
The unexpected mechanism of transformation from conventional room-temperature phosphorescence to TADF-type organic afterglow triggered by simple chemical modification†
Minjian
Wu
ab,
Jiuyang
Li
a,
Ju
Huang
a,
Xuepu
Wang
a,
Guangming
Wang
a,
Xiuzheng
Chen
a,
Xun
Li
a,
Xuefeng
Chen
a,
Shuhui
Ding
a,
Hefeng
Zhang
 *b and
Kaka
Zhang
*a
*b and
Kaka
Zhang
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, People's Republic of China. E-mail: zhangkaka@sioc.ac.cn
bDepartment of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, College of Science, Shantou University, Shantou, 515063, People's Republic of China. E-mail: hfzhang@stu.edu.cn
First published on 11th January 2023
Abstract
The study of transformation of photophysical behaviours in organic afterglow systems has emerged as an important topic, whereas the transformation in the reported studies only gives change of afterglow colour, duration or intensity. Herein, we report a serendipitous finding of the mechanism of afterglow transformation from conventional RTP to TADF-type organic afterglow triggered by simple chemical modification of coronene systems; usually, chemical modification can only lead to spectral shifts of luminescent systems. Coronene molecules show typical RTP behaviours when doped in organic matrices. After being substituted by difluoroboron β-diketonate moieties, the coronene-containing materials exhibit a TADF-type organic afterglow mechanism, which features a moderate kRISC to harvest triplet energies, enhance afterglow efficiency, and maintain long afterglow lifetimes. Interestingly, the TADF-type afterglow materials can be excited by visible lights, possess emission wavelength > 600 nm and PLQY > 40%, display excellent processability into desired patterns and aqueous dispersion, and function as high-contrast in vivo bioimaging agents. The present study provides a unique pathway for the manipulation of triplet excited states to fabricate high-performance organic afterglow materials.
 Kaka Zhang | Kaka Zhang was born in 1985 in Ningbo, Zhejiang Province, China. He received his BS degree in 2008 and PhD degree in 2013 from Fudan University under the supervision of Professor Daoyong Chen, Professor Ming Jiang and Professor Ping Yao. From 2013 to 2019, he did his postdoctoral work in The University of Hong Kong with Professor Vivian Wing-Wah Yam. In 2019, he was appointed as a principle investigator at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. His research interest includes luminescent materials and macromolecular self-assembly. |
Introduction
Control of the excited state properties is of fundamental importance for constructing novel and high-performance luminescent materials.1–3 In room-temperature phosphorescence (RTP) and organic afterglow systems, the understanding and manipulation of triplet excited states represent a central topic since the population and decay of triplet excited states determine the photophysical properties of organic systems.4–9 Pioneers in the fields showed that although phosphorescence emission is spin-forbidden, the introduction of n–π* transition and heavy atom effects can facilitate intersystem crossing and enhance the radiative decay of triplet excited states to allow the occurrence of organic RTP.10–13 Crystalline and glassy environments have also been reported to inhibit the nonradiative decay of triplet excited states for the fabrication of RTP and organic afterglow materials.14–16 A supramolecular assembly such as host–guest inclusion can be used to restrict intramolecular motions of luminescent guests to obtain efficient organic RTP materials.17–21 Recent studies exhibit the achievements of ultralong organic afterglow durations in donor–acceptor systems via photo-induced charge separation and slow charge recombination mechanisms.22–25 Elegant design of molecular systems and the control of the triplet excited states have been reported to lead to very high RTP and afterglow efficiency in organic systems.26Besides achieving high afterglow efficiency and long afterglow lifetimes, the study of the transformation of photophysical properties in organic RTP and afterglow systems has attracted increasing interest.27–41 Early studies on this topic showed the preparation of oxygen-, mechanical force, pH- and ion-responsive RTP and afterglow materials with intriguing photophysical transformation behaviours.27–35 Recently, it has been reported that colour-tunable RTP materials can be obtained by adjusting the doping concentration of luminescent molecules in organic matrices.36,37 Emission lifetimes of RTP systems have been found to be modulated by the triplet energy levels of organic matrices.38 The Diels–Alder reaction has been applied to control the RTP and afterglow behaviours in organic systems.39 Supramolecular recognition has also been reported to enhance the RTP properties of crown ether systems.40 A very recent study in our laboratory showed that the reaction between pyrylium salts and amine-containing compounds can be used as time-gated afterglow chemodosimeters.41 Despite these advancements in RTP systems, the mechanism of transformation of afterglow induced by specific stimuli or chemical reactions has been rarely reported. The study of the mechanism of transformation of afterglow not only has significant impact on the deep understanding of triplet excited state behaviours in organic systems, but also paves the way for high-performance organic afterglow materials.
Herein, we report a serendipitous finding of the mechanism of photophysical transformation from conventional RTP to TADF-type organic afterglow triggered by simple chemical modification of coronene systems; usually, chemical modification can only lead to spectral shifts of luminescent systems. Coronene molecules show the typical RTP mechanism when doped in 4-methoxybenzophenone matrices. After being substituted by difluoroboron β-diketonate moieties, the coronene-containing materials are found to exhibit the TADF-type organic afterglow mechanism, which features a moderate kRISC (rate constant of reverse intersystem crossing) to harvest triplet energies, enhance afterglow efficiency, and maintain afterglow lifetimes. The TADF afterglow materials can be excited by visible lights, possess emission wavelength > 600 nm and PLQY > 40%, display excellent processability into desired patterns and aqueous dispersion, and function as high-contrast in vivo bioimaging agents.
Results and discussion
By doping coronene (Cor) into 4-methoxybenzophenone (MeOBP) at 0.1 wt%, Cor-MeOBP-0.1% samples exhibited bright green organic afterglow under ambient conditions (Fig. 1A).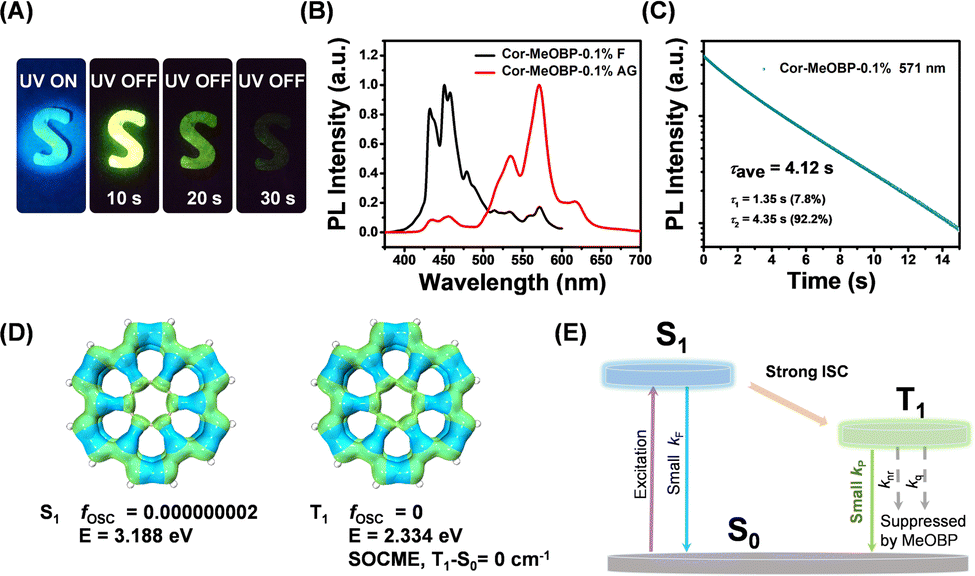 | ||
| Fig. 1 (A) Photographs of Cor-MeOBP-0.1% materials under 365 nm UV light and after removal of an UV lamp. (B) The steady-state emission spectra and delayed emission (1 ms delay) spectra of Cor-MeOBP-0.1% materials. (C) Room-temperature emission decay of Cor-MeOBP-0.1% materials monitored at 571 nm. (D) Isosurface maps of the electron-hole density difference of the lowest singlet excited states and lowest triplet excited states of the coronene molecule, where blue and green isosurfaces correspond to hole and electron distributions, respectively. (E) The proposed mechanism of room-temperature phosphorescence in the Cor-MeOBP system, which features small kF, small kP, and very small knr + kq. | ||
The Cor-MeOBP-0.1% materials displayed a fluorescence band in the range of 400 nm to 480 nm in their steady-state emission spectra, exhibited a phosphorescence band ranging from 500 to 650 nm in the delayed emission spectra (1 ms delay), and possessed a long phosphorescence lifetime up to 4.12 s (Fig. 1B, C and Fig. S1, ESI†). The afterglow of Cor-MeOBP-0.1% materials mainly originates from room temperature phosphorescence. At room temperature, the delayed emission spectra exhibit a small delayed fluorescence band ranging from 400 nm to 480 nm. Upon lowering temperature, this 400–480 nm delayed fluorescence band first decreases and then disappears at 243 K (Fig. S2, ESI†). TD-DFT calculation on the ORCA 4.2.1 program with the B3LYP/G functional and def2-TZVP(-f) basis set shows that coronene has very small oscillator strength (fosc) in its S1 state due to symmetry-forbidden S1–S0 transition (Fig. 1D), which would allow the occurrence of intersystem crossing to a large extent. It is noteworthy that T1–S0 transition shows very a small spin–orbit coupling matrix element (SOCME), which agrees well with the long phosphorescence lifetimes of Cor-MeOBP-0.1% materials. These, together with the reported studies on coronene systems,42–44 motivate us to propose that the coronene molecules dispersed in MeOBP matrices can be excited by UV or visible light to form singlet excited states, then undergo intersystem crossing to reach triplet excited states, and subsequently emit ultralong RTP due to the small kP (rate constant of phosphorescence decay) of coronene and very small knr + kq (rate constants of nonradiative decay and oxygen quenching) resulted from the rigid environment provided by MeOBP matrices (Fig. 1E).
