
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaching the 5% theoretical limit of fluorescent OLEDs with push–pull benzophospholes†
Nicolas
Ledos
a,
Denis
Tondelier
b,
Bernard
Geffroy
 c,
Denis
Jacquemin
*de,
Pierre-Antoine
Bouit
*a and
Muriel
Hissler
*a
c,
Denis
Jacquemin
*de,
Pierre-Antoine
Bouit
*a and
Muriel
Hissler
*a
aUniv Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: pierre-antoine.bouit@univ-rennes.fr; muriel.hissler@univ-rennes.fr
bLPICM, CNRS, Ecole Polytechnique, Institut Polytechnique de Paris, route de Saclay, 91128 Palaiseau, France
cUniversité Paris-Saclay, CEA, CNRS, NIMBE, LICSEN, 91191, Gif-sur-Yvette, France
dNantes Université, CNRS, CEISAM UMR 6230, F-44300, Nantes, France. E-mail: denis.jacquemin@univ-nantes.fr
eInstitut Universitaire de France, F-75005 Paris, France
First published on 20th February 2023
Abstract
We report the synthesis and characterization of benzophosphole oxides featuring an ethoxy substituent on the P atom and electro-donating amino groups on the lateral phenyl groups. The optical and redox properties of these compounds are studied experimentally and computationally (TD-DFT). In particular, we show how the nature of the donor allows fine-tuning the internal charge transfer (ICT) and, thus, the optical properties. Considering the intense fluorescence in the solid state (powder or thin film) and the favorable redox and thermal properties, these compounds were introduced in multilayered organic light emitting devices. All compounds display high efficiency and one compound even exhibits an external quantum efficiency (EQE) of 5%, which represents the theoretical limit of the EQE of purely fluorescent emitters. These results highlight the potential of this novel family of fluorophores to develop next generation optoelectronic/photonic devices.
Introduction
Organic light-emitting diodes (OLEDs) are optoelectronic devices that convert electricity into light. Since their development in the late 80s,1 they represent a sustainable lightning technology and have been applied to various displays (screens and phones) and lightning panels.2 The first generation of OLEDs was based on fluorescent emitters. This technology allowed the field to rapidly expand but suffered from its intrinsic internal quantum efficiency (IQE) limit of 25%. This limit was then overcome with the use of phosphorescent emitters or thermally activated delayed fluorescent emitters (IQE = 100%). These two technologies now represent the benchmark for OLED devices. However, the combination of fluorescent emitters with a TADF assisted dopant (the so called TADF sensitized fluorescence TSF) appears as an elegant alternative to produce efficient OLEDs with high color purity.3 In addition, purely fluorescent electroluminescent