
Equipping carbon dots in a defect-containing MOF via self-carbonization for explosive sensing†
Ling-Xiao
Li‡
a,
Shan
He‡
a,
Shanshan
Zeng
b,
Wan-Tao
Chen
c,
Jia-Wen
Ye
 *c,
Hao-Long
Zhou
*ad and
Xiao-Chun
Huang
ae
*c,
Hao-Long
Zhou
*ad and
Xiao-Chun
Huang
ae
aDepartment of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou, 515063, China. E-mail: hlzhou@stu.edu.cn
bCentral Laboratory, Shantou University, Shantou, 515063, China
cSchool of Biotechnology and Health Science, Wuyi University, Jiangmen, 529000, P. R. China. E-mail: wyuchemyjw@126.com
dKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, P. R. China
eChemistry and Chemical Engineering Guangdong Laboratory, Shantou, 515031, China
First published on 30th November 2022
Abstract
Convenient and controllable methods for preparing carbon dots (CDs) and their composites have attracted great interest. Metal–organic frameworks (MOFs), which consist of metal ions or clusters and organic ligands, are considered as ideal platforms for creating functional nanocarbon materials. Here, by facile thermal treatment, we successfully prepared CDs@MOF composites via MOF self-carbonization. By replacing 1,4-benzenedicarboxylic acid with 1,4-naphthalenedicarboxylic acid and adopting hydrochloric acid or benzoic acid as a modulator, the near defect-free and defect-containing forms of a luminescent UiO-66 type MOF (named UiO-66N) can be obtained through a solvothermal reaction. The synthesized products were characterized by powder X-ray diffraction, transmission electron microscopy and gas/dye sorption, so as to determine the existence of crystal defects. After calcination at different temperatures from 100 to 400 °C, the luminescence of the near defect-free UiO-66N had no significant change, while that of the defect-containing UiO-66N changed from blue to green. Experimental evidence indicated that the change of luminescence can be attributed to the formation of CDs and the existence of defects is favourable for self-carbonization in microporous MOFs under mild conditions. Moreover, the CDs@MOF composites exhibited exceptional luminescence sensing for picric acid with a high quenching constant (KSV = 4.0 × 105 M−1) and a low limit of detection (LOD: 6.54 × 10−7 M).
Introduction
Carbon dots (CDs) are zero-dimensional discrete quasi-spherical nanoparticles with a size of less than 10 nm.1–3 As a new class of carbon nanomaterials, they have been widely used in chemical sensing, biological imaging, drug delivery, catalysis, electrochemistry and other fields because of their simple preparation, easy modification, low toxicity, adjustable fluorescence and good water solubility.2–10 At present, the methods for synthesizing CDs can be divided into “top-down” and “bottom-up” methods.1,2,11 The top-down route yields CDs through “decomposing” larger carbon structures (graphite, graphene, carbon nanotubes, carbon black, coal, etc.) into smaller nano-sized ones under severe conditions, which may lead to low quantum yield of CDs.4,12 In contrast, the bottom-up method synthesizes CDs from small molecules or polymer precursors under relatively mild conditions. Although this method is simple and convenient, it occasionally produces amorphous carbon nuclei and the control of the chemical structure and particle size is difficult.13,14 Constructing CDs by the bottom-up method on templates can inhibit the aggregation of CDs during the synthesis process and control the size of CDs.15,16 However, the disadvantage is that the carbon source will also be unevenly distributed on the template, and even the escape of the carbon source in the preparation process of CDs will lead to the formation of CDs outside the template.11,16 Moreover, some templates cannot be easily separated from CDs after synthesis, and the luminescence performance of CDs may be affected in the process of acid–base etching or heating to remove templates.17,18 An ideal template is the key to the preparation of CDs using the template method.Metal–organic frameworks (MOFs), as a new type of crystalline organic–inorganic hybrid porous materials, are regarded as a promising platform for the construction of nanocarbon materials because of their structural diversity as well as regular and adjustable pores.19–29 The confinement effects of nano/sub-nanopores or specific binding sites in MOFs can effectively limit the aggregation of carbon structures and stabilize carbon nanostructures.16,30–34 The conventional method of preparing CDs with MOFs as templates is pyrolyzing organic solvents or guest molecules captured in MOFs.30,35 Considering that some MOF materials also have excellent optical properties,36–38 if they are used as templates, the products can be used as composites without removing the templates even after the CDs are prepared. MOFs can not only continue to provide protection for CDs, but also greatly improve the selectivity and efficiency of CDs in sensing and catalysis.32,35,39 However, in the conventional process of CD preparation with a carbon source introduced into MOFs, the uneven distribution and escape of the carbon source still exist.11,16,35,39 Compared with traditional inorganic crystalline porous materials, MOFs have organic linkers and can be self-carbonized.40–45 Taking advantage of the reticular chemistry and modifiable characteristic of MOFs,21,46 organic groups easy to be carbonized can be decorated into the host framework as carbon sources, so as to realize the orderly arrangement of carbon sources and prevent their escape (Scheme 1).
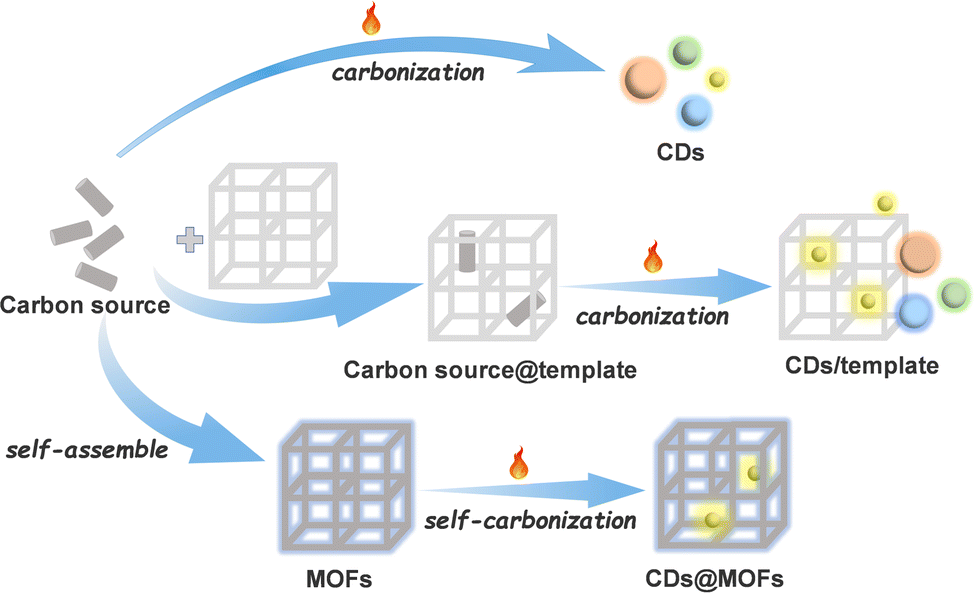 | ||
| Scheme 1 Schematic illustration of different bottom-up CD synthesis processes. | ||
However, for most MOF materials, no obvious carbonization of their host frameworks was observed below their decomposition temperature.22,25,26 It seems that, since the organic linkers in the MOFs are far away from each other so that they are uncooperative to rearrange and be carbonized, their carbonization is more difficult than that of the dense stacking aggregates of discrete molecules. In other words, it is still a great challenge to construct CDs@MOF composites via self-carbonization of MOFs. Determining the triggering factors of MOF self-carbonization is a key to solve this problem. Here, we propose to employ defect-containing MOFs for preparing CDs@MOF composites via self-carbonization. By adopting 1,4-naphthalenedicarboxylic acid (H2NDC) with a relatively large π conjugation system as an organic linker and hydrochloric acid or benzoic acid as a modulator, the near defect-free and defect-containing forms of a luminescent UiO-66 type MOF were obtained through a solvothermal reaction (Fig. 1). Through the facile thermal treatment without introducing additional CD precursors as carbon sources, CDs@MOF composites were successfully constructed from defect-containing ones rather than near defect-free ones. These CDs@MOF composites displayed a better explosive sensing performance than original MOFs and CDs.