Given that the long and bright afterglow of the coronene-MeOBP two-component system, we chemically modified the coronene to regulate the UV-vis and emission spectra of the materials. To this end, luminescent difluoroboron β-diketonate (BF2bdk) compounds were synthesized via the cascade reaction developed in our laboratory,45–47 then separated and purified by column chromatography and recrystallization successively (Fig. 2A). The structure of CorBF2 compounds was characterized by LRMS, HRMS, NMR, and FT-IR (see the ESI†). UV-vis spectra of CorBF2 in dichloromethane solutions showed broad absorption bands ranging from 360 nm to 500 nm with absorption maxima at 415 nm and a molar absorption coefficient (ε) of 18000 M−1 cm−1 (Fig. 2B). Upon excitation, CorBF2 in the dichloromethane solution displayed greenish yellow emission with structureless bands at 547 nm in its steady-state emission spectra (Fig. 2C; PLQY, 34.8%; fluorescence lifetime, 8.07 ns), which is much red-shifted compared to the coronene solution (Table 1). TD-DFT calculation showed that the S1 state of CorBF2 possessed intramolecular charge transfer (ICT) characters from the coronene group to the dioxaborine ring plus localized excitation (LE) characters (Fig. S3, ESI†), which agree with the positive solvatochromicity of CorBF2 solutions (Fig. S4, ESI†). The T1 state of CorBF2 also shows ICT (from the coronene group to the dioxaborine ring) plus LE (within the coronene group) characters. The CorBF2 in the solid state and in dichloromethane solution did not show afterglow upon switching off the UV lamp under ambient conditions (Fig. S5, ESI†).
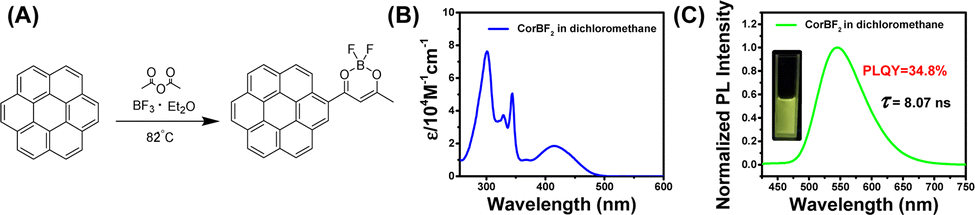 | ||
| Fig. 2 (A) Cascade synthesis of luminescent BF2bdk compounds. (B) UV-vis spectra of CorBF2 in the dichloromethane solution. (C) Steady-state emission spectra of CorBF2 in the dichloromethane solution. | ||
| Entry | λ abs/nm (ε × 10−4M−1 cm−1) | λ em/nm | τ/ns | Φ/% |
|---|---|---|---|---|
| Coronene in dichloromethane | 304 (14.0) | 445 | 1.7 (40%) | 4.8% |
| 325 (1.5) | 20.6 (60%) | |||
| 341 (3.2) | ||||
| CorBF2 in dichloromethane | 301 (7.6) | 547 | 8.07 | 34.8% |
| 329 (3.7) | ||||
| 344 (5.0) | ||||
| 415 (1.8) | ||||
| R1 in dichloromethane | 303 (9.2) | 640 | 1.58 | 27.8% |
| 341 (3.6) | ||||
| 547 (9.2) |
The introduction of organic matrices to control excited states of luminescent dopants has been demonstrated by us and other research groups to be significant for constructing organic room-temperature afterglow materials.22,23,48–51 Various organic matrices have been selected to accommodate CorBF2 dopants (Fig. S6, ESI†). When doping CorBF2 into MeOBP at 0.01%, the CorBF2-MeOBP-0.01% powders showed green emission under 365 nm UV light and performed green afterglow with duration longer than 4.5 s in a dark room after the removal of the UV lamp (Fig. 3A). The steady-state emission spectra of CorBF2-MeOBP materials have been found to be almost identical to the delayed emission (1 ms delay) bands (Fig. 3B). It is an unexpected finding because, in general, the delayed emission spectra of afterglow materials show a significant spectral red-shift compared to the steady-state emission spectra, for example, in the case of the coronene-matrix system (Fig. 1B). The steady-state emission spectra and delayed emission spectra of CorBF2-MeOBP-0.01% and CorBF2-MeOBP-0.001% materials, where CoBF2 molecules are molecularly dispersed in MeOBP matrices, showed broad and vibronic-structured emission bands ranging from 450 nm to 650 nm (Fig. 3B and Fig. S7, ESI†), which agree with TD-DFT calculation that CorBF2 possesses ICT plus LE characters (Fig. S3, ESI†).
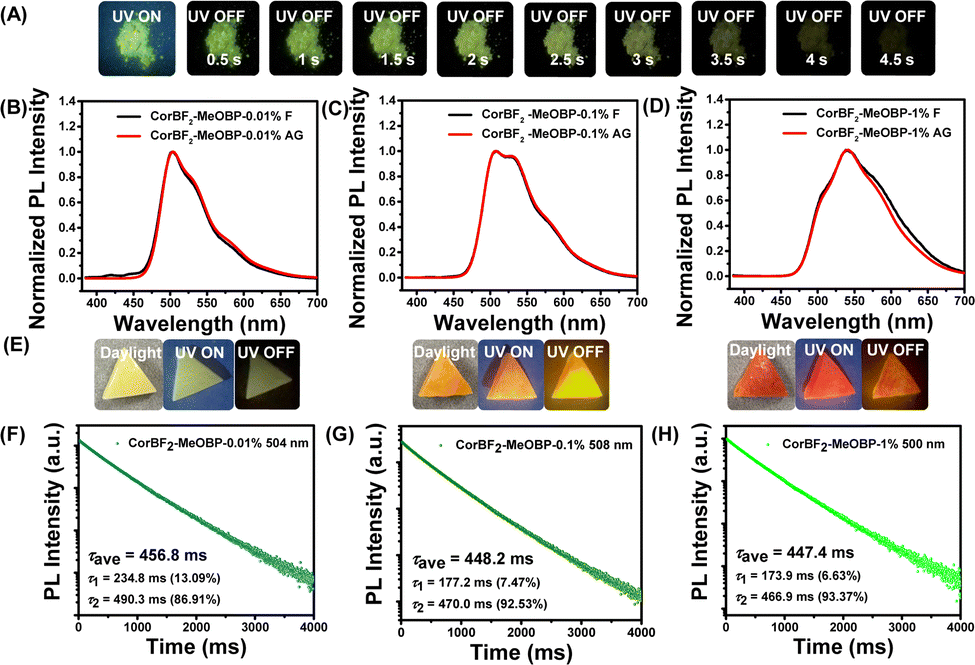 | ||
| Fig. 3 (A) Photographs of the CorBF2-MeOBP-0.01% powders under a UV lamp and after ceasing UV excitation. (B–D) Room-temperature steady-state and delayed emission (1 ms delay) spectra of CorBF2-MeOBP materials with different doping concentrations (B, 0.01%; C, 0.1%; D, 1%). (E) Photographs of the CorBF2-MeOBP melt-cast afterglow objects (left, CorBF2-MeOBP-0.01%; middle, CorBF2-MeOBP-0.1%; right, CorBF2-MeOBP-1%) under a 365 nm UV lamp and after ceasing UV excitation, respectively. (F–H) Room-temperature emission decay of CorBF2-MeOBP-0.01% materials monitored at 504 nm (F), CorBF2-MeOBP-0.1% melt-cast materials monitored at 508 nm (G) and CorBF2-MeOBP-1% materials monitored at 500 nm (H). These emission decay profiles were recorded using a Hitachi FL-4700 fluorescence spectrometer equipped with chopping systems. | ||
