
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-stimuli responsive (L-tartrato)oxovanadium(V) complex salt with ferroelectric switching and thermistor properties†
Marko
Dunatov
 ,
Andreas
Puškarić
and
Lidija
Androš Dubraja
*
,
Andreas
Puškarić
and
Lidija
Androš Dubraja
*
Ruđer Bošković Institute, Bijenička cesta 54, 10000 Zagreb, Croatia. E-mail: lidija.andros@irb.hr; Tel: +385 1 4561184
First published on 17th January 2023
Abstract
Molecular materials that respond to external triggers and the environment by changing certain properties are currently a very active area of research and are being considered for use in devices. The responsiveness of these highly organized structures is the result of flexible structural arrangements, which in most cases are controlled by non-covalent interactions. Here, we report the exceptional structural flexibility of a complex salt with tetraethylammonium cations, (L-tartrato)oxovanadium(V) anions and water molecules of crystallization. This compound undergoes multiple crystal transformations from the undecahydrate phase to the dehydrated phase triggered by changes in humidity and temperature. The structural transformations are characterized by single-crystal X-ray diffraction, powder X-ray diffraction and infrared spectroscopy at variable temperature and humidity and by thermal analysis. The dynamics of the structural transformations occurring in this tetraethylammonium (L-tartrato)oxovanadium(V) system indicates a low energy barrier for reversible release and uptake of water molecules from air. During these transformations, which occur over a very narrow temperature and humidity range, the compound changes from the non-polar P212121 structure to the polar P21, C2 and P2 structures. The structural transformations are accompanied by significant changes in the electrical behaviour of the material: from proton conductivity in the non-polar P212121 phase to ferroelectricity in the P21 phase at room temperature, and thermistor behaviour in the temperature range of 313–333 K. In addition, the optical properties of the material, which exhibits pleochroism and photoresponsive behaviour, were investigated using UV-vis diffuse reflectance spectroscopy.
Introduction
Compounds and complexes of tartaric acid have been studied for centuries, and yet the number of tartrato transition metal complexes is not very large (less than 300 deposits in the CSD).1,2 This may be related to the rather complex behaviour of this seemingly simple α-hydroxycarboxylic acid, which depends strongly on the experimental conditions, and to the stability problems that some of the tartrato complexes exhibit in the solid state.3–7 This is also one of the reasons why the solution chemistry of the tartrato complexes has been studied much more thoroughly than the solid state using various methods, i.e., UV-vis spectroscopy, conductometry, cryoscopy, infrared spectroscopy, NMR spectroscopy, polarography, and cyclic voltammetry.1,3–7 Considering its availability, low cost, nontoxicity, and natural occurrence in the enantiomeric form, L-(+)-tartaric acid (C4H6O6) could find much wider application in the development and preparation of inorganic–organic materials as chiral building units.The complexation of vanadium(V) with L-tartaric acid is pH dependent, and two different forms can be obtained (Scheme 1a and b): the tetranuclear [V4O8(L-tart)2]4− anion,7–9 and the less stable dinuclear [V2O4(L-tartH2)2]2− anion10 formed in acidic solutions [L-tart = (C4H2O6)4−; L-tartH2 = (C4H4O6)2−]. In addition to these two forms, the use of racemic DL-tartaric acid gives the [V2O2(D-tart)(L-tart)]2− anion,10 which was isolated in the solid state (Scheme 1c). Complexes of oxovanadium(IV) with L-, D-, or DL-tartaric acid occur only in the dimeric form as the [V2O2(tart)2]4− anion,11,12 in which each fully deprotonated tartrate residue forms two chelate rings to two vanadium(IV) ions, similar to the structure shown in Scheme 1c. Due to the diversity of coordination topologies and nuclearities of (L-tartrato)oxovanadium complex anions, they provide a good platform for chiral building blocks that can potentially be used to prepare compounds with polar crystal structures of interest for application in nonlinear optics, ferroelectrics, and other switchable dielectrics.8,13–15
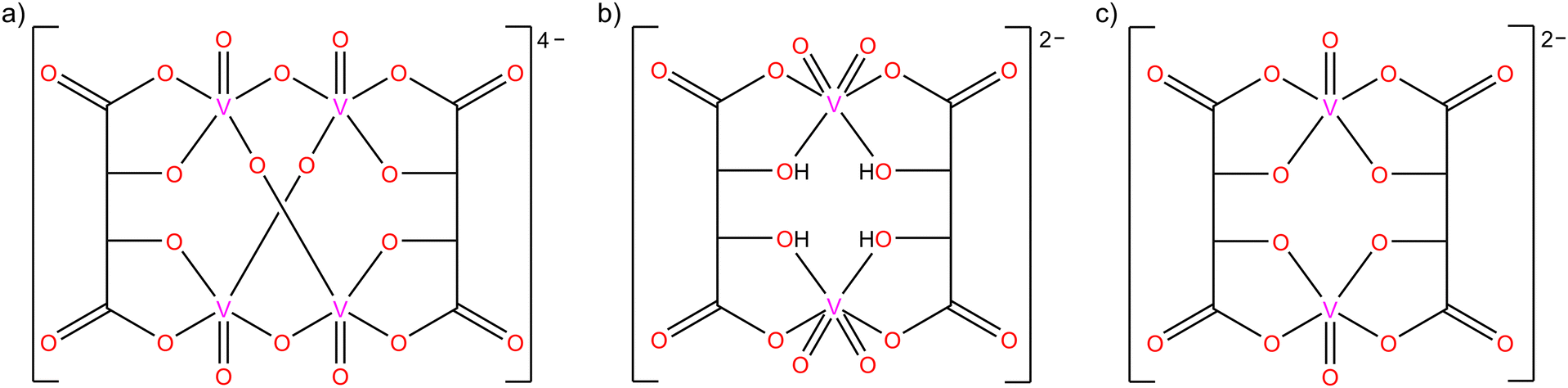 | ||
| Scheme 1 Different (tartrato)oxovanadium(V) anions: (a) [V4O8(L-tart)2]4−, (b) [V2O4(L-tartH2)2]2−, and (c) [V2O2(D-tart)(L-tart)]2−. | ||
In this work, we have focused on the preparation of a dominant and stable tetranuclear [V4O8(L-tart)2]4− anion that reacts with tetraethylammonium cations (TEA)+ to give a complex salt (TEA)4[V4O8(L-tart)2]. The investigated compound coexists in several hydrate phases and undergoes reversible crystal transformation from the undecahydrate phase to the completely dehydrated phase, through multiple solid state transformations triggered by changes in humidity and temperature. The structural transformations were followed by in situ infrared spectroscopy and powder X-ray diffraction, as well as by thermal analysis. The process of crystal transformation from the new undecahydrate phase (TEA)4[V4O8(L-tart)2]·11H2O (1·11H2O) to the previously known hexahydrate phase (TEA)4[V4O8(L-tart)2]·6H2O (1·6H2O)7 is followed by a symmetry breakdown from the non-polar orthorhombic P212121 space group to the polar monoclinic P21 space group. The polar phase 1·6H2O is ferroelectric and exhibits a switchable spontaneous polarization under an external electric field at room temperature. Interestingly, the first ferroelectric crystal discovered by Valasek in 1920 was also a tartrate-based system, potassium sodium tartrate tetrahydrate (Rochelle salt), but not many ferroelectric tartrate compounds have been mentioned since.16,17 One of the few ferroelectric tartrates is the supramolecular adduct of 1,4-diazabicyclo[2.2.2]octane-N,N′-dioxide and L-tartaric acid,18 and a hydrogen-bonded ionic co-crystal with imidazole and L-tartaric acid.19 Apart from the ferroelectric properties, the studied (TEA)4[V4O8(L-tart)2] compound exhibits other properties induced by external stimuli, such as the temperature- and humidity-induced electrical responses and optical properties dependent on irradiation with visible light. These properties were investigated using chronoamperometric measurements and UV-vis diffuse reflectance spectroscopy.
