
Modulating room temperature phosphorescence through intermolecular halogen bonding†
Dongyan
Jiang
,
Chunya
Du
,
Zhenyu
Yan
,
Shuyuan
Ge
,
Zijun
Feng
,
Liang
Wan
and
Ping
Lu
 *
*
State Key Laboratory of Supramolecular Structure and Materials, Department of Chemistry, Jilin University, Changchun 130012, China. E-mail: lup@jlu.edu.cn
First published on 20th February 2023
Abstract
Purely organic room temperature phosphorescence (RTP) luminogens have drawn much attention owing to their potential application in anti-counterfeiting, biological imaging and various sensors, etc. In this work, a series of halogenated derivatives (MPh-R, R = F, Cl, Br, I) based on 4-phenylmorpholine are designed and synthesized. The effect of halogen atoms on the crystal packing modes and RTP performance is systematically investigated. The experimental results, theoretical calculations and ESP analysis indicate that halogen bonding is successfully formed in MPh-Cl, MPh-Br, and MPh-I, which significantly accelerates the intersystem crossing (ISC) and enhances the solid-state phosphorescence quantum yields (QYs) by inhibiting the vibration and rotation of molecules. For MPh-F, only single emission of fluorescence is detected due to the absence of the halogen bonding. The phosphorescent QY is increased from 0.6% for MPh to 6.8% for MPh-Cl. The relatively long afterglow feature of MPh, MPh-Cl, and MPh-Br also realizes time-dependent anti-counterfeiting encryption. This work provides a new perspective for designing single-component organic RTP materials.
Introduction
Persistent room temperature phosphorescence (RTP) has drawn increasing interest in recent years because of its full utilization of the excited state and long lifetime. Compared to the widely used metal-containing phosphorescent dyes, purely organic compounds showing RTP offer the benefits of low-cost, weak toxicity, convenient preparation and high biocompatibility. Applications of organic RTP compounds spanning from organic optoelectronics, oxygen sensing, bio-imaging and security devices have been assessed.1–12 When the spin multiplicity of the singlet state excitons changes, through the ISC process to the triplet excited state, the triplet state undergoes a radiative transition process and then back to the ground state; this process releases energy to produce phosphorescence. In metal-containing phosphors, the heavy-atom effect of metals gives rise to strong spin–orbit coupling (SOC) which contributes to the generation and radiation of triplet excitons. In comparison, purely organic phosphors with RTP are extremely limited, which is mainly due to the very weak SOC and the instability of triplet excitons. Thus, two factors need to be fulfilled to obtain purely organic RTP systems. One is the enhancement of SOC, which induces the spin-flip transition to generate the triplet states via ISC.13–18 The other is the restriction of molecular motion to suppress nonradiative transition, which can usually be obtained by crystallization, polymerization and host–guest doping, since the triplet excitons are extremely influenced by environmental factors such as moisture and oxygen.19–32In principle, SOC is highly dependent on the nuclear charge, which is also referred to as the heavy-atom effect.33–38 The non-metal halogen atoms of chlorine, bromine and iodine are generally acknowledged to greatly modify the molecular photophysical behavior and improve the phosphorescence QY. In addition, halogen bonding is also an effective and reliable tool in crystal engineering at the supramolecular level, wherein the halogen atoms are directly involved in forming intermolecular interactions.39,40
In this context, we present the construction of a purely organic system with the introduction of a heavy halogen atom, which modulates RTP by promoting the SOC via the heavy atom effect and forming special aggregates to stabilize the triplet exciton through halogen bonding.41–45 Morpholine, a simple, small N,O-containing six-membered ring is selected as the basic constructing unit. A halogenated-phenyl ring is connected to morpholine to increase the rigidity of the molecules. According to the El-Sayed rule, ISC is more likely to occur between different kinds of orbital, i.e., triplet (n–π*) to singlet (π–π*) or singlet (n–π*) to triplet (π–π*).46 The lone pair electron on the nitrogen atom of morpholine linking with benzene will contribute to the enhancement of SOC by enabling the n–π* transition.47,48 The oxygen atom on morpholine may interact with the heavy halogen to form the halogen bonding, and the lone pairs of oxygen attracted by chloride, bromine or iodine would intensify the triplet generation (Fig. 1a). With this design strategy, a series of 4-phenylmorpholine (MPh) derivatives, namely 4-(4-fluorophenyl) morpholine (MPh-F), 4-(4-chlorophenyl) morpholine (MPh-Cl), 4-(4-bromophenyl) morpholine (MPh-Br) and 4-(4-iodophenyl) morpholine (MPh-I), are designed and synthesized. The RTP properties are significantly affected by halogen substitution in terms of the electron-withdrawing property, steric hindrance, and heavy atom effect (Fig. 1b).49,50 The crystal lifetimes of the corresponding luminogens are 208.53 ms (MPh-Cl), 10.73 ms (MPh-Br), and 2.60 ms (MPh-I). Single crystal analyses clearly demonstrate that compact molecular stacking is beneficial in producing longer RTP lifetime and higher phosphorescence QY. For MPh-F, only single emission of fluorescence is observed. The extremely strong electron-withdrawing property of the fluorine atom results in a distinct molecular arrangement without halogen bonding. Both experimental and theoretical investigations indicate that the RTP efficiency from 0.6% (MPh) to 6.8% (MPh-Cl) in a crystal is mainly attributed to the simultaneous enhancement of the SOC between the singlet and triplet states as well as ISC and RTP radiative rate by the chlorine substituent.
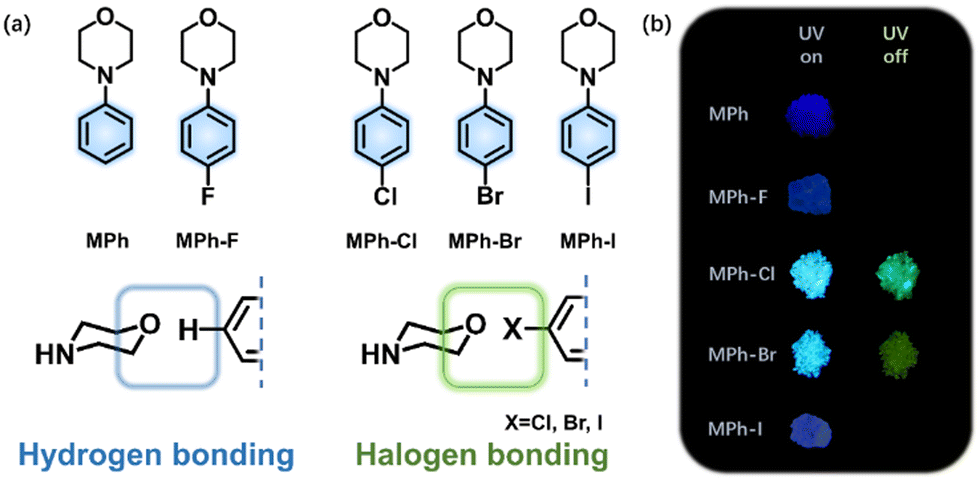 | ||
| Fig. 1 (a) Chemical structures of MPh, MPh-F, MPh-Cl, MPh-Br and MPh-I. (b) Photographs taken before and after irradiation (365 nm) under ambient conditions. | ||
Results and discussion
Herein, MPh-F, MPh-Cl, MPh-Br and MPh-I were all facilely prepared by a one-step Ullmann reaction. All target compounds were systematically characterized by 1H and 13C NMR and single crystal X-ray diffraction (Scheme S1 and Fig. S1–S10, ESI†). In the UV-Vis spectra in dilute tetrahydrofuran, the absorption peaks appearing at around 300 nm for MPh-F, MPh-Cl, MPh-Br and MPh-I were ascribed to the n–π* transition of the morpholine moiety (Fig. S11, ESI†).51,52 Meanwhile, MPh-Cl, MPh-Br and MPh-I exhibited the π–π* transition at around 260 nm, where the absorption of MPh-F was blue-shifted to 245 nm due to the relatively strong electron-withdrawing effect of fluorine.51 From the photoluminescence (PL) spectra in various polar solvents, MPh-F, MPh-Cl, MPh-Br and MPh-I showed similar red-shifted emission as compared with MPh, indicating that weak intramolecular charge transfer (ICT) existed in these compounds (Fig. S12, ESI†). Phosphorescence emission was not detected because the molecules were dispersed in the solvent and it was difficult for them to suppress the nonradiative transition.In contrast to the single emission peak in the solvent, MPh-Cl, MPh-Br and MPh-I displayed dual emission of fluorescence and phosphorescence in the crystal. As shown in Fig. 2a and b, MPh, MPh-F, MPh-Cl, MPh-Br and MPh-I showed main emission peaks at 345 nm, 355 nm, 367 nm, 360 nm and 362 nm, respectively, and they all exhibited nanosecond-level lifetimes indicating the typical fluorescence characteristics. Under the time-dependent phosphorescence measurement, MPh, MPh-Cl, MPh-Br, and MPh-I exhibited RTP emission at 504 nm (458.91 ms), 495 nm (208.53 ms), 505 nm (10.73 ms) and 529 nm (2.60 ms), respectively (Fig. 2c and d). However, there was no delayed emission signal detected for MPh-F at room temperature. It is worth mentioning that MPh-Cl exhibited the optimal intensity ratio of phosphorescence to fluorescence possibly stemming from the heavy atom effect and multiple intermolecular interactions, which facilitated the ISC process and the production of more triplet excitons for the phosphorescent emission. This was further verified by the phosphorescence QY measurement. The QY was calculated to be 0.6% for MPh and 6.8% for MPh-Cl, showing a nearly 10-fold increase with the incorporation of a chlorine atom. The lifetimes of MPh, MPh-Cl, MPh-Br and MPh-I were gradually reduced as the atomic volume increased from H to I, indicating that the heavy atom effect enhanced the radiative transition from the excited triplet state to the ground state. The PL spectra of the crystals at 77 K were measured, and a new contribution appeared, which belongs to the monomeric phosphorescence like in the solvent at 77 K (Fig. S13–S15, ESI†). The ratio between these two contributions strongly depends on the derivative. The monomeric phosphorescence in the crystal was only observed at low temperature, which further verified that the phosphorescence at room temperature originated from the aggregated state. When the crystals were ground into a powder, the sharp diffraction peaks in powder X-ray diffraction (PXRD) became much weaker, indicating that the originally regular and ordered molecular stacking was effectively disrupted (Fig. S16, ESI†). The process was accompanied by a significant reduction of the phosphorescence lifetime, which confirmed the importance of tight molecular stacking for RTP in these molecules (Fig. S17, ESI†).
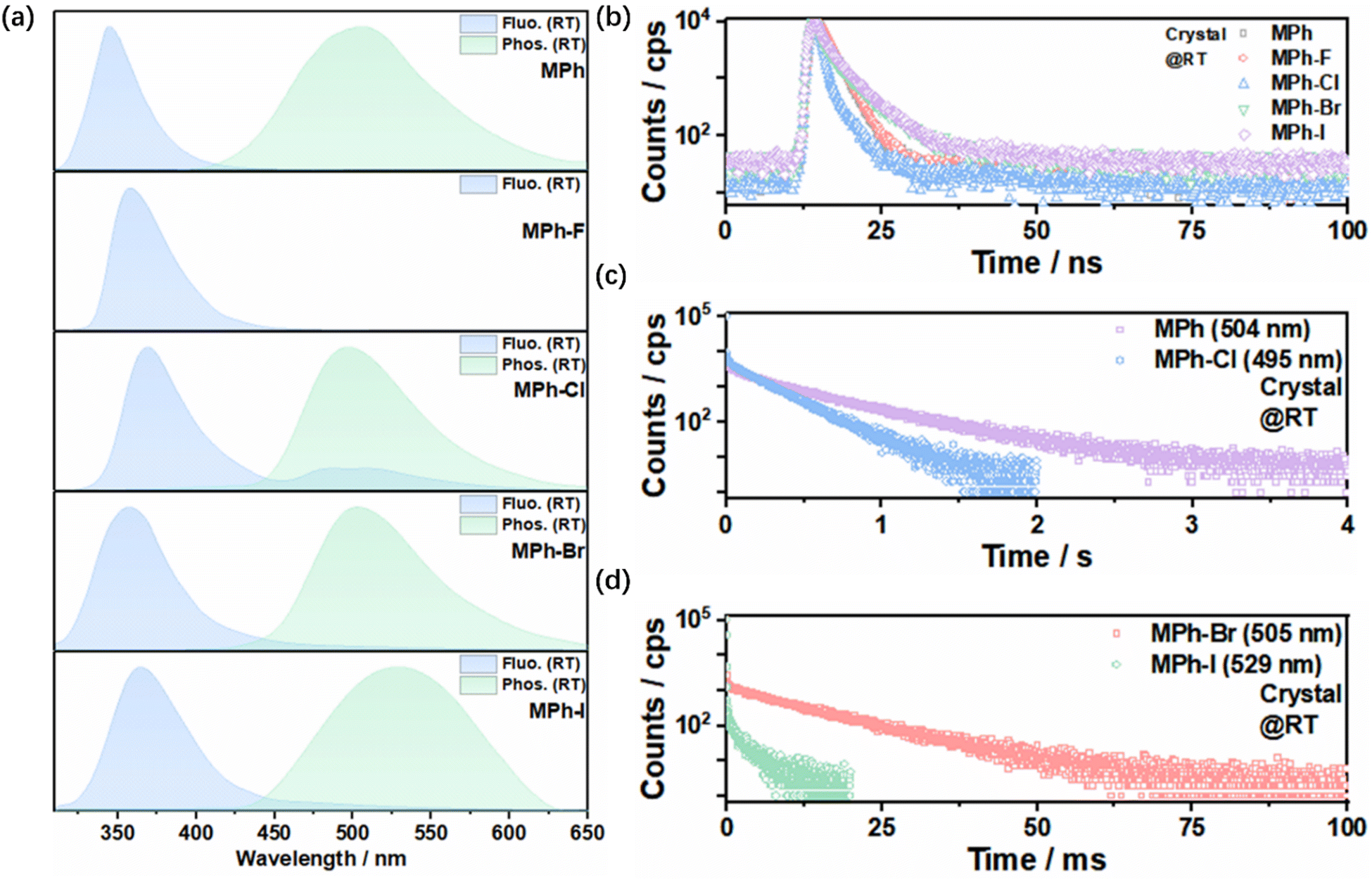 | ||
| Fig. 2 (a) Normalized fluorescence and phosphorescence spectra of MPh, MPh-F, MPh-Cl, MPh-Br and MPh-I crystals at room temperature. (b) The transient fluorescence decay spectra of MPh (345 nm), MPh-F (355 nm), MPh-Cl (367 nm), MPh-Br (360 nm) and MPh-I (362 nm) in the crystal. (c) Phosphorescence lifetimes of MPh (504 nm) and MPh-Cl (495 nm) at room temperature. (d) Phosphorescence lifetimes of MPh-Br (505 nm) and MPh-I (529 nm) at room temperature. | ||
The crystals of MPh, MPh-F, MPh-Cl, MPh-Br and MPh-I were prepared by crystallization from the mixed solution of dichloromethane and petroleum ether at room temperature, and their structural difference was investigated by single crystal X-ray diffraction.53,54 All the morpholine moieties linked to benzene took a relatively stable chair conformation (Fig. 3a). With the incorporation of various halogen atoms, MPh-Cl, MPh-Br and MPh-I exhibited edge-to-face stacking modes with subtle changes. In MPh without halogen substitution, six types of C–H⋯π interactions and two kinds of C–H⋯O interactions were observed (Fig. S18–S22 and Tables S2–S7, ESI†). The molecule was rigidly limited by a variety of different interactions, such as C–H⋯O hydrogen bonding, which restricted the non-radiative transition and prolonged the RTP lifetime, resulting in a lifetime of 458.91 ms. When the chlorine atom was introduced, MPh-Cl exhibited a edge-to-face stacking pattern and there existed eight intermolecular interactions, including 6 types of C–H⋯π (2.866–2.897 Å) interactions and 2 kinds of C–Cl⋯O (3.124 Å) halogen bonding (Fig. 3b). The multiple interactions enabled MPh-Cl to exhibit a lifetime of 208.53 ms. As the atomic number increased, the atomic radius gradually become larger while the molecules tended to slide along the long axis, which loosened the orderly arrangement of molecules and significantly reduced the intermolecular interactions. There were only five interactions in the MPh-Br crystal and four interactions in the MPh-I crystal with the adjacent molecules, respectively, and thus the nonradiative transitions could not be effectively suppressed. Viewing these crystals from the perspective of the stacking modes, the crystal arrangement of MPh-F was completely different. The MPh-F crystal, which lacked the RTP property, preferred to form an anti-parallel arrangement rather than the edge-to-face packing as observed in other molecules. There existed two kinds of C–H⋯π (2.879 Å) interactions, four types of C–H⋯O (2.437–2.659 Å) interactions, and two kinds of C–H⋯F (2.554 Å) interactions between MPh-F and the five neighboring molecules. Moreover, a more careful analysis of the MPh-F intermolecular interactions showed that there was no halogen bonding, and the minor atomic number of the fluorine atom would not show the same heavy-atom effect as Cl, Br and I. The undesirable stacking and the lack of halogen bonding might be mainly responsible for the absence of RTP emission in the MPh-F crystal.
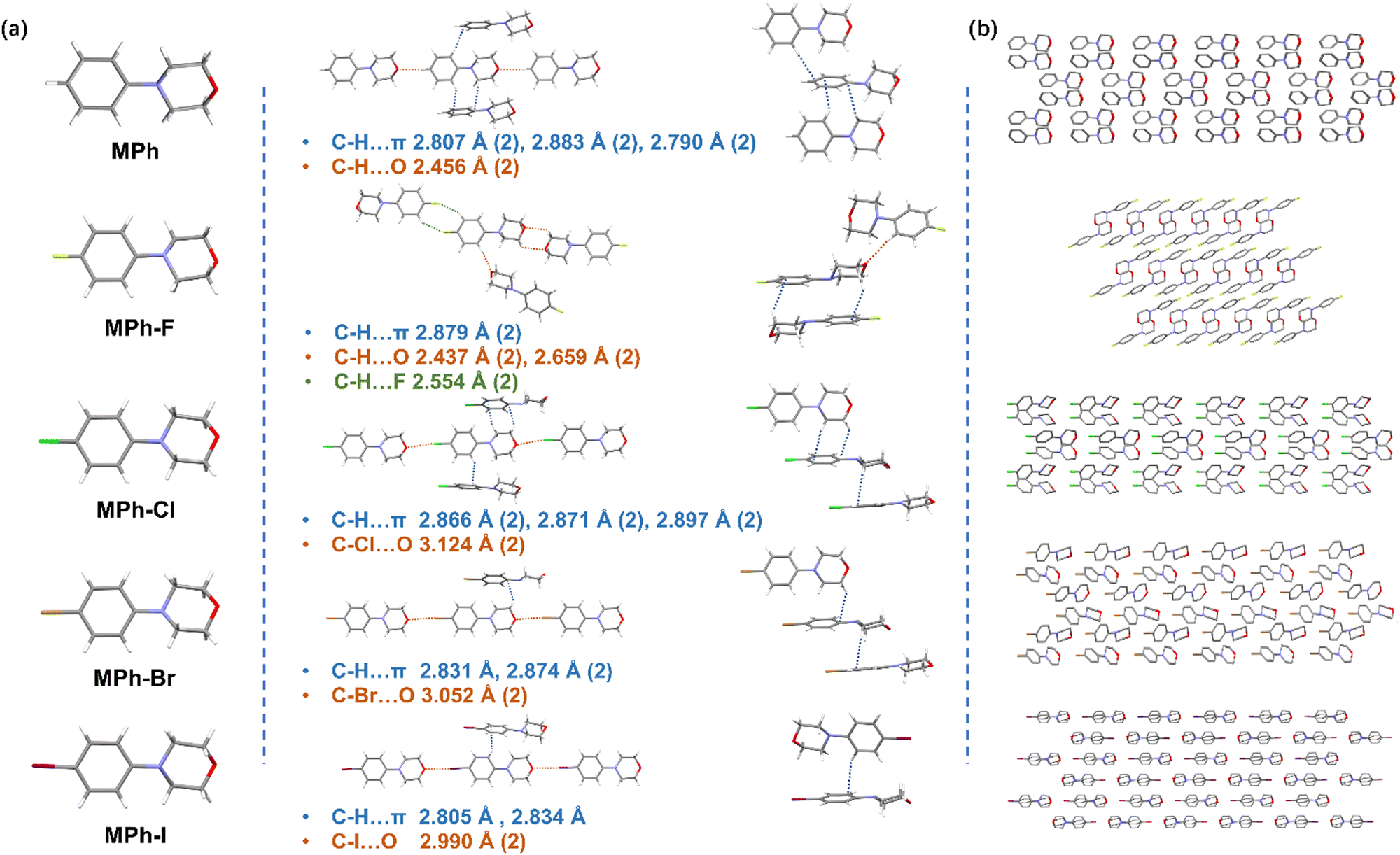 | ||
| Fig. 3 (a) Molecular arrangement viewed from different orientations and the intermolecular interactions with the adjacent molecules. (b) The packing modes of MPh, MPh-F, MPh-Cl, MPh-Br and MPh-I. | ||
To deepen the understanding of the high RTP efficiency of the MPh-Cl crystal, the radiative and non-radiative transition rate constants were calculated in Table S8 (ESI†). Compared to MPh, the phosphorescence QYs were increased to 6.8% for MPh-Cl, 4.2% for MPh-Br, and 2.3% for MPh-I, respectively. The highest QY of MPh-Cl was caused by multiple factors. As the atomic number increased, the radiative transition was accelerated along with the nonradiative transition, but the strong intermolecular interactions in the MPh-Cl crystal significantly suppressed the nonradiative transition process (knr is 5.03 s−1 in MPh-Cl crystal, 95.87 s−1 in MPh-Br crystal and 396.71 s−1 in MPh-I crystal). Also, the radiative transition rate of MPh-Cl had been improved by an order of magnitude compared to MPh (kr is 0.14 × 10−1 s−1 in MPh crystal and 3.67 × 10−1 s−1 in MPh-Cl).
In order to clarify the intrinsic mechanism of the improvement of phosphorescence QY, the TD-DFT calculations at the level of B3LYP/6-31G(d,p) were performed to optimize the geometries. The SOC coefficients were quantitatively estimated at the level of B3LYP/6-31G(d,p) by a Beijing density function (BDF) program. ISC is an important process between the single state and triplet state that could be directly reflected by the SOC constant. As shown in Fig. 4a, the coupling constant of MPh-Cl was significantly enhanced compared to MPh, with the related SOC value from T1 to S0 improving from 1.159 cm−1 (MPh) to 2.637 cm−1 (MPh-Cl) on account of the heavy atom effect. Meanwhile, although both MPh and MPh-Cl possessed two triplet energy levels below S1, the SOC of MPh-Cl was much higher than that of MPh. In particular, more triplet states close to S1 were produced in the dimer, indicating that the ISC process of MPh-Cl was further promoted (Fig. S23 and Tables S9 and S10, ESI†). The energy gap of S1 and T3 was only 0.0433 eV, corresponding to the SOC of 1.149 cm−1. For MPh-F without RTP properties, the energy gap between S1 and T2 reached 0.6022 eV in the dimer, which was not conducive to the ISC process (Fig. S24 and Table S11, ESI†). Theoretical calculations suggested that the SOC of T1 with both S0 and S1 in the dimer was 0 cm−1, verifying the hypothesis that this stacking mode would not give rise to the RTP effect.
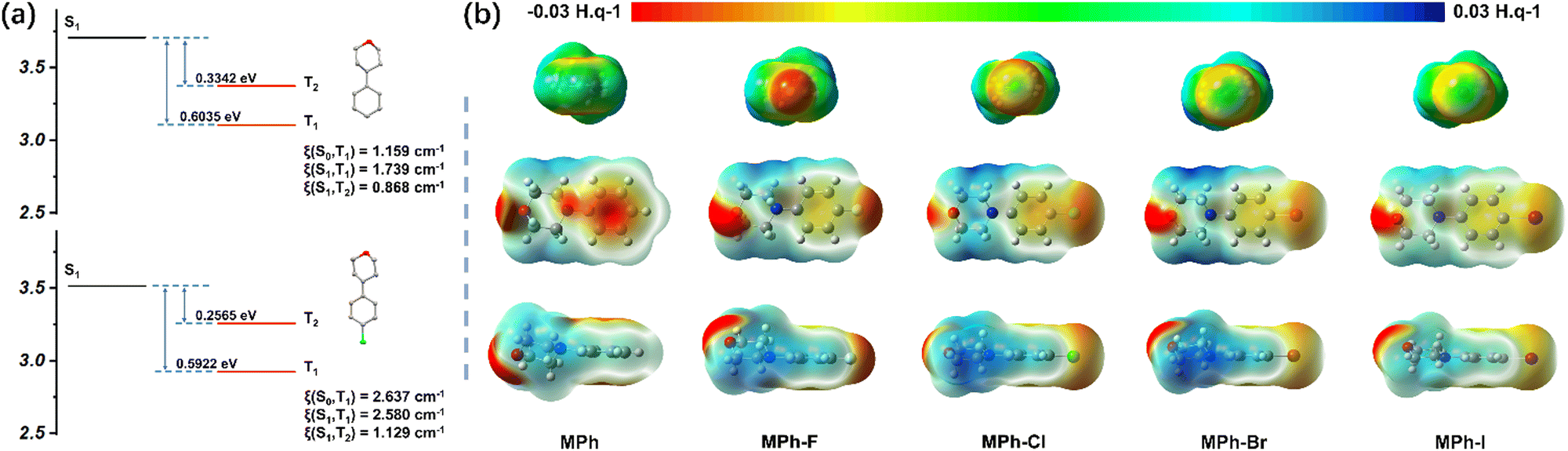 | ||
| Fig. 4 (a) Calculated excitation energies, and spin–orbit couplings (ξ) for MPh and MPh-Cl as isolated states. (b) Different electrostatic potential analyses of MPh, MPh-F, MPh-Cl, MPh-Br and MPh-I. The potential energy range is −0.03 to 0.03 H. q−1 for all surfaces shown. | ||
The effect of halogens on molecular stacking could be clearly seen through ESP analysis. As shown in Fig. 4b, the potential energy range was from −0.03 to 0.03 H. q−1 along with the color from red to blue. When the electrostatic potential was negative, it means that this position is attractive for positive electrons, like the red region around the fluorine atom (Fig. S25, ESI†). And when the electrostatic potential was positive, as in the case of the alkyl chain part of the morpholine group, it tended to be closer to the negative charge. Furthermore, with halogen atoms varying from F to Cl, Br or I with larger sizes, their bigger steric effect and reduced electron-withdrawing abilities resulted in the much looser stacking.50 The adjacent benzene units in the MPh-R could not form π–π stacking since the surface of each benzene showed an electron-rich characteristic. The fluorine atom remained entirely electronegative, whereas each of the other three halogen atoms displayed the occurrence of an electropositive crown along the C–X axis, surrounded by an electroneutral ring and a farther electronegative belt.39,40 The size of the electropositive crown also increased with the radius of the halogen, thus preferring to form halogen bonds. When the fluorine atom was introduced, F was more likely to form hydrogen bonds with H. The entirely electronegative fluorine atom exhibited the same electrostatic potential around the oxygen atom, and the electrostatic repulsion made them show no intermolecular interactions. This was in accordance with the distinct stacking pattern of MPh-F as observed in the crystal structure. Thus, the electropositive crowns of Cl, Br and I interacted with the electronegative atom of oxygen and formed halogen bonding.
The strong intermolecular interactions in the crystal facilitated the phosphorescence emission. However, it was not enough to make a high efficiency radiative transition of the triplet state excitons. The possible photophysical processes of the halogen substituted compounds under photoexcitation were proposed to further understand the mechanism of halogen bonding in the crystal (Fig. 5a). When halogen atoms were introduced in the MPh, the SOC among the excited-state electrons of the compound and the massive nucleus of the heavy atom were enhanced. Halogen bonding occurring between the molecules delocalized the electrons of the oxygen partially towards the neighboring halogen.42 The motion of the resulting molecules was restricted by the tight packing, which enhanced the radiative transition of phosphorescence while weakening the non-radiative transition, allowing triplets to decay emissively. Given the relatively long afterglow properties of phosphorescent materials, the compounds were well suited for time-resolved display models. As shown in Fig. 5b, when excited with an ultraviolet lamp, the three materials emitted light at the same time, the part of the pattern of raindrops was made of MPh, the part of clouds was made of MPh-Cl, and the part of the sun was MPh-Br. When the lights went out, the clouds and the sun would still last for a while, representing that the rain turned cloudy. Only clouds remained at the final stage, which could act as a rain or shine indicator. Thus, abundant codes could be obtained by various combinations of these materials with different locations, making them ideal and promising materials for data encryption.
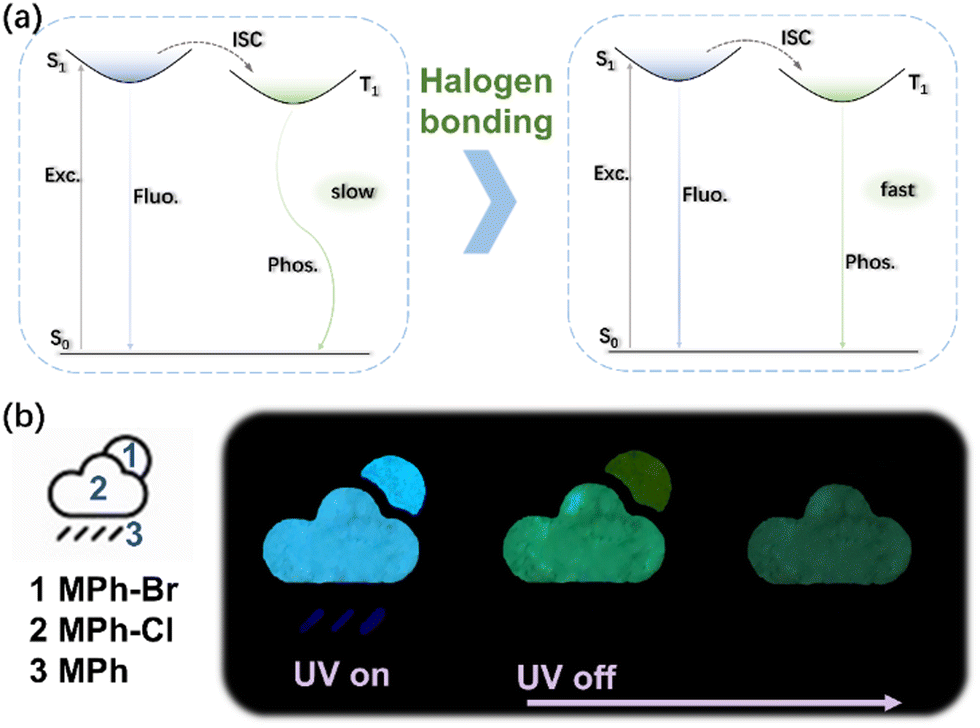 | ||
| Fig. 5 (a) Simplified Jablonski diagram to explain the halogen bonding induced phosphorescence emission and other photophysical processes. (b) Application of information encryption by using three kinds of components of MPh, MPh-Cl and MPh-Br. | ||
Conclusion
In summary, a systematic investigation on the photophysical properties of MPh and its halogenated derivatives (MPh-R, R = F, Cl, Br, I) is performed. A simple atomic change can be made to tune the RTP properties in terms of electron-withdrawing ability, heavy-atom effect and crystal packing. A single crystal XRD experiment reveals that the tightly arranged stacking mode is responsible for the formation of halogen bonding in MPh-Cl, MPh-Br, and MPh-I. The DFT calculations elucidate that the chloride atom facilitates the spin–orbit coupling and essentially increases the SOC value. The intermolecular coupling produces energy level splitting in the aggregated state of MPh-Cl, which generates more efficient ISC channels. An increase of the phosphorescent QY from 0.6% for MPh to 6.8% for MPh-Cl is realized. Moreover, the relatively long afterglow feature allows these halogenated derivatives to be used in the field of data encryption. This work not only provides a strategy to improve the QY of phosphorescence but also provides a better understanding of the relationship between halogen bonding and the RTP effect.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research is supported by the National Natural Science Foundation of China (22075100) and the Jilin Provincial Science and Technology Department (20220201082GX).References
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed
.
