
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of chain length in the physical properties and hydrophobicity of (CnH2n+1NH3)2PbX4 (n = 6, 8, 10, 12, 14, and 16; X = Br and I) 2D metal halide perovskites†
Camilla
Anelli
a,
Rossella
Chiara
a,
Marta
Morana
 f,
Andrea
Listorti
b,
Vincenza
Armenise
b,
Silvia
Colella
c,
Benedetta
Albini
d,
Chiara
Milanese
a,
Maria
Medina Llamas
a,
Barbara
Vigani
e,
Paolo
Quadrelli
a,
Silvia
Rossi
e,
Pietro
Galinetto
d and
Lorenzo
Malavasi
*a
f,
Andrea
Listorti
b,
Vincenza
Armenise
b,
Silvia
Colella
c,
Benedetta
Albini
d,
Chiara
Milanese
a,
Maria
Medina Llamas
a,
Barbara
Vigani
e,
Paolo
Quadrelli
a,
Silvia
Rossi
e,
Pietro
Galinetto
d and
Lorenzo
Malavasi
*a
aDepartment of Chemistry and INSTM, University of Pavia, Via Taramelli 16, Pavia, 27100, Italy. E-mail: lorenzo.malavasi@unipv.it; Tel: +39 382 987921
bDepartment of Chemistry, University of Bari “Aldo Moro”, Via Orabona 4, 70126, Bari, Italy
cNational Research Council, Institute of Nanotechnology (CNR-NANOTEC), c/o Department of Chemistry, 70125, Bari, Italy
dDepartment of Physics, University of Pavia, Via Bassi 6, Pavia, 27100, Italy
eDepartment of Drug Sciences, University of Pavia, Via Taramelli 12, Pavia, 27100, Italy
fDepartment of Earth Sciences, University of Firenze, Via G. La Pira 4, Firenze, 50121, Italy
First published on 15th February 2023
Abstract
We report here the preparation and characterization of two families of RP 2D perovskites including linear monoammonium cations, namely (CnH2n+1NH3)2PbBr4 and (CnH2n+1NH3)2PbI4 with n = 4, 6, 8, 10, 12, 14 and 16. Their structural and optical properties show some similarities with, however, distinct features related to the presence of phase transitions occurring when different ligands are present. The optical properties confirm a general blue-shift for the (CnH2n+1NH3)2PbBr4 system with respect to the (CnH2n+1NH3)2PbI4 family with the PL data showing two distinct variation paths due to their excitonic emission behavior, one directly related to the chain length and another one depending on the ammonium coordination to the halogen atoms. The water stability of (CnH2n+1NH3)2PbBr4 and (CnH2n+1NH3)2PbI4 has been assessed and the results show an improved hydrophobicity upon increasing the number of carbon atoms of the alkyl chain as well as by moving from iodide to bromide perovskites.
Introduction
In the last few years, there has been growing interest in layered or so-called two-dimensional (2D) metal halide perovskites (MHPs) that for their superior moisture and air stability, are suitable for application in photovoltaics to design 2D/3D solar cell architectures.1 The most investigated families of 2D perovskites are the Ruddlesden–Popper (RP) and Dion–Jacobson (DJ) phases with the general formula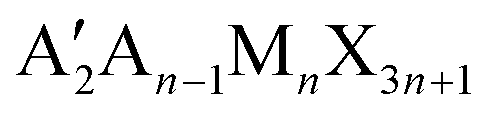 and A′An−1MnX3n+1, respectively, where A′ and A represent organic cations, and n represents the number of staggered inorganic layers made of metal M and halide X atoms.2 Lead-based members with n = 1 of the RP family, i.e. A2PbX4, have been thoroughly studied thanks to the extremely rich variety of organic spacers which can be incorporated into the layered structure giving origin to an impressive modulation of their optical, structural and moisture-resistance properties.2,3 2D RP perovskites containing linear monoammonium cations with the general formula (CnH2n+1NH3)2PbX4 have been among the first to be explored, even before the advent of MHP photovoltaics.4–9 These early studies mostly focused on the elucidation of the crystal structure as a function of the number of carbon atoms in the aliphatic chain as well as on the understanding of the phase transitions commonly found in these compounds. While the early investigation of the optical properties is quite limited, this important group of publications provides a very accurate and solid structural basis for current research.4–6,10,11
and A′An−1MnX3n+1, respectively, where A′ and A represent organic cations, and n represents the number of staggered inorganic layers made of metal M and halide X atoms.2 Lead-based members with n = 1 of the RP family, i.e. A2PbX4, have been thoroughly studied thanks to the extremely rich variety of organic spacers which can be incorporated into the layered structure giving origin to an impressive modulation of their optical, structural and moisture-resistance properties.2,3 2D RP perovskites containing linear monoammonium cations with the general formula (CnH2n+1NH3)2PbX4 have been among the first to be explored, even before the advent of MHP photovoltaics.4–9 These early studies mostly focused on the elucidation of the crystal structure as a function of the number of carbon atoms in the aliphatic chain as well as on the understanding of the phase transitions commonly found in these compounds. While the early investigation of the optical properties is quite limited, this important group of publications provides a very accurate and solid structural basis for current research.4–6,10,11