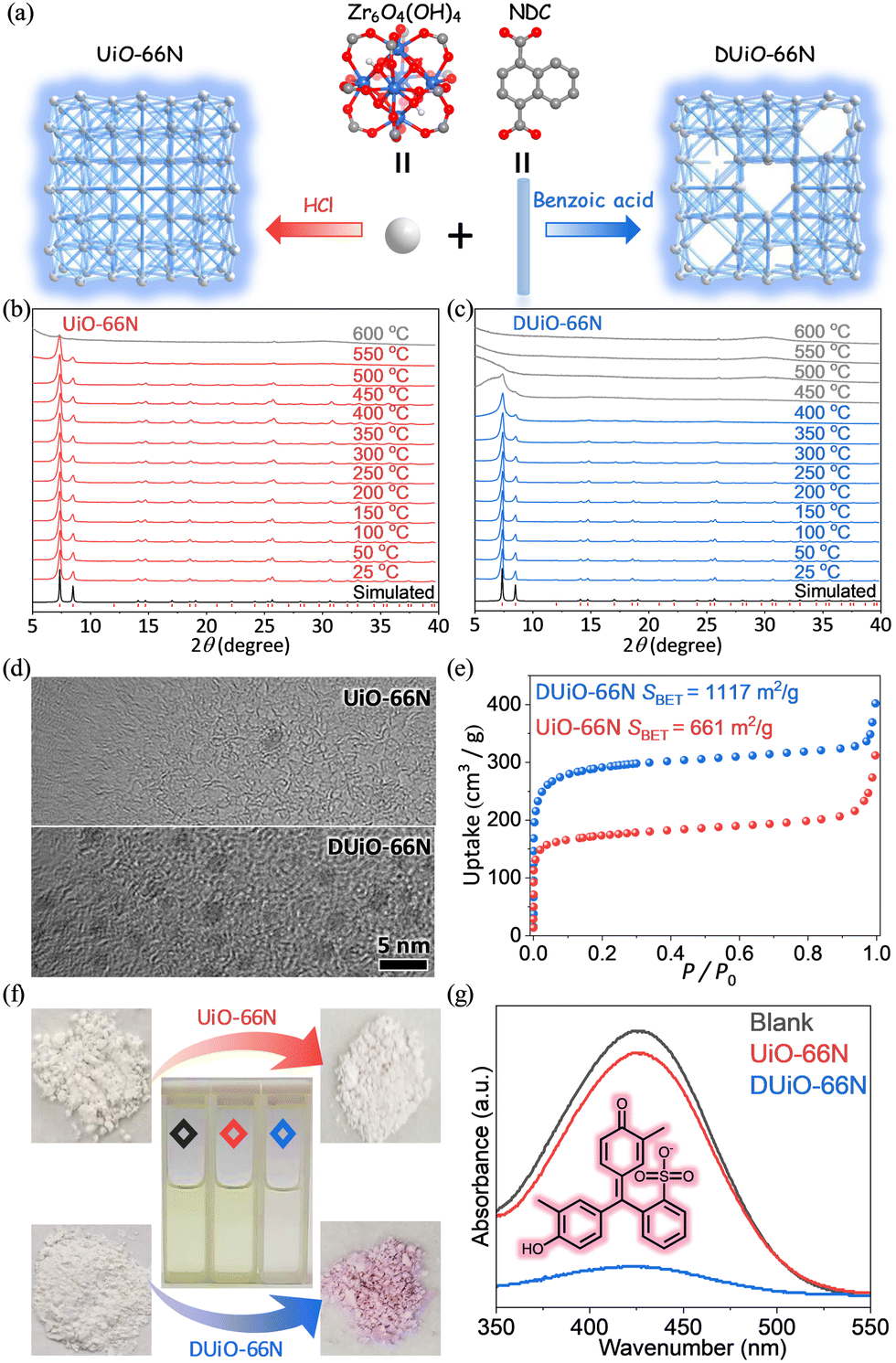 | ||
| Fig. 1 (a) Simplified structural view of UiO-66N and DUiO-66N. (b and c) VTPXRD of UiO-66N and DUiO-66N. (d) TEM images of UiO-66N and DUiO-66N. (e) N2 sorption isotherms at 77 K of UiO-66N and DUiO-66N. (f and g) Photographs and UV-vis spectra of the dye adsorption experiments. Blank: black; UiO-66N: red; DUiO-66N: blue. | ||
Experimental section
Materials
All reagents were commercially available and used without purification except otherwise specified.Physical measurements
Powder X-ray diffraction (PXRD) patterns were collected in 2θ the range of 5–40° on a Rigaku MiniFlex600 X-ray powder diffractometer (Cu Kα, λ = 1.5418 Å) at a scan rate of 10° min−1 at room temperature. Variable-temperature PXRD (VTPXRD) data were collected using a Rigaku Ultima IV diffractometer (Cu Kα, λ = 1.5418 Å) at a scan rate of 2° min−1 under N2 flow. The measured temperatures were set at 25 °C, 50 °C, 100 °C, 150 °C, 200 °C, 250 °C, 300 °C, 350 °C, 400 °C, 450 °C, 500 °C, 550 °C and 600 °C with a heating rate of 10 °C min−1 and held for 5 min before measurement. Thermalgravimetric (TG) data were obtained on a TA Q50 instrument at a heating rate of 30 °C min−1 under a nitrogen atmosphere. Elemental analyses (EA) data were obtained using an Elementar Vario EL Cube elemental analyzer. Potentiometric titrations were performed using a Yolk PHS-3C potentiometer equipped with a rechargeable pH composite electrode. Prior to titration, 50 mg of the MOF sample after simple grinding was dispersed in 60 mL of 0.01 mol L−1 NaNO3 aqueous solution for 18 h, and then the suspension was adjusted to pH = 3 using 0.1 mol L−1 HCl aqueous solution. The scanning electron microscopy (SEM) images were collected on a Zeiss Gemini-300 field emission microscope. The transmission electron microscopy (TEM) images were collected on a 200 kV JEOL JEM-F200 microscope. Gas sorption isotherms were collected on a BSD-PM specific surface & pore size analyzer. The photoluminescence (PL) measurements of solid samples or suspensions were performed using a PTI QM-TM spectrometer or Edinburgh FLS1000 spectrometer. Solid-state diffuse reflectance ultraviolet-visible (UV-vis) spectra of the samples were collected on a Lambda 950 UV-vis spectrometer from 320 to 800 nm. For dye sorption experiments, MOF samples (5 mg) were placed in 10 ppm dye methanol solutions (4 mL), and the solution absorbances were measured by using a SHIMADZU UV-1780 UV-vis spectrophotometer before adding MOF samples and after sorption equilibrium.Synthesis of UiO-66N
A mixture of ZrCl4 (0.378 g, 1.62 mmol) and H2NDC (1.051 g, 4.86 mmol) was dissolved in N,N-dimethyl formamide (DMF, 9.74 mL) with hydrochloric acid (HCl, 11.3 mol L−1, 0.286 mL, 3.24 mmol) by sonication. Then, the solution was sealed in a Telflon-lined stainless steel autoclave (15 mL) and heated at 220 °C for 12 h. After being cooled to room temperature, the precipitate was collected by centrifugation, washed with methanol and dried under vacuum. Yields: 408 mg, 75% based on ZrCl4.Synthesis of DUiO-66N
A mixture of ZrCl4 (1.189 g, 5.1 mmol) and H2NDC (1.103 g, 5.1 mmol) was dissolved in DMF (300 mL) with benzoic acid (37.369 g, 306 mmol) by sonication. Then the solution was sealed in a 500 mL reagent bottle and heated at 120 °C for 24 h. After being cooled to room temperature, the precipitate was collected by centrifugation, washed with methanol and dried under vacuum. Yields: 668 mg, 40% based on ZrCl4.Preparation of UiO-66N-X and DUiO-66N-X
UiO-66N and DUiO-66N were heated under vacuum at 100 °C, 200 °C, 300 °C and 400 °C for 12 h, respectively. After cooling to room temperature and backfilling with nitrogen, the products were collected and named UiO-66N-X and DUiO-66N-X (X = 100, 200, 300 or 400), respectively.Preparation of CDs
The samples of DUiO-66N-400 were completely digested with 1 M KOH aqueous solution. Then the pH of digestion solution was adjusted to 7 using hydrochloric acid. After removing the insoluble precipitate by centrifugation, the digestion solution was extracted several times with ethyl acetate. The yellow powder of CDs can be obtained from the organic phase by rotary evaporation under vacuum. EA found%: C 85.72, H 12.22.Explosive sensing
