also revealed a preference for larger halide ions to locate themselves in cages with Na2Ca species, when such cages are available.
3.6 Potential applications
Yellow photochromic sodalites function in the same way as their pink counterparts, in that their color deepens with increasing UV dose (Fig. 7a). They can also be colored by X-rays (Fig. 7a insert, Fig. 7b), though high doses are required to produce coloration, and contrast is low. | ||
| Fig. 7 (a) Rise of yellow photochromism intensity with respect to UVC fluence (dose). Insert: Photograph of the same sodalite colored by X-rays. The colored portion is visible inside the circle. (b) Dose-corrected fading of yellow photochromism under five different light sources. | ||
As discussed in Section 3.4, these yellow photochromic materials show particular sensitivity to blue light. Modern white light sources such as indoor LED lighting, computer screens and smartphones, strongly emit blue light, which can be damaging to the eyes and disruptive to the circadian rhythm.30 Blue light in the ranges of 400–450 nm is considered particularly dangerous to the retina.31 This wavelength range also happens to be close to the optimal bleaching wavelength of the yellow F-center of calcium-containing photochromic sodalites. Thus, we propose that these materials would be particularly suitable as blue light sensors.
Fig. 7b shows how the yellow color of the x = 0.18 material fades under different light sources. Table 6 presents the time constants of the bi-exponential decay curves. Of particular interest is how rapidly the material fades under the light of a WLED-backlit LCD computer screen: this light source has strong blue light emission (ESI,† Section S9), but also emits green light capable of bleaching the pink F-centers in the material. The relative proportions of blue and green light emitted by the screen are ideal for bleaching this material, and thus it would function very effectively as an indicator that someone has been looking at a computer screen for too long. Bleaching by cool white LED lighting typical of a modern office environment is comparatively slower, meaning the material could indicate to the user excessive exposure specifically to the blue light from the screen in front of them. The detector can easily be re-colored at the start of each working day with UV light and used to track a worker's exposure to blue light throughout the day. Alternatively, it could be used in a home environment, where a user could use it to monitor blue light exposure before going to sleep.
| Light source | τ 1 (h) | A(τ1) (%) | τ 2 (h) | A(τ2) (%) |
|---|---|---|---|---|
| 40 W Incandescent lamp | 0.164 ± 0.008 | 21 | 3.1 ± 0.4 | 79 |
| 2700 K Warm white LED | 0.127 ± 0.001 | 64 | 1.21 ± 0.02 | 36 |
| Cool white LED | 0.072 ± 0.002 | 30 | 1.27 ± 0.02 | 70 |
| Blue LED | 0.027 ± 0.003 | 39 | 0.73 ± 0.09 | 61 |
| WLED-LCD computer screen | 0.0043 ± 0.0003 | 44 | 0.041 ± 0.004 | 56 |
4. Conclusions
We have developed a photochromic sodalite displaying white-to-yellow tenebrescence, a new shade of photochromism in these materials. This discovery represents a significant breakthrough in the tuning of sodalite's photochromism. The mechanism of this color change in Na2−2xCax(Na,K)6(AlSiO4)6(Cl,S)2 has been confirmed and the degree of calcium substitution optimized to produce the strongest yellow color – namely 0.13 ≤ x ≤ 0.18. We have explored the effects of increasing the amount of calcium up to full substitution of two sodium atoms, and have found that above x ≥ 0.27, the presence of the by-product davyne becomes non-trivial. In fact, the increasing concentration of davyne has a significant effect on the photochromic property, even destroying it at x ≥ 0.50, which must be considered when choosing the Na:Ca ratio for a potential application. It was also observed that while the absorbance properties of pink F-centers in these materials match previous studies on the relationship between vacancy size and the absorbance maximum, we cannot be sure if the yellow F-centers behave similarly. This is due to the formation of davyne at increasing x preventing the study of a system containing only Na2Ca.
Furthermore, the unique combination and ratio of pink and yellow F-centers in these materials and their respective sensitivity to blue and green light make these materials particularly sensitive to fading under artificial lighting, most notably that of computer and phone screens. Thus, these unique materials are well suited to applications in detection and monitoring of blue light exposure, which is a topical issue in the modern age of technology. These materials are also able to function similarly to pink photochromic hackmanites in many other ways, including being sensitive to X-rays. However, the yellow color is still weaker than the pink color of typical photochromic sodalites, and steps could be taken to optimize the synthesis further to produce more chloride vacancies and hence a deeper color.