In particular, Billing reported the crystal structure and phase transition behavior of the (CnH2n+1NH3)2PbI4 perovskite series for 4 ≤ n ≤ 16 showing, in general, the presence of multiple reversible phase transitions due to changes in the hydrocarbon chains and the relative arrangement of the inorganic layers.4–6 In general, all these systems undergo two first order phase transitions, with the first one, when n = 6, 8, and 10, between 235 and 275 K, and the second one in the temperature interval from 313 to 340 K.6 When n is >10, both these phase transitions are above room temperature.5 The crystal structures of these two phases are the same and belong to the Pbca space group, while there is a change in the thermal motion of the organic chains placed between the inorganic layers.6 In particular, the two orthorhombic structures, from lower to higher temperature, have been named phase III and phase II and their main difference is in the alignment of the alkyl chains. A sketch of these two phases for n = 10 is shown in Fig. S1 of the ESI.† Some of these compositions, in the form of single crystals, have also been characterized in terms of their optical properties indicating that the photoluminescence (PL) spectra for 4 ≤ n ≤ 12 are very similar but without providing detailed quantitative data and putting emphasis on the temperature dependence of the optical transitions.8 More recently, thin films of compounds of the (CnH2n+1NH3)2PbI4 family for n = 4, 5, 7, 8 and 9 have been characterized by low-temperature PL reporting a variation of the excitonic structure along with the number of carbon atoms and showing the formation of a fine structure made of three levels for temperatures below 100 K.7 Recently, the optical properties as a function of the number of carbon atoms in (CnH2n+1NH3)2PbI4 perovskites for 4 ≤ n ≤ 18, in the form of thin films, have been reported, and the trend of the optical band-gap systematically correlated with the structural effects induced by the variation of the chain length on the PbI6 octahedra.12
A limited amount of work has been carried out on the analogous system containing Br instead of I, namely the (CnH2n+1NH3)2PbBr4 perovskites. To the best of our knowledge, only the crystal structure of (C4H9NH3)2PbBr4, together with the room-temperature characterization of optical properties, has been recently reported.13 A partial systematic exploration of the effect of the hydrocarbon chain length on the (CnH2n+1NH3)2BrI4 systems has been carried out for n = 4, 5, 7, and 12, providing general evidence of the role of n in the excitonic structure.9
One of the most appealing aspects of 2D MHPs is their improved air and moisture resistance compared to 3D perovskites. This specific aspect has been considered only in one recent publication, where the hydrophobicity of the (C16H33NH3)2PbI4 phase has been not only demonstrated by collecting X-ray diffraction (XRD) data before and after water treatment, but also effectively employed in photoredox catalysis.14,15
The relevance of 2D MHPs in the current research, particularly due to their improved moisture resistance, calls for systematic and complete studies of their structural and optical properties, including their phase transition behavior, especially for the Br-containing systems, and stability investigation as a function of hydrocarbon chain length. In the present paper we address these issues by studying the (CnH2n+1NH3)2PbX4 systems for X = Br and I and n = 4, 6, 8, 10, 12, 14 and 16. In addition to the aspects reported previously, this work provides the first detailed investigation of the (CnH2n+1NH3)2PbBr4 series and a comparative study of the impact of the number of carbon atoms on two analogous systems characterized by a different halide (Br and I). The results presented below allowed defining a complete picture of the impact of the hydrocarbon chain length on the properties of 2D MHPs containing linear monoammonium cations which can be of relevance to other analogous organic spacers and help in the further design of layered perovskites with tailored properties.
Results and discussion
The room temperature X-ray diffraction (XRD) patterns of the compounds of the (CnH2n+1NH3)2PbI4 and (CnH2n+1NH3)2PbBr4 systems (n = 6, 8, 10, 12, 14 and 16) are reported in Fig. 1a and b, respectively. The patterns of both series are dominated by the (00l) reflections of the long axis typical of the low-dimensional perovskites and related to their plate-like morphology. Representative Scanning Electron Microscopy (SEM) images of n = 6 and 16 samples are reported in Fig. S2 (ESI†) showing micro-sized grains with a pronounced lamellar morphology.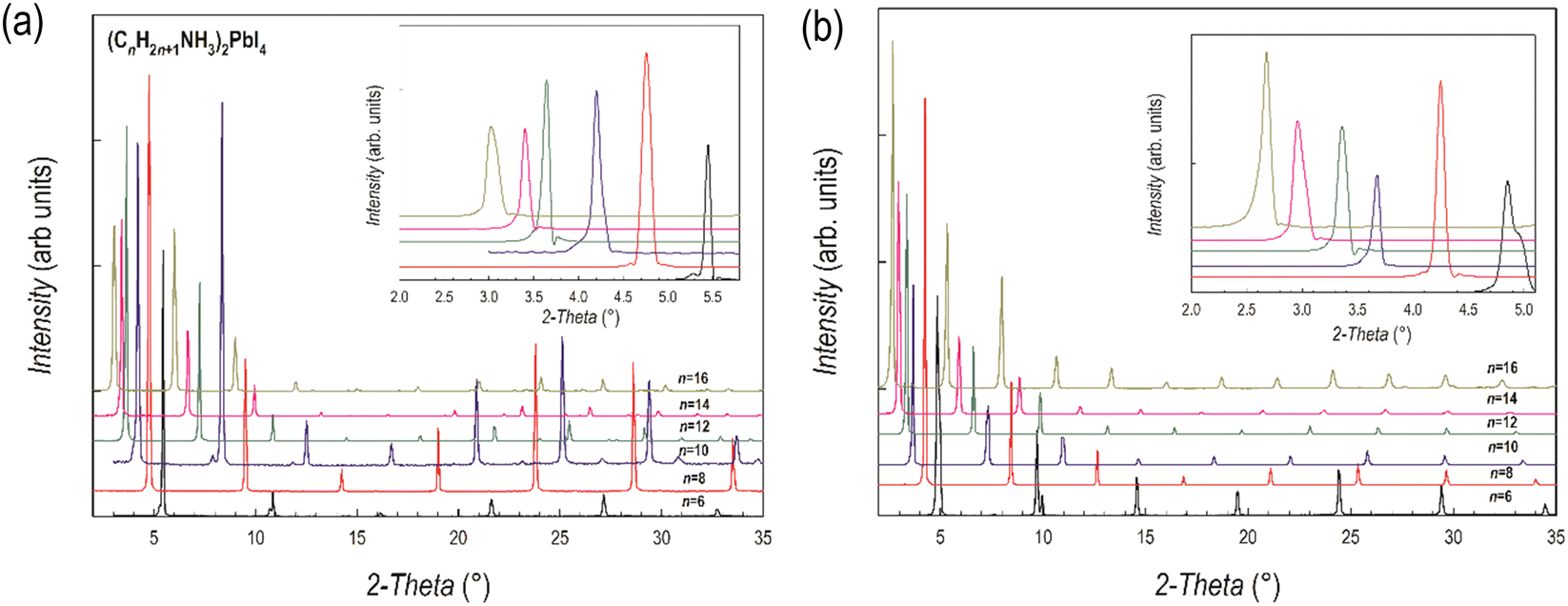 | ||
| Fig. 1 XRD patterns of (a) (CnH2n+1NH3)2PbI4 and (b) (CnH2n+1NH3)2PbBr4 (n = 6, 8, 10, 12, 14 and 16). Insets: Enlargement of the low-angle part of the patterns highlighting the first peak. | ||
The crystal structures of the (CnH2n+1NH3)2PbI4 (n = 6, 8, 10, 12, 14 and 16) compositions are known in the current literature and match with the present diffraction patterns of Fig. 1a.4–6 The patterns of the analogous bromide series have been treated using the Le Bail method starting from the crystal structures of the I-containing analogues, providing good fits to the data. From the patterns of Fig. 1a and b, a progressive shift of the main peaks ((00l) reflections) to lower angles upon increasing the length of the organic ligand is evident, indicating the progressive expansion of the unit cell. The trend of the c-axis dimension vs. the number of carbon atoms of the amine ligand (n = 6, 8, 10, 12, 14 and 16) is reported in Fig. 2 for the two series of samples.