Experimental
Synthetic procedure
Tetraethylammonium hydroxide (TEAOH) solution 35 wt% in water and L-(+)-tartaric acid were purchased from Alfa Aesar. V2O5 was prepared by decomposition of NH4VO3 at 773 K, purchased from Sigma Aldrich. The method described in the literature for the preparation of (TEA)4[V4O8(L-tart)2]·6H2O (1·6H2O)7 was modified according to the procedure described below, giving a 1·11H2O phase as a reaction product.General procedure for the synthesis of phase 1·11H2O
Freshly prepared V2O5 (0.045 g, 0.25 mmol) was dissolved in an aqueous solution of TEAOH (0.5 mL of a 2.34 mol L−1 solution, 1.22 mmol), and then an aqueous solution of L-(+)-tartaric acid (0.5 mL of a 1 mol L−1 solution, 0.5 mmol) was added to the reaction mixture. The pH of the red solution was 6.5. After this, ethanol (2.5 mL) and acetone (10 mL) were added to the reaction mixture to promote crystallization. From the reaction mixture that was kept in a refrigerator at 280 K, large red rod-like crystals of (TEA)4[V4O8(L-tart)2]·11H2O (1·11H2O) appeared after 12 h. Anal. Calcd % for C40H106N4O31V4 (found, %): C, 35.77 (34.91); H, 7.96 (8.30); N, 4.17 (4.01); V, 15.17 (15.09). IR bands (4000–400 cm−1): 3460 s, br, 3380 s, br, 3270 s, br [νs,as(OH)]; 1663 vs [νas(COO−)]; 1595 vs, 1350 m [νs(COO−)]; 1296 m, 1173 s, 1097 s [ν(C–Oh)]; 1070 m [ν(C–Oh)]; 1001 s, 966 vs [ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot)]; 931 w, 852 m, 822 m, 780 w, 745 s [νas(VOV)]; 656 m [νs(VOV)]; 634 w, 602 w, 546 s [ν(V–Otart)]; 511 sh, 480 m, 435 m, 403 m.
Ot)]; 931 w, 852 m, 822 m, 780 w, 745 s [νas(VOV)]; 656 m [νs(VOV)]; 634 w, 602 w, 546 s [ν(V–Otart)]; 511 sh, 480 m, 435 m, 403 m.
Spectroscopic measurements
Attenuated total reflectance (ATR) spectra were recorded in the 4000–400 cm−1 range using a PerkinElmer FT-IR Frontier spectrometer. Humidity-dependent ATR measurements were recorded in situ using a chamber with a manual device for monitoring the relative humidity (RH) that was attached to the ATR module of the spectrometer. The relative humidity of the device was adjusted with a flow of dry or wet gas in the chamber. Temperature-dependent ATR spectra were recorded in situ using a hot air gun for heating. UV-vis diffuse reflectance spectra were recorded using a Shimadzu UV-Vis-NIR spectrometer (model UV-3600) equipped with an integrated sphere. The MAX-303-Compact Xenon Light source with a 300 W lamp and UV filters was used to irradiate the samples.Thermogravimetric measurements
Thermal analysis (TG/DTA) was performed using a Shimadzu DTG-60H analyser, in the range from room temperature to 1073 K, in a stream of synthetic air at a heating rate of 2 K min−1.Electrical measurements
The current response to changes in relative humidity and temperature associated with structural transformations was monitored using a PalmSens4 potentiostat on a compressed powder samples. Ferroelectric tests were performed using a TF Analyzer 1000 at room temperature in the frequency range of 0.1–10 Hz.Crystallographic measurements
The single-crystal X-ray diffraction data were collected by ω-scans using Cu-Kα radiation (λ = 1.54179 Å, microfocus tube, mirror monochromator) using a Rigaku XtaLAB Synergy diffractometer at 160 K. The crystal data, experimental conditions and final refinement parameters are summarized in Table S1 in the ESI.† Data reduction, including the multiscan absorption correction, was performed using the CrysAlisPRO software package.20 The molecular and crystal structures were solved by direct methods using the program SIR201921 and refined by the full-matrix least-squares method based on F2 with anisotropic displacement parameters for all non-hydrogen atoms (SHELXL-2014/7).22 Both programs were operating under the WinGX program package.23 Hydrogen atoms attached to the C atoms of the (TEA)+ cation and tartrate anions were treated as riding in idealized positions, with the C–H distances of 0.93 Å and displacement parameters assigned as Uiso(H) = 1.2Ueq(C). Hydrogen atoms of water molecules were identified based on different Fourier maps [O–H distances were restrained to a target value of 0.85(2) Å, and the H–O–H angle to 104°]. Geometrical calculations were carried out using PLATON24 and the figures were generated using CCDC-Mercury programs.25 The powder X-ray diffraction (PXRD) data were collected in reflection mode using Cu-Kα radiation (λ = 1.54060 Å) using a Malvern Panalytical Empyrean diffractometer with a step size of 0.001° in a 2θ range between 5° and 60°. For in situ PXRD measurements, high temperature camera was used. The peak search and indexing were carried out using the TOPAS software.26 Unit cell and profile refinements were carried out using the Pawley method.27Results and discussion
Crystal transformations
The target complex salt (TEA)4[V4O8(L-tart)2] was prepared using a modified literature method, giving the undecahydrate complex (1·11H2O), whereas a hexahydrate phase (1·6H2O) was reported in a previous publication (ref. code EDAZOZ).2,7 The two solvate phases crystallise in different crystal systems: 1·11H2O adopts the orthorhombic structure P212121, while 1·6H2O has a monoclinic polar space group P21. Fig. 1, which shows a comparison between the two solvate phases, suggests that the (TEA)+ cations and the [V4O8(L-tart)2]4− anions in both compounds have essentially the same geometry, with the (TEA)+ cations in the 1·6H2O phase exhibiting a higher degree of thermal motion. The tetranuclear anion [V4O8(L-tart)2]4− consists of a {V4O8} core, with four oxygen atoms bridging vanadium(v) ions (single bonded), four terminal oxygen atoms (double bonded) and two fully deprotonated (L-tart)4− anions that adopt a tetradentate coordination mode to bridge metal ions and to chelate them with two hydroxyl oxygen and two carboxylic oxygen atoms. The coordination polyhedra around the vanadium(V) metal centres have a shape of a distorted square pyramid, and the overall complex anion has a boat conformation. The metal–oxygen bond lengths in the {V4O8} core are in the range of 1.590(3)–1.601(2) Å for terminal oxo atoms and in the range of 1.775(2)–1.876(2) Å for bridging oxo atoms. One oxo bridge in the {V4O8} core is flatter than the others, with a V–O–V angle of 170.7(1)° compared to 127.9(1), 113.6 (1) and 114.3(1)° of the other oxo bridges. The other bonds are comparable to those of the corresponding bonds in other structurally reported [V4O8(L-tart)2]4− anions.2,7–10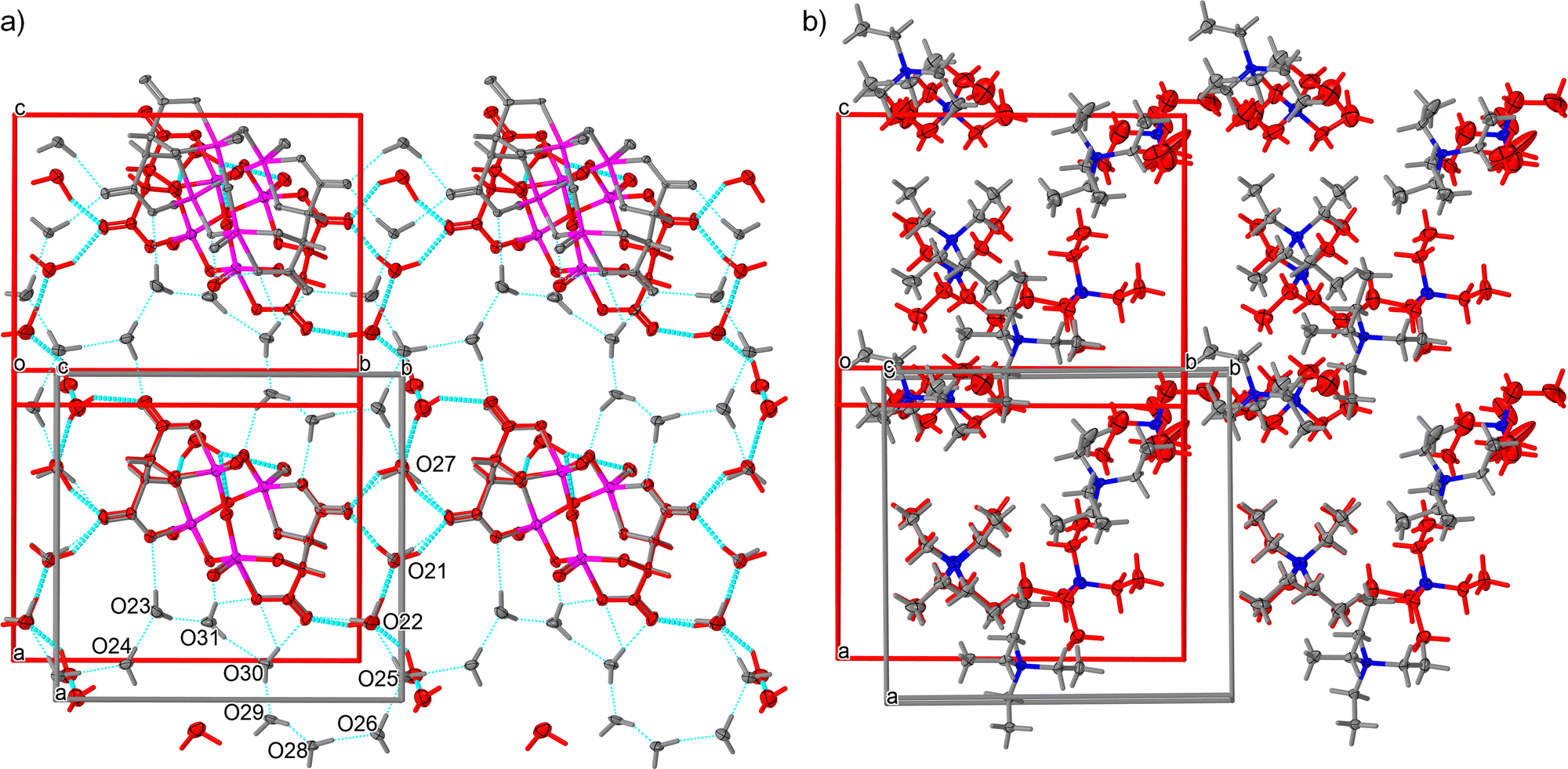 | ||
| Fig. 1 Difference in crystal packing between the structure of 1·11H2O (grey) and 1·6H2O (red), showing (a) a layer with [V4O8(L-tart)2]4− anions and water molecules connected by hydrogen bonds (cyan sticks), and (b) a layer with hydrophobic (TEA)+ cations. | ||
A comparison of the two phases from single-crystal XRD measurements at low temperatures shows that the 1·11H2O phase has an evidently larger unit cell than 1·6H2O (∼380 Å3, 6% volume of the unit cell), as expected due to 5 more water molecules in the crystal structure. The water molecules together with the [V4O8(L-tart)2]4− anions form a two-dimensional hydrogen bonded layer in both structures (Fig. 1a). Geometrical parameters describing hydrogen bonding are given in the ESI† (Tables S3 and S4) for both phases. When the relative humidity level is below 60%, water molecules O23, O24, O28, O30 and O31 are released from the structure of 1·11H2O (Fig. 1a). The positions of water molecules O21, O22, O25 and O27 only slightly change. During transformation, the water molecule O29 is being moved for about 3.2 Å to a new position very close to the [V4O8(L-tart)2]4− anion in the structure of 1·6H2O. The difference between two structures in terms of hydrogen bonding is that in 1·11H2O all water molecules participate in the supramolecular 2D hydrogen bonding layer (12 bonds of the donor–donor–acceptor type), forming an 8-membered cooperative infinite O–H⋯O–H⋯O–H chain. In 1·6H2O, there are no cooperative hydrogen bonding chains, and most of the interaction are the donor–donor–acceptor type, except one water molecule that is forming a discreet donor–donor type of bond with one [V4O8(L-tart)2]4− anion. Both structures contain two hydrophobic layers; in 1·11H2O, they are moderately twisted, whereas in 1·6H2O one layer is almost planar and the other forms a zigzag wrinkled sheet. In this type of structural rearrangement, the distances between the nearest (TEA)+ cations are shorter in 1·11H2O than those in the 1·6H2O phase. A detailed study and comparison of the two structures show that the arrangement of the cations and anions is very similar and that even water molecules occupy very close positions with respect to the complex anions.