materials are also expected to have great potential in the field of electrically pumped organic lasers, where triplet states are highly detrimental,4 or in visible light communications (VLC) that need an ultrafast response excluding the use of triplet emitters (or TADF).5 These recent developments triggered renewed interest for the development of highly efficient fluorescent emitters for OLEDs. In this field, some of us pioneered the use of π-conjugated phospholes6 such as A–B (Fig. 1) as purely fluorescent emitters for both colored and white OLEDs,7 as well as for the development of hybrid-LEDs.8 This P-based fluorophore takes advantage of an extended π-system and a trigonal P-atom limiting the stacking in the solid-state and, thus, promoting an intense solid-state luminescence. Furthermore, the specific reactivity in the P-atom also allows fine-tuning the emission wavelength.9 Recently, the discovery of efficient metal-mediated synthesis of benzophospholes and the intense fluorescence associated with this core has led to the preparation of new libraries of fluorophores.10 Among them, Tang et al. reported purely fluorescent OLEDs based on C (Fig. 1), which combine a strong electron donor (triarylamine) with the benzophosphole electron acceptor.11 These devices displayed a satisfying efficiency with an EQE of 2%. In the present work, we report the synthesis of three new benzophosphole-based fluorophores having an ethoxy substituent on the P-atom (compounds D in Fig. 1) including different electron-donating groups, their full physico-chemical characterization based on a combined experimental–theoretical approach and their introduction in OLEDs. In particular, we show that one compound reaches an efficiency limit (EQE) of 5% for a purely fluorescent emitter, making it a very appealing derivative for further use in ultra-fast OLEDs or organic lasers. | ||
| Fig. 1 Examples of phospholes (A)–(C) used as emitters in OLEDs and (D) the structure of the compounds considered in this study. | ||
Synthesis
The synthetic pathway to access fluorophores 2a–c is illustrated in Scheme 1. The first step to obtain different arylamine-substituted alkynes is based on a Hartwig-Buchwald reaction between 1,2-bis(4-bromophenyl)ethyne and the corresponding H-substituted arylamines. Then, benzophospholes were obtained by applying the procedure of Chen and coworkers,10 that is using Ag2O, ethyl phosphininate, and the corresponding alkynes via oxidative cyclization. Compounds 2a–c were obtained in moderate yields between 31 and 40%. These compounds were fully characterized by multinuclear NMR spectroscopy, high-resolution mass spectrometry and elemental analyses (see the ESI†). In addition, they display good solubility in classical organic solvents.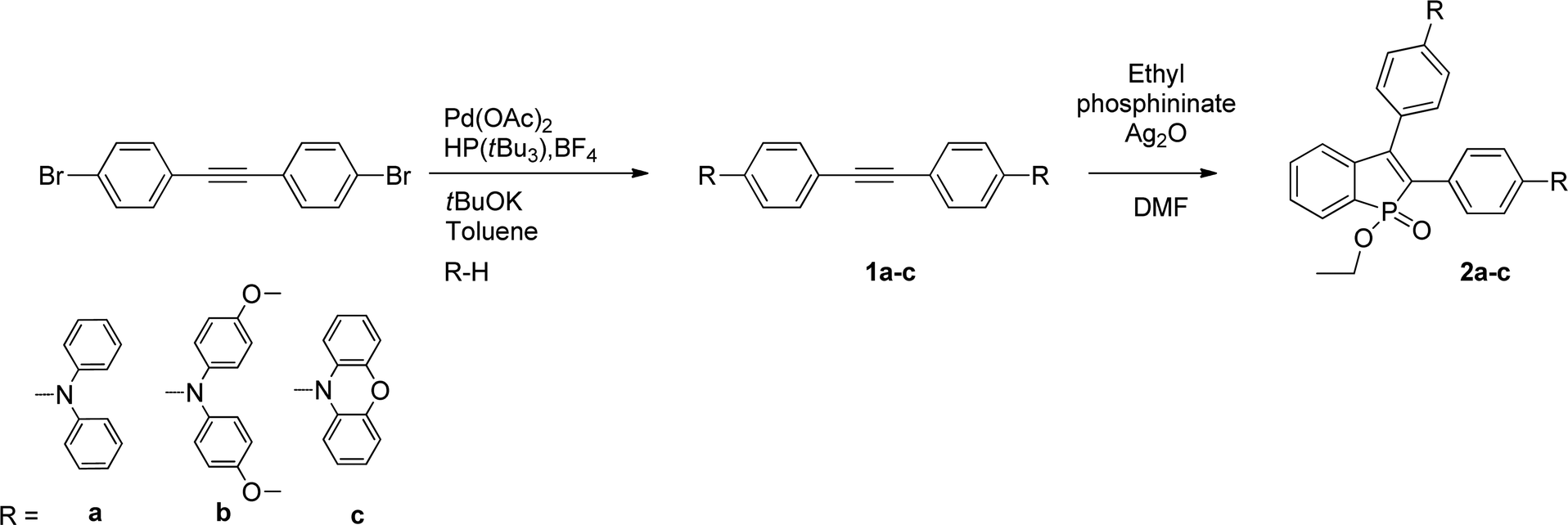 | ||