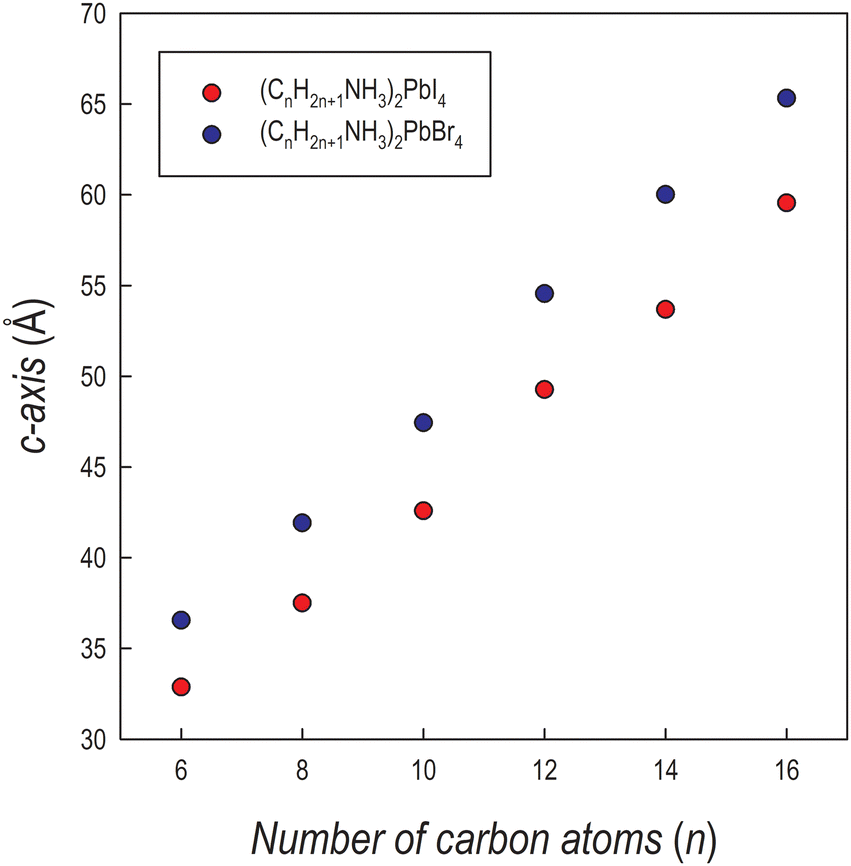 | ||
| Fig. 2 Trend of the c-axis as a function of the number of carbon atoms of the amine for (CnH2n+1NH3)2PbI4 (red dots) and (CnH2n+1NH3)2PbBr4 (blue dots). | ||
The expansion of the lattice parameter c for both series of 2D perovskites is roughly linear with an increase of the long axis with the number of carbon atoms of about 5 Å in the range 6 ≤ n ≤ 10, while a jump of ∼7 Å is found in both series when passing from 10 to 12, and then the difference returns to be around 5.5 Å. Such behavior can be explained based on the room temperature (RT) crystal structure as a function of n. In the (CnH2n+1NH3)2PbI4 system, for n = 6, 8, and 10, the stable unit cell is orthorhombic (S.G. no. 61 Pbca). According to previous works, in this phase (named by Billing et al. as phase II), a bidimensional arrangement of two layers of interdigitated ammonium cations embedded between two consecutive inorganic [PbI6] sheets is found, forming an alternated inorganic–organic layered structure.6 On the other hand, according to the sequence of phase transition found in these 2D perovskites, when moving to n = 12, the stable phase at RT, still orthorhombic (S.G. Pbca), is phase III.4,5 In this novel phase, two main modifications occur: in the inorganic framework, in terms of bond angles (see later in the text), and in the tilt angle of the ammonium group which may account for the observed increase of the long axis of about ∼7 Å.4–6 As a matter of fact, the transition from phase II to phase III leads to an increase, for example in (C10H21NH3)2PbI4, of about 1.5 Å.6 phase III is then the stable RT arrangement for the n = 14 and 16 members of the (CnH2n+1NH3)2PbI4 series, thus further maintaining the change in the c-axis around 5 Å. Interestingly, such behavior is also found for the (CnH2n+1NH3)2PbBr4 series where the sequence of phase transitions is analogous to that of the iodide-counterpart even though slightly shifted to lower temperatures, as determined by differential scanning calorimetry (see Fig. S3 and S4 of the ESI†). This result will be discussed later when presenting the results of the optical properties of the investigated samples.