Similarities in the crystal structures of 1·11H2O and 1·6H2O are a direct indication that these two compounds are linked by certain solid-state transformations. Indeed, in situ PXRD measurements (Fig. 2) have shown that 1·11H2O is stable only in a humid atmosphere (RH ∼ 60%), while a decrease in humidity leads to the elimination of 5 water molecules and consequently a symmetry breaking to the polar phase 1·6H2O. Furthermore, heating of 1·6H2O leads to the formation of three different crystalline phases at 313, 341 and 373 K, respectively. According to the PXRD data and profile fitting analysis, these phases are the result of the elimination of water molecules from the crystal structure (see Fig. S1–S5, ESI†). The relevant structural parameters (unit cell and space groups) of the five different hydrates and the dehydrated phase of the studied complex salt are listed in Table 1 (more detailed in Table S2, ESI†). The schematic representation of the structural transformations under specific conditions is shown in Scheme 2. Under humid conditions, the 1·6H2O phase shows an affinity for the uptake of water molecules from air, undergoing a structural transformation from the monoclinic P21 phase to the orthorhombic P212121 phase. Mild heating from room temperature to 314 K leads to a new structural transformation within the same space group triggered by the release of a water molecule, indicating a very flexible structural rearrangement of the constituting units in this tetraethyammonium (L-tartrato)oxovanadium(V) system. Water molecules also play a role in the next structural transformation, which occurs upon heating to 341 K, when the release of three water molecules triggers the structural change from the P21 phase to the C2 phase. Upon further heating to 373 K, the complete loss of the water molecules occurs and the dehydrated phase 1 is formed. After thermal treatment at 423 K, the compound becomes dark brown in colour and based on the PXRD pattern appears to be an amorphous phase, showing only three broad humps (Fig. S6, ESI†). Interestingly, the amorphous phase can be recovered to a 1·6H2O crystal phase when exposed to air with a RH of > 40% at room temperature. After about 2 days in humid air, the amorphous phase absorbs water vapour from the air, causing it to dissolve and then recrystallize (see Fig. S6, ESI†).
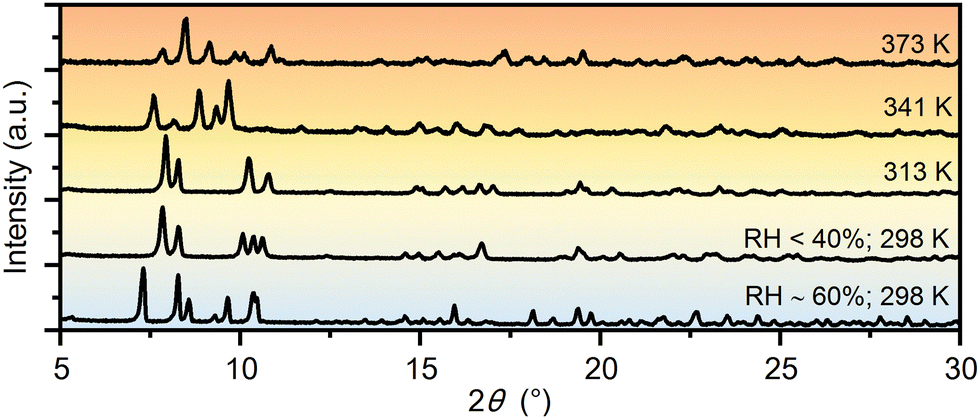 | ||
| Fig. 2 PXRD patterns of the different hydrate phases and the dehydrated phase of the complex salt (TEA)4[V4O8(L-tart)2] (1) measured during heating and exposure to different relative humidities. | ||
| Phase | 1·11H2O | 1·6H2O | 1·5H2O | 1·2H2O | 1 |
|---|---|---|---|---|---|
| T/K | 298 | 298 | 313 | 341 | 373 |
| M r/g mol−1 | 1343.04 | 1252.97 | 1234.95 | 1180.90 | 1144.87 |
| Space group | P212121 | P21 | P21 | C2 | P2 |
| a/Å | 13.331(1) | 12.292(1) | 12.051(1) | 23.946(3) | 11.720(2) |
| b/Å | 14.150(1) | 14.023(1) | 13.983(2) | 12.401(1) | 14.095(3) |
| c/Å | 34.466(2) | 18.377(1) | 18.600(2) | 19.470(2) | 16.786(3) |
| α/° | 90 | 90 | 90 | 90 | 90 |
| β/° | 90 | 108.929(4) | 109.430(5) | 103.387(10) | 94.720(14) |
| γ/° | 90 | 90 | 90 | 90 | 90 |
| Z | 4 | 2 | 2 | 4 | 2 |
| V/Å3 | 6501.2(6) | 2996.1(4) | 2955.8(5) | 5624.5(1) | 2763.6(10) |
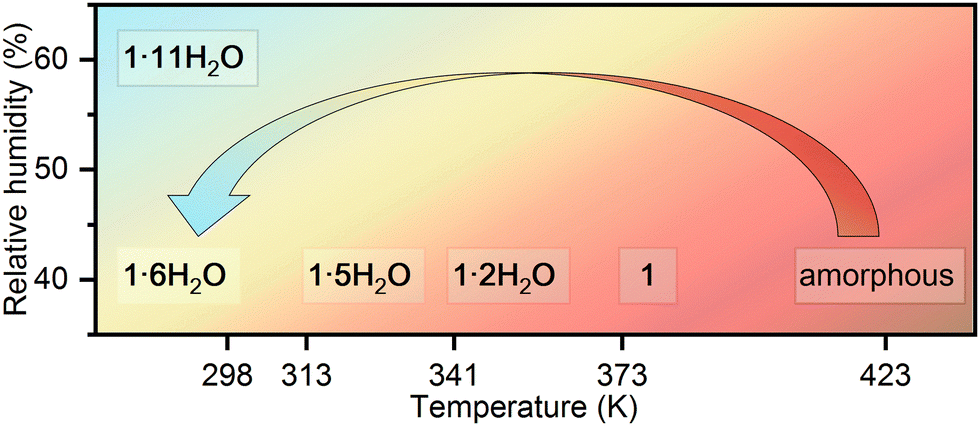 | ||
| Scheme 2 Schematic representation of the structural transformations in the complex salt (TEA)4[V4O8(L-tart)2] (1) triggered by the change in humidity and temperature. | ||
Our findings on the dynamics of structural transformations in this (L-tartrato)oxovanadium(V) complex salt are somewhat consistent with previous observations on the stability of tartrates,1,10,14i.e., how difficult it is to find and control the experimental conditions under which these materials exhibit adequate stability, uniform composition and consequently reproducible properties.
These results are also confirmed by the alternating isothermal and dynamic TG/DTA analysis (Fig. S8, ESI†), which show that phase 1·6H2O loses water molecules in three successive steps during heating. While phase 1·6H2O is heated to 313 K, one water molecule is eliminated (Fig. S8, ESI†), leaving the 1·5H2O phase (weight loss for 1H2O: wcalc = 1.44%; wexp = 1.29%). The next step, corresponding to the elimination of three water molecules, begins with further heating and ends at 341 K, resulting in a 1·2H2O phase (weight loss for 3H2O: wcalc = 4.32%; wexp = 5.15%). Further heating to 373 K gives the dehydrated phase 1 (weight loss for 2H2O: wcalc = 2.87%; wexp = 2.08%).
The reversible structural transformations in the complex salt (TEA)4[V4O8(L-tart)2] triggered by humidity and temperature changes, in addition to PXRD, were monitored in situ by ATR spectroscopy (Fig. 3). The intensity of the bands associated with the O–H stretching vibration [ν(OH)] of the lattice water molecules (range 3650–3250 cm−1)28 decreases as the structure is transformed from 1·11H2O to 1·6H2O, and further from 1·6H2O to 1·2H2O. The position of these bands also changes: in 1·11H2O there are three medium broad bands at 3460, 3380 and 3270 cm−1; in 1·6H2O two broad bands at 3390 and 3280 cm−1; and in 1·2H2O three bands at 3517, 3368 and 3264 cm−1. The tartrate ligands form hydrogen bonds with the surrounding water molecules, so the intensity and positions of the bands related to the vibrations of [νas(CO)] (range 1680–1590 cm−1) and [νs(CO)] (range 1390–1330 cm−1) are also affected by these structural transformations. For example, in 1·11H2O, a strong band related to the asymmetric C–O stretching vibration of the tartrate ligands, located at 1663 cm−1, shifts to lower energies at 1628 and 1623 cm−1, in 1·6H2O and 1·2H2O, respectively. In contrast, the positions and intensities of the bands associated with the symmetric and asymmetric stretching vibrations of the –CH2 and –CH3 groups of the (TEA)+ cations appearing at 2989, 2956, 2923 and 2903 cm−1 show no change with temperature or humidity. This is also true for other deformation vibrations originating from the (TEA)+ cations, such as the bending of the –CH3 group or the scissoring of the –CH2 group, which appear as bands at 1483, 1455 and 1439 cm−1.29 This is consistent with the structural studies that have shown that the cations arrange in separate hydrophobic layers and form only weak interactions with the anions. Another indication of the structural transformations is the red shift of the strongest band in the spectra corresponding to the vibrations of the terminal VO bond, from 966 to 951 cm−1.30
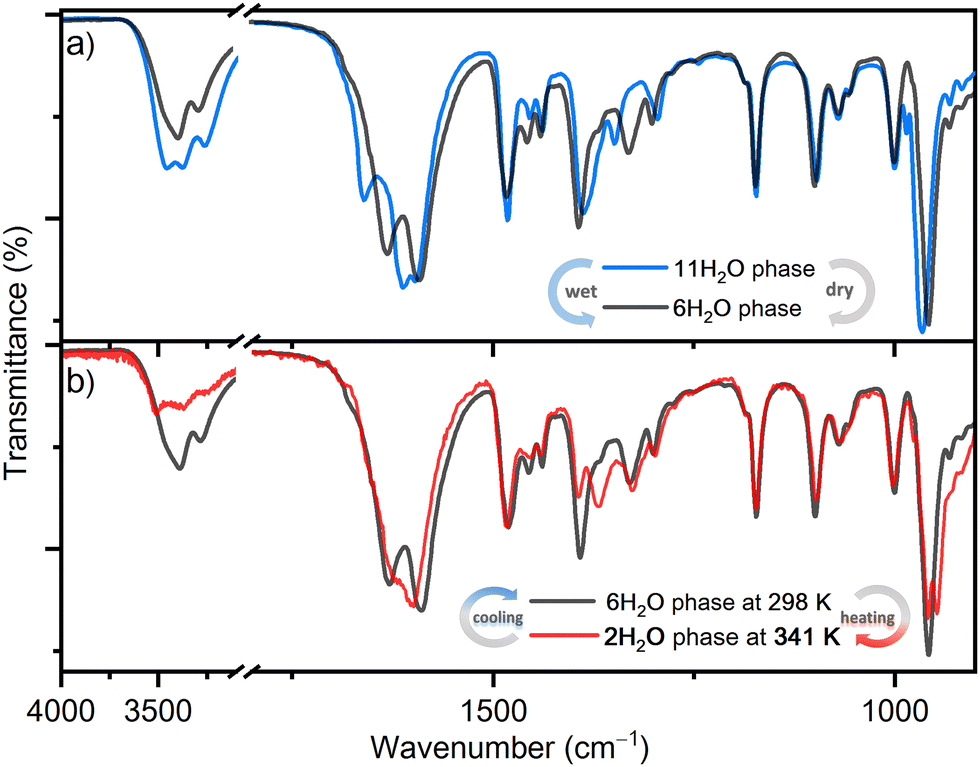 | ||
| Fig. 3 ATR spectra recorded in situ following transformations: (a) from the initial 1·11H2O phase to the 1·6H2O phase and backward induced by a change in the humidity at room temperature; (b) from the 1·6H2O phase to the 1·2H2O phase and backward induced by a change in the temperature. | ||
Electrical properties
The observed structural transformations in the complex salt (TEA)4[V4O8(L-tart)2] involving dehydration/rehydration processes have a significant impact on the electrical properties of the material. The reversible transformation from the 1·11H2O phase to the 1·6H2O phase is dependent on the specific RH conditions and shows a prominent current response to a change in the RH (range from 5% to 60%). Humidity sensing experiments were performed by passing alternately dry and humid air at room temperature (293–298 K) over a 0.22 μm thick pellet attached to silver wires with a silver paste. The current response to changes in the relative humidity is shown in Fig. 4. As can be seen, the current response is less than 40 nA at low humidity (below 10% RH). An increase in humidity results in an order of magnitude increase in the current response, up to a maximum value of 0.22 μA at 60% RH. The observed humidity-induced electrical responses are related to the formation of phase 1·6H2O under dry conditions, and phase 1·11H2O at a RH of ∼ 60%. The formation of these phases was confirmed by in situ PXRD and ATR measurements (Fig. 2 and 3). Exposure of the 1·6H2O phase to humid air at room temperature induces the uptake of 5 additional water molecules, leading to a solid-state transformation from the polar P21 phase to the non-polar P212121 phase (1·11H2O). The higher current response of the non-polar 1·11H2O phase is a consequence of the cooperative and infinite hydrogen bonding chains in the structure, which promote the proton conductivity of the material. The small hysteresis that exists between the drying and wetting processes is related to the dynamics of the solid-state transformations. The reproducibility between drying/wetting cycles is very good, and multiple cycles can be performed on a single pressed pellet. However, the pellet is not stable for measurements at a RH above 70% due to hygroscopicity. The observed hygroscopicity at high RH can be used as a self-recovery process, as the material dissolves at high RH and recrystallizes when the RH falls below 50% (see Fig. S6 with PXRD patterns in the ESI†).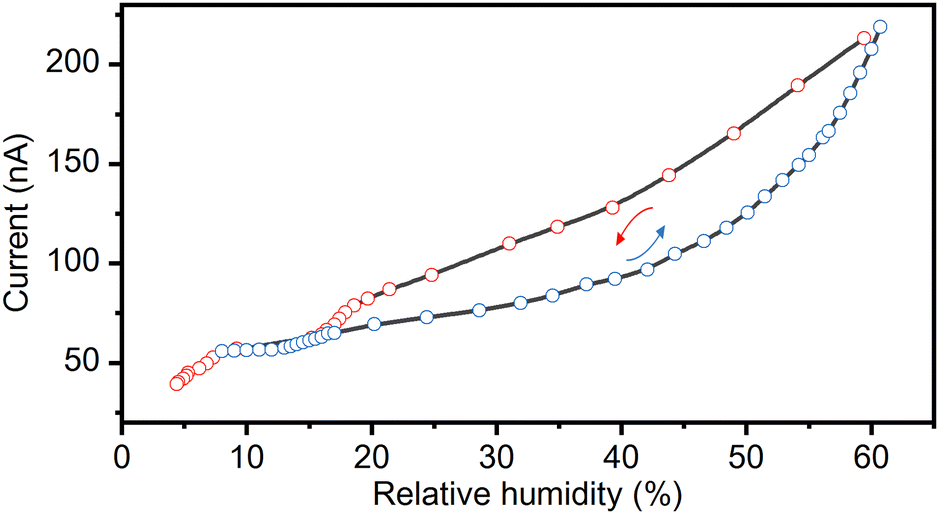 | ||
| Fig. 4 Changes in the current response (conductivity) at 295 K and 1 V with an increase and a decrease in the relative humidity, induced by the reversible structural conversion of 1·11H2O to 1·6H2O. | ||
As already mentioned, the complex salt of (TEA)4[V4O8(L-tart)2] has a room temperature hexahydrate phase, 1·6H2O,belonging to the polar P21 space group, which indicates that it is a potential ferroelectric material. The polarization–electric field (P–E) measurements at room temperature (295 K) and a RH of < 5% in a low-frequency electric field of 0.1–0.3 Hz confirmed the occurrence of the ferroelectricity in the 1·6H2O phase. To exclude the non-switching contributions (dielectric capacitance and leakage current) in this compressed ferroelectric powder sample, a positive up-negative-down (PUND) method with a double triangular voltage pulses was used.31–34 The typical rectangular polarization hysteresis loop as a function of the electric field (P–E) is shown in Fig. 5 along with the current density versus the electric field (J–E) peak, confirming the macroscopic ferroelectric response due to the intrinsic spontaneous polarization. The saturation of the spontaneous polarization (Ps) value under these conditions is about 0.20 μC cm−2 with a low coercive electric field (Ec) of 0.7 kV cm−1. These results are very similar to those of single-crystal potassium sodium L-tartrate tetrahydrate (Ps = 0.25 μC cm−2; Ec = 0.28 kV cm−1) measured at 276 K and a frequency of 50 Hz.35 However, it has been reported that the spontaneous polarization is an one order of magnitude smaller and the coercive field is slightly larger in the compressed potassium sodium L-tartrate tetrahydrate powders compared with single-crystals.36 Considering that our measurements were also performed on compressed polycrystalline samples, we can conclude that the overall ferroelectric performance of 1·6H2O is far better than that of a potassium sodium L-tartrate tetrahydrate. Both compounds are hydrogen-bonded tartrate-based materials with a single axis of spontaneous polarization, so a correlation can be made between them to some extent. The origin of the ferroelectricity of potassium sodium L-tartrate tetrahydrate has long been the subject of experimental and theoretical debate,17,37,38 and it has recently been proposed that the ferroelectric-paraelectric phase transition from the monoclinic P21 to the orthorhombic P212121 structure is not of the pure order–disorder type or the displacement type, but lies between the two types. The ferroelectric–paraelectric phase transition for 1·6H2O was not observed due to the limited thermal stability of the compound. Instead of transition to the non-polar paraelectric phase with increasing temperature, 1·6H2O loses water molecules of crystallization and undergoes a structural phase transition to the phase 1·5H2O at 313 K, which is transformed into the phase 1·2H2O at 341 K, and further into dehydrated phase 1 at 373 K, all belonging to the polar space groups, P21, C2, and P2, respectively.
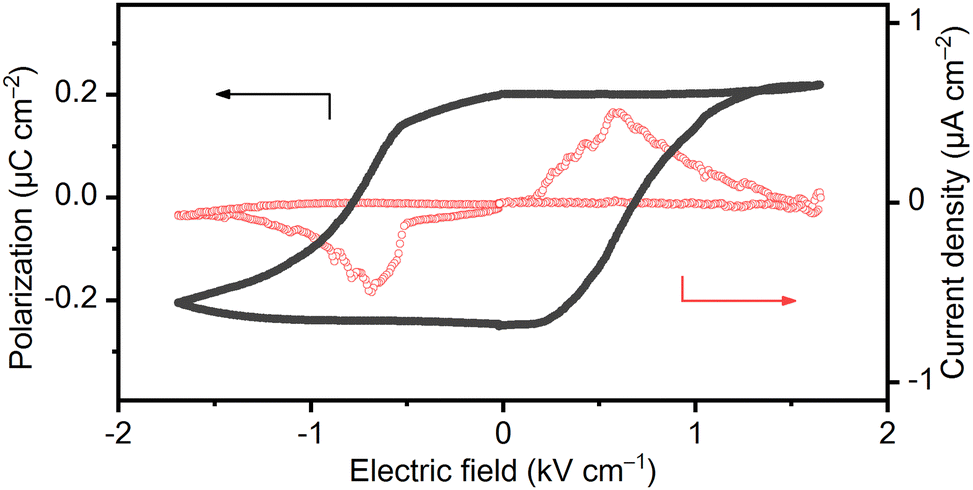 | ||
| Fig. 5 Ferroelectric polarization at 295 K and RH < 5% for 1·6H2O showing P–E hysteresis loops and the corresponding current response at a 0.1 Hz switching frequency. | ||
The pentahydrate phase of the complex salt (TEA)4[V4O8(L-tart)2], 1·5H2O is formed after heating to 313 K. The temperature dependence of the resistivity of this phase is not linear, as can be seen in Fig. S9 (ESI†). During rapid (t = 20 s) heating to 333 K and cooling (t = 40 s) to 313 K cycles, this phase shows a significant change in resistance, as shown in Fig. 6. The experiment was performed under a constant dry air flow to maintain the relative humidity below 20% during the measurement and also to allow more efficient cooling. The sample, in the form of a 0.22 μm thick pellet attached to silver wires with a silver paste, was heated with a hot air gun. It was found that the current response increased sharply when heated and decreased when cooled. The maximum current increases from 0.062 μA at 313 K to 0.502 μA at 333 K, showing that the 1·5H2O phase has a negative temperature coefficient. To evaluate the sensitivity of a thermal response, the value of the temperature coefficient of resistance (TCR) was calculated using the formula TCR = [(R – R0)/R0]/ΔT, where R is the instantaneous resistance at a measured temperature and R0 is the original resistance.39 The measured TCR of –4.38% K−1 is similar to inkjet-printed Cs2SnI6 thermistors (TCR = –4.95% K−1),40 and highly crystalline ferroelectric Bi4Ti3O12 nanoparticles (TCR = –5.76% K−1),41 whose performance is comparable to commercial negative-temperature-coefficient thermistors fabricated on solid substrates. Phase 1·5H2O also exhibited a high thermal index of 4856 K, and a temperature sensing range of 20 K. For compound [Pb7I14(bcbp)2]n with a metal–organic framework structure, a similar thermal index value (4671 K) was reported but with a lower temperature coefficient of resistivity (0.72% K−1) and a wider operating temperature range (180 K).42
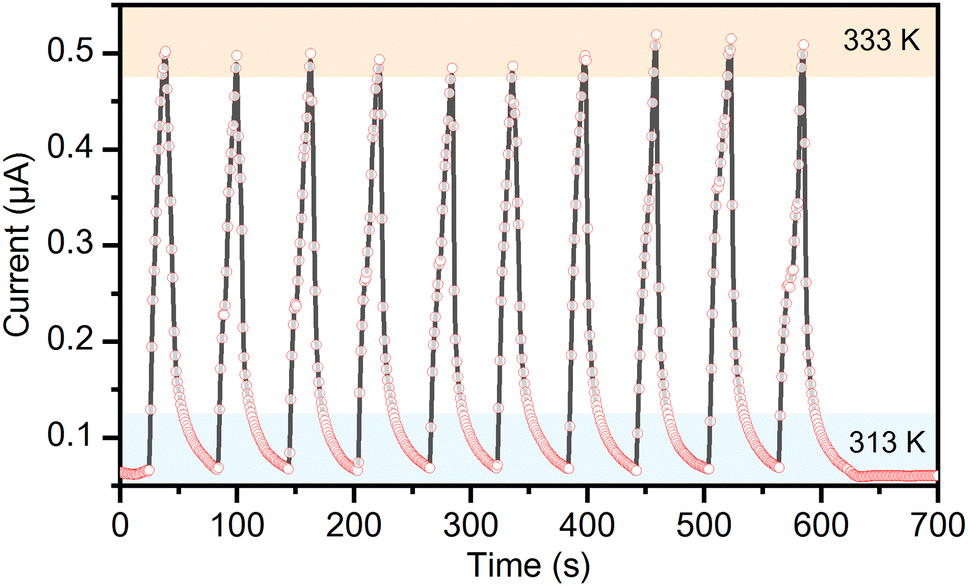 | ||
| Fig. 6 Current response of the 1·5H2O phase at 10 V while switching between two heating stages at 313 K and at 333 K. | ||
The 1·5H2O phase exhibits very good stability in the repeated temperature responses and the overall performance is superior to other metal–organic thermistor systems. The reason for the observed thermistor behaviour of the 1·5H2O phase could be the thermal activation of the carriers, (TEA)+ cations and bulky [V4O8(L-tart)2]4− anions in this flexible molecular solid. The tartrate anions that coordinate, chelate, and bridge the vanadium ions from the inorganic {V4O8} core are responsible for most of the non-covalent intermolecular interactions that govern the crystal packing of the compound. The flexibility of the crystal structure, controlled by the tartrate anions, could be one of the reasons for the observed excellent thermoelectric properties. The low energy barrier for the reversible release and uptake of water molecules from the crystalline solid-state product is a direct consequence of the flexibility of the crystal packing. Such soft materials, whose properties and sensing are based on external triggers and environments, are attracting more attention recently.43–45