Author contributions
Hannah Byron – investigation, data curation, formal analysis, funding acquisition, writing – original draft, writing – review & editing. Teppo Kreivilä – investigation, data curation, writing – review & editing. Pauline Colinet – investigation, writing – review & editing. Tangui Le Bahers – investigation, data curation, formal analysis, supervision, writing – review & editing. Mika Lastusaari – conceptualization, funding acquisition, supervision, writing – review & editing. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
University of Turku Graduate School (UTUGS) Doctoral Programme in Exact Sciences (EXACTUS), Turku, Finland, and the French agency ANR (TeneMod project ANR-17-CE29-0007-21) are thanked for financial support. Thanks go to Dr Ian Pompermayer Machado for fruitful discussions and feedback during the writing of this manuscript.References
- R. Pardo, M. Zayat and D. Levy, Photochromic Organic–Inorganic Hybrid Materials, Chem. Soc. Rev., 2011, 40(2), 672–687, 10.1039/C0CS00065E.
- W. Marckwald, Ueber Phototropie, Zeitschrift für Physikalische Chemie, 1899, 30U(1), 140–145, DOI:10.1515/zpch-1899-3007.
- Y. Badour, V. Jubera, I. Andron, C. Frayret and M. Gaudon, Photochromism in Inorganic Crystallised Compounds, Opt. Mater. X, 2021, 12, 100110, DOI:10.1016/j.omx.2021.100110.
- G. P. Smith, Photochromic Glasses: Properties and Applications, J. Mater. Sci., 1967, 2(2), 139–152, DOI:10.1007/BF00549573.
- Y. Ohko, T. Tatsuma, T. Fujii, K. Naoi, C. Niwa, Y. Kubota and A. Fujishima, Multicolour Photochromism of TiO2 Films Loaded with Silver Nanoparticles, Nat. Mater., 2003, 2(1), 29–31, DOI:10.1038/nmat796.
- K. Naoi, Y. Ohko and T. Tatsuma, TiO2 Films Loaded with Silver Nanoparticles: Control of Multicolor Photochromic Behavior, J. Am. Chem. Soc., 2004, 126(11), 3664–3668, DOI:10.1021/ja039474z.
- S. K. Deb, Optical and Photoelectric Properties and Colour Centres in Thin Films of Tungsten Oxide, Philos. Mag., 1973, 27(4), 801–822, DOI:10.1080/14786437308227562.
- J. Wei, X. Jiao, T. Wang and D. Chen, The Fast and Reversible Intrinsic Photochromic Response of Hydrated Tungsten Oxide Nanosheets, J. Mater. Chem. C, 2015, 3(29), 7597–7603, 10.1039/C5TC01350J.
- S. Wang, W. Fan, Z. Liu, A. Yu and X. Jiang, Advances on Tungsten Oxide Based Photochromic Materials: Strategies to Improve Their Photochromic Properties, J. Mater. Chem. C, 2018, 6(2), 191–212, 10.1039/C7TC04189F.
- S. Nishio and M. Kakihana, Evidence for Visible Light Photochromism of V2O5, Chem. Mater., 2002, 14(9), 3730–3733, DOI:10.1021/cm0204270.
- Y. Han, M. Hamada, I.-Y. Chang, K. Hyeon-Deuk, Y. Kobori and Y. Kobayashi, Fast T-Type Photochromism of Colloidal Cu-Doped ZnS Nanocrystals, J. Am. Chem. Soc., 2021, 143(5), 2239–2249, DOI:10.1021/jacs.0c10236.
- G. Ju, Y. Hu, L. Chen and X. Wang, Photochromism of Europium and Gadolinium Co-Doped Barium Haloapatite, ECS Solid State Lett., 2012, 1(1), R1, DOI:10.1149/2.001201ssl.