The coincidence of steady-state and delayed emission spectra has been reported in organic systems of triplet-to-singlet excited state energy transfer from the RTP donor to the fluorescence acceptor.52–56 It is known that the triplet excited state of benzophenone (BP) derivatives can be easily populated due to their highly efficient intersystem crossing (ISC).11 The excited state energy transfer can only occur when the matrices are sufficiently excited. In the present study, when excited at longer wavelength such as 405 nm and 420 nm visible lights, CorBF2-MeOBP materials still showed significant afterglow (Fig. S8 and S9, ESI†). MeOBP matrices possess insignificant absorption at 420 nm, so we reason that the excited state energy transfer from MeOBP to CorBF2 may exist when the samples are excited at 365 nm but is not necessary for the emergence of organic afterglow and the coincidence of steady-state and delayed emission spectra in the present CorBF2-MeOBP system. In the literature,22,23 the donor–acceptor system of intermolecular charge transfer characters exhibits that the organic long persistent luminescence (OLPL) mechanism can also show identical spectra between steady-state and delayed emission spectra according to recent reported studies. Cyclic voltammetry of CorBF2 and MeOBP has been performed. MeOBP matrices (HOMO, −6.24 eV; LUMO, −2.56 eV) possess low-lying HOMOs and high-lying LUMOs when compared to CorBF2 dopants (HOMO, −5.80 eV; LUMO, −2.82 eV), which suggests the absence of an exciplex between MeOBP and CorBF2. UV-vis absorption spectra and excitation spectra of CorBF2-MeOBP-0.01% materials also showed absorption bands with maxima similar to the UV-vis absorption spectra of CorBF2 in dichloromethane solutions (Fig. S10, ESI†). On the other hand, the two-photon ionization mechanism has been reported to give rise to OLPL.57,58 Since the two-photon ionization process involves two sequential excitation steps, the brightness and duration of OLPL are strongly dependent on excitation power. In the present study, power-dependent delayed fluorescence intensity measurements showed a quasi-linear relationship between delayed fluorescence intensity and excitation power (Fig. S11, ESI†). These results can rule out the two-photon ionization mechanism in the CorBF2-MeOBP system. In addition, the afterglow decay profiles of CorBF2-MeOBP materials follow exponential decay (rather than power law decay observed in some OLPL systems). Impurity has a vital contribution to organic afterglow reported in the reported studies.59 There are recent studies showing that the existence of impurities has critical impact on some organic afterglow systems. Considering the similar maxima of excitation spectra and solid UV-vis absorption spectra of CorBF2-MeOBP-0.01% materials (Fig. S10, ESI†) and the relatively high purity of CorBF2 confirmed by HPLC (Fig. S12A, ESI†), the afterglow was not likely to be originated from the impurity mechanism. Furthermore, by doping higher purity of CorBF2 (separated and collected fractions through HPLC) into MeOBP, we have prepared CorBF2-MeOBP (HPLC) materials whose room temperature emission spectra and emission decay profile showed similar afterglow properties compared to CorBF2-MeOBP materials (Table 2, Fig. 3B, F and Fig. S12, ESI†). We have also purified MeOBP matrices via two cycles of recrystallization. The purified MeOBP matrices showed very weak delayed emission signals and the absence of room-temperature afterglow (Fig. S13, ESI†). These results powerfully proved that the impurity mechanism can be ruled out in the present study.
| Sample | λ F/nm | λ AG/nm | τ AG (ms) | Φ (%) |
|---|---|---|---|---|
| Cor-MeOBP-0.1% | 450 | 571 | 4113 | — |
| CorBF2-MeOBP-0.01% | 504 | 504 | 456.8 | — |
| 534 | 534 | 456.0 | ||
| 590 | 590 | 499.2 | ||
| CorBF2-MeOBP-0.1% | 508 | 508 | 448.2 | 42.5 |
| 534 | 534 | 436.1 | ||
| 595 | 595 | 466.4 | ||
| CorBF2-MeOBP-1% | 500 | 500 | 447.4 | — |
| 541 | 541 | 429.1 | ||
| 590 | 590 | 462.7 | ||
| R1-MeOBP-0.002% | 610 | 610 | 31.77 | — |
| R1-MeOBP-0.01% | 619 | 619 | 26.03 | 43.7 |
| R1-MeOBP-0.02% | 619 | 619 | 49.30 | — |
| R1-MeOBP-0.2% | 619 | 619 | 81.10 | — |
To study the afterglow mechanism of the CorBF2-MeOBP system, we performed low temperature delayed emission experiments and observed CorBF2-MeOBP-0.01% powders showing orange afterglow after switching off the UV lamp at 77 K (Fig. 4A). Temperature-dependent delayed emission spectra from 77 K to 300 K have been recorded to study the triplet excited state of CorBF2-MeOBP-0.01%, where the phosphorescence (λP = 584 nm) is dominant in the emission spectra at 77 K, and a significant delayed emission band ranging from 450–550 nm gradually appears with the increase of the experimental temperature (Fig. 4B). Especially when the temperature has been set at 298 K, the delayed emission spectra nearly coincide with the steady-state emission spectra. In this case, the afterglow mechanism could be either triplet–triplet annihilation (TTA) or thermally activated delayed fluorescence (TADF). The TTA mechanism that is a bimolecular mechanism should have insignificant contribution to the organic afterglow due to the low doping concentration, 0.01% and even 0.001%. Temperature-dependent delayed emission lifetime measurements (monitored at 504 nm) of the CorBF2-MeOBP-0.01% materials have been performed. Upon increasing the temperature, the delayed fluorescence lifetimes have been found to decrease (Fig. S14, ESI†). These observations suggest that kRISC increases with temperature, which supports the TADF-type afterglow mechanism in the present study. Furthermore, power-dependent delayed fluorescence intensity measurements showed a quasi-linear relationship between delayed fluorescence intensity and excitation power (Fig. S11, ESI†). These experiments together with TD-DFT calculation (vide infra) all supported that CorBF2-MeOBP-0.01% materials undergo a TADF-type organic afterglow mechanism under ambient conditions.