| Scheme 1 Synthetic access to 2a–c. | ||
Optical and redox properties
The spectroscopic properties of 2a–c were investigated in diluted DCM solution (see Fig. 2A). All compounds display transitions in the UV-visible range up to 500 nm. 2a and 2b exhibit almost the same absorption properties with a first broad band around 300 nm. These two compounds also display a larger band in the visible range (λmax(2a) = 406 nm; λmax(2b) = 415 nm, see Table 1 and Fig. 2A). This large band is attributed to internal charge transfer (ICT) from the arylamine donor to the benzophosphole acceptor as it has been reported for similar compounds.112c shows a maximal absorption wavelength at 327 nm and broad shoulder up to 480 nm (vide infra for theoretical rationalization).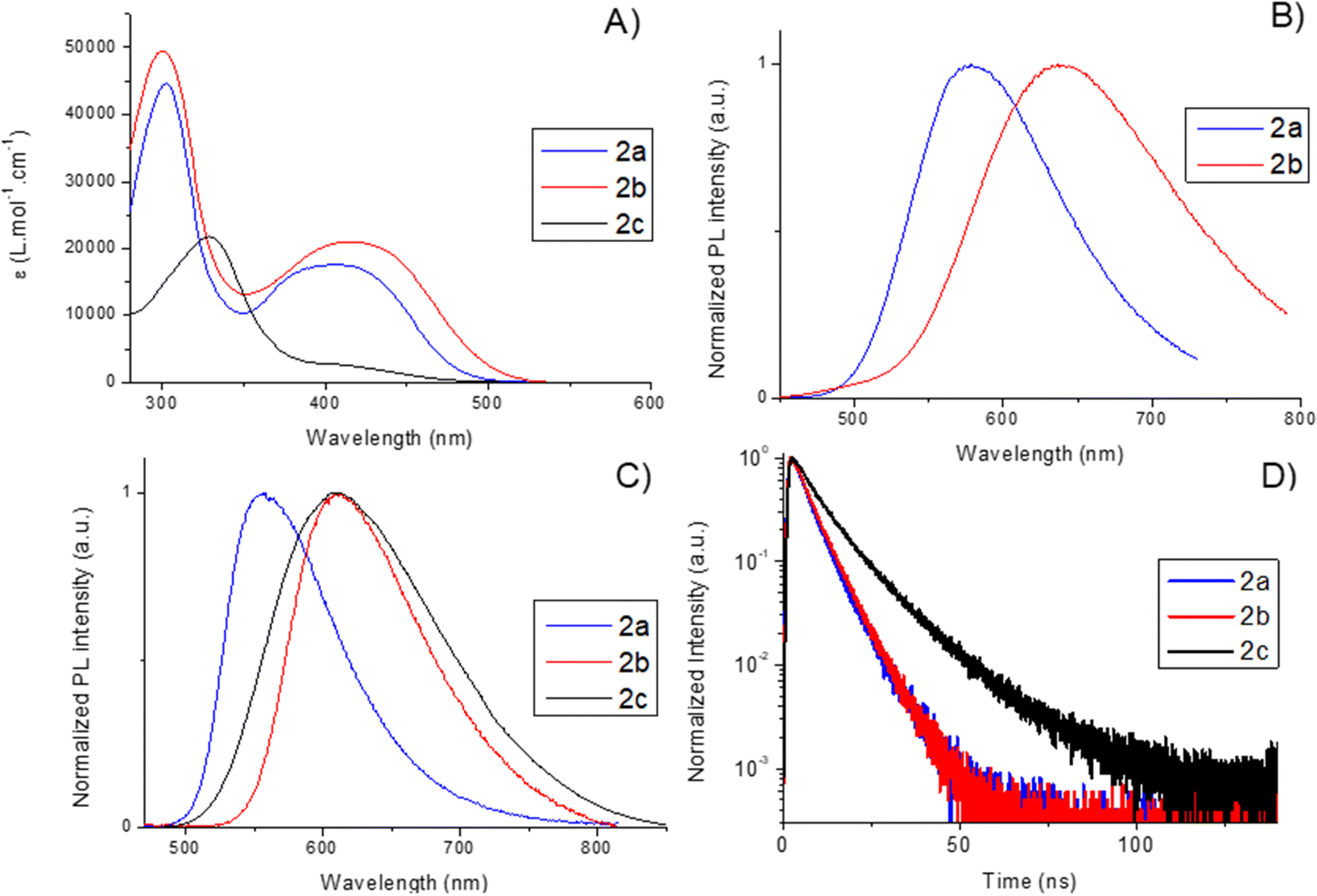 | ||
| Fig. 2 (A) Absorption spectra of 2a–c in DCM solution (c ≈ 1 × 10−6 M). (B) Normalized emission spectra of 2a–c in DCM solution (c ≈ 1 × 10−6 M). (C) Normalized emission spectra of 2a–c in powder. (D) Normalized transient PL decay of 2a–c in powder. | ||
| λ abs (nm) | ε (L mol−1 cm−1) | λ em (nm) | Φ DCM | λ em powder (nm) | Φ powder | Φ film | τ (ns) powder | k r (107 s−1) powder | k nr (107 s−1) powder | |
|---|---|---|---|---|---|---|---|---|---|---|
| a In DCM solution (10−5 M). b Calculated relative to fluorescein (NaOH, 0.1 M), ϕref = 0.79. c Measured using a calibrated integration sphere. d Films prepared by vacuum sublimation. e k r = radiative relaxation rate (kr = Φ/τ); knr = non-radiative relaxation rate (knr = (1 − Φ)/τ). | ||||||||||
| 2a | 302, 406 | 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 18000 000, 18000 |
578 | 0.34 | 556 | 1 | 0.90 | 4.4(72%) | 17.9 | — |
| 8.6(28%) | ||||||||||
| 2b | 301, 415 | 50000, 21000 |
637 | 0.09 | 610 | 0.65 | 0.54 | 4.7(61%) | 11.1 | 6.0 |
| 7.6(39%) | ||||||||||
| 2c | 329, 410 | 22000, 2600 |
— | — | 609 | 0.14 | 0.16 | 6.3 (48%) | 1.3 | 8.2 |
| 14.3(52%) | ||||||||||
2a and 2b display fluorescence in diluted solution (DCM) with their maximum wavelengths at 578 nm and 637 nm, respectively (Fig. 2B and Table 1). A stronger redshift (Δλ = 59 nm) is observed compared to the absorption, which is typical for molecules showing ICT. A solvent dependence study on 2a indicates a positive solvatofluorochromism confirming the ICT nature of this transition (Fig. S13, ESI†). Both 2a and 2b show moderate photoluminescence quantum yields (PLQYs) of 0.34 and 0.09, respectively. This difference in PLQYs follows the energy-gap rule and is therefore unsurprising. 2c does not luminesce in diluted DCM solution. All compounds display luminescence lifetime in the range of ns, typical of purely fluorescent emitters, even in degassed toluene (see the ESI†).