The luminescent responses of MOFs, CDs and their composites towards explosive solutions of various concentrations were monitored using PL spectra. Powder samples (5 mg) of CDs, UiO-66N-X and DUiO-66N-X were placed in 10 mL explosive methanol solutions of different concentrations ranging from 0 to 200 μM, respectively. Then these solutions were sonicated for 5 min before taking the PL measurements.The luminescence quenching constants (Ksv) were quantitatively calculated using the Stern–Volmer equation:
| I0/I = 1 + KSV × C, | (1) |
The limit of detection (LOD) was estimated by using the standard equation:
| LOD = 3σ/K, | (2) |
Results and discussion
Synthesis and characterization of DUiO-66N
A classical MOF material, UiO-66, with a 12-connected fcu topological network based on Zr6O4(OH)4 clusters, is favoured for its excellent stability and adjustable crystalline defects.47–53 Modulators (such as benzoic acid, acetic acid, trifluoroacetic acid, etc.) are generally introduced into the synthesis of UiO-66, which is conducive to the crystallization of UiO-66.48,54 The products obtained by this synthesis method are generally considered to have crystal defects. Recently, the synthesis method of UiO-66 to avoid a large number of defects by using HCl has been reported.55,56 Considering the weak fluorescence intensity of UiO-66 (Fig. S1, ESI†), 1,4-naphthalene dicarboxylic acid with a larger conjugation system was used to replace terephthalic acid for constructing a luminescent UiO-66 type MOF named UiO-66N (Fig. 1a). Then near defect-free UiO-66N and defect-containing UiO-66N (named DUiO-66N) were synthesized via a solvothermal reaction by adding HCl or benzoic acid as a modulator, respectively.PXRD refinement showed that UiO-66N and DUiO-66N were isostructural with the same unit-cell parameters and pure (Fig. S2 and Table S1, ESI†), while VTPXRD indicated that UiO-66N can maintain its crystallinity until 550 °C and DUiO-66N can only be stable till 400 °C (Fig. 1b and c). This difference in thermal stability was also confirmed by TG analysis under nitrogen (Fig. S3, ESI†). TG curves under oxygen revealed that the components of UiO-66N matched the theoretical formula Zr6O4(OH)4(NDC)6, and DUiO-66N had components missing (Fig. S4, ESI†).55 SEM images displayed that the crystals of UiO-66N and DUiO-66N were octahedral with uniform morphology (Fig. S5, ESI†), in which the crystal size of UiO-66N was about 150 nm and that of DUiO-66N was about 1 μm. TEM observation for DUiO-66N showed clearly the isolated and well-distributed crystal defects with the size of ca. 2 nm but not for UiO-66N (Fig. 1d). Potentiometric acid–base titration data also identified the existence of defect sites in DUiO-66N (Fig. S6, ESI†).57 For crystalline porous materials, the existence of crystal defects will significantly affect their porosity. N2 sorption isotherms at 77 K (Fig. 1e and Table S2, ESI†) revealed that UiO-66N possessed a saturated uptake at P/P0 = 0.2 of 173 cm3 g−1 and a pore volume of 0.31 cm3 g−1 matched with the theoretical values calculated from its crystal structure,58 as well as Brunauer–Emmett–Teller (BET) and Langmuir specific surface areas of 661 and 853 m2 g−1, respectively. For DUiO-66N, a higher saturated uptake at P/P0 = 0.2 of 291 cm3 g−1 and a higher pore volume of 0.50 cm3 g−1 were observed, and BET and Langmuir specific surface areas were calculated to be 1117 and 1359 m2 g−1, respectively. To visually characterize the difference of their pore channels induced by defects, Cresol Red (CR), a dye with a molecular size (8.3 × 11.3 × 13.2 Å3) larger than the aperture size of UiO-66N, was selected as a molecular probe for the dye adsorption experiments (Fig. 1f). UV-Vis light absorption spectra showed that no obvious change of the CR solution was observed in the presence of UiO-66N, while the absorbance of the CR solution decreased 87% for DUiO-66N (Fig. 1g), attributed to the larger pore width of DUiO-66N (Fig. S7, ESI†). In a word, MOF samples with a similar chemical composition but different defect extents and pore sizes were successfully prepared.