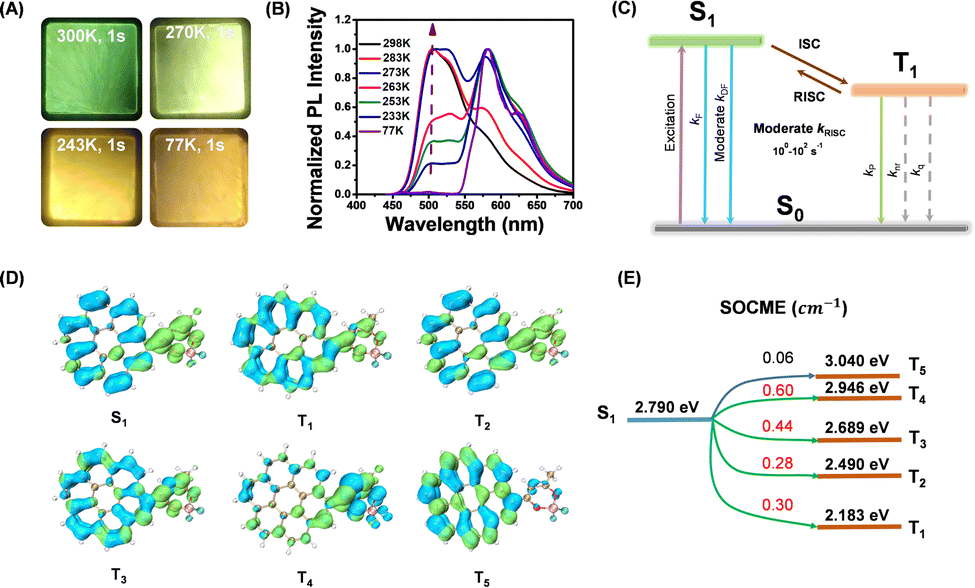 | ||
| Fig. 4 (A) Photographs of the afterglow of the CorBF2-MeOBP-0.01% materials at different temperatures. (B) Variable temperature delayed emission (1 ms delay) spectra of CorBF2-MeOBP-0.01% materials (excited at 420 nm). (C) Schematic illustration of the photophysical processes in the TADF-type organic afterglow system which features a moderate kRISC. (D and E) TD-DFT-calculated electron density difference of singlet and triplet excited states of CorBF2. TD-DFT calculations were performed on the ORCA 4.2.1 program with the B3LYP/G functional and def2-TZVP(-f) basis set (D) and energy levels and SOCME between S1 and triplet excited states (E). | ||
By increasing doping concentrations, the CorBF2-MeOBP-0.1% and CorBF2-MeOBP-1% materials also exhibited the coincidence between steady-state and delayed emission spectra (Fig. 3C and D). The lower-energy vibronic signals at around 534 nm and 590 nm were found to increase with CorBF2 doping concentrations, which should be caused by the aggregation of the planar CorBF2 molecules in MeOBP matrices; similar photophysical behaviours of vibronic signal change upon aggregation have also been reported in the literature.60,61 The CorBF2 aggregation gave rise to the red shifts of afterglow colours from green to orange (Fig. 3E). The lifetime of excited states of CorBF2-MeOBP-0.1% materials can be fit into double exponential decay with τ1 = 53.66 ms (5.92%) and τ2 = 471.15 ms (94.08%) and the PLQY had also been measured to be up to 42.5% (Fig. S15 and S16, ESI†). The lifetimes of the τ1 part that can be attributed to prompt fluorescence measured by a microsecond flash lamp were largely overestimated, similar to the reported studies.62,63 Since the room-temperature afterglow can be attributed to the τ2 part (delayed fluorescence) in the decay profile, the afterglow quantum yield of CorBF2-MeOBP-0.1% materials can be estimated to be 40.0%. The kRISC value of the CorBF2-MeOBP-0.1% materials can be estimated from their delayed fluorescence lifetimes (471.15 ms) to be on the order of 100 to 101 s−1. The moderate kRISC value enables the TADF mechanism to harvest triplet energies and maintain emission lifetimes >0.1 s, which is the key design factor of the present study to significantly improve afterglow quantum yields (Fig. 4C). It is noteworthy that the kRISC values in the CorBF2-MeOBP-0.01% materials are much smaller than those in TADF-type OLED systems64–67 (kRISC, 103 to 106 s−1) that possess large kRISC values leading to short emission lifetimes. TADF emitters with large kRISC values of 103–106 s−1 are necessary to construct efficient OLED devices. We understand that it is difficult to achieve large kRISC values of 103–106 s−1 in organic systems with ΔEST of 0.3 eV or 0.4 eV or above.68 Usually, organic molecular systems should simultaneously meet two requirements to realize large kRISC: (1) small ΔEST, for example, smaller than 0.2 eV; (2) large SOCME.64–66,69 In the absence of the heavy atom effect, relatively large SOCME can be realized by incorporating triplet excited states of different symmetries from the lowest singlet excited states according to the El-Sayed rule. We reason that if organic molecular systems meet requirement (1), or meet requirement (2), or partially meet requirements (1) and (2), a moderate kRISC value of 10−1–102 s−1 can be obtained. Given that kP values in ordinary organic systems in the absence of the heavy atom effect are in the range of 10−2–103 s−1, such a moderate kRISC value would be suitable to initiate a TADF mechanism to harvest triplet energy. Most conventional organic LE systems have very small kRISC values due to their large ΔEST and small SOCME. CorBF2 molecules possess a much smaller singlet–triplet splitting energy than LE systems, which enhances intersystem crossing to a large extent. Based on the rational molecular design of CorBF2 and a two-component strategy, the TADF-type afterglow materials with the afterglow quantum yield of 40.0% and lifetimes of 471.15 ms have been prepared by doping CorBF2 compounds into MeOBP matrices.
Polymers like poly(methyl methacrylate) (PMMA) have also been selected as organic matrices to prepare CorBF2-matrix afterglow materials. The CorBF2-PMMA-0.01% materials (PLQY = 48.0%) have been found to show blue-shifted steady-state emission spectra (λF = 490 nm, Fig. S17A, ESI†) when compared to CorBF2-MeOBP-0.01% materials (λF = 504 nm); PMMA has a smaller dipole moment than MeOBP. These results agree well with the description of the dipole effect,51 that is, the dipole–dipole interactions between dopants’ 1ICT states and organic matrices can reduce dopants’ S1 levels. The room-temperature delayed emission spectra of CorBF2-PMMA-0.01% materials mainly consist of room-temperature phosphorescence signals, as well as a small amount of delayed fluorescence signals (Fig. S17B, ESI†); at 77 K, the delayed emission spectra exhibit a phosphorescence band with maxima at 577 nm and shoulder at 620 nm (Fig. S17C, ESI†). In contrast, the room-temperature delayed emission spectra of CorBF2-MeOBP-0.01% materials showed predominant delayed fluorescence (Fig. 3B). These observations suggest the decrease of kRISC in PMMA matrices, since the relative intensity of delayed fluorescence to RTP is proportional to kRISC/kP. These results also agree well with the description of the dipole effect,51 that is, the dipole–dipole interactions between dopants’ 1ICT states and organic matrices can reduce ΔEST and facilitate dopants’ ISC and RISC; it has been reported that the decrease of ΔEST by 0.05 eV would enhance kRISC by around 10 times.64 Therefore, the selection of MeOBP matrices is very important for fabricating TADF-type organic afterglow materials in the present study.
TD-DFT calculations (ORCA 4.2.1 program with the B3LYP/G functional and def2-TZVP basis set) of CorBF2 molecules show that there are rich ISC and RISC channels with SOCME values around 0.3 cm−1 and above (Fig. 4D and E). Compared to coronene, the values of ΔEST of CorBF2 are relatively small and the SOCME values of CorBF2 are much larger (Fig. S18, ESI†). CorBF2 also possess excited states with different ICT and LE components. As a result, some triplet excited states with different electronic configurations from the lowest singlet excited state (S1) would show increased kISC and kRISC values to a moderate level according to the El-Sayed rule. Furthermore, since CorBF2-MeOBP-0.01% materials have been found to exhibit apparent TADF-type afterglow behaviours, the T1 level was estimated to be 2.12 eV from their phosphorescence maxima at 77 K, and the S1 level of CorBF2-MeOBP-0.01% materials was estimated to be 2.46 eV from the fluorescence maxima at room temperature collected in the steady-state emission mode (Fig. 4B). The ΔEST of CorBF2-MeOBP-0.01% materials can be estimated to be moderate at 0.34 eV. In view of the relatively small ΔEST and rich ISC/RISC channels, it is understandable that the present CorBF2-MeOBP system can obtain a moderate kRISC value on the order of 100 to 101 s−1 and undergo a TADF-type organic afterglow mechanism; kP values of the CorBF2-MeOBP systems should be much smaller than kRISC at room temperature. The relative intensity of delayed fluorescence to RTP is proportional to kRISC/kP of the CorBF2-matrix system. In the case of CorBF2-PMMA-0.01% materials, the room-temperature delayed emission spectra suggest that kP could compete or surpass kRISC (Fig. S17B, ESI†). In the case of CorBF2-MeOBP-0.01% materials, because MeOBP matrices can significantly enhance kRISC, the room-temperature delayed emission spectra showed predominant delayed fluorescence (Fig. 3B). The kRISC/kP of CorBF2-MeOBP-0.01% materials is also dependent on temperature; kRISC/kP increases with temperature.
We also studied the impact of oxygen on afterglow properties of CorBF2-MeOBP-0.01% materials, and the emission lifetime of CorBF2-MeOBP-0.01% materials under degassed conditions is slightly longer than that under ambient conditions (Fig. S19, ESI†). The observation showed that the rigid matrix efficiently protected the triplet excited state of CorBF2 against oxygen quenching by the encapsulation under ambient conditions. Moreover, the fluorescence intensities of steady-state emission spectra of CorBF2 in the dichloromethane solution under degassed conditions have also been recorded, which showed negligible changes compared to air conditions (Fig. S20, ESI†). Such behaviours are different from conventional TADF emitters of kRISC on the order of 103–106 s−1. In the present system, even though the triplet excited state of CorBF2via intersystem crossing could be populated, it can also be quenched by the active nonradiative decay process in solution states due to the small kRISC. These observations about the relationship between oxygen and the TADF-type afterglow materials have also been reported in our previous study.62,70
A further chemical modification of CorBF2 has been performed to obtain R1 of the donor–acceptor–donor molecular design to achieve more red-shifted emission by aldol condensation between α-methyl of CorBF2 and aldehyde group (Fig. 5A). R1 has been separated and purified successfully by column chromatography and recrystallization, and the structure was determined through NMR, LRMS, HRMS, and FT-IR, respectively (see the ESI†). No afterglow has been observed for R1 molecules in solid or solution states under ambient conditions (Fig. S21, ESI†). Both the UV-vis spectra and steady-state emission spectra of R1 in the dichloromethane solution showed significant red shifts compared to those of CorBF2, which showed the success of rational molecular design (Fig. 5B and C). To study the UV-vis spectra of R1 molecules, the emergence of the absorption band at 547 nm can be attributed to 1ICT transition, and UV-vis spectra of R1 in the dichloromethane solution exhibit an intense ICT absorption at 547 nm with the molar absorption coefficient (ε) as high as 92000 M−1 cm−1. These agree with the TD-DFT calculations where S0 to S1 transition possesses ICT characters and large oscillator strength (Fig. S22, ESI†). The steady-state emission spectra of the R1 dilute solution in dichloromethane displayed a broad fluorescence band ranging from 550 nm to 800 nm with the emission maxima at 640 nm (Fig. 5C). A high PLQY of 27.8% and a nanosecond lifetime of 1.58 ns of R1 in the dichloromethane dilute solution have been measured (Table 1) and a significant red-shifted emission with positive solvatochromicity has also been observed (Fig. S23, ESI†).