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS
- J. Ren, Y. Wang, Y. Tian, Z. Liu, X. Xiao, J. Yang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 12335–12340 CrossRef CAS PubMed
- J. Wang, B. Liang, J. Wei, Z. Li, Y. Xu, T. Yang, C. Li and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 15335–15339 CrossRef CAS PubMed
- M. Gao, Y. Tian, J. Yang, X. Li, M. Fang and Z. Li, J. Mater. Chem. C, 2021, 9, 15375–15380 RSC
- Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef PubMed
- Y. Zhou, W. Qin, C. Du, H. Gao, F. Zhu and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 12102–12106 CrossRef CAS PubMed
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed
- S. Cai, Z. Sun, H. Wang, X. Yao, H. Ma, W. Jia, S. Wang, Z. Li, H. Shi, Z. An, Y. Ishida, T. Aida and W. Huang, J. Am. Chem. Soc., 2021, 143, 16256–16263 CrossRef CAS PubMed
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS PubMed
- H. Shi, L. Zou, K. Huang, H. Wang, C. Sun, S. Wang, H. Ma, Y. He, J. Wang, H. Yu, W. Yao, Z. An, Q. Zhao and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 18103–18110 CrossRef CAS PubMed
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed
- A. Martin, W. Kiarie, B. Chang and M. Thuo, Angew. Chem., Int. Ed., 2020, 59, 1 CrossRef CAS
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. Wu, P. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed
- Z. Ma, Z. Yang, L. Mu, L. Deng, L. Chen, B. Wang, X. Qiao, D. Hu, B. Yang, D. Ma, J. Peng and Y. Ma, Chem. Sci., 2021, 12, 14808–14814 RSC
- Y. Tian, J. Yang, Z. Liu, M. Gao, X. Li, W. Che, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 20259–20263 CrossRef CAS PubMed
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS PubMed
- X. Yao, J. Wang, D. Jiao, Z. Huang, O. Mhirsi, F. Lossada, L. Chen, B. Haehnle, A. J. C. Kuehne, X. Ma, H. Tian and A. Walther, Adv. Mater., 2021, 33, 2005973 CrossRef CAS PubMed
- Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li, Z. Cai, Y. Zhao and C. Yang, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed
- L. Xiao, Y. Wu, J. Chen, Z. Yu, Y. Liu, J. Yao and H. Fu, J. Phys. Chem. A, 2017, 121, 8652–8658 CrossRef CAS PubMed
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Matter, 2020, 3, 449–463 CrossRef
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 6–18 CrossRef
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed
- Z. Chen, M. Li, W. Qiu, W. Xie, Q. Gu and S. Su, J. Mater. Chem. C, 2021, 9, 15998–16005 RSC
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS PubMed
- S. Garain, S. Kuila, B. C. Garain, M. Kataria, A. Borah, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 12323–12327 CrossRef CAS PubMed
- Y. Wen, H. Liu, S. Zhang, Y. Gao, Y. Yan and B. Yang, J. Mater. Chem. C, 2019, 7, 12502–12508 RSC
- T. Zhu, T. Yang, Q. Zhang and W. Z. Yuan, Nat. Commun., 2022, 13, 2658 CrossRef CAS PubMed
- K. Kanosue and S. Ando, ACS Macro Lett., 2016, 5, 1301–1305 CrossRef CAS PubMed
- G. Farias, C. A. M. Salla, M. Aydemir, L. Sturm, P. Dechambenoit, F. Durola, B. de Souza, H. Bock, A. P. Monkman and I. H. Bechtold, Chem. Sci., 2021, 12, 15116–15127 RSC
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed
- K. E. Riley and K. M. Merz, J. Phys. Chem. A, 2007, 111, 1688–1694 CrossRef CAS PubMed
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CAS
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC
- H. Liu, W. Liu, N. Ando, S. Yamaguchi and H. Zhang, J. Mater. Chem. C, 2021, 9, 2738–2743 RSC
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Pavanello, A. Previtali, S. Righetto and E. Cariati, Angew. Chem., Int. Ed., 2017, 56, 16302–16307 CrossRef CAS PubMed
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed
- J. Ren, Y. Tian, Y. Wang, J. Yang, M. Fang and Z. Li, J. Mater. Chem. C, 2022, 10, 13741–13746 RSC
- Q. Zhang, Y. Fan, Q. Liao, C. Zhong, Q. Li and Z. Li, Sci. China: Chem., 2022, 65, 918–925 CrossRef CAS
- J. Yuan, Y. Wang, L. Li, S. Wang, X. Tang, H. Wang, M. Li, C. Zheng and R. Chen, J. Phys. Chem. C, 2020, 124, 10129–10134 CrossRef CAS
- J. Yuan, S. Wang, Y. Ji, R. Chen, Q. Zhu, Y. Wang, C. Zheng, Y. Tao, Q. Fan and W. Huang, Mater. Horiz., 2019, 6, 1259–1264 RSC
- D. Jiang, T. Lu, C. Du, F. Liu, Z. Yan, D. Hu, A. Shang, L. Gao, P. Lu and Y. Ma, Sci. China: Chem. DOI:10.1007/s11426-022-1507-2
- F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, L. Midgley, L. J. Bourhis, A. Genoni, L. A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkötter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Chem. Sci., 2021, 12, 1675–1692 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2224489 (MPh-F), 2224503 (MPh-Cl), 2224534 (MPh-Br) and 2224504 (MPh-I). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc05237g |
| This journal is © The Royal Society of Chemistry 2023 |