Interestingly, the c-axis is always longer for the (CnH2n+1NH3)2PbBr4 series, notwithstanding the smaller ionic radius of the bromide ion with respect to iodide, and that the average size of the inorganic slab is about 6.5 Å for lead iodide perovskites and about 6 Å for the lead bromides (as determined from the available crystal structures). However, such an effect has already been observed in other series of 2D perovskites where moving from the iodide to the bromide ion, by retaining the same ligand, leads to an expansion of the long axis (of about 5–6 Å, depending on the amine) as a result of a change in the staggering of the organic cations, which move apart from the halides, thus reducing the tilting of the organic cations. The origin of this effect is mostly related to the strength of the hydrogen bonding of the protonated amine with the apical halides which is well known and discussed in many recent papers.16–19
Further insight into the structural properties of the two series of 2D perovskites has been obtained by RT micro-Raman experiments. The RT Raman spectra of the (CnH2n+1NH3)2PbI4 and (CnH2n+1NH3)2PbBr4 systems (n = 6, 8, 10, 12, 14 and 16) are shown in Fig. 3a and b, respectively. We limited our observation to the low energy part of the spectrum, in the region between 40 and 240 cm−1.
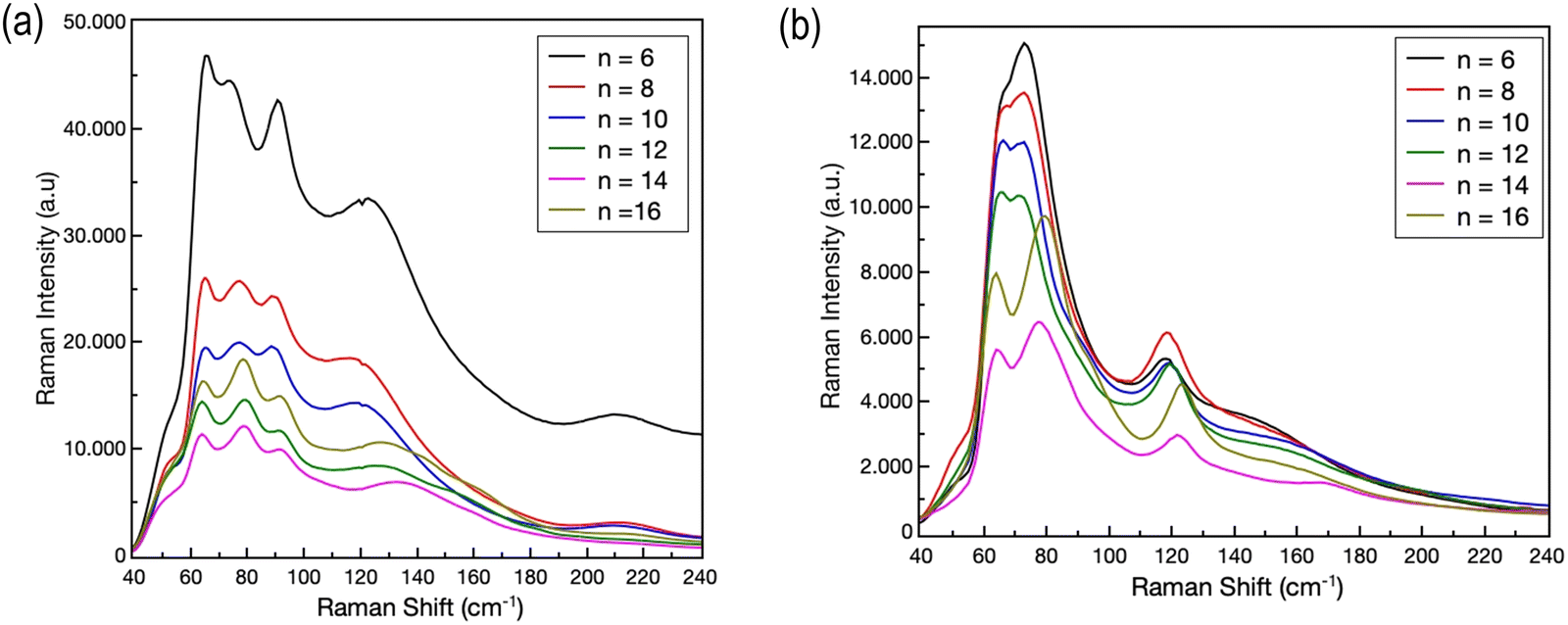 | ||
| Fig. 3 RT Raman spectra of (a) (CnH2n+1NH3)2PbI4 and (b) (CnH2n+1NH3)2PbBr4 (n = 6, 8, 10, 12, 14 and 16). | ||
According to ref. 18 and 19, in this region the Raman activity is mainly associated with the vibrational reservoir of the inorganic cage, i.e. by the motion of the [PbX6]4− unit, even if the organic cations play at least an indirect role in particular for the modes above 100 cm−1.20–22 The Raman yield is directly associated to the organic bonds, i.e. C–C and C–N stretching modes, since CHn and (NH3)+ group modes are detected above 700 cm−1.23 For both systems intense Raman features are observed between 60 and 140 cm−1. For (CnH2n+1NH3)2PbI4, four Raman bands are clearly evidenced in this region, while for (CnH2n+1NH3)2PbBr4 three Raman bands are recorded. Regardless of the length of the organic chain, for both systems, at higher energies, the Raman features appear less intense and broadened. All the observed Raman bands should be the convolution of several Raman modes which can be resolved by lowering the temperature.20,21 However, it is commonly accepted that for the modes below 80 cm−1 the main contributions come from the Pb–Br bond bending and twisting, and from Br–Pb–Br scissoring in the octahedral plane, while above 90–100 cm−1 (depending on the presence of bromide or iodide) the in- and out-of-plane Pb–Br bond stretching contributes dominantly to the Raman yield. Through analyses of the Raman bands at different chain lengths, some indication about the role of the organic moieties can be derived. We performed a series of best-fitting procedures in order to derive band parameters, first of all the peak position, for all the reported spectra. As an illustrative example, Fig. 4a shows the best fitting curve (R2 = 0.996) obtained for the Raman spectrum of the (C8H17NH3)2PbBr4 sample.