- Y. Lv, Z. Li, Y. Jin, H. Wu, C. Wang, G. Ju, L. Chen, Z. Hu and Y. Hu, A Novel Photochromic Material Based on Halophosphate: Remote Light-Controlled Reversible Luminescence Modulation and Fluorescence Lifetime Regulation, Ceram. Int., 2019, 45(5), 5971–5980, DOI:10.1016/j.ceramint.2018.12.067.
- Y. Jin, Y. Hu, Y. Fu, L. Chen, G. Ju and Z. Mu, Reversible Colorless-Cyan Photochromism in Eu2+-Doped Sr3YNa(PO4)3F Powders, J. Mater. Chem. C, 2015, 3(36), 9435–9443, 10.1039/C5TC01797A.
- Z. Yang, J. Du, L. I. D. J. Martin and D. Poelman, Reversible Yellow-Gray Photochromism in Potassium-Sodium Niobate-Based Transparent Ceramics, J. Eur. Ceram. Soc., 2021, 41(3), 1925–1933, DOI:10.1016/j.jeurceramsoc.2020.10.046.
- E. R. Williams, A. Simmonds, J. A. Armstrong and M. T. Weller, Compositional and Structural Control of Tenebrescence, J. Mater. Chem., 2010, 20(48), 10883–10887, 10.1039/c0jm02066d.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Titanium and Zinc Oxide Nanoparticles Are Proton-Coupled Electron Transfer Agents, Science, 2012, 336(6086), 1298–1301, DOI:10.1126/science.1220234.
- The Periodic Table of Endangered Elements. American Chemical Society. https://www.acs.org/greenchemistry/research-innovation/endangered-elements.html (accessed 2022-12-16).
- E. F. Williams, W. G. Hodgson and J. S. Brinen, Synthetic Photochromic Sodalite, J. Am. Ceram. Soc., 1969, 52(3), 139–144, DOI:10.1111/j.1151-2916.1969.tb11200.x.
- S. Vuori, P. Colinet, I. Norrbo, R. Steininger, T. Saarinen, H. Palonen, P. Paturi, L. C. V. Rodrigues, J. Göttlicher, T. Le Bahers and M. Lastusaari, Detection of X-Ray Doses with Color-Changing Hackmanites: Mechanism and Application, Adv. Opt. Mater., 2021, 2100762, DOI:10.1002/adom.202100762.
- S. Vuori, P. Colinet, J.-P. Lehtiö, A. Lemiere, I. Norrbo, M. Granström, J. Konu, G. Ågren, P. Laukkanen, L. Petit, A. J. Airaksinen, L. Goethem, T. L. van; Bahers and M. Lastusaari, Reusable Radiochromic Hackmanite with Gamma Exposure Memory, Mater. Horiz., 2022, 9, 2773–2784, 10.1039/D2MH00593J.
- I. Norrbo, J. M. Carvalho, P. Laukkanen, J. Mäkelä, F. Mamedov, M. Peurla, H. Helminen, S. Pihlasalo, H. Härmä, J. Sinkkonen and M. Lastusaari, Lanthanide and Heavy Metal Free Long White Persistent Luminescence from Ti Doped Li-Hackmanite: A Versatile, Low-Cost Material, Adv. Funct. Mater., 2017, 27(17), 1606547, DOI:10.1002/adfm.201606547.
- I. Norrbo, A. Curutchet, A. Kuusisto, J. Mäkelä, P. Laukkanen, P. Paturi, T. Laihinen, J. Sinkkonen, E. Wetterskog, F. Mamedov, T. Le Bahers and M. Lastusaari, Solar UV Index and UV Dose Determination with Photochromic Hackmanites: From the Assessment of the Fundamental Properties to the Device, Mater. Horiz., 2018, 5(3), 569–576, 10.1039/C8MH00308D.
- I. Norrbo, P. Gluchowski, I. Hyppänen, T. Laihinen, P. Laukkanen, J. Mäkelä, F. Mamedov, H. S. Santos, J. Sinkkonen, M. Tuomisto, A. Viinikanoja and M. Lastusaari, Mechanisms of Tenebrescence and Persistent Luminescence in Synthetic Hackmanite Na8Al6Si6O24(Cl,S)2, ACS Appl. Mater. Interfaces, 2016, 8(18), 11592–11602, DOI:10.1021/acsami.6b01959.