Optical properties
Electronic spectra in the solid state, as shown in Fig. 7, were recorded for the most stable phase under ambient conditions (temperature 295 K and 40% relative humidity), the hexahydrate phase 1·6H2O. The phase 1·6H2O contains vanadium(V) centres with a d0 electronic configuration. The strongest band in the visible part of the spectrum at 480 nm, which is also responsible for the intense red colour of the compound, is due to the ligand-to-metal charge transfer (LMCT) of the bridging oxo groups. The LMCT band decreases when the compound is exposed to visible light (generated by the 300 W xenon light source with UV filter), and crystals turn to dark brown colour. In parallel with the decrease in the intensity of the oxo-LMCT band, new absorptions can be observed in the 600–1300 nm range. The bands in this range are often associated with d–d transitions of compounds containing the oxovanadium(IV) group.46,47 Thus, this observation suggests that vanadium(V) in the initial complex is partially reduced to vanadium(IV). When an irradiated compound is left in the dark for 2 days, it partially returns to the initial state. The recovery process can be observed first visually, by the change in the colour of the crystal from dark brown to red, and also from the electronic spectra where an increase in the LMCT band at 480 nm is observed. The self-recovery process can be sped up by exposing the irradiated material to air with high relative humidity, which induces its dissolution and recrystallization.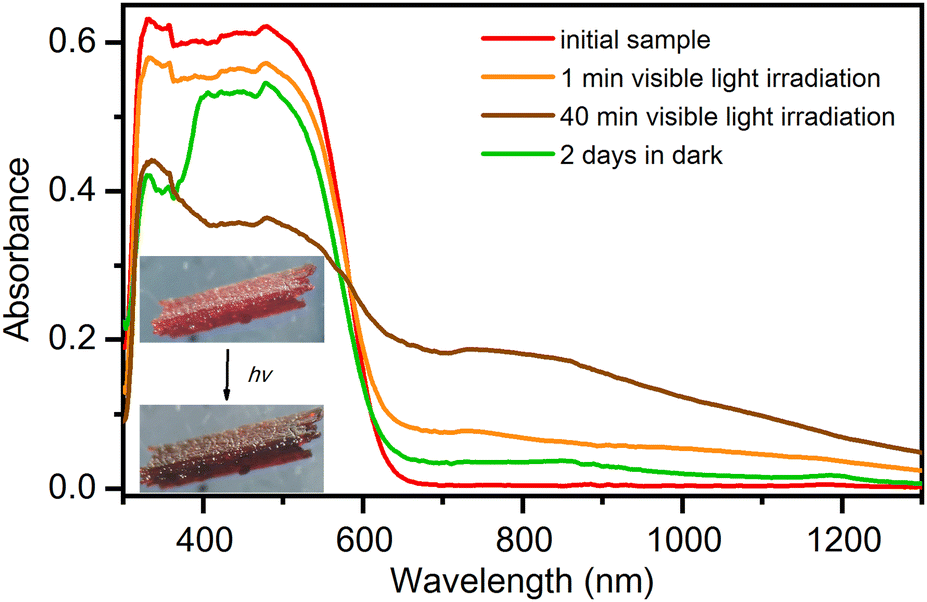 | ||
| Fig. 7 Irradiation-time dependence of the UV-vis diffuse reflectance spectra of 1·6H2O. Spectra were recorded at 295 K ex situ at 0, 1 and 40 minutes of irradiation with a Xenon lamp (300 W, UV filter) and after a recovery period of 2 days in the dark. | ||
Another interesting feature of the obtained single-crystals of 1·11H2O is pleochroism. If the crystal is rotated for 90° under the polarised light, its colour changes from yellow to red, depending on the direction of light polarization with respect to the crystallographic axes (see Fig. S7, ESI†). This optical property is the result of crystal anisotropy and it is a rather rare phenomenon in metal–organic compounds. A similar effect is observed in recently reported bis(oxalato)chromium(III) complex salts, where crystals change the colour from purplish red to orange.45
Conclusion
A straightforward synthesis of the undecahydrate phase of (TEA)4[V4O8(L-tart)2] is reported, giving the targeted compound in high yield and a single-crystal form. The investigated compound exhibits multiple solid-state transformations triggered by humidity and temperature changes in a very narrow range, leading to the formation of different (hexa-, penta- and di-) hydrate phases and the dehydrated phase. The structural transformations were followed by in situ PXRD and ATR measurements, demonstrating their fast dynamics, reversibility and reproducibility. The different hydrate phases exhibit different electrical properties. The undecahydrate phase belongs to a non-polar orthorhombic P212121 structure, comprising a cooperative infinite chain of hydrogen-bonded water molecules. However, the undecahydrate phase is stable only under humid conditions (RH ∼ 60%), and a decrease in humidity leads to the loss of 5 water molecules and the formation of the polar monoclinic hexahydrate phase. This phase shows a room temperature ferroelectric polarization switching of about 0.20 μC cm−2 at a low coercive electric field of 0.7 kV cm−1, comparable to that of an archetypal ferroelectric Rochelle salt. In addition to the ferroelectric properties, the hexahydrate phase also exhibits photoresponsive behaviour, associated with the partial reduction of V(V) to V(IV) ions under visible light. Mild heating to 313 K leads to the formation of the pentahydrate phase, which has a high thermal index and a large value of the temperature coefficient of resistance, comparable to those of commercial negative-temperature-coefficient thermistors. Further heating to 341 and 373 K leads to the successive elimination of water molecules, and the formation of dihydrate and dehydrated phases, respectively. Interestingly, the material becomes amorphous after heating to 423 K and can be returned to the hexahydrate phase by exposure to humid air.Altogether, we have shown that the system of tetraethylammonium cations and complex anions consisting of an inorganic {V4O8} core chelated with fully deprotonated L-tartaric acid is a strikingly soft material whose properties and sensing can be fine-tuned by external triggers and environments, which may be of interest for use in devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Croatian Science Foundation (UIP-2019-04-7433) is gratefully acknowledged.Notes and references
- M. Biver, Inorg. Chem., 2021, 60, 18360–18369 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, B72, 171–179 Search PubMed.