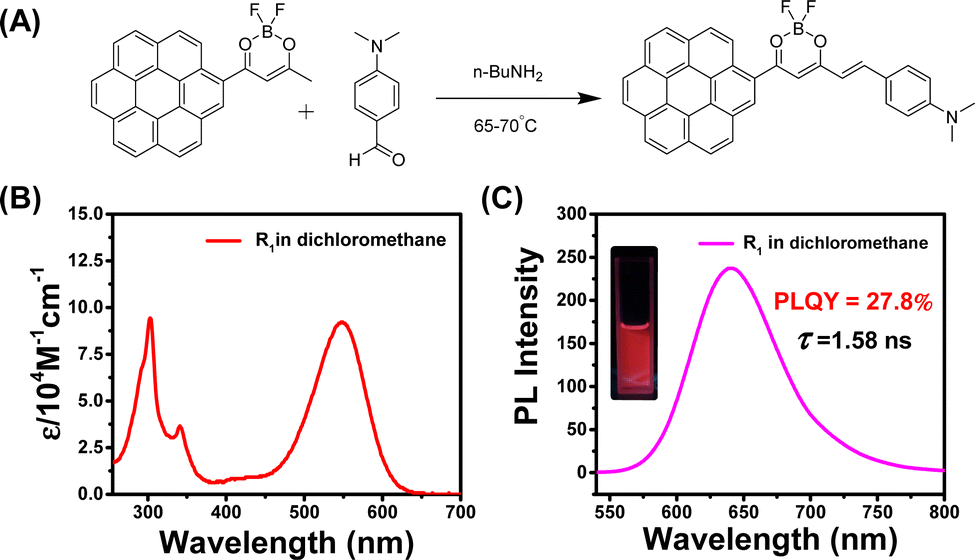 | ||
| Fig. 5 (A) Synthesis of R1 BF2bdk luminescent compounds via aldol condensation. (B) UV-vis spectra of R1 in the dichloromethane solution. (C) Steady-state emission spectra of R1 in the dichloromethane solution. | ||
The selection of organic matrices to accommodate luminescent dopants and to facilitate intersystem crossing of excited states of dopants have been considered to be significant to prepare organic room-temperature organic afterglow materials using a two-component strategy. Various matrices have been tested as the second component to control the triplet excited state of R1 in the R1-matrix system (Fig. S24, ESI†). When doping R1 into MeOBP at 0.002% to 0.2%, the R1-MeOBP powders have been found to exhibit bright red afterglow after the removal of the UV lamp (Fig. 6). The afterglow colour is very similar to the fluorescence colour, and the steady-state emission spectra of R1-MeOBP-0.002% are almost identical to the delayed emission spectra with the emission maxima of 610 nm excited by a 365 nm UV lamp. Interestingly, significant delayed emission spectra of R1-MeOBP-0.002% powders can also be obtained by 520 nm excitation (Fig. S25, ESI†) and the red afterglow can be achieved by 532 nm laser excitation (Fig. S26, ESI†). Again, these suggest that even though the process of energy transfer from MeOBP to R1 exists when the samples are excited by a UV lamp, it is not necessary for the emergence of red organic afterglow in the R1-MeOBP system. The formation of an exciplex via intermolecular charge transfer that has been reported to give rise to organic afterglow can be ruled out in the R1-MeOBP system because the MeOBP matrix possesses low-lying HOMOs and high-lying LUMOs when compared to R1 dopants (Fig. S27, ESI†). Besides, solid UV-vis spectra and excitation spectra of R1-MeOBP-0.002% possess similar maxima to the UV-vis spectra of R1 in dichloromethane solutions (Fig. S27, ESI†). The HPLC experiment has been performed to exclude the impurity effects in afterglow materials. However, the reverse-phase HPLC profile of R1 has been found to be very broad with significant tailing, which complicates the purity measurement. It is found that other boron difluoride hemicurcuminoid compounds in our lab also showed a very broad peak with significant tailing under reverse-phase HPLC measurements. We also tried normal-phase HPLC. However, because of the low polarity of R1, we did not capture the elution profile of R1 in the normal-phase HPLC measurement. About whether the impurity mechanism is responsible for the red afterglow, we reason that if there are impurities in R1 compounds, these possible impurities and R1 molecules would be well separated by MeOBP matrices after low concentration doping. The interaction between these possible impurities and R1 molecules would be negligible. If these possible impurities can give rise to the significant organic afterglow when doped into MeOBP matrices, the excitation spectra would be different from the UV-vis absorption spectra. In the present system, it has been found that the UV-vis absorption and excitation spectra of R1-MeOBP-0.002% and UV-vis spectra of R1 in dichloromethane solutions showed similar maxima (Fig. S27, ESI†). These can rule out the impurity mechanism in the R1-MeOBP system.
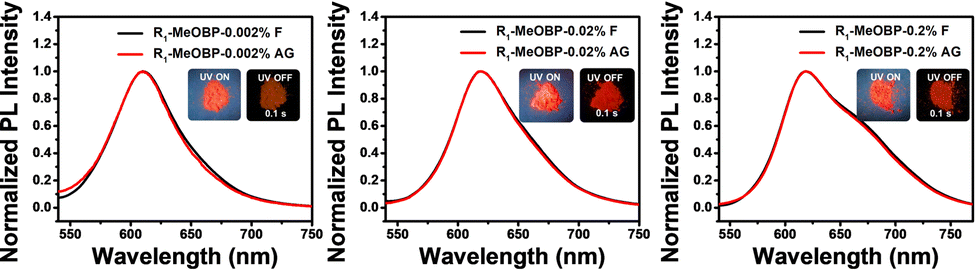 | ||
| Fig. 6 The room-temperature steady-state and delayed emission (1 ms delay) spectra of R1-MeOBP afterglow materials at doping concentrations of 0.002%, 0.02% and 0.2%, respectively. | ||
Low temperature experiments have been performed to study the afterglow mechanism of R1 molecules, and we have observed that the R1-MeOBP-0.01% powders showed red afterglow excited by a 532 nm laser at 77 K (Fig. S28, ESI†). Delayed emission spectra obtained with multiple temperature points from 77 K to 300 K have been recorded to study the triplet excited states of R1 molecules (Fig. 7A). The delayed emission spectra showed that an emission band ranging from 650 to 900 nm with the emission maxima of 701 nm at 77 K originates from triplet excited states of R1-MeOBP-0.01% samples (Fig. 7A). With the temperature gradually raised to 298 K, the delayed emission spectra nearly coincide with the steady-state emission spectra and show emission bands ranging from 540 nm to 650 nm with an emission maximum of 619 nm. The TTA afterglow mechanism can be ruled out due to the low concentration of R1 molecules doped into MeOBP. Power-dependent delayed fluorescence intensity measurements show a quasi-linear relationship between delayed fluorescence intensity and excitation power (Fig. S29, ESI†). These observations support that the R1-MeOBP-0.01% materials undergo a TADF afterglow mechanism.