In the solid state, all derivatives display fluorescence emission, and their PLQY is enhanced compared to the diluted solution (Fig. 2C and Table 1). In powders, the emission wavelengths are blue-shifted compared to the solution, with maxima centered at 556 nm instead of 578 nm for 2a and 610 nm instead of 637 nm for 2b. 2a even shows a PLQY of 1 in powder instead of 0.34 in the solution. This trend is the same when the PLQY is measured on thin films prepared by vacuum sublimation (Table 1). Interestingly, this value measured in the solid state is higher than the corresponding value of the analog molecule with an exocyclic phenyl moiety (PLQY = 0.68) instead of ethoxy.11 It is clear that the modification of the phosphorus center can enhance the PLQY. 2b displays a PLQY of 0.65 in powder, which is much higher than that in the solution (0.09). Interestingly, 2c, which was poorly luminescent in solution, displays solid-state emission with a PLQY of 0.14. This enhancement of the emission in the solid state is known for related benzophosphole derivatives whose aggregation-induced emission (AIE) properties were already reported.11
The fluorescence lifetimes (τ) of these emitters were also measured at room temperature for the powder samples (Fig. 2D and Table 1). All these emitters display a biexponential decay in the ns range, typical of fluorescence. This is in marked contrast to the TADF behavior observed using dibenzophosphole oxides substituted with similar amino groups,12 highlighting the strong differences between benzophosphole and dibenzophosphole cores, as TADF has never been observed on substituted benzophospholes. The radiative (kr) and non-radiative (knr) constants of these fluorophores have been calculated using their PLQY and the lifetime of their most contributive component. 2a has a kr value of 17.9 × 107 s−1 and no accurate value of knr can be given as the PLQY reaches 1. 2b displays a lower kr value of 11.1 × 107 s−1 and the appearance of a non-trifling knr of 6.0 × 107 s−1 reflecting the energy gap law. 2c displays a considerably lower kr value (1.3 × 107 s−1), consistent with a small oscillator strength for the transition due to the poor conjugation between the donor and the acceptor driven by the twisted conformation of the phenoxazine group, as well as a rather large knr value (8.2 × 107 s−1).
The electrochemical behavior of 2a–c was investigated by cyclic voltammetry in DCM solution (Table 2 and Fig. S19–S23, ESI†). All three compounds display similar reduction processes, with the potential characteristic of the benzophosphole oxide unit: Ered(2a) = −1.59 V vs. FeCp*2+/0, Ered(2b) = −1.70 V vs. FeCp*2+/0 and Ered(2c) = −1.46 V vs. FeCp*2+/0. Moreover, 2a and 2b display an irreversible process for the reduction while 2c shows a quasi-reversible process. Regarding oxidation, all compounds display oxidation waves typical of arylamines. These processes are quasi-reversible for 2a and 2b, and reversible for 2c. All the oxidation waves are doubled in intensity compared to the reductions, due to the presence of two arylamine groups. The values of Eox nicely reflect the electron-donating ability of different amines: Eox (2a) = +1.06 V vs. FeCp*2+/0, Eox (2b) = +0.83 V vs. FeCp*2+/0 and Eox (2c) = +0.87 V vs. FeCp*2+/0.