Calcination-induced luminescence change
Considering that both UiO-66N and DUiO-66N can be stabilized at 400 °C (Fig. 1b and c), they were calcinated at a series of temperatures under vacuum for 12 hours to obtain the samples of UiO-66N-X and DUiO-66N-X, where X represents their thermal treatment temperature. Interestingly, the luminescence of DUiO-66N-X under a UV light with a wavelength of 365 nm changed from blue to green with the increase of thermal treatment temperatures, while that of UiO-66N-X had no significant change (Fig. 2a). To evaluate the difference in optical properties induced by thermal treatment, the UV-vis absorption and PL emission spectra of UiO-66N-X and DUiO-66N-X were monitored. The UV-vis spectra displayed that the absorption of all samples was concentrated in the range of 340–360 nm (Fig. S8, ESI†). With an excited wavelength at 360 nm, both UiO-66N-100 and DUiO-66N-100 exhibited the same blue emission peak at 430 nm. With the increase of thermal treatment temperature from 100 to 400 °C, the emission peak exhibited a slight red shift from 430 to 438 nm for UiO-66N-X, and the emission peak of DUiO-66N-X was dramatically red shifted from 430 to 494 nm (Fig. 2). The Commission Internationale de l’Eclairage (CIE) coordinates calculated from PL emission spectra (Fig. 2) were used to display their luminescence alteration of UiO-66N-X and DUiO-66N-X. The coordinates of DUiO-66N-X shifted monotonically from the blue light region to the yellow light region with the increase of thermal treatment temperature. In contrast, the coordinates of UiO-66N-X were concentrated in the blue light region.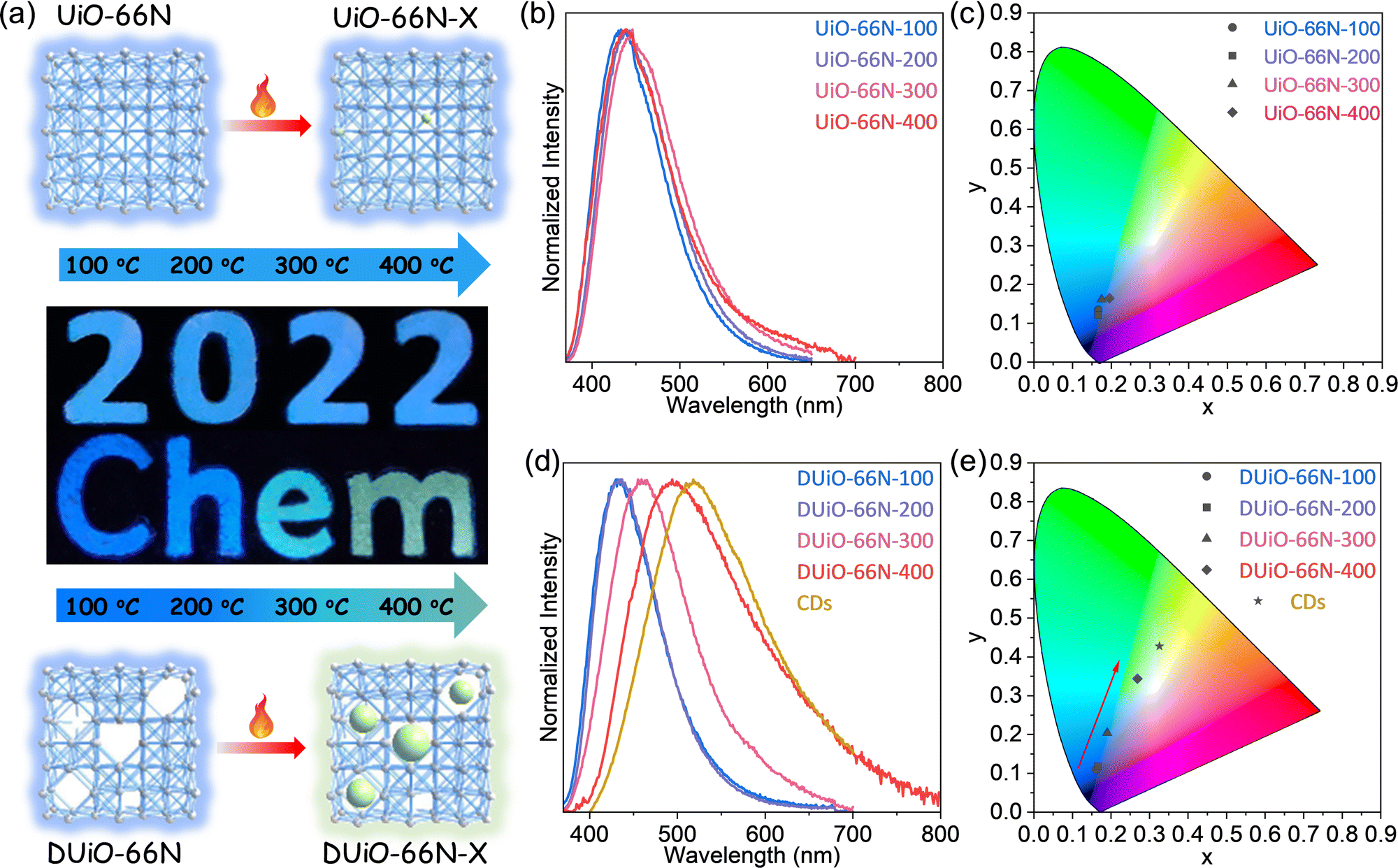 | ||
| Fig. 2 (a) Photographs of calcination-induced luminescence change of UiO-66N-X and DUiO-66N-X. (b and c) PL emission spectra and CIE coordinates of UiO-66N-X. (d and e) PL emission spectra and CIE coordinates of DUiO-66N-X and CDs. | ||
Synthesizing CDs in defect-containing MOF crystals
To clarify the origin of their luminescence difference caused by thermal treatment, UiO-66N-X and DUiO-66N-X were characterized by electron microscopy and nitrogen sorption. The SEM micrographs of UiO-66N-X and DUiO-66N-X showed that their crystal morphologies had no change and no other particles were observed on the external surface of their crystals (Fig. S5, ESI†). The TEM micrographs showed that the effect of thermal treatment temperature on UiO-66N-X was not significant, while the crystal defects in DUiO-66N-X increased with the increase of thermal treatment temperature, suggesting the occurrence of “thermolysis” within the crystals of DUiO-66N-X (Fig. 3). N2 sorption isotherms at 77 K were measured for UiO-66N-X and DUiO-66N-X to evaluate their porosities (Fig. 3 and Fig. S9–S12 and Table S2, ESI†). With the thermal treatment temperature increasing from 100 to 400 °C, the BET specific surface areas of UiO-66N-X were almost unchanged, while that of DUiO-66N-X decreased from 1117 to 177 m2 g−1. Apparently, some substances formed inside the crystals of DUiO-66N after thermal treatment, which not only blocked its pores but also changed its luminescence.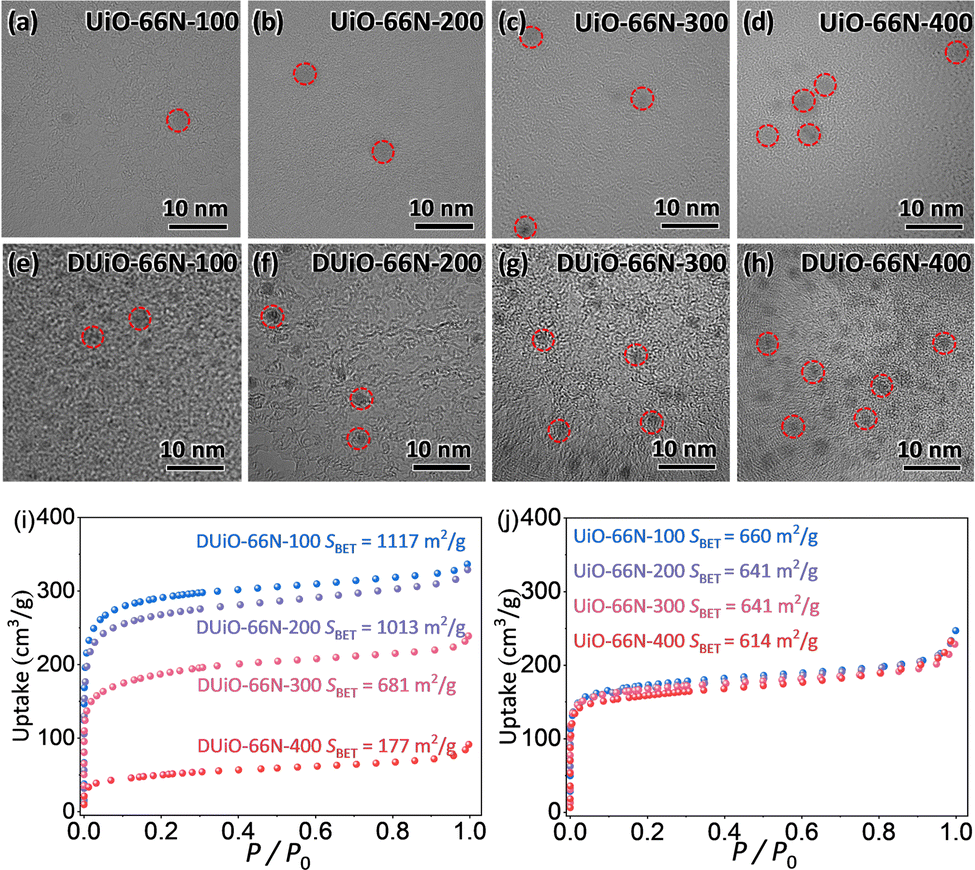 | ||
| Fig. 3 (a–d) TEM images of UiO-66N-X. (e–h) TEM images of DUiO-66N-X. (i and j) N2 sorption isotherms at 77 K of UiO-66N-X and DUiO-66N-X. Circles in TEM images highlight potential crystal defects or CDs. | ||