 | ||
| Fig. 4 (a) The result from the best-fitting procedure obtained for the Raman spectrum of (CnH2n+1NH3)2PbBr4 (n = 8). (b) The energies of the peak around 120 cm−1 associated to the Pb–X stretching mode (see the text) for (CnH2n+1NH3)2PbI4 (blue squares) and (CnH2n+1NH3)2PbBr4 (red circles). | ||
We used in this case a superposition of five Lorentzian curves. Since at RT these bands are the convolution of several vibrational modes, the derived parameters can be used only for qualitative analysis. This is particularly true for the low energy part of the spectrum where bending and twisting modes can give origin to several contributions. We focused our attention on the behavior of the Raman band peaked at around 120 cm−1. This Raman band is due to the symmetric Ag mode caused by the stretching – both in and out of plane – of the Pb–I and Pb–Br bond.20,21 The energy of the phonon mode remains approximately constant for n = 6, 8 and 10. A red shift is observed when increasing the chain length above 10. This trend seems to be consistent with the transition from phase II to phase III observed (cf. the diffraction results) for both systems passing from n = 10 to n = 12. The shift in the Raman mode energy appears sharper for the iodide system; this fact could be associated to an indirect effect of the observed jump of ∼7 Å of the lattice parameter c as reported above.
The optical properties of the (CnH2n+1NH3)2PbBr4 and (CnH2n+1NH3)2PbI4 samples have been investigated by UV-Vis absorption and photoluminescence spectroscopies at RT. Fig. 5a and b show the UV-Vis spectra of the two series, while the Tauc plots, used to extract the bandgap values, are reported in Table S1 (ESI†).
 | ||
| Fig. 5 Absorption spectra for (a) (CnH2n+1NH3)2PbI4 and (b) (CnH2n+1NH3)2PbBr4. | ||
The spectra are characterized by narrow absorption peaks on top of the band edge, consistent with a stable excitonic population, typical of the quantum confinement effect.24 In both cases a general blue shift is observed upon increasing the chain length of the organic cation. The bandgaps (Table S1, ESI†) show small variation along with the increase of the chain lengths except for the two jumps from n = 12 to 14 for (CnH2n+1NH3)2PbI4 and from n = 8 to 10 for (CnH2n+1NH3)2PbBr4 (Fig. 5).
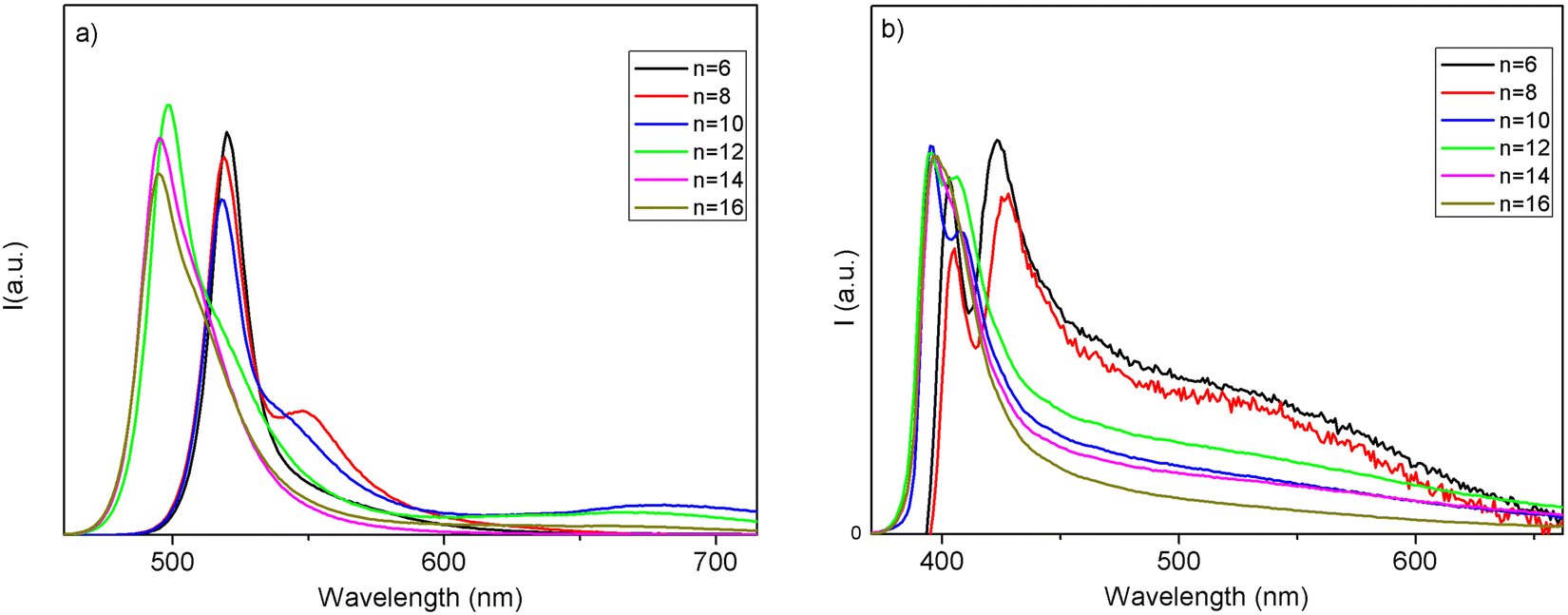 | ||
| Fig. 6 PL spectra for (a) (CnH2n+1NH3)2PbI4 and (b) (CnH2n+1NH3)2PbBr4. | ||
The corresponding PL spectra are shown in Fig. 6. The PL spectra well reflect the absorption features in terms of sample compositions where a discontinuity in the shift is found, as well as in the presence of a structured emission involving two main contributions. The structuring of the 2D MHP emission in 2 main contributions (S1 and S2) has been debated and, with fluence and temperature experiments, most likely attributed to a phonon replica of the main line.25 Additionally, in our samples, this band structuring is more evident for the specimens embedding small alkyl chains. As we introduce longer chains, we also observe an abrupt blue shift of the emissions, of about 120 meV for the main peak passing from n = 10 to n = 12 in iodide samples and of about 60 meV moving from n = 8 to n = 10 in bromide samples (consistent with the absorption measurements). The shift is therefore more pronounced in iodide-based samples (see values of the emission peak maximum in Table S2, ESI†). In the past, several studies have shown, experimentally and theoretically, that, in 2D perovskites, the quantum confinement of the electron and hole, and thus the emission wavelength, can be tuned by varying the thickness of the inorganic part, i.e. the quantum well thickness.26,27 It is evident that, in our case, it is not the length of the alkyl chain variation which induces the abrupt emission blue shift along the sample series, as this length linearly varies across the series (Fig. 2). The transition from phase II to phase III in the iodine samples is accompanied by a relatively large abrupt change in the I–Pb–I angle, from around 156° for n ≤ 10 to around 150° for n ≥ 12. As already observed by some of us, this change leads to a decrease in the contributions of Pb orbitals to the CBM and to a band-gap increase and a concomitant blue shift of the PL.28,29 We prove therefore the reason for the thermochromism observed in these materials being induced by a polymorphic transition, from phase II to phase III, involving a change in the ammonium cation positioning impacting the halogen/lead coordination sphere, in turn affecting the halogen/lead atomic orbital interaction.
As mentioned in the Introduction, a peculiar characteristic of 2D perovskites is their enhanced moisture and even water stability.2,30–35 To date, no studies on the effect of the alkyl chain length on such a phenomenon have been performed. To address this issue, we subjected three selected members of the (CnH2n+1NH3)2PbBr4 and (CnH2n+1NH3)2PbI4 series, namely with n = 6, 10, and 16, to two kinds of tests. In the first experiment, we put the perovskite powdered sample directly in deionized water and left it for 4 hours under stirring and recovered the material after the time following water evaporation under the hood. For these samples, we collected XRD patterns and compared them to those of the pristine, as-prepared materials. The second test was the determination of the contact angle using water as a solvent as a function of time on thin film samples.
Fig. 7a–f show the patterns of the lead bromide and iodide 2D perovskites with n = 6, 10, and 16 perovskites after water treatment, compared to the as-prepared batches.
 | ||
| Fig. 7 XRD patterns before (black line) and after (blue line) water treatment (a–c) for the (CnH2n+1NH3)2PbI4 perovskites and (d–f) for the (CnH2n+1NH3)2PbBr4 perovskites. | ||
In general, for both series of samples, complete degradation of the materials is not observed even after immersion in water and sample recovery. However, the patterns show an improvement in structural stability upon increasing the length of the hydrocarbon chain. In more detail, for the (CnH2n+1NH3)2PbI4 series, extra peaks are found for n = 6 and 10, while the patterns before and after the water treatment are superimposable when n = 16. For the bromide analogues, while significant differences are found when n = 6, a nearly impressive similarity between the patterns before and after the water treatment is found for n = 10 and 16. This difference in water resistance between the two series of samples could be ascribed to the more polar nature of the inorganic framework when the iodide anion is present in the perovskite.
Further insight into the hydrophobic characteristics of the 2D perovskites has been achieved through the measurement of the contact angle of the same samples reported above. For the measurements, films of about 500 nm have been prepared on a quartz substrate using a spin-coating method (see the Experimental section). Fig. 8a and b show the trend of the contact angle as a function of time for the (CnH2n+1NH3)2PbI4 and (CnH2n+1NH3)2PbBr4 samples, respectively, for n = 6, 10, and 16, while Fig. S5 and S6 (ESI†) show the corresponding images at the beginning and at the end of the experiments.
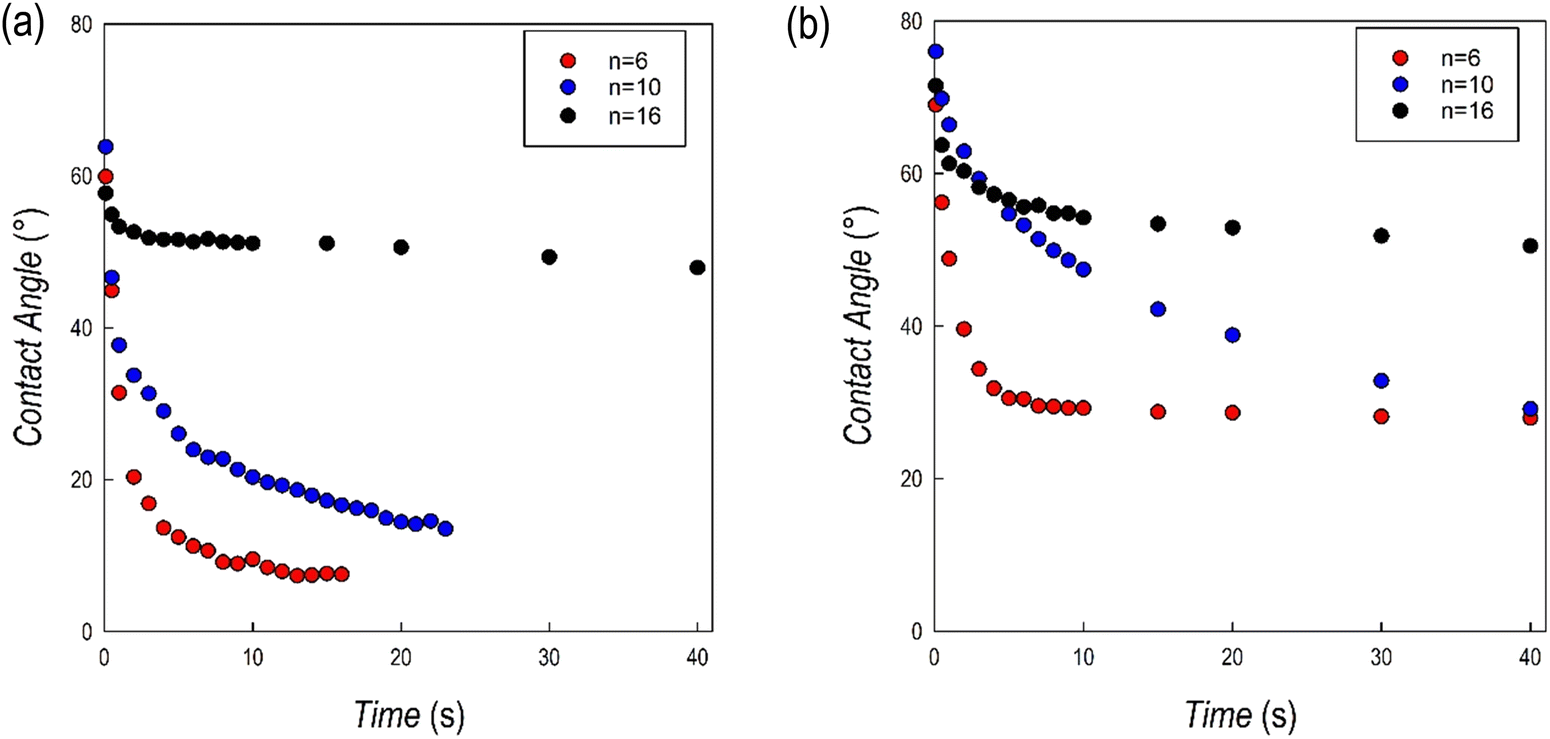 | ||
| Fig. 8 Contact angle values as a function of time for the (a) (CnH2n+1NH3)2PbI4 and (b) (CnH2n+1NH3)2PbBr4 samples. | ||
For the (CnH2n+1NH3)2PbI4 series, the starting value of the contact angle is roughly around 60°, without relevant differences as a function of n. For n = 6 and 10, the angle value rapidly drops within the first few seconds of measurements and after about 20 seconds the measurement is not reliable anymore. On the opposite, for n = 16, after an initial slight reduction of the angle, the value remains constant at around 50°. This result confirms the evidence of a high hydrophobicity for the (C16H33NH3)2PbI4 composition as reported recently in ref. 13 where the perovskite has been used in photocatalytic applications.13 For the (CnH2n+1NH3)2PbBr4 series the starting values of the contact angles are higher with respect to the iodide analogues (cf.Fig. 8a and b). In this case, the most relevant initial drop was observed for n = 6 but also for this sample and differently from the iodide analogue, it was possible to measure the sample up to 40 s. The drop for the n = 10 composition was less pronounced, but a significant reduction of the contact angle was observed during the measurements. Also in this case, differently with respect to the (CnH2n+1NH3)2PbI4 series, the measurement could be performed up to the maximum time interval selected. Finally, for n = 16, the value of the contact angle remains constant, around 60°, for the whole measurement, confirming the strong hydrophobicity also for the (C16H33NH3)2BrI4 composition. The contact angle measurements well correlate with the results of the tests reported in Fig. 7a–f, indicating a generally improved hydrophobicity upon extending the hydrocarbon chain length but also moving from the iodide to the bromide anion.