| E ox (V vs. FeCp*2+/0) | E red (V vs. FeCp*2+/0) | HOMOb (eV) | LUMOb (eV) | E g redox (eV) | E g optical (eV) | ||
|---|---|---|---|---|---|---|---|
| a (c = 10−3 M) recorded in CH2Cl2 with Bu4N+PF6− (0.2 M) at a scan rate of 200 mV s−1. b HOMO (eV) = −Eox(onset) (−4.8 + 0.545)//LUMO (eV) = −Ered(onset) (−4.8 + 0.545). E (1/2)FeCp2+/0vs. FeCp*2+/0 in DCM = 0.545 V.13,14 | |||||||
| 2a | +1.06 | −1.59 | −5.21 | −2.74 | 2.47 | 2.58 | |
| 2b | +0.83 | −1.70 | −4.97 | −2.64 | 2.33 | 2.48 | |
| 2c | +0.87 | −1.46 | −5.06 | −2.86 | 2.20 | 2.58 | |
To gain deeper insights into the optical properties of the fluorophores, DFT and TD-DFT calculations were carried out (see the ESI† for details). By considering the various systems, a notable difference appears in absorption. Indeed, while for 2a and 2b, TD-DFT yields a strongly dipole-allowed first transition with respective f larger than 0.400, the two first excitations of 2c show very weak oscillator strengths of 0.027 and 0.017, and it is only the third transition that develops a sizeable f (0.317) in this compound. The computed vertical absorptions for the first transition are 402 and 415 for 2a and 2b, respectively. Although the vertical absorption values are not λmax,14 one can see a reasonable qualitative match between experiment and theory in terms of the position of the first strong absorption. For 2c, the two weakly allowed transitions are positioned at 374 and 367 nm, and the main strong band at 324 nm. This is well in line with the experimental finding: a strong absorption between 300 and 350 nm, followed by a long tail at longer wavelengths for 2c (Fig. 2A).
In Fig. 3, we illustrate the lowest excited states of 2a and 2b. The topology of these transitions is obviously similar, with NPh2 units acting as donor groups and the P-containing core of the dye acting as the acceptor. The ICT seems a bit stronger in the group located at the 2-position of phosphole, whereas the impact of methoxy substituents is rather limited, which is unsurprising given the already strong donating character of the NPh2 groups.
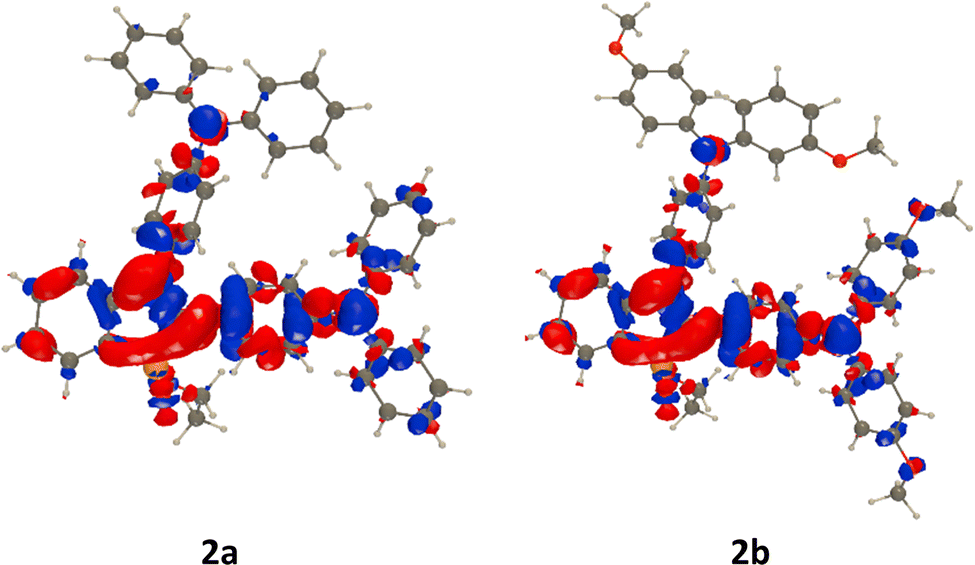 | ||
| Fig. 3 Electron density difference (EDD) plots for the lowest excited states of 2a and 2b. The blue and red regions correspond to the decrease and increase of density upon excitation, respectively. Contour threshold: 0.001 au. | ||
Let us now focus on 2c, see Fig. 4: the two weakly allowed transitions correspond to pure ICT states from phenoxazine to the dye core, but the almost perpendicular nature of the two π-conjugated (sub)systems makes these absorptions nearly dark. The strong absorption at higher energy is localized on the dye core. This means that the difference in absorption spectra between the various dyes is mainly a consequence of the geometrical constraints: while diphenylamine is electronically connected to the P-bearing cycles thanks to its propeller shape, the orthogonality of phenoxazine cuts such direct connection and only through-space ICT takes place.