For determining the substances generated by thermal treatment, the samples of UiO-66N-X and DUiO-66N-X were completely digested with a 1 M KOH aqueous solution. Then the digestion solution was modulated to be neutral with hydrochloric acid. After removing the insoluble precipitate, the digestion solution was extracted several times with ethyl acetate, and the luminescence of the organic phase was observed under the 365 nm UV light (Fig. S13, ESI†). UiO-66N-X only produced a small amount of luminescent substances when the thermal treatment temperature reached 400 °C. For DUiO-66N-X, an appreciable amount of luminescent substances generated when the heat treatment temperature reached 300 °C or above. The solid sample of luminescent substances was obtained by rotary evaporation and characterized by elemental analyses, which displayed that the carbon content exceeds 85% (Fig. S14, ESI†). The TEM micrographs showed that the average size of these particles was ca. 2 nm, with a rather narrow size distribution (Fig. 4). The high resolution TEM micrograph revealed the lattice spacing of these particles was ca. 0.22 nm, consistent with lattice spacing of the (100) plane of graphene (Fig. 4).1,17,43,59
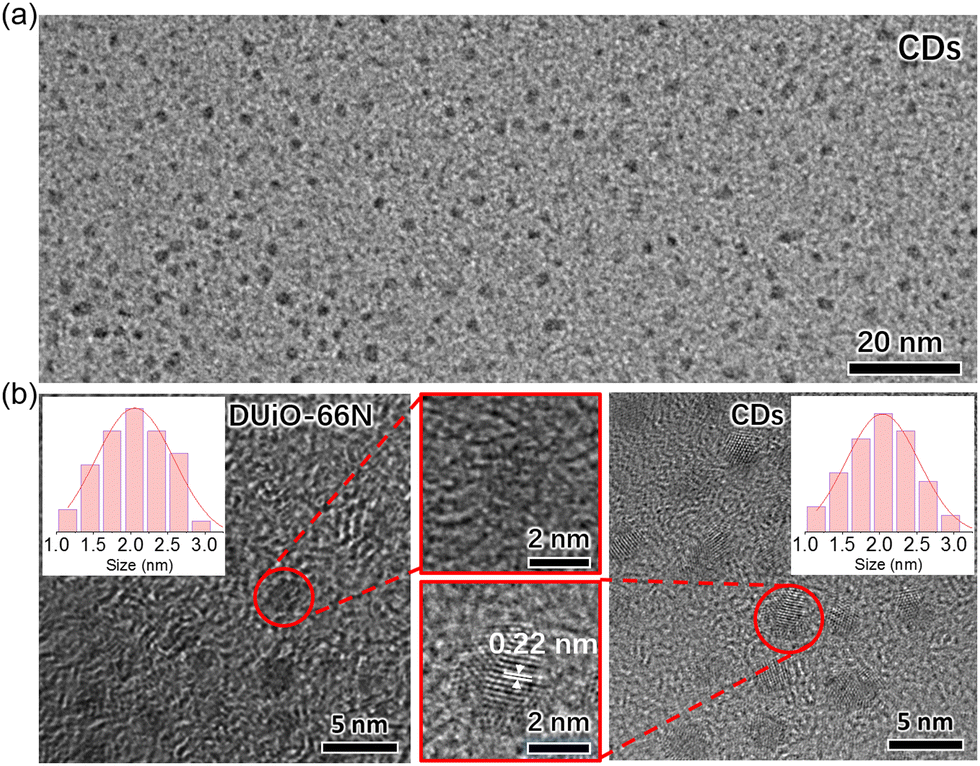 | ||
| Fig. 4 (a) The TEM micrograph of separated CDs. (b) The high resolution TEM micrographs of DUiO-66N and CDs. Inset shows the size distribution for crystal defects and CDs. | ||
Convincingly, the luminescent substances generated in the DUiO-66N-X crystals were CDs. In other words, taking advantage of the local self-carbonization behaviours of MOFs, we successfully equipped CDs in MOFs by a simple thermal treatment method without introducing extra carbon sources and destroying the crystallinity of MOFs. This phenomenon was observed in the defect-containing MOF DUiO-66N and its occurrence was difficult in the near defect-free MOF UiO-66N, suggesting that the crystal defects of MOFs play a key role in MOF self-carbonization. In view of the difference in their porosities, it is speculated that the larger pore size of the defect-containing MOF DUiO-66N, allowing the migration and rearrangement of organic molecules, facilitated the self-carbonization of MOFs. In addition, the uniform CD size was consistent with the defect size (Fig. 4), indicating that the defect-containing MOFs also act as the template. It could be expected that the sizes/morphologies of CDs can be confined by the defects of MOFs.
The PL spectrum of CDs showed that its emission peak was further red shifted to 518 nm in comparison with that of DUiO-66N-X (Fig. 2). The shift trend of the emission peaks and CIE coordinates also confirmed that the source of the thermal treatment induced luminescence change in DUiO-66N-X was the formation of CDs (Fig. 2). With increasing thermal treatment temperature, more CDs formed and the emission peak was gradually red-shifted. Similar to CDs@MOF composites, all samples of DUiO-66N-X adopted single-peak emission behaviours, indicating that the energy transfer between MOFs and CDs was efficient. This benefits the synergistic response of CDs and MOFs, thus exploiting their sensing advantages simultaneously.
Luminescence sensing towards picric acid
For exploring whether the obtained CDs@MOF composites combine the advantages of CDs and MOFs to achieve the “one plus one is greater than two” effect, the luminescence sensing performance of CDs, UiO-66N-X and DUiO-66N-X towards a classical explosive, picric acid, was tested.37 UiO-66N-X, DUiO-66N-X and CDs were placed in series of picric acid methanol solutions of different concentrations ranging from 0 to 200 mM and the PL emission spectra of their suspensions were detected (Fig. 5 and Fig. S15–S17, ESI†). In 200 mM picric acid solutions, 92% (I0/I = 11.9) and 79% (I0/I = 4.7) emission intensities were quenched for CDs and UiO-66N-100, respectively (Fig. S15 and S16 and