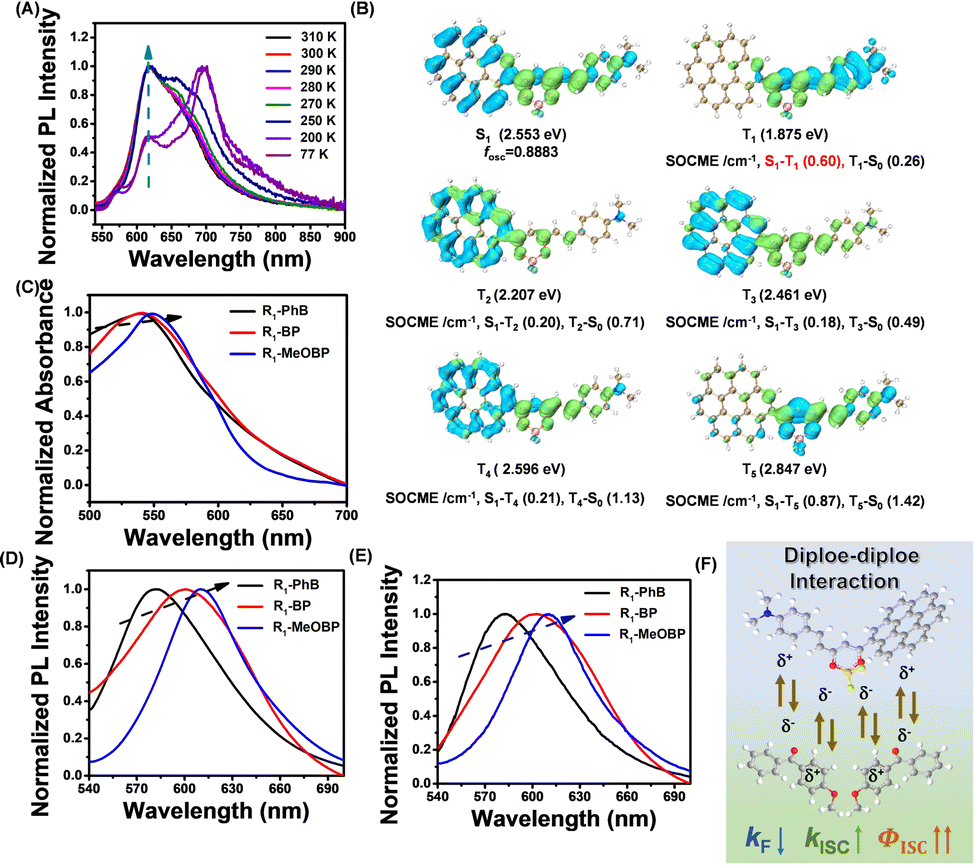 | ||
| Fig. 7 (A) Variable temperature delayed emission (1 ms delay) spectra of R1-MeOBP-0.01% materials (excited at 520 nm). (B) TD-DFT-calculated electron density difference of the singlet and triplet excited states of R1. TD-DFT calculations were performed on the ORCA 4.2.1 program with the B3LYP/G functional and def2-TZVP(-f) basis set. (C–E) UV-vis spectra (C), steady-state emission spectra (D), and delayed emission spectra (E) of the R1-matrix-0.002% samples. (F) Schematic illustration of dipole–dipole interactions between the S1 states of R1 and MeOBP matrices that can significantly enhance intersystem crossing of R1. | ||
The PLQY of R1-MeOBP-0.01% materials was measured to be 43.7% (Fig. S30, ESI†). The excited decay profile of R1-MeOBP-0.01% materials monitored at 619 nm showed tri-exponential decay that can be fit into τ1 = 2.27 ms (4.63%), τ2 = 16.73 ms (61.37%), and τ3 = 48.87 ms (31.00%) (Fig. S31, ESI†). In solid samples, there are heterogeneous microenvironments.71 The R1-MeOBP system has smaller S1–T1 and T1–S0 energy gaps than that of the CorBF2-MeOBP system. Because of the energy gap law, the photophysical processes would be enhanced in systems with low S1 and T1 levels. Therefore, the effect of heterogeneous microenvironments on excited state decay would be enhanced in the R1-MeOBP system, which possibly results in the triple-exponential decay behavior. The τ1 part and τ2 + τ3 part can be assigned to prompt fluorescence and delayed fluorescence, respectively. The delayed fluorescence (that is responsible for the afterglow property) quantum yield can be estimated to be 41.6% according to the PLQY of R1-MeOBP-0.01% materials and the emission decay profile with a high proportion of delayed fluorescence (τ2 + τ3, 95.37%) (Fig. S31, ESI†). The kRISC value of the R1-MeOBP-0.02% materials can be estimated from their delayed fluorescence lifetimes (τaverage = 25.41 ms) to be on the order of 101 to 102 s−1, which is smaller than those of TADF-type OLED materials. This is the key design of the work to harvest the triplet excited state and maintain a relatively longer emission lifetime.
TD-DFT calculations (ORCA 4.2.1 program with the B3LYP/G functional and def2-TZVP) show that the S1 state of R1 mainly possesses ICT characters from coronene and hemicurcuminoid groups to the dioxaborine ring (Fig. 7B). The T1 state of R1 shows ICT characters from the hemicurcuminoid group to the dioxaborine ring (Fig. 7B). Because of the different excited state nature between S1 and T1 states, the ISC channel of S1–T1 possesses a relatively large SOCME value of 0.60 cm−1 (Fig. 7B). The ΔEST can be estimated to be 0.23 eV from emission maxima; the T1 level is estimated to be 1.77 eV from phosphorescence maxima at 77 K and the S1 level is estimated to be 2.00 eV from fluorescence maxima at room temperature (Fig. 6 and 7A). The relatively small ΔEST together with relatively large SOCME values of 0.60 cm−1 for T1–S1 RISC suggests that the R1-MeOBP-0.01% materials possess a moderate kRISC value. Oxygen impacts on afterglow properties have also been taken into consideration, and we have observed an inactive relationship between oxygen and R1-MeOBP-0.01% afterglow materials because of the triplet excited states of R1 molecules well protected by the rigid matrix. The steady-state emission spectra of R1 in different solvents under degassed conditions show an insignificant change after exposure to air conditions (Fig. S32, ESI†). We reason that even though triplet excited states of R1 can be populated via intersystem crossing, these can be easily competed by nonradiative decay due to relatively larger knr and small kRISC in solution. These observations show that oxygen has little influence on the photoluminescence intensity of R1 in a dilute solution.
The UV-vis spectra and room-temperature emission spectra of the R1-matrix have been recorded to study the effects of dipole moments of organic matrices on excited states of the R1 dopant. With the dipole moments of organic matrices increased, the UV-vis spectra of the R1-matrix show slightly red-shifted absorption (Fig. 7C), while the steady-state emission spectra and delayed emission spectra of the R1-matrix revealed red-shifted emission which indicated the reduction of the S1 level of the R1 dopant (Fig. 7D and E). The S1 levels are estimated from their fluorescence emission maxima. Given that most of the organic systems possess T1 levels that are insensitive to the medium or environment, the T1 levels (1.77 eV) are estimated from the phosphorescence maxima of R1-MeOBP-0.01% materials at 77 K. The ΔEST values of R1-PhB (2.13 eV), R1-BP (2.06 eV) and R1-MeOBP (2.03 eV) can be estimated from their fluorescence emission maxima and T1 level of R1-MeOBP-0.01% materials. These observations indicate that dipole–dipole interaction between R1 dopants and organic matrices can stabilize single excited states and effectively reduce the ΔEST.51 The emission decay profile of prompt fluorescence (τPF) and delayed fluorescence (τDF) of the R1-matrix-0.002% samples has also been collected (Fig. S33, ESI†). When BP derivatives have been served as organic matrices, the τPF values of R1-BP-0.002% (2.14 ns) and R1-MeOBP-0.002% (2.20 ns) powders have been found to decrease compared to R1-PhB-0.002% (2.26 ns) samples (Fig. S33, ESI†). In the literature,72–74kF of 1CT states of BF2bdk compounds has been reported to decrease with the increase of dipole moments of the medium or environment. Since τPF is inversely proportional to (kF + knr+ kISC) of the 1CT states, and kF would decrease with regard to a relatively larger dipole of BP and MeOBP matrices, the decrease of τPF in R1 suggests a significant enhancement of kISC of R1 excited states in MeOBP and BP matrices. These observations suggest that the BP and MeOBP matrices can enhance ISC and decrease the kF values of R1's S1 states via dipole–dipole interactions, leading to a remarkable population of the triplet excited states of R1, which contributes to excellent afterglow properties of R1-MeOBP-0.002% materials (Fig. 7F). The MeOBP matrices in the present study not only suppress knr and kq of dopants’ T1 by a rigid crystalline microenvironment but also enhance dopants’ ISC and RISC by the dipole effect.51 This dipole effect in enhancing both ISC and RISC has also been demonstrated by Gillett and coworkers in a very recent study.75
Many small-molecule RTP and afterglow materials show poor processability. In the present study, MeOBP matrices have a melting point of 60–63 °C, which allowed them to be constructed into desired objects with the aid of silicone moulds. Cat paw and bone-shaped objects of Cor-MeOBP-0.1%, CorBF2-MeOBP-0.1% and R1-MeOBP-0.01% materials were prepared to exhibit bright yellowish green afterglow with the duration of 30 s, yellow afterglow with the duration of 11 s and red afterglow with the duration of 3 s, respectively (Fig. 8A and Fig. S34, ESI†). Thanks to the low melting point of MeOBP, large-area films have been prepared by sandwiching hot dopants-MeOBP melts between a pair of 10 cm × 10 cm glass plates followed by cooling under ambient conditions where diverse afterglow patterns, such as fancy Christmas, sea turtle and rabbit, were obtained through predesigned masks after the removal of a UV lamp (Fig. 8B and C). The CorBF2-MeOBP-0.01% and R1-MeOBP-0.002% large-area afterglow films also allowed direct writing by 405 nm and 532 nm lasers to exhibit afterglow characters like “9”, “1”, “3” and “ ”, respectively (Fig. 8D and E). These suggest that the afterglow films can be employed as afterglow writing panels for data security.
”, respectively (Fig. 8D and E). These suggest that the afterglow films can be employed as afterglow writing panels for data security.
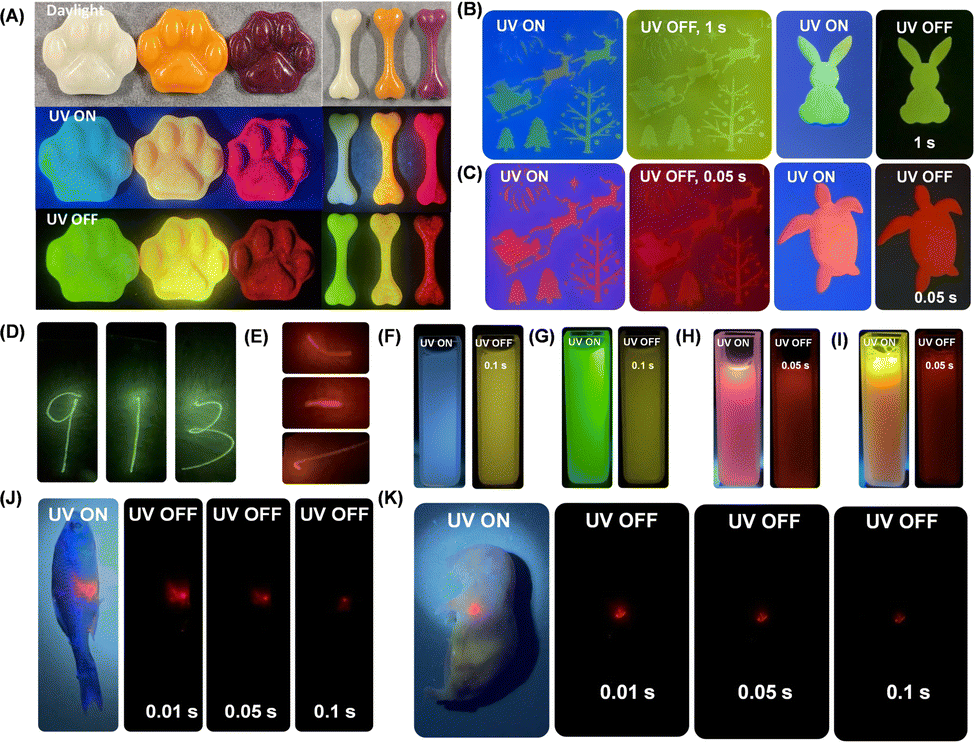 | ||
Fig. 8 (A) Photographs of Cor-MeOBP-0.1%, CorBF2-MeOBP-0.1%, and R1-MeOBP-0.01% afterglow objects under daylight, under 365 nm UV light, and after the removal of UV excitation, respectively. (B and C) Photographs of diverse afterglow patterns obtained through predesigned masks on a CorBF2-MeBOP-0.01% film (B) and R1-MeOBP-0.002% film (C) under 365 nm UV light and after the removal of a UV excitation source. (D and E) Afterglow writing of “9”, “1”, “3” and “ ” letters on CorBF2-MeBOP-0.01% (D) and R1-MeOBP-0.002% films (E) (10 cm × 10 cm) by 405 nm and 532 nm laser pointers (50 mW). (F and G) Photographs of the CorBF2-MeOBP-0.1% aqueous dispersion under UV light and after the removal of UV light (F) and the CorBF2-MeOBP-0.1% aqueous dispersion in the presence of fluorescein sodium dyes under 365 nm UV light and after the removal of UV light (G). (H and I) Photographs of the R1-MeOBP-0.02% aqueous dispersion under UV light and after the removal of UV light (H) and the R1-MeOBP-0.02% aqueous dispersion in the presence of rhodamine 6G dyes under 365 nm UV light and after the removal of UV light (I). (J and K) Preliminary bioimaging studies of the aqueous dispersion of R1-MeOBP-0.02% afterglow in fish (J) and mice (K). ” letters on CorBF2-MeBOP-0.01% (D) and R1-MeOBP-0.002% films (E) (10 cm × 10 cm) by 405 nm and 532 nm laser pointers (50 mW). (F and G) Photographs of the CorBF2-MeOBP-0.1% aqueous dispersion under UV light and after the removal of UV light (F) and the CorBF2-MeOBP-0.1% aqueous dispersion in the presence of fluorescein sodium dyes under 365 nm UV light and after the removal of UV light (G). (H and I) Photographs of the R1-MeOBP-0.02% aqueous dispersion under UV light and after the removal of UV light (H) and the R1-MeOBP-0.02% aqueous dispersion in the presence of rhodamine 6G dyes under 365 nm UV light and after the removal of UV light (I). (J and K) Preliminary bioimaging studies of the aqueous dispersion of R1-MeOBP-0.02% afterglow in fish (J) and mice (K). | ||
Afterglow materials that can maintain afterglow properties when dispersed in an aqueous medium have shown promising biomedical applications. Generally, the majority of the reported studies showed that organic RTP or afterglow materials lose afterglow properties when being transferred in aqueous dispersions. Here, the afterglow dispersions can be obtained after the molten CorBF2-MeOBP-0.1% droplets were added into an aqueous solution of the Pluronic F127 surfactant at 80 °C under sonication, then the hot dispersion was immediately frozen by liquid nitrogen and the dispersion recovered to room temperature finally (Fig. S35, ESI†). The CorBF2-MeOBP-0.1% aqueous dispersion has been found to exhibit yellow afterglow after the removal of the UV lamp (Fig. 8F). Strong background fluorescence interference studies have been conducted, and the CorBF2-MeOBP-0.1% aqueous dispersion can maintain afterglow properties in the presence of fluorescein sodium after the removal of the UV excitation source (Fig. 8G). The R1-MeOBP-0.02% aqueous dispersion has also been prepared by following the similar procedure above, which exhibits bright red afterglow after the removal of 365 nm UV light at room temperature (Fig. 8H). The orange fluorescence of the R1-MeOBP-0.02% aqueous dispersion was observed in the presence of rhodamine 6G and only red afterglow was observed after turning off the UV excitation source (Fig. 8I). These observations indicated that both CorBF2-MeOBP-0.1% and R1-MeOBP-0.02% aqueous afterglow dispersions have strong capability to eliminate the interference of strong background fluorescence by making use of their long-lived excited states.
The aqueous suspension of R1-MeOBP-0.02% afterglow materials can be prepared using a grinding method with the assistance of the Pluronic F-127 surfactants, which also can exhibit red afterglow after the removal of an UV excitation source in a dark room. The diameter of the R1-MeOBP-0.02% afterglow dispersion has been found to be 3.5 μm by fluorescence microscope studies (Fig. S36, ESI†). The aqueous afterglow suspension can be readily drawn by a plastic syringe and easily injected into living fish and mice to perform preliminary biological imaging experiments. The biological imaging in the afterglow modes can avoid the interference from background fluorescence and possible scattering from the excitation lights to display very clean backgrounds in the photographs captured (Fig. 8J and K).
Conclusions
In summary, this study presents the unexpected mechanism of transformation from conventional RTP to efficient TADF-type organic afterglow in coronene-containing dopant-matrix systems. All the room-temperature photophysical studies, variable-temperature delayed emission experiments, the understanding of the ICT molecular design and dopant-matrix strategy to achieve kRISC of 100–102 s−1, TD-DFT calculations, and the discussion to rule out the mechanism of excited state energy transfer and other delayed fluorescence mechanism, as well as the supports from our previously reported TADF-type afterglow systems,41,62,70 point to the fact that CorBF2-MeOBP and R1-MeOBP systems at room temperature exhibit TADF-type organic afterglow. Despite their serendipitous nature, detailed studies on the underlying photophysics reveal the strong potential of ICT molecular design to achieve intriguing photofunctional materials. ICT molecules possess much smaller singlet–triplet splitting energy than LE systems, which enhances intersystem crossing to a large extent. The incorporation of more than one electron-donating group enriches excited state characters where some Tn states can mediate intersystem crossing. Furthermore, the dipole–dipole interactions between dopants’ 1ICT states and organic matrices facilitate intersystem crossing. Meanwhile, the organic matrices suppress nonradiative decay and oxygen quenching of 3ICT states. This advanced ICT technology would provide a general strategy for constructing efficient TADF-type organic afterglow materials, which features a moderate kRISC to harvest triplet energies, enhance afterglow efficiency, and maintain long afterglow lifetimes (the advantages of TADF-type afterglow materials have been detailed in Text S1, ESI†). The novel design strategy extracted from the present study would have significant impact on the development of high-performance organic afterglow materials and their promising applications in diverse fields.Author contributions
Minjian Wu, Jiuyang Li, Ju Huang, Xuepu Wang, Guanming Wang, Xiuzheng Chen, Xun Li, Xuefeng Chen and Shuhui Ding performed the studies. Minjian Wu and Jiuyang Li drafted the manuscript. Hefeng Zhang supervised this work. Kaka Zhang supervised this work and drafted and revised the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial supports from the National Natural Science Foundation of China (22175194), the Shanghai Scientific and Technological Innovation Project (20QA1411600, 20ZR1469200), and the Hundred Talents Program from the Shanghai Institute of Organic Chemistry (Y121078).Notes and references
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- D. Li, F. Lu, J. Wang, W. Hu, X. M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed.
- Z. Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- X. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- K. Jinnai, R. Kabe, Z. Lin and C. Adachi, Nat. Mater., 2022, 21, 338–344 CrossRef CAS PubMed.
- P. Alam, N. L. C. Leung, J. Liu, T. S. Cheung, X. Zhang, Z. He, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, C. C. S. Chan, K. S. Wong, Q. Peng and B. Z. Tang, Adv. Mater., 2020, 32, 2001026 CrossRef CAS PubMed.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed.
- Y. Zhou, W. Qin, C. Du, H. Gao, F. Zhu and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 12102 CrossRef CAS PubMed.
- L. Gu, X. Wang, M. Singh, H. Shi, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2020, 11, 6191 CrossRef CAS PubMed.
- H. Wu, W. Chi, G. Baryshnikov, B. Wu, Y. Gong, D. Zheng, X. Li, Y. Zhao, X. Liu, H. Agren and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4328–4333 CrossRef CAS PubMed.
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174 CrossRef CAS PubMed.
- P. Long, Y. Feng, C. Cao, Y. Li, J. Han, S. Li, C. Peng, Z. Li and W. Feng, Adv. Funct. Mater., 2018, 28, 1800791 CrossRef.
- L. Huang, B. Chen, X. Zhang, C. O. Trindle, F. Liao, Y. Wang, H. Miao, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16046 CrossRef CAS PubMed.
- H. Wu, L. Gu, G. V. Baryshnikov, H. Wang, F. Minaev, H. B. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 20765 CrossRef CAS PubMed.
- Y. Katsurada, S. Hirata, K. Totani, T. Watanabe and M. Vacha, Adv. Opt. Mater., 2015, 3, 1726 CrossRef CAS.
- Y. Liu, Z. Ma, J. Liu, M. Chen, Z. Ma and X. Jia, Adv. Opt. Mater., 2021, 9, 2001685 CrossRef CAS.
- Y. Su, Y. Zhang, Z. Wang, W. Gao, P. Jia, D. Zhang, C. Yang, Y. Li and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 9967–9971 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, C. Wang, X. Zheng, Y. Zheng, L. Gao, C. Yang, Y. Li, L. Qu and Y. Zhao, Adv. Mater., 2020, 32, e1907355 CrossRef PubMed.
- Z. Xie, X. Zhang, H. Wang, C. Huang, H. Sun, M. Dong, L. Ji, Z. An, T. Yu and W. Huang, Nat. Commun., 2021, 12, 3522 CrossRef CAS PubMed.
- X. Lin, J. Wang, B. Ding, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 3459–3463 CrossRef CAS PubMed.
- P. Wei, X. Zhang, J. Liu, G. G. Shan, H. Zhang, J. Qi, W. Zhao, H. H. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9293–9298 CrossRef CAS PubMed.
- G. Wang, J. Li, X. Li, X. Wang, Y. Sun, J. Liu and K. Zhang, Chem. Eng. J., 2022, 431, 134197 CrossRef CAS.
- M. Wu, X. Wang, Y. Pan, J. Li, X. Li, Y. Sun, Y. Zou, H. Zhang and K. Zhang, J. Phys. Chem. C, 2021, 125, 26986–26998 CrossRef CAS.
- H. Mieno, R. Kabe and C. Adachi, Commun. Chem., 2018, 1, 27 CrossRef.
- H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Adv. Opt. Mater., 2016, 4, 1015–1021 CrossRef CAS.
- X. Li, G. Wang, J. Li, Y. Sun, X. Deng and K. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 1587–1600 CrossRef CAS PubMed.
- J. Li, X. Wang, Y. Pan, Y. Sun, G. Wang and K. Zhang, Chem. Commun., 2021, 57, 8794–8797 RSC.
- Y. Sun, G. Wang, X. Li, B. Zhou and K. Zhang, Adv. Opt. Mater., 2021, 9, 2100353 CrossRef CAS.
- I. Bhattacharjee and S. Hirata, Adv. Mater., 2020, 32, 2001348 CrossRef CAS PubMed.
- S. Guo, W. Dai, X. Chen, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS.
- J. Li, G. Wang, X. Chen, X. Li, M. Wu, S. Yuan, Y. Zou, X. Wang and K. Zhang, Chem. – Eur. J., 2022, 28, e202200852 CAS.
- Y. Sun, J. Liu, J. Li, X. Li, X. Wang, G. Wang and K. Zhang, Adv. Opt. Mater., 2021, 10, 2101909 CrossRef.
- D. Li, M. Wu, X. Chen, J. Liu, Y. Sun, J. Huang, Y. Zou, X. Wang, D. Chen and K. Zhang, J. Phys. Chem. Lett., 2022, 13, 5030–5039 CrossRef PubMed.
- S. Xu, W. Wang, H. Li, J. Zhang, R. Chen, S. Wang, C. Zheng, G. Xing, C. Song and W. Huang, Nat. Commun., 2020, 11, 4802 CrossRef CAS PubMed.
- J.-X. Wang, H. Zhang, L.-Y. Niu, X. Zhu, Y.-F. Kang, R. Boulatov and Q.-Z. Yang, CCS Chem., 2020, 2, 1391–1398 CrossRef CAS.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- Q. Dang, Y. Jiang, J. Wang, J. Wang, Q. Zhang, M. Zhang, S. Luo, Y. Xie, K. Pu, Q. Li and Z. Li, Adv. Mater., 2020, 32, 2006752 CrossRef PubMed.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Adv. Mater., 2020, 32, 2003911 CrossRef CAS PubMed.
- X. Liang, Y.-X. Zheng and J.-L. Zuo, Angew. Chem., Int. Ed., 2021, 60, 16984–16988 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175 CrossRef CAS PubMed.
- M. Monarul Islam, Z. Hu, Q. Wang, C. Redshaw and X. Feng, Mater. Chem. Front., 2019, 3, 762–781 RSC.
- R. Zhang, H. Zheng and J. Shen, Synth. Met., 1999, 105, 49–53 CrossRef CAS.
- X. Wang, Y. Sun, G. Wang, J. Li, X. Li and K. Zhang, Angew. Chem., Int. Ed., 2021, 60, 17138 CrossRef CAS PubMed.
- J. Jin, H. Jiang, Q. Yang, L. Tang, Y. Tao, Y. Li, R. Chen, C. Zheng, Q. Fan, K. Y. Zhang, Q. Zhao and W. Huang, Nat. Commun., 2020, 11, 842 CrossRef CAS PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J. L. Bredas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- H. Noda, X. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J. Bredas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51(9), 2215–2224 CrossRef CAS PubMed.
- H. Noda, H. Nakanotani and C. Adachi, Sci. Adv., 2018, 4, eaao6910 CrossRef PubMed.
- Y. Pan, J. Li, X. Wang, Y. Sun, J. Li, B. Wang and K. Zhang, Adv. Funct. Mater., 2021, 32, 2110207 CrossRef.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129(29), 8942–8943 CrossRef CAS PubMed.
- P.-Z. Chen, J.-X. Wang, L.-Y. Niu, Y. Z. Chen and Q. Z. Yang, J. Mater. Chem. C, 2017, 5, 12538 RSC.
- S. Wang, J. Cai, R. Sadygov and E. C. Lim, J. Phys. Chem., 1995, 99, 7416 CrossRef CAS.
- C.-T. Poon, W. H. Lam, H.-L. Wong and V. W.-W. Yam, Chem. – Eur. J., 2015, 21, 2182 CrossRef CAS PubMed.
- A. J. Gillett, A. Pershin, R. Pandya, S. Feldmann, A. J. Sneyd, A. M. Alvertis, E. W. Evans, T. H. Thomas, L.-S. Cui, B. H. Drummond, G. D. Scholes, Y. Olivier, A. Rao, R. H. Friend and D. Beljonne, Nat. Mater., 2022, 21, 1150–1157 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc05261j |
| This journal is © The Royal Society of Chemistry 2023 |