- M. Bobtelsky and J. Jordan, J. Am. Chem. Soc., 1947, 69, 2286–2290 CrossRef CAS.
- H. Sakurai, S. Funakoshi and Y. Adachi, Pure Appl. Chem., 2005, 77, 1629–1640 CrossRef CAS.
- H. Nakamura, M. Fujii, Y. Sunatsuki, M. Kojima and N. Matsumoto, Eur. J. Inorg. Chem., 2008, 1258–1267 CrossRef CAS.
- T. G. Hörner and P. Klüfers, Eur. J. Inorg. Chem., 2016, 1798–1807 CrossRef.
- P. Schwendt, A. S. Tracey, J. Tatiersky, J. Gáliková and Z. Žák, Inorg. Chem., 2007, 46, 3971–3983 CrossRef CAS PubMed.
- J. Cao, Y. Xiong, X. Luo, L. Chen, J. Shi, M. Zhoua and Y. Xu, Dalton Trans., 2018, 47, 6054–6058 RSC.
- P. Antal, P. Schwendt, J. Tatiersky, R. Gyepes and M. Drábik, Transition Met. Chem., 2014, 39, 893–900 CrossRef CAS.
- J. Gáliková, P. Schwendt, J. Tatiersky, A. S. Tracey and Z. Žák, Inorg. Chem., 2009, 48(17), 8423–8430 CrossRef PubMed.
- J. G. Forrest and C. K. Prout, J. Chem. Soc. A, 1967, 1312–1317 RSC.
- J. T. Wrobleski and M. R. Thompson, Inorg. Chim. Acta, 1988, 150, 269–277 CrossRef CAS.
- Z. Sun, T. Chen, C. Ji, S. Zhang, S. Zhao, M. Hong and J. Luo, Chem. Mater., 2015, 27, 4493–4498 CrossRef CAS.
- P.-F. Li, Y. Ai, Y.-L. Zeng, J.-C. Liu, Z.-K. Xu and Z.-X. Wang, Chem. Sci., 2022, 13, 657–664 RSC.
- J. Wu, T. Takeda, N. Hoshino and T. Akutagawa, J. Mater. Chem. C, 2020, 8, 10283–10289 RSC.
- J. Valasek, Phys. Rev., 1921, 17, 475–481 CrossRef CAS.
- F. Jona and G. Shirane, Ferroelectric Crystals, Dover Publications, New York, 1993 Search PubMed.
- H.-Y. Ye, Y. Zhang, S. Noro, K. Kubo, M. Yoshitake, Z.-Q. Liu, H.-L. Cai, D.-W. Fu, H. Yoshikawa, K. Awaga, R.-G. Xiong and T. Nakamura, Sci. Rep., 2013, 3, 2249 CrossRef PubMed.
- M. Szafrański, Angew. Chem., Int. Ed., 2013, 52, 7076–7078 CrossRef PubMed.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
- F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- TOPAS version 4.2, Bruker-AXS, Karlsruhe, Germany, 2009.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley, New York, 6th edn, 2009 Search PubMed.
- S. Hajlaoui, I. Chaabane, A. Oueslati, K. Guidara and A. Bulou, Spectrochim. Acta, Part A, 2014, 117, 225–233 CrossRef CAS PubMed.
- M. F. Davis, W. Levason, J. Paterson, G. Reid and M. Webster, Eur. J. Inorg. Chem., 2008, 802–811 CrossRef CAS.
- S. Martin, N. Baboux, D. Albertini and B. Gautier, Rev. Sci. Instrum., 2018, 88, 023901 CrossRef PubMed.
- L. Androš Dubraja, R. Kruk and T. Brezesinski, Adv. Electron. Mater., 2019, 5, 1800287 CrossRef.
- U. Chowdhury, S. Goswami, D. Bhattacharya, A. Midya and P. Mandal, Appl. Phys. Lett., 2016, 109, 092902 CrossRef.
- A. Puškarić, M. Dunatov, I. Jerić, I. Sabljić and L. Androš Dubraja, New J. Chem., 2022, 46, 3504–3511 RSC.
- M. Topić, Croat. Chem. Acta, 1965, 37, 245–253 Search PubMed.
- A. Mansingh and S. S. Bawa, Phys. Status Solidi A, 1974, 21, 725–731 CrossRef CAS.
- M. I. Khan and T. C. Upadhyay, Eur. Phys. J. D, 2021, 75, 211 CrossRef CAS.
- E. Suzuki and Y. Shiozaki, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 5217 CrossRef CAS PubMed.
- C. Yan, J. Wang and P. S. Lee, ACS Nano, 2015, 9, 2130–2137 CrossRef CAS PubMed.
- S. Li, A. Kosek, M. N. Jahangir, R. Malhotra and C.-H. Chang, Adv. Funct. Mater., 2021, 31, 2006273 CrossRef CAS.
- N. P. M. J. Raj, N. R. Alluri, A. Chandrasekhar, G. Khandelwal and S.-J. Kim, Nano Energy, 2019, 62, 329–337 CrossRef.
- J. Liu, X. Zhang, J. Zhang, S. Zhang, Y. Chen, H. Chen, H. Chen and M. Lin, ACS Appl. Mater. Interfaces, 2022, 14(21), 24575–24582 CrossRef CAS PubMed.
- M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem. – Eur. J., 2019, 25, 5105–5112 CrossRef CAS PubMed.
- P. P. Ferreira da Rosa, Y. Kitagawa, S. Shoji, H. Oyama, K. Imaeda, N. Nakayama, K. Fushimi, H. Uekusa, K. Ueno, H. Goto and Y. Hasegawa, Nat. Commun., 2022, 13, 3660 CrossRef CAS PubMed.
- M. Dunatov, A. Puškarić, L. Pavić, Z. Štefanić and L. Androš Dubraja, J. Mater. Chem. C, 2022, 10, 8024–8033 RSC.
- A. L. Godiksen and S. Birk Rasmussen, Catal. Today, 2019, 336, 45–49 CrossRef CAS.
- S. N. Choing, A. J. Francis, G. Clendenning, M. S. Schuurman, R. D. Sommer, I. Tamblyn, W. W. Weare and T. Cuk, J. Phys. Chem. C, 2015, 119, 17029–17038 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Single-crystal XRD data (Tables S1, S3 and S4); PXRD data (Table S2 and Fig. S1–S6); TG/DTA results (Fig. S8); pleochroism in crystals (Fig. S7); and resistivity measurements (Fig. S9). CCDC 2221444. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc05064a |
| This journal is © The Royal Society of Chemistry 2023 |