Conclusions
In the present paper we investigated two families of RP 2D perovskites including linear monoammonium cations, namely (CnH2n+1NH3)2PbBr4 and (CnH2n+1NH3)2PbI4 with n = 4, 6, 8, 10, 12, 14, and 16. While previous investigations addressed some structural and optical data of the iodide-containing system, this work provides the first detailed investigation of the (CnH2n+1NH3)2PbBr4 series, also including a comparative study between the two series. XRD and Raman analyses evidenced similar structural features for the two systems at room temperature, with the bromide compounds characterized by a series of transitions occurring at lower temperatures, as evidenced by DSC measurements. The optical properties confirm a general blue-shift for the (CnH2n+1NH3)2PbBr4 system with respect to (CnH2n+1NH3)2PbI4. The PL data show that, interestingly, the investigated 2D perovskites possess two distinct variation paths due to their excitonic emission behavior, one directly related to the chain length, influencing the interplane interaction between organic layers, and another one depending on the ammonium coordination to the halogen atoms (polymorphism) which directly depend on the temperature of the system. Finally, the water stability of (CnH2n+1NH3)2PbBr4 and (CnH2n+1NH3)2PbI4 has been assessed by two types of tests, whose results are in agreement with each other and allow confirmation of an improved hydrophobicity by increasing the number of carbon atoms of the alkyl chain as well as by moving from iodide to bromide perovskites.This comparative study between two analogue 2D perovskites including linear monoammonium cations, characterized by different halides, provided novel information about their structural, optical, and water-stability characteristics. Considering the key interest in expanding the scope of low-dimensional perovskites well beyond the photovoltaics field, the present data provide relevant results for further exploitation of these RP systems. Future work can exploit the wide range of absorption/emission properties of the present samples in diverse applicative fields taking advantage of the superior stability of compositions including long-chain organic spacers. These could be potentially useful phases for engineering 2D/3D perovskite solar cells as well as suitable candidates for organic photocatalysis. Finally, starting from the present results, the design of other long chain monoammonium ligands including functional groups in the main organic skeleton can provide additional functionalities and interactions with the inorganic framework leading to novel 2D perovskites of interest for optoelectronic and photocatalytic applications.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
A. L. acknowledges the Puglia Regional Council (Grant name: Perseo, CUP: H95F20000890003).References
- A. Krishna, S. Gottis, M. K. Nazeeruddin and F. Sauvage, Adv. Funct. Mater., 2019, 29, 1806482 CrossRef.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- D. G. Billing and A. Lemmerer, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 735–747 CrossRef CAS PubMed.
- D. G. Billing and A. Lemmerer, New J. Chem., 2008, 32, 1736 RSC.
- A. Lemmerer and D. G. Billing, Dalton Trans., 2012, 41, 1146–1157 RSC.
- N. Kitazawa, M. Aono and Y. Watanabe, Thin Solid Films, 2010, 518, 3199–3203 CrossRef CAS.
- T. Ishihara, J. Takahashi and T. Goto, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 11099–11107 CrossRef CAS PubMed.
- N. Kitazawa, M. Aono and Y. Watanabe, J. Phys. Chem. Solids, 2011, 72, 1467–1471 CrossRef CAS.
- S. Barman, N. V. Venkataraman, S. Vasudevan and R. Seshadri, J. Phys. Chem. B, 2003, 107, 1875–1883 CrossRef CAS.
- F. Kang, Y. Du, Z. Yang, P. Boutinaud, M. Wubs, J. Xu, H. Ou, D. Li, K. Zheng, A. T. Tarekegne, G. Sun, X. Xu and S. Xiao, Laser Photonics Rev., 2023, 17, 2200166 CrossRef CAS.
- J. A. Sichert, A. Hemmerling, C. Cardenas-Daw, A. S. Urban and J. Feldmann, APL Mater., 2019, 7, 041116 CrossRef.
- T. Sheikh and A. Nag, J. Phys. Chem. C, 2019, 123, 9420–9427 CrossRef CAS.
- Z. Hong, W. K. Chong, A. Y. R. Ng, M. Li, R. Ganguly, T. C. Sum and H. S. Soo, Angew. Chem., Int. Ed., 2019, 58, 3456–3460 CrossRef CAS PubMed.
- F. Kang, G. Sun, P. Boutinaud, H. Wu, F.-X. Ma, J. Lu, J. Gan, H. Bian, F. Gao and S. Xiao, Chem. Eng. J., 2021, 403, 126099 CrossRef CAS.
- A. Pisanu, M. Coduri, M. Morana, Y. O. Ciftci, A. Rizzo, A. Listorti, M. Gaboardi, L. Bindi, V. I. E. Queloz, C. Milanese, G. Grancini and L. Malavasi, J. Mater. Chem. A, 2020, 8, 1875–1886 RSC.
- L. Z. Tan, F. Zheng and A. M. Rappe, ACS Energy Lett., 2017, 2, 937–942 CrossRef CAS.
- F. El-Mellouhi, A. Marzouk, E. T. Bentria, S. N. Rashkeev, S. Kais and F. H. Alharbi, ChemSusChem, 2016, 9, 2648–2655 CrossRef CAS PubMed.
- K. L. Svane, A. C. Forse, C. P. Grey, G. Kieslich, A. K. Cheetham, A. Walsh and K. T. Butler, J. Phys. Chem. Lett., 2017, 8, 6154–6159 CrossRef CAS PubMed.
- B. Dhanabalan, Y.-C. Leng, G. Biffi, M.-L. Lin, P.-H. Tan, I. Infante, L. Manna, M. P. Arciniegas and R. Krahne, ACS Nano, 2020, 14, 4689–4697 CrossRef CAS PubMed.
- D. Spirito, Y. Asensio, L. E. Hueso and B. Martín-García, J. Phys. Mater., 2022, 5, 034004 CrossRef.
- D. B. Straus and C. R. Kagan, J. Phys. Chem. Lett., 2018, 9, 1434–1447 CrossRef CAS PubMed.
- N. V. Venkataraman, S. Bhagyalakshmi, S. Vasudevan and R. Seshadri, Phys. Chem. Chem. Phys., 2002, 4, 4533–4538 RSC.
- X. Li, X. Lian, J. Pang, B. Luo, Y. Xiao, M.-D. Li, X.-C. Huang and J. Z. Zhang, J. Phys. Chem. Lett., 2020, 11, 8157–8163 CrossRef CAS PubMed.
- K. Gauthron, J.-S. Lauret, L. Doyennette, G. Lanty, A. Al Choueiry, S. J. Zhang, A. Brehier, L. Largeau, O. Mauguin, J. Bloch and E. Deleporte, Opt. Express, 2010, 18, 5912 CrossRef CAS PubMed.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed.
- V. A. Hintermayr, A. F. Richter, F. Ehrat, M. Döblinger, W. Vanderlinden, J. A. Sichert, Y. Tong, L. Polavarapu, J. Feldmann and A. S. Urban, Adv. Mater., 2016, 28, 9478–9485 CrossRef CAS PubMed.
- S. Bonomi, I. Tredici, B. Albini, P. Galinetto, A. Rizzo, A. Listorti, U. A. Tamburini and L. Malavasi, Chem. Commun., 2018, 54, 13212–13215 RSC.
- M. R. Filip, G. E. Eperon, H. J. Snaith and F. Giustino, Nat. Commun., 2014, 5, 5757 CrossRef CAS PubMed.
- M. Aamir, Z. H. Shah, M. Sher, A. Iqbal, N. Revaprasadu, M. A. Malik and J. Akhtar, Mater. Sci. Semicond. Process., 2017, 63, 6–11 CrossRef CAS.
- A. Latini, S. Quaranta, F. Menchini, N. Lisi, D. Di Girolamo, O. Tarquini, M. Colapietro, L. Barba, N. Demitri and A. Cassetta, Dalton Trans., 2020, 49, 2616–2627 RSC.
- D. Ricciarelli, W. Kaiser, E. Mosconi, J. Wiktor, M. W. Ashraf, L. Malavasi, F. Ambrosio and F. De Angelis, ACS Energy Lett., 2022, 1308–1315 CrossRef CAS.
- L. Romani, A. Bala, V. Kumar, A. Speltini, A. Milella, F. Fracassi, A. Listorti, A. Profumo and L. Malavasi, J. Mater. Chem. C, 2020, 8, 9189–9194 RSC.
- L. Romani, A. Speltini, F. Ambrosio, E. Mosconi, A. Profumo, M. Marelli, S. Margadonna, A. Milella, F. Fracassi, A. Listorti, F. De Angelis and L. Malavasi, Angew. Chem., Int. Ed., 2021, 60, 3611–3618 CrossRef CAS PubMed.
- L. Romani, A. Speltini, C. N. Dibenedetto, A. Listorti, F. Ambrosio, E. Mosconi, A. Simbula, M. Saba, A. Profumo, P. Quadrelli, F. De Angelis and L. Malavasi, Adv. Funct. Mater., 2021, 31, 2104428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Differential calorimetry and contact angle measurements. Experimental details. See DOI: https://doi.org/10.1039/d3tc00044c |
| This journal is © The Royal Society of Chemistry 2023 |