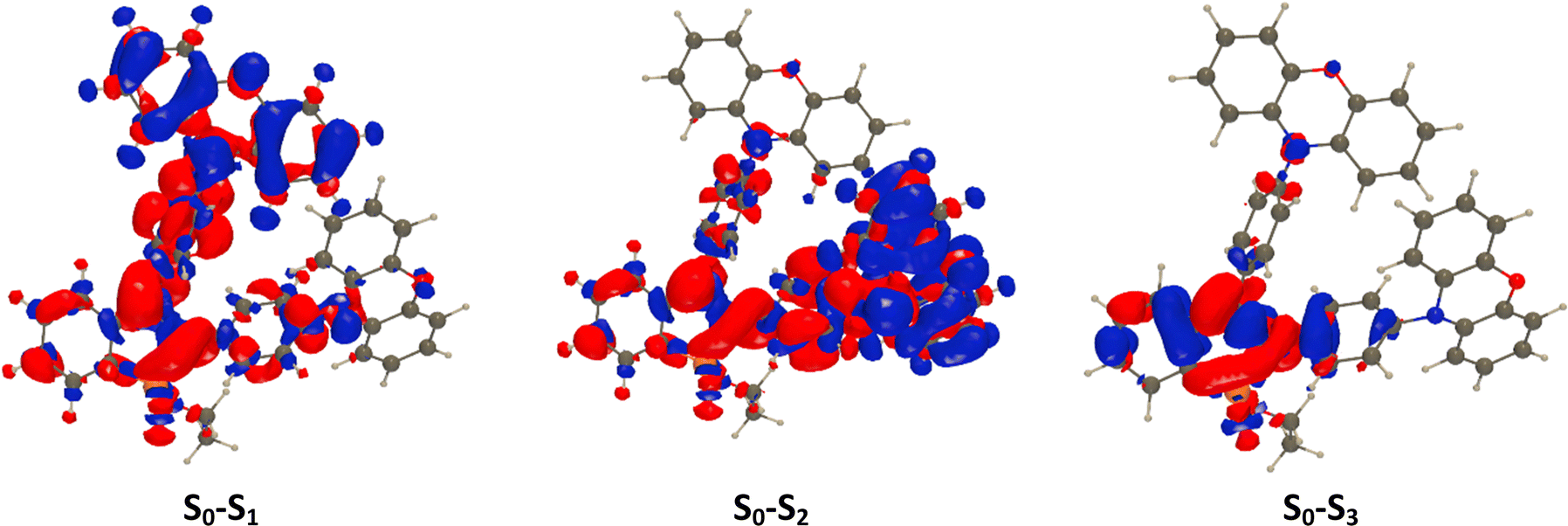 | ||
| Fig. 4 Electron density difference (EDD) plots for the three lowest excited state of 2c. | ||
We have determined the 0–0 energies of the three emissive dyes in the solution; as these can be straightforwardly compared between experiment and theory, we obtained the following values: 481 nm (2a) and 495 nm (2b), which are close to the experimental absorption–emission crossing point, with a +14 nm redshift between 2a and 2b, also fitting experiment (+17 nm). These results confirm the robustness of the selected theoretical level.
Finally, theoretical calculations were performed to determine the singlet–triplet (S–T) gaps at the optimal S1 geometries of 2a, 2b, and 2c (see the ESI† for details). For 2a, only one triplet state is lower than the S1 state, with a large gap of 0.73 eV. The results are similar for both 2b and 2c, with respective S1–T1 gaps of 0.71 eV and 0.66 eV, respectively. This indicates an unfavorable situation for TADF to appear, in full agreement with the experimental observations.
OLED devices
Taking advantage of the excellent luminescence properties of 2a–c and their high thermal stability (325 °C < Td(2a–c) < 330 °C), compounds 2a–c were introduced as emitters into multilayered OLED devices (see the ESI† for details). Because of their suitable HOMO–LUMO levels and their high solid-state luminescence, 2a–c were used as pure emitters and inside a host 4,4-bis(2,2-diphenylvinyl)-1,1-diphenyl (DPVBi) matrix. The performances of the OLED devices are reported in Table 3 for 2a–c used as pure emitters or at a doping ratio of 5% in the DPVBi matrix. This doping ratio of 5% has been set after a study of EQE vs. doping ratio on 2a (see Fig. S24, ESI†).| Device | Emitter | Doping ratio (%) | V on (V) | EQEb (%) | λ max EL (nm) | CEb (cd A−1) | PEb (lm W−1) | Maximal brightness (cd m−2) (@mA cm−2) |
|---|---|---|---|---|---|---|---|---|
| a Recorded at a luminance of 0.1 cd m−2. b External quantum efficiency, current efficiency (CE) and power efficiency (PE) recorded at 30 mA cm−2. | ||||||||
| A | 2a | 5 | 3.7 | 5.1 | 532 | 17.06 | 8.20 | 27010 (220) |
| B | 2b | 5 | 3.6 | 4.2 | 556 | 14.28 | 6.86 | 19200 (200) |
| C | 2c | 5 | 4.2 | 2.3 | 556 | 6.42 | 2.75 | 5958 (140) |
| D | 2a | Pure | 2.8 | 3.6 | 552 | 13.55 | 7.98 | 17500 (300) |
| E | 2b | Pure | 2.6 | 2.0 | 588 | 5.30 | 3.38 | 8586 (340) |
| F | 2c | Pure | 3.4 | 0.8 | 588 | 1.52 | 0.90 | 3600 (340) |
All emitters display blue-shifted electroluminescence compared to the photoluminescence in the solid state. This blue shift is even stronger when these emitters are used in the DPVBi matrix: λmaxEL (2a 5% in dPVBi) = 532 nm; λmaxEL (2a pure) = 552 nm; λmaxem (2a powder) = 556 nm. As DPVBi is an apolar matrix (Fig. S25, ESI†), this hypsochromic shift was expected. In all cases, the values of EQE follow the trend of the PLQY of the emitters in powder: EQE(2c) < EQE(2b) < EQE(2a). The theoretical value of 5% EQE for a fluorescent OLED is even reached with compound 2a in device A. It should be noted that such a theoretical limit of EQE is calculated for an outcoupling efficiency of 20%. However, the outcoupling might be higher by horizontal orientations of the emissive transition dipole moment.15 Moreover, device A displays a small roll-off efficiency illustrated by its very high brightness of 27010 cd m−2 at a high current density of 220 mA cm−2. These values represent a considerable increase compared to the previously reported related compounds and thus highlight the dramatic impact of small molecular variations on the properties of the device.11,16
Devices containing pure emitters in the emissive layer exhibit less efficient properties than devices containing DPVBi (EQE(A) = 5.1%; EQE(D) = 3.6%). Interestingly, the Von values of devices D, E, and F are significantly lower than those of devices A, B, and C (by ca. 1 V), indicating that the injection of charge inside the emissive layer is easier when there is no DPVBi. This property leads to better stability of devices D, E, and F: the maximal brightness is reached at a higher value of current density (300 to 340 mA cm−2) than that of devices A, B, and C (140 to 220 mA cm−2). To rationalize the difference of EQE, the photoluminescence of emissive layers (deposited on a glass substrate using evaporation) was studied (Table S2, ESI†). DPVBi has almost no impact on the PLQY of 2a (ΦPL(5% in DPVBi) = 0.93 and ΦPL(pure) = 0.90) while the impact on 2b and 2c is much larger (ΦPL(2b 5% in dPVBi)= 0.85; ΦPL(2b pure) = 0.54; ΦPL(2c 5% in DPVBi) = 0.36; ΦPL(2c pure) = 0.16). This effect is related to the ICT character of the emissive transition of 2a–c: the apolar environment of DPVBI enhances the contribution of the locally excited state, higher in energy and more emissive than the ICT state. Such behavior explains both the hypsochromic shift and the higher EQE for devices A, B and C using a host matrix.
Conclusions
In this article, we report the synthesis and full characterization of a family of benzophopsholes 2a–c substituted by electro-donating amino groups. The nature of the donor allows fine-tuning the ICT and thus the optical properties. In particular, these compounds display intense fluorescence in the solid state (powders/thin films). Considering the favorable redox and thermal properties, the compounds were introduced in multilayered OLED devices displaying high EQE reaching 5% for compound 2a. This is indeed the theoretical limit of the efficiency of purely fluorescent emitters. Ethoxy-substituted benzophopshole oxides thus represent a new class of highly efficient emitters for OLEDs. The introduction of these emitters into organic lasers or fast OLEDs will be investigated in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministère de la Recherche et de l’Enseignement Supérieur, the CNRS, the Région Bretagne, the French National Research Agency (ANR Fluohyb ANR-17-CE09-0020) and the China-French associated international laboratory in “Functional Organophosphorus Materials”, the GDR Phosphore. NL thanks the Région Bretagne for PhD grant. PAB thanks C. Quinton for assistance with lifetime measurements. DJ is indebted to the CCIPL computational center installed in Nantes for generous allocation of computational time.References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- (a) K. Müllen and U. Scherf, Organic Light Emitting Devices, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) B. Geffroy, P. Le Roy and C. Prat, Polym. Int., 2006, 55, 572 CrossRef CAS; (c) G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- (a) H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed; (b) M. Cai, D. Zhang and L. Duan, Chem. Rec., 2019, 19, 1611 CrossRef CAS PubMed.