Table S3, ESI†), as the electron-deficient nature of nitroaromatic analytes.37 It can be seen that, relative to MOFs, CDs are more sensitive to the picric acid luminescence sensing, which should be attributed to its larger p conjugation system. Interestingly, the emission intensity of the CDs@MOF composite DUiO-66N-200 was quenched by 98.5%, corresponding to I0/I = 64.3 (Fig. 5a and Table S3, ESI†). This implies that the combination of CDs and MOFs does lead to more efficient luminescence sensing. With the integration of CDs into MOFs, the enrichment and mass transfer effect of MOFs allow CDs to contact the picric acid more efficiently.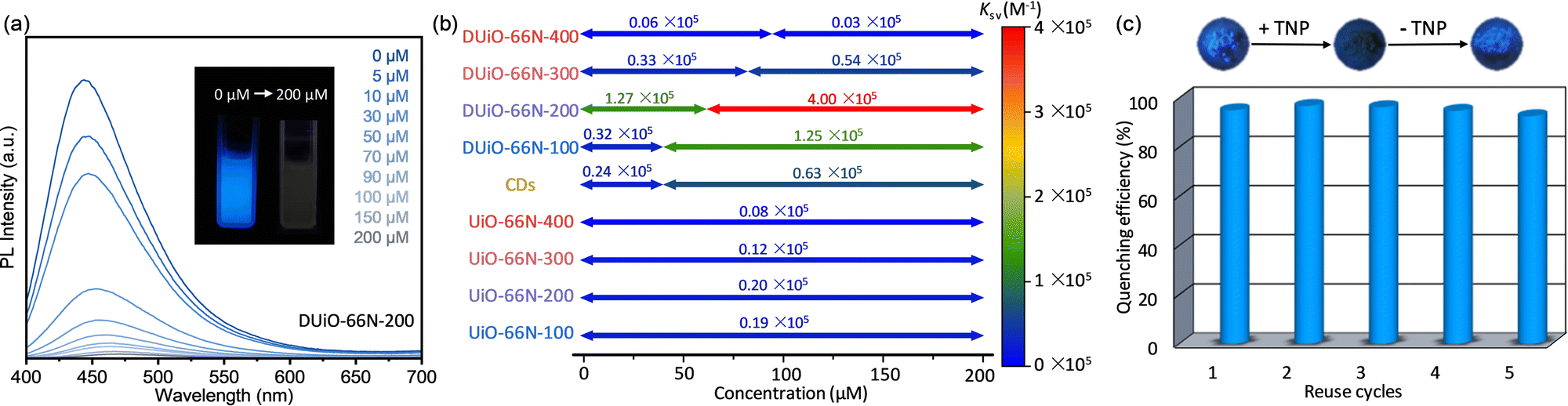 | ||
| Fig. 5 (a) PL emission spectra of DUiO-66N-200 in picric acid solutions. Inset shows the photographs of suspension luminescence change without/with picric acid. (b) Quenching constants Ksv and the corresponding analyte concentration ranges of CDs, UiO-66N-X and DUiO-66N-X. (c) Luminescence sensing cycling performance of DUiO-66N-200. | ||
To further quantify and elucidate the luminescence sensing performance of CDs, UiO-66N-X and DUiO-66N-X, the quenching data at different concentrations were analyzed using the linear Stern–Volmer equation. Linear fitting gave the quenching constants Ksv of 0.19/0.20/0.12/0.08 × 105 M−1 for UiO-66N-100/200/300/400, respectively (Fig. 5b and Fig. S16, ESI†). The amount of CDs generated in UiO-66N crystals was quite low; therefore, UiO-66N-X can be regarded as almost a single component, maintaining its intrinsic sensing performance. Due to the complex structure, the Stern–Volmer plots of CDs displayed a significant nonlinearity. The segmented fitting gave Ksv of 0.24/0.63 × 105 M−1 for the low/high concentration range, respectively (Fig. 5b and Fig. S15, ESI†). DUiO-66N-X also showed a dual-site sensing model after equipping with CDs via thermally-induced self-carbonization. The inflection points of their Stern–Volmer plots shifted toward higher concentration with increasing thermal treatment temperature, consistent with their two-component luminescence and CD loading increase. It is worth noting that for DUiO-66N-400, which generated the most CDs, its Ksv was lower than those of both UiO-66N-X and CDs due to its distinct carbonization and the CDs blocking the pore channels (Fig. 5b and Fig. S17, ESI†). This also reaffirmed that the enrichment and mass transfer effects of the porous structures play an important role in achieving effective contact between the analyte and the probe, and thus increase sensitivity. For DUiO-66N-200, which is equipped with a small amount of CDs but substantially maintained its porosity, its Ksv reached 1.27/4.00 × 105 M−1 for the low/high concentration range, respectively (Fig. 5b and Fig. S17, ESI†), with a low limit of detection of 6.54 × 10−7 M (Fig. S18, ESI†). To our knowledge, this is one of the best performing MOF-based materials for picric acid sensing (Table S4, ESI†).60–62
In addition, the luminescence sensing cycling performance of the CDs@MOF composites was confirmed by five reuse cycles (Fig. 5c). After the luminescence of DUiO-66N-200 was quenched by picric acid, its luminescence could be easily recovered by a simple methanol-washing operation. This indicated that the luminescence quenching is achieved by physical adsorption or contact, rather than chemical reaction. The quenching efficiency of DUiO-66N-200 had no significant change after five reuse cycles. This phenomenon also illustrated that CDs can be effectively protected and retained in microporous MOFs.
Conclusions