- W. Phillips, Properties of Cathodochromic Sodalite, J. Electrochem. Soc., 1970, 117(12), 1557–1561, DOI:10.1149/1.2407383.
- D. Reinen and G.-G. Lindner, The Nature of the Chalcogen Colour Centres in Ultramarine-Type Solids, Chem. Soc. Rev., 1999, 28(2), 75–84, 10.1039/a704920j.
- A. A. Finch, H. Friis and M. Maghrabi, Defects in Sodalite-Group Minerals Determined from X-Ray-Induced Luminescence, Phys. Chem. Miner., 2016, 43(7), 481–491, DOI:10.1007/s00269-016-0816-7.
- H. Byron, I. Norrbo and M. Lastusaari, A Zeolite-Free Synthesis of Luminescent and Photochromic Hackmanites, J. Alloys Compd., 2021, 872, 159671, DOI:10.1016/j.jallcom.2021.159671.
- G. M. Johnson, P. J. Mead and M. T. Weller, Synthesis of a Range of Anion-Containing Gallium and Germanium Sodalites, Microporous Mesoporous Mater., 2000, 38(2–3), 445–460, DOI:10.1016/S1387-1811(00)00169-4.
- Z.-C. Zhao, Y. Zhou, G. Tan and J. Li, Research Progress about the Effect and Prevention of Blue Light on Eyes, Int. J. Ophthalmol., 2018, 11(12), 1999–2003, DOI:10.18240/ijo.2018.12.20.
- G. Tosini, I. Ferguson and K. Tsubota, Effects of Blue Light on the Circadian System and Eye Physiology, Mol Vis, 2016, 22, 61–72 CAS.
- P. Colinet, H. Byron, S. Vuori, J.-P. Lehtiö, P. Laukkanen, L. Van Goethem, M. Lastusaari and T. Le Bahers, The Structural Origin of the Efficient Photochromism in Natural Minerals, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(23), e2202487119, DOI:10.1073/pnas.2202487119.
- F. J. Garlick and F. Gibson, The Electron Trap Mechanism of Luminescence in Sulphide and Silicate Phosphors, Proc. Phys. Soc., 1948, 60, 574–590 CrossRef.
- R. Dovesi; V. R. Saunders; C. Roetti; R. Orlando; C. M. Zicovich-Wilson; F. Pascale; B. Civalleri; K. Doll; N. M. Harrison; I. J. Bush; P. D’Arco; M. Llunell; M. Causà; Y. Noël; L. Maschio; A. Erba; M. Rerat and S. Casassa, CRYSTAL17, 2017 Search PubMed.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110(13), 6158–6170, DOI:10.1063/1.478522.
- P. Colinet, A. Gheeraert, A. Curutchet and T. Le Bahers, On the Spectroscopic Modeling of Localized Defects in Sodalites by TD-DFT, J. Phys. Chem. C, 2020, 124(16), 8949–8957, DOI:10.1021/acs.jpcc.0c00615.
- R. Nada, J. B. Nicholas, M. I. McCarthy and A. C. Hess, Basis Sets for Ab Initio Periodic Hartree—Fock Studies of Zeolite/Adsorbate Interactions: He, Ne, and Ar in Silica Sodalite, Int. J. Quantum Chem., 1996, 60(4), 809–820, DOI:10.1002/(SICI)1097-461X(1996)60:4
![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) 809::AID-QUA3
809::AID-QUA3![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) 3.0.CO;2-0.
3.0.CO;2-0. - R. Demichelis, Y. Noel, B. Civalleri, C. Roetti, M. Ferrero and R. Dovesi, The Vibrational Spectrum of α-AlOOH Diaspore: An Ab Initio Study with the CRYSTAL Code, J. Phys. Chem. B, 2007, 111(31), 9337–9346, DOI:10.1021/jp072501d.
- M. Corno, C. Busco, B. Civalleri and P. Ugliengo, Periodic Ab Initio Study of Structural and Vibrational Features of Hexagonal Hydroxyapatite Ca10(PO4)6(OH)2, Phys. Chem. Chem. Phys., 2006, 8(21), 2464–2472, 10.1039/B602419J.
- P. C. Hariharan and J. A. Pople, The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies, Theor. Chim. Acta, 1973, 28(3), 213–222, DOI:10.1007/BF00533485.
- G. Sophia; P. Baranek; C. Sarrazin; M. Rerat and R. Dovesihttps://www.crystal.unito.it/Basis_Sets/sodium.html.
- D. Vilela Oliveira, J. Laun, M. F. Peintinger and T. Bredow, BSSE-Correction Scheme for Consistent Gaussian Basis Sets of Double- and Triple-Zeta Valence with Polarization Quality for Solid-State Calculations, J. Comput. Chem., 2019, 40(27), 2364–2376, DOI:10.1002/jcc.26013.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino; B. G. Janesko; R. Gomperts; B. Mennucci; H. P. Hratchian; J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg; D. Williams-Young; F. Ding; F. Lipparini; F. Egidi; J. Goings; B. Peng; A. Petrone; T. Henderson; D. Ranasinghe; V. G. Zakrzewski; J. Gao; N. Rega; G. Zheng; W. Liang; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell; J. A. Montgomery Jr.; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman and D. J. Fox. Gaussian 16 Revision C.01, 2016 Search PubMed.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32(5), 751–767, DOI:10.1107/S0567739476001551.
- R. J. Angel, G. D. Gatta, T. B. Ballaran and M. A. Carpenter, THE MECHANISM OF COUPLING IN THE MODULATED STRUCTURE OF NEPHELINE, Can. Mineral., 2008, 46(6), 1465–1476, DOI:10.3749/canmin.46.6.1465.
- V. N. Baumer, S. S. Galkin, L. V. Glushkova, T. P. Rebrova and Z. V. Shtitelman, Solubility of Al2O3 in Some Chloride−Fluoride Melts, Inorg. Chem., 2006, 45(18), 7367–7371, DOI:10.1021/ic060181r.
- W. P. Davey, Precision Measurements of Crystals of the Alkali Halides, Phys. Rev., 1923, 21(2), 143–161, DOI:10.1103/PhysRev.21.143.
- R. D. Kirk, Role of Sulfur in the Luminescence and Coloration of Some Aluminosilicates, J. Electrochem. Soc., 1954, 101(9), 461–465, DOI:10.1149/1.2781301.
- W. G. Hodgson, J. S. Brinen and E. F. Williams, Electron Spin Resonance Investigation of Photochromic Sodalites, J. Chem. Phys., 1967, 47(10), 3719–3723, DOI:10.1063/1.1701527.
- I. Norrbo, P. Gluchowski, P. Paturi, J. Sinkkonen and M. Lastusaari, Persistent Luminescence of Tenebrescent Na8Al6Si6O24(Cl,S)2: Multifunctional Optical Markers, Inorg. Chem., 2015, 54(16), 7717–7724, DOI:10.1021/acs.inorgchem.5b00568.
- T. E. Warner and J. Hutzen Andersen, The Effects of Sulfur Intercalation on the Optical Properties of Artificial ‘Hackmanite’, Na8[Al6Si6O24]Cl1.8S0.1; ‘Sulfosodalite’, Na8[Al6Si6O24]S; and Natural Tugtupite, Na8[Be2Al2Si8O24](Cl,S)2−δ, Phys. Chem. Miner., 2012, 39(2), 163–168, DOI:10.1007/s00269-011-0471-y.
- P. Colinet, Tenebrescent Minerals by in Silico Modelling. These de doctorat, Lyon, 2022. https://www.theses.fr/2022LYSEN002 (accessed 2022-10-20).
- G. D. Gatta, P. Lotti, G. Nénert and V. Kahlenberg, On the Crystal Structure and Low-Temperature Behaviour of Davyne: A Single-Crystal X-Ray and Neutron Diffraction Study, Microporous Mesoporous Mater., 2014, 185, 137–148, DOI:10.1016/j.micromeso.2013.10.027.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray fluorescence results, reference diffractograms and additional information obtained by Rietveld refinement, simulated structures and transitions for Ca2 and Na3Ca species around VCl, thermotenebrescence fading, additional spectra of light sources used in tenebrescence fade measurements, additional reflectance and XRPD data (PDF). See DOI: https://doi.org/10.1039/d3tc00116d |
| This journal is © The Royal Society of Chemistry 2023 |