- (a) A. S. D. Sandanayaka, T. Matsushima, F. bencheickh, S. Terakawa, W. J. Postcavage Jr, C. Qin, T. Fujihara, K. Goushi, J. C. Ribierre and C. Adachi, Appl. Phys. Express, 2019, 12, 061010 CrossRef CAS; (b) Q. Zhang, W. Tao, J. Huang, R. Xia and J. Cabanillas-Gonzalez, Adv. Photonics Res., 2021, 2, 2000155 CrossRef.
- K. Yoshida, P. P. Manousiadis, R. Bian, Z. Chen, C. Murawski, M. C. Gather, H. Haas, G. A. Turnbull and I. D. W. Samuel, Nat. Commun., 2020, 11, 1171 CrossRef CAS PubMed.
- (a) F. Mathey, Chem. Rev., 1988, 88, 429 CrossRef CAS; (b) R. Réau and T. Baumgartner, Chem. Rev., 2006, 106, 4691 Search PubMed; (c) Y. Matano and H. Imahoria, Org. Biomol. Chem., 2009, 7, 1258 RSC.
- (a) C. Fave, T. Y. Cho, M. Hissler, C. W. Chen, T. Y. Luh, C. C. Wu and R. Réau, J. Am. Chem. Soc., 2003, 125, 9254 CrossRef CAS PubMed; (b) O. Fadhel, M. Gras, N. Lemaitre, V. Deborde, M. Hissler, B. Geffroy and R. Réau, Adv. Mater., 2009, 21, 1261 CrossRef CAS; (c) H. Chen, W. Delaunay, L. Yu, D. Joly, Z. Wang, J. Li, Z. Wang, C. Lescop, D. Tondelier, B. Geffroy, Z. Duan, M. Hissler, F. Mathey and R. Réau, Angew. Chem., Int. Ed., 2012, 51, 214 CrossRef CAS PubMed; (d) C. Romero-Nieto, M. Marcos, S. Merino, J. Barbera, T. Baumgartner and J. Rodriguez-Lopez, Adv. Funct. Mater., 2011, 21, 4088 CrossRef CAS.
- (a) J. Phelipot, N. Ledos, T. Dombray, M. P. Duffy, M. Denis, T. Wang, Y. Didane, M. Gaceur, Q. Bao, X. Liu, M. Fahlman, P. Delugas, A. Mattoni, D. Tondelier, B. Geffroy, P.-A. Bouit, O. Margeat, J. Ackermann and M. Hissler, Adv. Mater. Technol., 2022, 2100876 CrossRef CAS; (b) J. Kuno, N. Ledos, P.-A. Bouit, T. Kawai, M. Hissler and T. Nakashima, Chem. Mater., 2022, 34, 9111–9118 CrossRef CAS; (c) J. Phelipot, P. Manzhi, N. Ledos, D. Tondelier, B. Geffroy, P.-A. Bouit, J. Ackermann, M. Hissler and O. Margeat, New J. Chem., 2022, 46, 22574–22580 RSC.
- M. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296 RSC.
- (a) Y.-R. Chen and W. -L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed; (b) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975 CrossRef CAS PubMed; (c) V. Quint, F. Morlet-Savary, J.-F. Lohier, J. Lalevée, A.-C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436–17441 CrossRef CAS PubMed; (d) C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 127, 15428 CrossRef.
- Z. Zhuang, F. Bu, W. Luo, H. Peng, S. Chen, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 1836 RSC.
- J. I. Nishida, Y. Kawakami, S. yamamoto, Y. Matsui, H. Ikeda, Y. Hirao and T. Kawase, Eur. J. Org. Chem., 2019, 3735 CrossRef CAS.
- J. R. Aranzaes, M. C. Daniel and D. Astruc, Can. J. Chem., 2006, 84, 288 CrossRef.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334 RSC.
- F. Tenopala-Carmona, O. S. Lee, E. Crovini, A. M. Neferu, C. Murawski, Y. Olivier, E. Zysman-Colman and M. C. Gather, Adv. Mater., 2021, 33, 2100677 CrossRef CAS PubMed.
- In addition to the structural change in the emitter, the structure of the OLED stack also slightly differs, see ref. 11.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc00245d |
| This journal is © The Royal Society of Chemistry 2023 |