In summary, the near defect-free and defect-containing forms of a luminescent UiO-66 type MOF UiO-66N have been prepared and well characterized to elucidate the triggering factors of MOF self-carbonization. After calcination at different temperatures from 100 to 400 °C, the luminescence of the defect-containing one changed from blue to green with CDs generated inside crystals, while the near defect-free one had no significant luminescence change. The results of our investigation demonstrated that the crystal defect as well as the larger pore size it caused, allowing the migration and rearrangement of carbon sources, facilitated the self-carbonization of MOFs. The CDs@MOF composites exhibited not only a better sensing performance than the individual components CDs and MOFs but also one of the best performances of MOF-based materials towards picric acid sensing, which provided a positive example for gaining a deeper insight into composite construction.Author contributions
H. L. Z. and J. W. Y. designed the research. L. X. L and S. H. performed the material synthesis and characterisation measurements. S. Z. helped to collect electron microscopy images. J. W. Y. and W. T. C. helped to perform photoluminescence measurements. H. L. Z., J. W. Y. and X.-C. H. analyzed the data and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22071142, 22001160 and 22101210), Chemistry and Chemical Engineering Guangdong Laboratory (1922003), Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education (KFJJ2022007) and Li Ka Shing Foundation Cross-Disciplinary Research Project (2020LKSFG09A and 2020LKSFG10A).References
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- S. Y. Lim, W. Shen and Z. Q. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- Y. Yan, J. Gong, J. Chen, Z. Zeng, W. Huang, K. Pu, J. Liu and P. Chen, Adv. Mater., 2019, 31, 1808283 CrossRef PubMed.
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS PubMed.
- H. Li, X. Yan, D. Kong, R. Jin, C. Sun, D. Du, Y. Lin and G. Lu, Nanoscale Horiz., 2020, 5, 218–234 RSC.
- W. Liu, M. Li, G. Jiang, G. Li, J. Zhu, M. Xiao, Y. Zhu, R. Gao, A. Yu, M. Feng and Z. Chen, Adv. Energy Mater., 2020, 10, 2001275 CrossRef CAS.
- B. Zhao and Z. Tan, Adv. Sci., 2021, 8, 2001977 CrossRef CAS PubMed.
- X. Li, S. Zhao, B. Li, K. Yang, M. Lan and L. Zeng, Coord. Chem. Rev., 2021, 431, 213686 CrossRef CAS.
- L. Tian, Z. Li, P. Wang, X. Zhai, X. Wang and T. Li, J. Energy Chem., 2021, 55, 279–294 CrossRef CAS.
- S. Chung, R. A. Revia and M. Zhang, Adv. Mater., 2021, 33, 1904362 CrossRef CAS PubMed.
- C. Xia, S. Zhu, T. Feng, M. Yang and B. Yang, Adv. Sci., 2019, 6, 1901316 CrossRef CAS PubMed.
- L. Ai, Y. Yang, B. Wang, J. Chang, Z. Tang, B. Yang and S. Lu, Sci. Bull., 2021, 66, 839–856 CrossRef CAS.
- C. Long, Z. Jiang, J. Shangguan, T. Qing, P. Zhang and B. Feng, Chem. Eng. J., 2021, 406, 126848 CrossRef CAS.
- M. L. Liu, B. B. Chen, C. M. Li and C. Z. Huang, Green Chem., 2019, 21, 449–471 RSC.
- D. Zhou, D. Li, P. Jing, Y. Zhai, D. Shen, S. Qu and A. L. Rogach, Chem. Mater., 2017, 29, 1779–1787 CrossRef CAS.
- H. Zhang, B. Wang, X. Yu, J. Li, J. Shang and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19390–19402 CrossRef CAS PubMed.
- J. Chen, G. Xiao, G. Duan, Y. Wu, X. Zhao and X. Gong, Chem. Eng. J., 2021, 421, 127743 CrossRef CAS.
- J. C. Liu, N. Wang, Y. Yu, Y. Yan, H. Y. Zhang, J. Y. Li and J. H. Yu, Sci. Adv., 2017, 3, e1603171 CrossRef PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- J. P. Zhang, Y. B. Zhang, J. B. Lin and X. M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- L. Feng, K. Y. Wang, G. S. Day, M. R. Ryder and H. C. Zhou, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS PubMed.
- C. Wang, J. Kim, J. Tang, M. Kim, H. Lim, V. Malgras, J. You, Q. Xu, J. Li and Y. Yamauchi, Chemistry, 2020, 6, 19–40 CrossRef CAS.
- G. Cai, P. Yan, L. Zhang, H. C. Zhou and H. L. Jiang, Chem. Rev., 2021, 121, 12278–12326 CrossRef CAS PubMed.
- C. Wang, Y. Yao, J. Li and Y. Yamauchi, Acc. Mater. Res., 2022, 3, 426–438 CrossRef CAS.
- M. Kim, X. Xu, R. Xin, J. Earnshaw, A. Ashok, J. Kim, T. Park, A. K. Nanjundan, W. A. El-Said, J. W. Yi, J. Na and Y. Yamauchi, ACS Appl. Mater. Interfaces, 2021, 13, 52034–52043 CrossRef CAS PubMed.
- M. Kim, K. L. Firestein, J. F. S. Fernando, X. Xu, H. Lim, D. V. Golberg, J. Na, J. Kim, H. Nara, J. Tang and Y. Yamauchi, Chem. Sci., 2022, 13, 10836–10845 RSC.
- M. Kim, R. Xin, J. Earnshaw, J. Tang, J. P. Hill, A. Ashok, A. K. Nanjundan, J. Kim, C. Young, Y. Sugahara, J. Na and Y. Yamauchi, Nat. Protoc., 2022, 17, 2990–3027 CrossRef CAS PubMed.
- Z. G. Gu, D. J. Li, C. Zheng, Y. Kang, C. Woell and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 6853–6858 CrossRef CAS PubMed.
- C. Yao, Y. Xu and Z. Xia, J. Mater. Chem. C, 2018, 6, 4396–4399 RSC.
- L. Giri, S. R. Rout, R. S. Varma, M. Otyepka, K. Jayaramulu and R. Dandela, Nanotechnol. Rev., 2022, 11, 1947–1976 CrossRef CAS.
- Y. Li, X. X. Jiang, J. X. Xie and Y. K. Lv, Chem. – Asian J., 2022, 17, e202200283 CAS.
- J. S. Li, Y. J. Tang, S. L. Li, S. R. Zhang, Z. H. Dai, L. Si and Y. Q. Lan, CrystEngComm, 2015, 17, 1080–1085 RSC.
- Y. Zhang, M. Sun, M. Peng, E. Du, X. Xu and C. C. Wang, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.04.076.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Z. C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- Y. J. Cui, B. Li, H. J. He, W. Zhou, B. L. Chen and G. D. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- B. Li, T. Suo, S. Xie, A. Xia, Y. J. Ma, H. Huang, X. Zhang and Q. Hu, TrAC, Trends Anal. Chem., 2021, 135, 116163 CrossRef CAS.
- S. Gadipelli, W. Travis, W. Zhou and Z. Guo, Energy Environ. Sci., 2014, 7, 2232–2238 RSC.
- A. J. Amali, H. Hoshino, C. Wu, M. Ando and Q. Xu, Chem. – Eur. J., 2014, 20, 8279–8282 CrossRef CAS PubMed.
- H. Xu, S. Zhou, L. Xiao, H. Wang, S. Li and Q. Yuan, J. Mater. Chem. C, 2015, 3, 291–297 RSC.
- Y. Wang, B. Wang, H. Shi, C. Zhang, C. Tao and J. Li, Inorg. Chem. Front., 2018, 5, 2739–2745 RSC.
- L. Feng, S. Yuan, L. L. Zhang, K. Tan, J. L. Li, A. Kirchon, L. M. Liu, P. Zhang, Y. Han, Y. J. Chabal and H. C. Zhou, J. Am. Chem. Soc., 2018, 140, 2363–2372 CrossRef CAS PubMed.
- T. A. Ablott, R. Webby, D. R. Jenkinson, A. Nikolich, L. Liu, H. Amer Hamzah, M. F. Mahon, A. D. Burrows and C. Richardson, Inorg. Chem., 2022, 61, 1136–1144 CrossRef CAS PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Y. Bai, Y. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- L. Feng, K. Y. Wang, G. S. Day and H. C. Zhou, Chem. Soc. Rev., 2019, 48, 4823–4853 RSC.
- N. Assaad, G. Sabeh and M. Hmadeh, ACS Appl. Nano Mater., 2020, 3, 8997–9008 CrossRef CAS.
- W. Xiang, J. Ren, S. Chen, C. Shen, Y. Chen, M. Zhang and C.-J. Liu, Appl. Energy, 2020, 277, 115560 CrossRef CAS.
- K. Tan, H. Pandey, H. Wang, E. Velasco, K. Y. Wang, H. C. Zhou, J. Li and T. Thonhauser, J. Am. Chem. Soc., 2021, 143, 6328–6332 CrossRef CAS PubMed.
- G. Jajko, J. J. Gutiérrez-Sevillano, A. Sławek, M. Szufla, P. Kozyra, D. Matoga, W. Makowski and S. Calero, Microporous Mesoporous Mater., 2022, 330, 111555 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- K. Wang, C. Li, Y. Liang, T. Han, H. Huang, Q. Yang, D. Liu and C. Zhong, Chem. Eng. J., 2016, 289, 486–493 CrossRef CAS.
- R. C. Klet, Y. Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- V. V. Butova, A. P. Budnyk, A. A. Guda, K. A. Lomachenko, A. L. Bugaev, A. V. Soldatov, S. M. Chavan, S. Øien-Ødegaard, U. Olsbye, K. P. Lillerud, C. Atzori, S. Bordiga and C. Lamberti, Cryst. Growth Des., 2017, 17, 5422–5431 CrossRef CAS.
- M. X. Li, T. Chen, J. J. Gooding and J. Q. Liu, ACS Sens., 2019, 4, 1732–1748 CrossRef CAS PubMed.
- X. X. Jia, R. X. Yao, F. Q. Zhang and X. M. Zhang, Inorg. Chem., 2017, 56, 2690–2696 CrossRef CAS PubMed.
- L. Chen, Z. Cheng, X. Peng, G. Qiu and L. Wang, Anal. Methods, 2022, 14, 44–51 RSC.
- X. Liu, Y. Han, Y. Shu, J. Wang and H. Qiu, J. Hazard. Mater., 2022, 425, 127987 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional PXRD patterns, TG curves, SEM images, and PL spectra. See DOI: https://doi.org/10.1039/d2tc04513c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |