
Disentangling the complex photodynamics of mixed-linker Zr-MOFs – efficient energy and charge transfer processes†
Mario
Gutiérrez‡
 a,
Maria Rosaria
Di Nunzio‡
a,
Elena
Caballero-Mancebo
a,
Félix
Sánchez
b,
Boiko
Cohen
*a and
Abderrazzak
Douhal
*a
a,
Maria Rosaria
Di Nunzio‡
a,
Elena
Caballero-Mancebo
a,
Félix
Sánchez
b,
Boiko
Cohen
*a and
Abderrazzak
Douhal
*a
aDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, INAMOL, Universidad de Castilla-La Mancha, Avenida Carlos III, S. N., 45071, Toledo, Spain. E-mail: boyko.koen@uclm.es; abderrazzak.douhal@uclm.es
bInstituto de Química Orgánica, CSIC, Juan de la Cierva, 3, 28006, Madrid, Spain
First published on 28th November 2022
Abstract
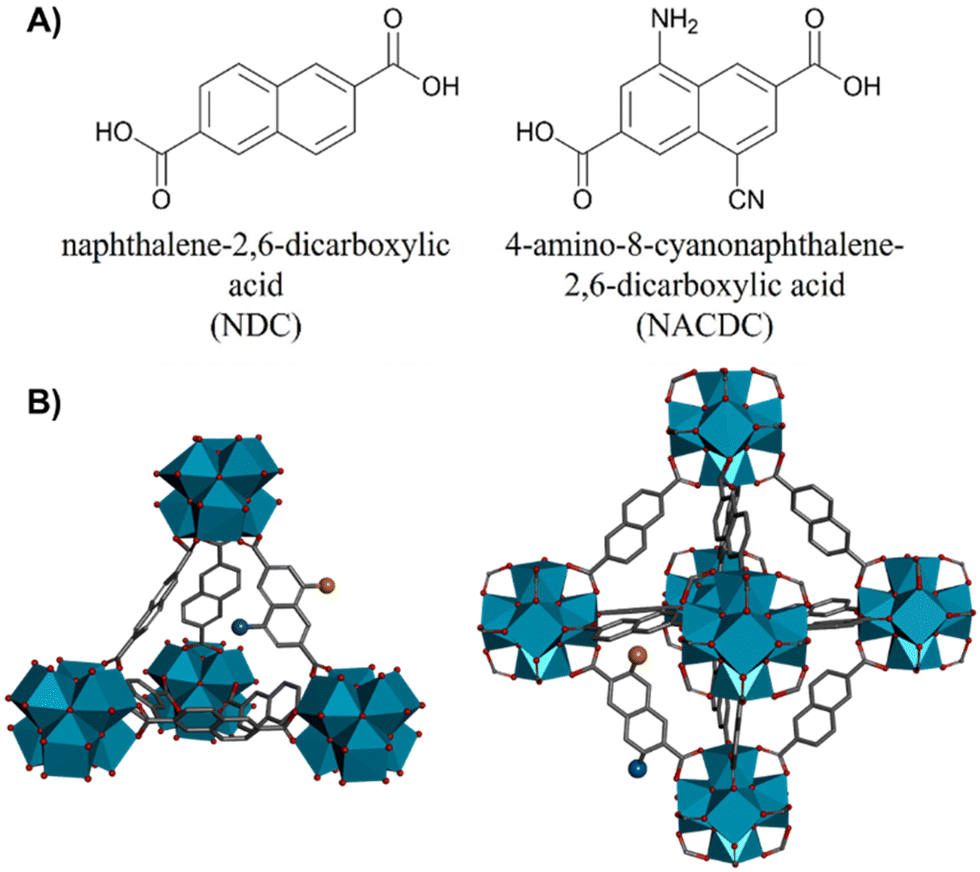
Controlling the composition, as well as the number and type of defects in metal–organic frameworks (MOFs) helps in the understanding of the interactions that govern their behaviours, and in consequence, their potential employment in a number of photonic applications. In this work, we report on the spectroscopy and photodynamics of two heterolinker (naphthalene-2,6-dicarboxylic acid, NDC, and 4-amino-8-cyanonaphthalene-2,6-dicarboxylic acid, NACDC) Zr-based MOFs that differ in the modulator used during the synthesis, thus leading to possible defects in the frameworks. We observed an inter-particle energy transfer (ET) that occurs from <10 to 160 ps in competition with an ultrafast intra-particle ET event from excited NDC to NACDC. In addition, due to the heterogeneity of the system, we found two different populations of the push–pull NACDC linker emitting from the charge transfer (CT) state. Single crystal fluorescence microscopy experiments further demostrated that the photobehavior of the studied MOFs is dominated by NACDC. Nanosecond (ns)-flash photolysis also revealed different photobehaviours related to the presence of defects, while the femtosecond (fs)-dynamics is independent of the modulators used for the MOFs’ synthesis. These results may help in engineering of MOFs for their application in photon-based science and technology and may lead to the construction of optoelectronic devices and better photocatalysts based on these materials.
Introduction
Metal–organic frameworks (MOFs) are porous crystalline hybrid materials with an outstanding relevance in material science. Owing to their advanced properties like high specific surface areas, tunable cavities/pore apertures, and large accessibility to many molecular guests, MOFs have been applied in several technological fields such as photonics,1,2 chemical sensors,3–5 optoelectronic devices,6–8 gas storage,9,10 catalysis,11,12 drug delivery,13–15 and, more recently, as solid-state flexible supercapacitors.16,17Luminescent and photoactive MOFs have attracted great interest for their unique photophysical properties making these materials excellent candidates in lighting, sensing, drug photodelivery/imaging, light-harvesting/energy transfer (ET) and photocatalysis applications.18–32 Their photophysical properties can be manipulated and tuned by a proper selection of photoactive organic linkers or metal clusters that may confer the MOF material with the desired spectroscopic and photodynamical properties or may induce additional photophysical pathways involving linker/linker or linker/metal interactions such as excimer/exciplex formation, ET, antennae effects and ligand-to-metal (or metal-to-ligand) charge transfer (LMCT or MLCT, respectively).33–36 Additionally, the MOFs’ structure might contain randomly generated defects (generally missing linkers or missing clusters) or they can be synthetically engineered.37–39 The defects might have a significant impact on the spectroscopic and photophysical properties of MOFs, in particular for their implementation in optoelectronic devices (light emitting diodes, LEDS) and catalysis or photocatalytic technologies.39–43 Recently, we reported on the photodynamical behaviour of a Zr-based MOF with naphthalene linkers (NDC, naphthalene-2,6-dicarboxylic acid), the Zr-NDC.36 The material showed rich photodynamics, revealing a great variety of photoreactions, such as ligand-to-cluster charge transfer (LCCT), excimers formation or intramolecular charge transfer (ICT) reactions.36 The understanding of these photoproperties was essential for developing the first LED in which a Zr-based MOF acted as the electroluminescent layer.40 In a subsequent work, we demonstrated how the addition of a secondary ligand, with an amine group added to the naphthalene core (NADC, 4-amino-naphthalene-2,6-dicarboxylic acid) enriches the photobehavior of the MOF, leading to a competition between the excimer formation and an ET reaction between the two ligands.44 Moreover, we observed that the LCCT process and the subsequent formation of long-lived charge separated states – both mechanisms being essential for photocatalysis applications45,46 – were retained in the mixed-linker MOF. In addition, we demonstrated that the functionalization of the linker and the partial doping of the MOF with this linker drives its absorption close to the visible region of the electromagnetic radiation, which is highly desired for photocatalytic applications.44
Herein, we present the synthesis, and the steady-state and time-resolved photophysical characterizations of two Zr-based MOFs composed by a mixture of NDC and a push–pull organic linker, (NACDC, 4-amino-8-cyanonaphthalene-2,6-dicarboxylic acid) with a ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight. The use of the push–pull organic linker is expected to steer even more the absorption to the visible region and to trigger the LCCT mechanism through an ICT within NACDC. The synthesis of both MOFs was performed under the same experimental conditions but using different modulators (benzoic acid and acetic acid) aiming to get knowledge on the possible presence of defects and their impact on the MOFs photoproperties. In dichloromethane (DCM) suspensions, we observed a small spectral shift in the emission spectra of the investigated materials explained in terms of presence of defects due to the use of different modulators. Pico- to microsecond (ps–μs) time-resolved spectroscopy and single crystal fluorescence microscopy experiments revealed multi-exponential photobehaviors due to the presence of different conformations and environments of the ligands and the presence of defects. An inter-particle ET occurs at the ps-time scale, together with an efficient ultrafast intra-particle ET process from the excited NDC monomers to the NACDC chromophores. For both materials, the global emission lifetimes of inter-particle ET process are 20–150 ps, depending on the excitation wavelength and the concentration of the MOFs. In addition, two different populations of NACDC were detected, increasing the heterogeneity of the system. Femtosecond transient absorption (fs-TA) spectroscopy was also employed in order study the non-radiative ultrafast processes at short time scale. The comparable fs-TA dynamics for both MOFs indicates that the ultrafast processes are independent of the modulators used in the synthesis. An ultrafast component of 110 fs was assigned to the LCCT process. The electron–hole (e−–h+) recombination occurs from two different trap states, in the case of ZrMOF-630, or just one trap state, in the case of ZrMOF-640. The different behaviour at the ns-time scale is ascribed to the different nature/number of defects present in the studied MOFs. Single crystal fluorescence microscopy revealed that in the ensemble solids, the inter-particle ET plays a significant role in the de-activation of the excited NDC linker. These results provide key knowledge on the photodynamics of these kinds of mixed-linker MOFs, confirming their potential to be implemented as photocatalysts.
:1 by weight. The use of the push–pull organic linker is expected to steer even more the absorption to the visible region and to trigger the LCCT mechanism through an ICT within NACDC. The synthesis of both MOFs was performed under the same experimental conditions but using different modulators (benzoic acid and acetic acid) aiming to get knowledge on the possible presence of defects and their impact on the MOFs photoproperties. In dichloromethane (DCM) suspensions, we observed a small spectral shift in the emission spectra of the investigated materials explained in terms of presence of defects due to the use of different modulators. Pico- to microsecond (ps–μs) time-resolved spectroscopy and single crystal fluorescence microscopy experiments revealed multi-exponential photobehaviors due to the presence of different conformations and environments of the ligands and the presence of defects. An inter-particle ET occurs at the ps-time scale, together with an efficient ultrafast intra-particle ET process from the excited NDC monomers to the NACDC chromophores. For both materials, the global emission lifetimes of inter-particle ET process are 20–150 ps, depending on the excitation wavelength and the concentration of the MOFs. In addition, two different populations of NACDC were detected, increasing the heterogeneity of the system. Femtosecond transient absorption (fs-TA) spectroscopy was also employed in order study the non-radiative ultrafast processes at short time scale. The comparable fs-TA dynamics for both MOFs indicates that the ultrafast processes are independent of the modulators used in the synthesis. An ultrafast component of 110 fs was assigned to the LCCT process. The electron–hole (e−–h+) recombination occurs from two different trap states, in the case of ZrMOF-630, or just one trap state, in the case of ZrMOF-640. The different behaviour at the ns-time scale is ascribed to the different nature/number of defects present in the studied MOFs. Single crystal fluorescence microscopy revealed that in the ensemble solids, the inter-particle ET plays a significant role in the de-activation of the excited NDC linker. These results provide key knowledge on the photodynamics of these kinds of mixed-linker MOFs, confirming their potential to be implemented as photocatalysts.
Experimental section
Synthesis of MOF-630 and MOF-640
General MOFs preparation: ZrCl4 (0.4 g, 1.7 mmol) was dispersed in dimethylformamide (DMF) (75 mL) by ultrasonication at 50–60 °C. A DMF solution (25 mL) of naphthalene-2,6-dicarboxylic acid (NDC, 323 mg, 1.5 mmol) and 4-amino-8-cyanonaphthalene-2,6-dicarboxylic acid47 (NACDC, 43 mg, 0.17 mmol) was added to previous prepared ZrCl4 solution and, then modulator (acetic acid: 5.2 mL, 92 mmol, for ZrMOF-630, or benzoic acid: 6.3 g, 52 mmol, for ZrMOF-640) and water (0.120 mL, 0.007 mmol) was added to the mixture. The tightly capped flask was sonicated at 60 °C and kept in a bath at 120 °C under static conditions for 24–48 h. Then, the solution was slowly cooled to room temperature and the precipitate was isolated by centrifugation. The solid was washed with DMF (10 mL). After standing at room temperature for 2 h, the suspension was centrifuged, and the solvent was decanted off. The obtained powders were washed several times with tetrahydrofuran (THF) in order to completely eliminate any rest of the modulator or other impurities. Finally, the solid was dried under reduced pressure (80 °C, 3 h). Powder X-ray diffraction was used to confirm the crystalline structure of both MOFs.– ZrMOF-630: Zr-based MOF with 90% of NDC and 10% of NACDC ligands was prepared with acetic acid (Scheme 1).
 | ||
| Scheme 1 (A) Molecular structures of the organic linkers that composed the ZrMOF-630 and ZrMOF-640. (B) Schematic representation of the Zr-MOF-630 and ZrMOF-640, where it is shown the typical tetrahedral (left) and octahedral (right) pores of UiO-type isostructural MOFs. Zr clusters = blue polyhedron, C = gray sticks, O = red balls, –CN and –NH2 groups of the NACDC = blue and orange balls respectively. | ||
– ZrMOF-640: Zr-based MOF with 90% of NDC and 10% of NACDC ligands was prepared with benzoic acid (Scheme 1).
Methods and materials characterization
The vibrational modes of the MOF samples have been characterized by Fourier Transform Infrared Transmission spectroscopy (FTIR) using a PerkinElmer Spectrum 100 equipped with a Universal ATR (attenuated total reflection). This instrument allows the collection of FTIR spectrum in a range between 370 and 7800 cm−1, with a spectral resolution of 0.5 cm−1. The scanning electron microscopy (SEM) images of ZrMOFs 630 and 640 were collected using a Zeiss GeminiSEM 500 microscope operating in high vacuum mode at an accelerating voltage of 2 kV. The samples have been placed on a carbon adhesive tab mounted on an aluminium holder and coated with a thin gold layer. The steady-state UV-visible absorption and fluorescence spectral measurements were carried out using JASCO V-670 and FluoroMax-4 (Jobin-Yvone) spectrophotometers, respectively. The diffuse reflectance spectra were measured using a 60 mm integrating sphere (ISN-723) and converted using the Kubelka–Munk function. The ps-time-resolved emission experiments have been recorded employing a time-correlated single-photon counting (TSCPC) system. The samples were excited by 40 ps-pulsed (∼1 mW, 40 MHz repetition rate) diode-lasers (PicoQuant) centred at 371 nm and 433 nm. The instrumental response function (IRF) was ∼70 ps. The decays were deconvoluted and fitted to multi-exponential global functions using the FLUOFIT package (PicoQuant). The quality of the fits as well as the number of exponentials were carefully selected based on the reduced χ2 values (which were always below <1.2) and the distributions of the residuals. The fs-TA experiments were done using a chirped pulse amplification (CPA) setup that consists of a Ti:Sapphire oscillator (TISSA 50, CDP Systems) pumped by a 5 W diode laser (Verdi 5, Coherent). The seed pulse (30 fs, 450 mW at 86 MHz) centred at 800 nm is directed to a CPA system (Legend-USP, Coherent). The amplified fundamental beam (50 fs, ∼3.1 W at 1 kHz) is then split by a beam splitter and the main portion (2.7 W) is directed through an optical parametric amplifier (OPA) for wavelength conversion (TOPAS, Light Conversion). The OPA output at 360 nm or 420 nm was used as the pump. The pump pulse intensity was kept below 500 μW. A small portion of the remaining fundamental beam (∼200 μW) goes through a delay line (7.8 fs step and 2 ns of maximum delay) and is focused on a 3 mm thick sapphire crystal for white light continuum (WLC) generation. The produced WLC is split into two parts to form probe and reference beams, which are directed to the sample, where the probe and the pump beams are overlapped. The polarization of the pump is set to the magic angle in respect to the probe. The transmitted light is focused to light guides, directed to a spectrograph, and collected by a pair of photodiode arrays (1024 elements, for spectral measurements). The TA measurements were performed in the spectral ranges of 560–1050 (vis-NIR region). To avoid photo degradation and re-excitation by consecutive pulses, the samples were placed in a 0.8 mm thick rotating quartz cell. The IRF was measured in terms of change in the optical density (ΔOD) for DCM following excitation at 360 and 420 nm to give ∼100 fs. Data were analysed using a multi-exponential fit. The quality of the fit was evaluated by examining the residual distributions. The setup used for the ns-flash photolysis experiments has been described elsewhere.48 In summary, it comprises a LKS.60 laser flash photolysis spectrometer (Applied Photophysics) and a Vibrant (HE) 355 II laser (Opotek). The excitation source was the third harmonic (355 nm) output whose original pump fluence, ∼80 mJ cm−2, was reduced to ∼2 mJ cm−2 before reaching the sample cuvette. The probing source was the output of a 150 W Xenon arc lamp. The light coming from the sample was later dispersed through a monochromator and detected by a photomultiplier (Applied Photophysics R928), combined with a digital oscilloscope (Agilent Infiniium DS08064A, 600 MHz, 4 GSa s1). The IRF of the system was ∼8 ns. All the experiments were carried out at 293 K.Results and discussion
Chemical, structural and morphological characterization
Based on the molecular formula of the pristine Zr-NDC MOF, Zr6O4(OH)4(NDC)6·6H2O,49 we calculated the theoretical mass% of the ZrMOF-630 and ZrMOF-640 with formula: Zr6O4(OH)4(NDC-NACDC)6·6H2O (considering a linker ratio NDC/NACDC as 9/1), which gives: Zr 26%, O 29%, C 41%, H 3%, and N 1%. The obtained experimental values for ZrMOF-630 were: C 34%, H 3%, and N 1%; while for ZrMOF-640 the results were: C 39%, H 3%, and N 0.6%. Two important facts can be extracted from those values: (i) the accurate ratio between the calculated and experimental content of N indicates that the linker proportion in the MOFs is 9:1 (NDC:NACDC) as expected; and (ii) the lower content of C found for both MOFs suggests the presence of linker defects that are more pronounced for ZrMOF-630. This can be explained attending to the pKa of the used modulators that are 4.8 for the acetic acid and 4.2 for the benzoic acid. It was demonstrated that acid modulators with a higher pKa generate more missing linkers in the structure.50 Hence, this agrees with our observations, where the lower C content in the ZrMOF-630 is caused by the presence of more missing linkers in the structure because of using acetic acid as modulator.
Fig. S1A (ESI†) shows the FTIR spectra of ZrMOF-630 and ZrMOF-640, whose band positions are similar to those observed for the Zr-NDC system.51 Moreover, both MOFs displayed characteristic powder X-ray diffraction (PXRD) profiles of unfunctionalized Zr-NDC MOF (Fig. S1B, ESI†), indicating that the functionalization with the secondary linker does not affect the long-range periodicity of the materials. However, the PXRD of ZrMOF-640 shows an additional small peak at ∼6.2° that could be attributed to the (110) reflection of the reo topology of Zr-MOF-640. This reo phase has been previously observed for UiO-66 MOF (isostructural to the MOFs reported here) synthesized using acid modulators.52,53 In this phase, the clusters are eight-connected with the linkers, instead of the typical twelve connections, leading to the so-called “missing cluster” defects. This is an additional indication of the difference in the number and type of defects presented in these two MOFs. Fig. 1 and Fig. S2 (ESI†) display the SEM images of the synthesized ZrMOF-630 and ZrMOF-640 materials. From these micrographs, we can conclude that the morphology of these materials has the typical octahedral shape of isostructural UiO-type MOFs, and that there are no remarkable differences either in the morphology or size of both MOFs.
 | ||
| Fig. 1 Scanning electron microscopy (SEM) images of (A and B) ZrMOF-630 and (C and D) ZrMOF-640. | ||
Steady-state of ZrMOF-630 and ZrMOF-640
To begin with, the steady-state UV-visible absorption and emission spectra of ZrMOF-630 and ZrMOF-640 in DCM suspensions are shown in Fig. 2A. Both materials present a vibrationally-resolved band between 350 and 375 nm, which corresponds to the absorption of π-conjugated naphthalene ligands (NDC).36 In addition to that, a broader band spanning from ∼400–475 nm is recorded. This band is attributed to the absorption of the NACDC linkers. MOFs formed by a mixture of NDC and its amino derivative, NADC linkers, have shown similar band located at 390 nm arising from the NADC linker.44 The composition of ZrMOF-630 and ZrMOF-640 with a push–pull organic linker (NACDC) with an electron donor (–NH2) and acceptor (–CN) pair in its molecular unit, causes a red-shift in the absorption spectra compared to the Zr-NADC MOF (having a monofunctionalized linker with only an –NH2 group) due to stronger charge transfer (CT) character of the former.47 | ||
| Fig. 2 (A) Normalized (to the maximum of intensity) UV-visible absorption and emission (λexc = 355 nm) spectra of ZrMOF-630 (red solid lines) and ZrMOF-640 (blue dashed lines). (B) Normalized (to the maximum of intensity) magic-angle ps-emission decays of (1) ZrMOF-630/DCM (red circles) and (2) ZrMOF-640/DCM (blue triangles) upon excitation at 371 nm and observation at 425 nm and 650 nm. The solid lines are from the best fit of the experimental data. (C) Comparison of the normalized (to the maximum of intensity) magic-angle ps-emission decays of ZrMOF-630 in DCM suspensions upon excitation at (1) 371 nm (orange dots) and (2) 433 nm (blue triangles) and observation at 475 nm and 650 nm. The solid lines are from the best fit of the experimental data. (D) Normalized (to the maximum of intensity) ps-time-resolved emission spectra (TRES) of ZrMOF-630 in DCM suspensions upon excitation at 371 nm at different delays as indicated in the inset. The dashed spectrum corresponds to the steady-state emission upon excitation at 355 nm. The symbol (*) indicates that in this spectral region and at the shortest time delays the total signal is a sum of sample emission and Raman scattering from DCM, having the latter its maximum at ∼430 nm. | ||
The emission spectrum exciting at 355 nm consists of a broad band with no-vibrational resolution and with maximum intensity located at 530 and 540 nm for ZrMOF-630 and ZrMOF-640, respectively. Since both MOFs have the same chemical composition, morphology, and crystalline structure, it is plausible to attribute this difference in the band position to the presence of different number of defects created in the framework because of the used modulator in the synthesis, as confirmed by elemental analysis (see above). Exciting at 400 nm, which corresponds to the absorption of the NACDC linker, results in emission spectra centred at 544 nm comparable to that exciting at 355 nm (Fig. S3, ESI†). Most notably, we did not see the characteristic emission arising from the excited NDC linkers. For Zr-NDC and Zr-NADC MOFs, highly vibrational-resolved emission spectra located at ∼400 nm were reported and assigned to the emission of the NDC monomer (with contribution of NDC excimers as well),36,44 and only when the proportion of the NADC linkers was 35%, the emission of NDC became almost negligible, because of an ET from the NDC to the NADC linkers.44 Hence, we suggest that in the present materials, the emission band at 530–540 nm is due to the emission of the NACDC ligands that might be populated via efficient ET (when exciting the NDC linkers at higher energies, 355 nm) or by direct excitation. We have previously demonstrated that the emission of NACDC linker arises from a CT state, due to an ultrafast ICT.47 Moreover, it should be noted that upon irradiation at 371 nm, the NACDC CT state can be populated either following direct excitation, since the linker presents a broad absorption spectrum between 300 and 500 nm,47 or through an efficient ET photoprocess from the photoexcited NDC linker to the NACDC one. It is also worth to note that the maximum of emission for both ZrMOFs in DCM suspension is significantly red-shifted (∼530–540 nm) in comparison to the emission spectrum of the pristine NACDC linker in DCM (506 nm).47 On the other hand, it is similar to the one reported for NACDC in more polar solvents, such as acetonitrile (ACN) (542 nm) and methanol (MeOH) (559 nm),47 which could be explained by two facts: (i) in the studied materials, the linker might experience local environments with polarity similar to ACN and MeOH, especially attending the Zr-oxo-hydroxo composition of the secondary building units (SBUs) and; (ii) the NACDC might undergo an efficient LCCT interaction with the metal clusters, which produces a red-shift in the emission spectrum as previously demonstrated.34,36 In order to shed light on the working mechanism, we have explored the possible correlation between the MOFs’ spectroscopic properties and the solvent polarity, by recording the emission spectra of ZrMOF-630 in diethyl ether (DE) and MeOH suspensions. Neither the absorption nor the emission spectra or QY values showed appreciable changes with the used solvent (Table S1 and Fig. S4A, ESI†). Even though this observation does not completely exclude the polarity environment as the cause of the emission red shift of the MOFs compared to the linkers, it suggests that the most probable cause is a strong LCCT phenomenon.
The excitation spectra of both MOFs (Fig. S5, ESI†) provide essential clues about the spectroscopic properties of these materials. On one hand, there is an increase in the intensity of the NACDC-band (maximum at ∼440 nm) at observation wavelengths above 490 nm, confirming that the emission mainly arises from the NACDC linkers. On the other hand, and very importantly, when the excitation spectrum was collected at 570 nm, we still observed the vibrationally-resolved band at 360 nm corresponding to the NDC linkers, which do not emit above 500 nm.36 This observation is an unequivocal evidence of an efficient ET from the photoexcited NDC to NACDC and will be discussed in more details within the time-resolved emission experiments section (vide infra).
The emission quantum yields (QYs) exciting at 355 nm in DCM were very similar for the two MOFs, being 0.04 ± 0.01 and 0.05 ± 0.01 for ZrMOF-630 and ZrMOF-640, respectively (Table S1, ESI†). These values were reduced to 0.02 ± 0.01 and 0.03 ± 0.01 when the excitation was changed to 430 nm. The wavelength-dependence of the QYs is explained in terms of a more efficient LCCT interaction with the Zr clusters when the excited linker is NACDC (430 nm) instead of NDC (355 nm) one. Similar results were reported for the Zr-NDC and Zr-NADC MOFs containing different amounts (from 2 to 35%) of the NADC linker with respect to the NDC one in DCM suspensions.44 In particular, the QY for the pristine Zr-NDC MOF (100% NDC linker) was 0.30 following excitation at 355 nm and it gradually decreased to 0.012 upon increasing the NADC content (similar trend was observed for the 400 nm excitation) due to more efficient LCCT reactions.44
In the solid state, the reflectance (converted to Kubelka–Munk, K–M) spectra of the studied ZrMOFs show a vibrationally-structured band with an intensity maximum at 355 nm and a second broad one ranging from ∼400 to 500 nm corresponding to the absorption of NDC and NACDC linkers, respectively (Fig. S6, ESI†). The emission spectra of ZrMOF-630 are centred at 555 nm and do not depend on the excitation wavelength, while for ZrMOF-640 we observed a 5 nm red-shift (from 550 to 555 nm) going from λexc = 355 nm to λexc = 390/433 nm (Fig. S6, ESI†). Compared with the emission spectra recorded in DCM suspensions, those in the solid-state are red-shifted by 25 and ∼15 nm for ZrMOF-630 and ZrMOF-640, respectively, suggesting a stronger stabilization of the NACDC linkers in the powder samples. The excitation spectra recorded at 550 nm confirm that, even in the solid-state, the emission is arising predominantly from the NACDC linkers (Fig. S6, ESI†). Moreover, the excitation spectra for both materials also show behaviour that is similar to the one observed in DCM suspensions indicating that ET process is still operational in the solid state.
Picosecond time-resolved fluorescence study of ZrMOF-630 and ZrMOF-640
Aiming to understand the photodynamics of the two MOFs, we carried out time-resolved emission experiments with ps-resolution upon excitation at three discrete wavelengths: 340 nm (almost selective light absorption by the NDC linkers, Fig. S7A, ESI†), 371 nm (light absorption by both NDC and NACDC linkers, Fig. S7B, ESI†), and 433 nm (selective light absorption by the NACDC linkers, Fig. S7C, ESI†). Fig. 2B exhibits representative emission decays of the materials in DCM suspensions upon excitation at 371 nm and observing at the bluest (425 nm) and reddest (650 nm) wavelengths (additional decays are presented in Fig. S7B, ESI†), while Table 1 gives the fitting parameters obtained from the best analysis of the signals. The decays were satisfactorily fit by a four-exponential function, which reflects the complex photobehaviour of these materials in their electronically excited states and indicates the presence of several deactivation pathways. We could not obtain an accurate fit using a model with three exponential functions. Upon excitation at 371 nm, the fit of the emission decays of ZrMOF-630 in DCM gives components of: τ1 = 150 ps, τ2 = 780 ps, τ3 = 2.7 ns, and τ4 = 7.5 ns (Table 1). The shortest one decays at the blue emission region and rises (negative amplitude) when the observation wavelength is 550 nm or longer. Nevertheless, the other three components decay along the whole observation range, showing their maximum contribution at 450 nm (τ2), 490 nm (τ3), and 575–600 nm (τ4). For the previously reported pristine Zr-NDC and Zr-NADC with 10% of the NADC linker (Zr-NADC (10%)), a tri-exponential behaviour was observed with the shortest, sub-ns, component assigned to intra-particle excimer formation between neighbouring naphthalene organic linkers (Zr-NDC) or a combination of excimers and ET from NDC to NADC linkers (Zr-NADC).36,44 On the other hand, the intermediate and longest components were differently assigned for the two MOFs. For Zr-NDC, they correspond to the monomer (4.2 ns) and excimer lifetimes (13.2 ns), respectively, while in the case of Zr-NADC (10%) they were related to emissions of different NADC populations. However, for the systems studied in this work, we observed a completely different photobehaviour, which is much more complex.| Sample | λ obs (nm) | τ 1 ± 20 (ps) | a 1 (%) | c 1 (%) | τ 2 ± 50 (ps) | a 2 (%) | c 2 (%) | τ 3 ± 0.2 (ns) | a 3 (%) | c 3 (%) | τ 4 ± 0.3 (ns) | a 4 (%) | c 4 (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 630/DCM λexc = 340 nm | 405 | 80 | 81 | 37 | 400 | 16 | 36 | 1.6 | 2 | 18 | 6.0 | 1 | 9 |
| 425 | 72 | 27 | 24 | 43 | 3 | 26 | 1 | 4 | |||||
| 450 | 57 | 14 | 34 | 41 | 8 | 36 | 1 | 9 | |||||
| 475 | 30 | 3 | 43 | 21 | 23 | 44 | 4 | 32 | |||||
| 490 | 12 | 1 | 42 | 13 | 36 | 41 | 10 | 45 | |||||
| 510 | 1 | <1 | 34 | 7 | 46 | 36 | 19 | 57 | |||||
| 550 | −100 | −100 | 21 | 3 | 48 | 28 | 31 | 69 | |||||
| 600 | −100 | −100 | 13 | 2 | 49 | 25 | 38 | 73 | |||||
| 630/DCM λexc = 371 nm | 425 | 150 | 82 | 36 | 780 | 16 | 36 | 2.7 | 1 | 8 | 7.5 | 1 | 20 |
| 450 | 73 | 26 | 23 | 42 | 3 | 17 | 1 | 15 | |||||
| 475 | 46 | 8 | 38 | 34 | 14 | 41 | 2 | 17 | |||||
| 490 | 31 | 3 | 42 | 25 | 23 | 48 | 4 | 24 | |||||
| 510 | 13 | 1 | 42 | 16 | 35 | 47 | 10 | 36 | |||||
| 530 | 4 | 1 | 39 | 11 | 42 | 44 | 15 | 44 | |||||
| 550 | −100 | −100 | 36 | 9 | 45 | 42 | 19 | 49 | |||||
| 575 | −100 | −100 | 31 | 7 | 47 | 41 | 22 | 52 | |||||
| 600 | −100 | −100 | 29 | 7 | 49 | 41 | 22 | 52 | |||||
| 625 | −100 | −100 | 29 | 7 | 49 | 42 | 22 | 51 | |||||
| 650 | −100 | −100 | 31 | 8 | 49 | 44 | 20 | 48 | |||||
| 630/DCM λexc = 433 nm | 475 | 140 | 56 | 10 | 800 | 33 | 35 | 2.9 | 10 | 40 | 8.0 | 1 | 15 |
| 490 | 43 | 5 | 37 | 27 | 17 | 46 | 3 | 22 | |||||
| 520 | 33 | 3 | 33 | 15 | 25 | 41 | 9 | 41 | |||||
| 550 | 26 | 2 | 30 | 10 | 29 | 36 | 15 | 52 | |||||
| 600 | 22 | 1 | 27 | 8 | 32 | 34 | 19 | 57 | |||||
| 650 | 21 | 1 | 28 | 8 | 32 | 35 | 19 | 56 | |||||
For further discussion, we focus on the origin of the 150 ps component, which is present as a decay at the blue and as a rise at the red side of the emission spectrum. Although its value is similar to the one found for the excimer formation in Zr-NDC MOF (250–840 ps), its spectral behaviour differs significantly, which suggests that it corresponds to a different process. For the Zr-NDC MOF, this component turns into a rise at 475 nm, coincident with the emission region of NDC excimers, while for the systems under study, this component becomes a rise at 550 nm (Table 1), a wavelength into the red from the emission of the NDC excimers, thus excluding the possibility that this component might be attributed to the excimer formation. Since it is decaying in the blue region and rising in the red part, it indicates the existence of an excited-state process in the MOF. Taking into account this behavior and the large spectral overlap between the emission of the NDC linker and the absorption of the NACDC one (Fig. S8, ESI†), it is clear the occurrence of an ET from the photoexcited NDC linker to the NACDC one, and thus, we attribute the first component (150 ps) to the time constant of this ET process. To better characterize this ET process, we recorded emission decays of suspensions with different ZrMOF-630 concentrations (from 1 mg/5 mL to 0.01 mg/5 mL) in DCM following excitation at 371 nm (Table S2 and Fig. S9A, B, ESI†). While the ns-decay components remained largely unaffected by the change in the concentration, the value of the time constant assigned to the ET event decreases significantly from ∼150 ps in the more concentrated sample to 30 ps for the intermediate one (0.25 mg mL−1), and <10 ps (below system resolution) in the more diluted one (0.01 mg mL−1). The speeding up of the ET process is concomitant with an increase in the relative contribution of the NDC band in the steady-state emission spectra (Fig. S9C and D, ESI†). Thus, the observed behaviour indicates the presence of an inter-particle ET taking place in tens to hundreds of picoseconds in addition to an ultrafast (few picoseconds) intra-particle ET. Additionally, we expect that the LCCT process from the excited NDC linkers that takes place on ultrafast timescales (vide infra) could also compete with the ET events.34
Next, we attribute the second component (780 ps) to the locally excited state of the photoexcited (either directly or through ET) NACDC, while the third (2.7 ns) and fourth (7.5 ns) ones to the emission of different CT populations of NACDC produced following an ultrafast ICT event (Scheme 2). These CT species experience different local environment giving rise to the observed decay components. In a recent study on the excited state dynamics of NACDC linker in solutions, a dependence on the solvent polarity was observed for the emission lifetime of CT species ranging from 8.9 ns in MeOH to 17.6 ns in DE.47 The significant decrease in the lifetime of the CT state for NACDC linker in ZrMOF-630 is explained in terms of efficient LCCT process. Note that we cannot gauge whether the LCCT occurs preferentially from a NACDC population in a specific local environment or that both contribute equally.
 | ||
| Scheme 2 Schematic representation (not in scale) of the photo-processes occurring in ZrMOF-630 and ZrMOF-640 in DCM suspensions following excitation of NDC at 340/371 nm (A and B), and of NACDC at 433 nm (C and D). Note that (A and B) show the processes occurring in the excited NDC linker, while (C and D) show those related to NACDC excited either directly or through inter- or intra-particle ET. LE corresponds to locally excited state, LCCT – ligand to cluster charge transfer, CT-charge transfer, ICT – intramolecular charge transfer. | ||
To further study the origin of the time component observed for ZrMOF-630 in DCM suspensions, the system was also interrogated upon excitation at 340 and 433 nm (Fig. S7A and C, ESI†). At 340 nm excitation wavelength, the photodynamics is still characterized by four-exponential decays with time components: τ1 = 80 ps, τ2 = 400 ps, τ3 = 1.6 ns, and τ4 = 6.0 ns (Table 1). The trend is similar to the one upon excitation at 371 nm, with the 80 ps lifetime decaying in the blue side and rising starting from 550 nm. These results further indicate the presence of an efficient ET from the NDC to NACDC linkers, considering that at 340 nm we are exciting almost exclusively the NDC linkers (Fig. S4B, ESI†). The shortening of τ1 from 150 to 80 ps is due to a faster relaxation after pumping the system with an excess energy at 340 nm. By tuning the excitation wavelength to 433 nm we predominantly excite the NACDC ligands. Fig. 2C shows a comparison of the emission decays for ZrMOF-630 in DCM suspensions upon excitation at 371 and 433 nm and observation at two different emission wavelengths, while Table 1 collects the obtained parameters from the multi-exponential fit. The decays were also fitted by a model of four components giving: τ1 = 140 ps, τ2 = 800 ps, τ3 = 2.9 ns, and τ4 = 8.0 ns. Most notably, here we did not observe a rising component, which further demonstrates that the rising component following excitation of the system at 340 nm and 371 nm has its origin in the NDC linker (ET from NDC to NACDC). Although in the emission transients upon excitation at 433 nm we got a time component (∼140 ps) similar to the one assigned to the inter-particle ET, it behaves as a decay through the whole observation spectral range. This behavior suggests that this component has a different origin from the one obtained following excitation at 340 and 371 nm. In similarity with the assignments for the decays following excitation at 371 nm, we expect to see the contribution of two different NACDC populations having CT character produced as a result of an ultrafast ICT in the directly excited the NACDC linker as a consequence of different local environments with different polarities (Scheme 2). Thus, we assign τ1 and τ2 (maxima contributions at the blue side of the emission spectrum) to the lifetimes of the LE states of the two NACDC species, while τ3 and τ4 (maxima contributions at 490 and 600 nm, in that order) correspond to the emission lifetime of the NACDC species with CT character.
To get more insight in the ps-dynamics and further assess of our assignments, we recoded ps time-resolved emission spectra (TRES) of ZrMOF-630 in DCM suspensions upon excitation at 371 (Fig. 2D) and 433 nm (Fig. S10, ESI†). To begin with 371 nm-excitation TRES (Fig. 2D), the observed red shift from 430 to 530 nm, on increasing the gating time, supports the explanation of the occurrence of the ET process between the NDC and NACDC linkers, along with the emission of NACDC from the LE state (∼500 nm) and the longer-lived emission from the CT state/s. Upon excitation at 433 nm (Fig. S10, ESI†), remarkably no spectral shift is observed in the recorded spectra, further confirming the presence of ET upon excitation of NDC linkers, and not from NACDC ones.
Next, we also studied the photodynamics of the ZrMOF-640 in DCM suspensions upon excitations at 340, 371, and 433 nm (Fig. S11, ESI†). The decays were also fitted using a four-exponential model giving time constants: τ1 = 160 ps, τ2 = 910 ps, τ3 = 3.2 ns, and τ4 = 8.6 ns. The observed components behave as those of ZrMOF-630; thus, we assign τ1 to the NDC-NACDC ET event, τ2 to the emission of the NACDC linker from the LE state, τ3 and τ4 to NACDC emission lifetimes from the CT states after experiencing an ultrafast ICT. The differences in the lifetimes between ZrMOF-630 and ZrMOF-640 are within the experimental and fit errors, which indicates that the modulator used during the synthesis does not affect significantly the photodynamics of the resulting materials at this timescale. This is further supported by the similar behaviour of both materials upon excitation at 340 and 433 nm (Tables 1 and 2).
| Sample | λ obs (nm) | τ 1 ± 20 (ps) | a 1 (%) | c 1 (%) | τ 2 ± 50 (ps) | a 2 (%) | c 2 (%) | τ 3 ± 0.2 (ns) | a 3 (%) | c 3 (%) | τ 4 ± 0.3 (ns) | a 4 (%) | c 4 (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 640/DCM λexc = 340 nm | 405 | 70 | 69 | 4 | 410 | 9 | 3 | 1.7 | 3 | 3 | 6.4 | 19 | 90 |
| 425 | 72 | 6 | 14 | 6 | 3 | 5 | 11 | 83 | |||||
| 450 | 59 | 6 | 28 | 17 | 7 | 18 | 6 | 59 | |||||
| 475 | 23 | 1 | 42 | 15 | 27 | 39 | 8 | 45 | |||||
| 490 | 8 | 1 | 40 | 9 | 38 | 37 | 14 | 53 | |||||
| 510 | −100 | −100 | 30 | 5 | 45 | 30 | 25 | 65 | |||||
| 550 | −100 | −100 | 15 | 2 | 44 | 21 | 41 | 77 | |||||
| 600 | −100 | −100 | 5 | 1 | 43 | 17 | 52 | 82 | |||||
| 640/DCM λexc = 371 nm | 425 | 160 | 60 | 4 | 910 | 6 | 2 | 3.2 | 15 | 22 | 8.6 | 19 | 72 |
| 450 | 61 | 7 | 23 | 16 | 5 | 11 | 11 | 66 | |||||
| 475 | 41 | 5 | 40 | 28 | 15 | 38 | 4 | 29 | |||||
| 490 | 28 | 3 | 43 | 24 | 23 | 44 | 6 | 29 | |||||
| 510 | 15 | 1 | 40 | 15 | 34 | 44 | 11 | 40 | |||||
| 530 | 7 | 1 | 35 | 10 | 40 | 41 | 18 | 48 | |||||
| 550 | −100 | −100 | 32 | 8 | 45 | 39 | 23 | 53 | |||||
| 575 | −100 | −100 | 24 | 5 | 46 | 35 | 30 | 60 | |||||
| 600 | −100 | −100 | 21 | 4 | 48 | 35 | 31 | 61 | |||||
| 625 | −100 | −100 | 21 | 5 | 49 | 36 | 30 | 59 | |||||
| 650 | −100 | −100 | 25 | 6 | 48 | 37 | 27 | 57 | |||||
| 640/DCM λexc = 433 nm | 475 | 220 | 54 | 10 | 1100 | 33 | 32 | 3.4 | 9 | 25 | 8.8 | 4 | 33 |
| 490 | 44 | 7 | 35 | 26 | 16 | 39 | 5 | 28 | |||||
| 520 | 32 | 3 | 32 | 16 | 25 | 39 | 11 | 42 | |||||
| 550 | 25 | 2 | 28 | 11 | 30 | 36 | 17 | 51 | |||||
| 600 | 21 | 1 | 24 | 8 | 33 | 34 | 22 | 57 | |||||
| 650 | 21 | 1 | 26 | 9 | 34 | 36 | 19 | 54 |
Excitation of solid ZrMOF-630 and ZrMOF-640 at 371 and 433 nm gives rise to emission decays that were fitted to four decaying components at all the observation wavelengths. Representative transients are shown in Fig. S12 (ESI†), while the respective fit parameters are reported in Table S3 (ESI†). For the ZrMOF-630 powder irradiated at 371 nm, we obtained: τ1 = 110 ps and τ2 = 600 ps, with maximum contribution at the blue side of the emission spectrum 450–500 nm; τ3 = 2.5 ns and τ4 = 8.6 ns, with maximum contribution at wavelengths > 550 nm (Table S3, ESI†). By exciting the sample at 433 nm, the analysis gives: τ1 = 170 ps, τ2 = 890 ps, and τ3 = 3.0 ns, with c% max at 500 nm; τ4 = 8.7 ns, with c% max at 600 nm (Table S3, ESI†). It is worth to note that we did not observe a rising component following excitation at 371 nm.
We suggest that due to the close packing between the MOF particles, the time constant related to the inter-particle ET becomes comparable to the one associated with the intra-particle ET. Based on these considerations and the similarity of the behaviour of the transients collected in solid and in DCM suspensions, we assign τ1 and τ2, following excitation at both 371 nm and 433 nm, to the LE lifetimes of NACDC. The notable exception is that the value of τ1 following excitation at 371 nm in solid state is most probably affected by the ultrafast inter- and intra-particle ET. Finally, τ3 and τ4 correspond to emission lifetimes of different CT-character NACDC populations either directly excited or coming from the ET event (the latter following excitation at 371 nm), after having experienced an ICT reaction. The study of ZrMOF-640 led to comparable results and interpretation, as it can be seen from Fig. S12 and Table S3 (ESI†).
Scheme 2 resumes the photophysical processes in the investigated compounds in DCM suspensions following excitation at 340 nm, 371 nm and 433 nm.
Single crystal fluorescence microscopy
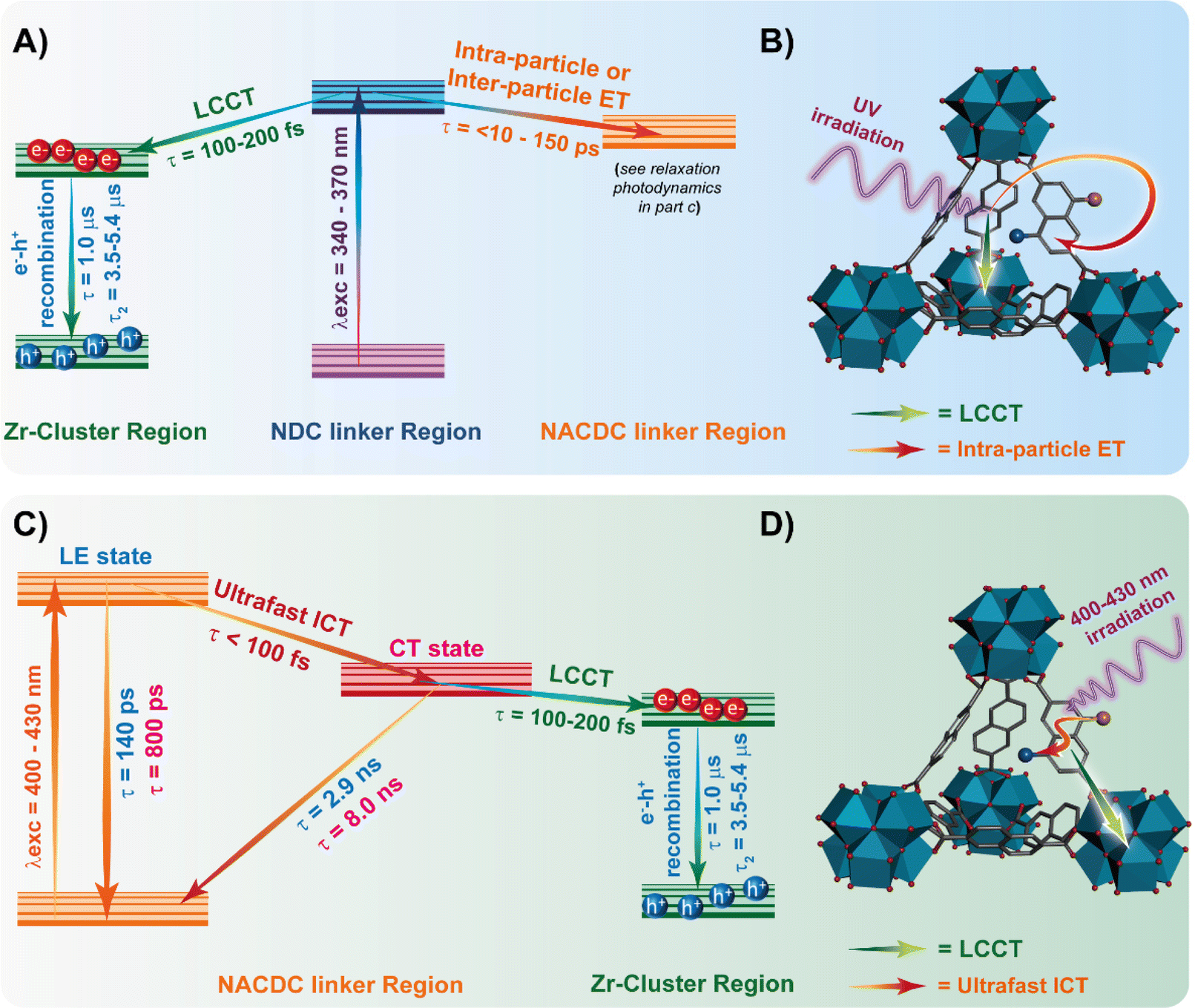
To disentangle the photoinduced processes, we also studied the intra-particle interactions in terms of single-crystal emission. To this end, we dispersed and spin-coated dilute suspensions of ZrMOF-630 and ZrMOF-640 on a quartz coverslip and performed fluorescence lifetime imaging microscopy (FLIM), and collected the emission spectra of the isolated crystals exciting at 390 nm. To begin with, Fig. 3A and B show the average emission spectra of the corresponding ZrMOFs after averaging between 30 and 40 spectra collected for isolated single crystals (Fig. S13, ESI† shows selected representative individual spectra of both MOFs). The average spectra consist of two bands centred on 464 nm and on 554 nm, with the latter being the dominant one. The one at 554 nm coincides with the one observed for the steady-state ensemble average solid-state spectra. Thus, we assign it to the contribution of the NACDC linkers in the MOF single crystals. On the other hand, the band at 464 nm, which is clearly detected in the spectra of the isolated crystals, is comparable with the one observed for very diluted ZrMOF-630 in DCM suspensions and is not present in the emission spectra of the solid-state samples. Hence, we assign this band to the emission of the excited NDC linkers (note that due to the cut-off filter (HQ420LP) we cannot collect the emission below 430 nm). The presence of this band in the emission spectra of the single crystals clearly indicates that the NDC linkers undergo additional quenching when the inter-particle interactions are present in the ensemble solids and in concentrated DCM suspensions due to efficient inter-particle ET as evidenced by the ensemble steady state and time-resolved measurements.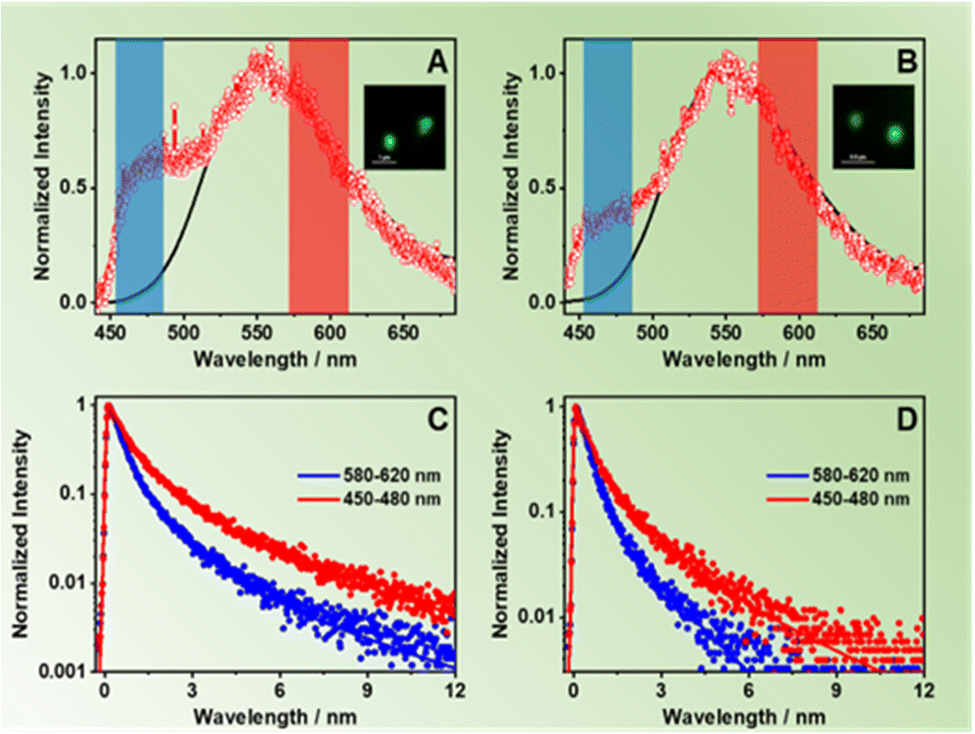 | ||
| Fig. 3 Average (of 50) emission spectrum (open red circles) of single particle of (A) ZrMOF-630 and (B) ZrMOF-640. The excitation wavelength was 390 nm. The solid black line corresponds to the emission spectrum of the ensemble solid. Representative emission decays of single particle of (C) ZrMOF-630 and (D) ZrMOF-640 collected for two discrete spectral regions as indicated by the shaded areas in panels A and B. The excitation wavelength was 390 nm. The solid lines correspond to the best bi-exponential fit. | ||
To further characterize the behaviour of the single ZrMOF crystals, we applied spectral filtering and recorded the emission decays at two discrete spectral regions, corresponding to the emission of the NDC (450–480 nm) and NACDC (580–620 nm) linkers. Between 40 and 50 decays were collected for each sample and analysed separately. Fig. 3C and D show representative decays on a logarithmic scale for both MOFs along with the respective multiexponential fits at the selected observation regions, while Table 3 gives the corresponding fit parameters. When the emission was monitored at the blue emission band, we obtained two-time components of 0.44 ns (90%) and 2.15 ns (10%) for ZrMOF-630; and 0.45 ns (86%) and 1.92 ns (14%) for ZrMOF-640. The emission at the reddest region decays tri-exponentially with values of the obtained time constants of 0.70 ns (61%), 2.78 ns (36%) and 7.31 ns (3%) for ZrMOF-630; and 0.83 ns (60%), 2.85 ns (33%) and 6.78 ns (7%) for the ZrMOF-640. In agreement with the ensemble experiments and previous reports, the two components for the blue- and red-emission decays arise from populations of NDC and NACDC linkers, respectively, having different intra-MOF environments.34,44 We expect that the MOF structure and the presence of defects should allow for different conformations of the linkers and environments, giving rise to the observed multiexponential and wavelength-dependent photobehavior. Since the time constant associated with the intra-particle ET process is below the time resolution of the experimental system (∼50 ps) we could not observe evidence for ET from the excited NDC to NACDC in the single crystals. However, the time components at the blue side of the emission spectra that correspond to the NDC monomer (0.45 and ∼2 ns) are significantly shorter than the previously reported ones for pristine NDC ZrMOF (2.5–4.2 ns). Thus, we explain these results as a result of the presence of efficient intra-particle ET from NDC to NACDC within the ZrMOF structure, which decreases the value of the fluorescence lifetime. On the other hand, the fit of the decays collected at the red side of the emission spectra (NACDC emission) shows comparable behaviour to the one observed for the ensemble average time-resolved measurements. Thus, we assign the longer components (∼2.8 ns and ∼7.5 ns) to the emission of NACDC (excited directly or through ET) CT state in local environments with different polarity, while the shorter component of ∼0.8 ns arises from the LE state prior to the ICT reaction. Notably, we did not observe the 0.14–0.16 ns component observed in the ensemble measurements, which we explain in terms of the lower resolution (∼0.20 ns) of the experimental set-up.
| System | 450–480 nm | 580–620 nm | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| τ 1 ± 0.05 (ns) | a 1 (%) | τ 2 ± 0.30 (ns) | a 2 (%) | τ 1 ± 0.05 (ns) | a 1 (%) | τ 2 ± 0.30 (ns) | a 2 (%) | τ 3 ± 0.50 (ns) | a 3 (%) | |
| ZrMOF-630 | 0.44 | 90 | 2.15 | 10 | 0.70 | 61 | 2.78 | 36 | 7.31 | 3 |
| ZrMOF-640 | 0.45 | 86 | 1.92 | 14 | 0.83 | 60 | 2.85 | 33 | 6.78 | 7 |
Femtosecond-transient absorption (fs-TA) study of ZrMOF-630 and ZrMOF-640
We also studied the involvement of non-radiative processes by using fs-TA following excitation of the NDC (360 nm) and NACDC (420 nm) linkers. No significant differences were observed in the spectral dynamics with the excitation wavelength. We also did not observe differences in the behaviour between the two MOFs exciting at both wavelengths. The comparable TA dynamics for both MOFs indicates that the ultrafast processes are independent of the used modulators to prepare the two MOFs. To begin with, the TA spectra in the interrogated spectral region (560–1050 nm) present a weak negative broad band at ∼600 nm, associated with the stimulated emission, and a more intense, positive one, centred at ∼900 nm that spans from 700 nm to 1050 nm (Fig. S14, ESI†). In agreement with previous studies on Zr-NDC/NADC MOFs, we assign the positive band to the signal of the charge separated state (CSS).34Fig. 4 and Fig. S15 (ESI†) show the fs-TA decay of ZrMOF-630 and ZrMOF-640, respectively, in DCM upon excitation at (A and B) 360 nm and (C and D) at 420 nm probed in the NIR (920 nm) region. | ||
| Fig. 4 Fs-TA decay at 920 nm in terms of changes in the optical density (ΔmOD) of ZrMOF-630 in DCM following excitation at 360 nm: (A) short and (B) long time windows; and following excitation at 420 nm: (C) short and (D) long time windows. The solid lines are from the best multi-exponential fits, and the IRF is the instrumental response function. Note that the initial decay of the signal, following excitation at 420 nm (C and D), is affected by the ultrafast solvent response at this excitation wavelength. | ||
The transients were fitted using multi-exponential functions with two ultrafast components of 110 ± 40 fs and 2.48 ± 0.25 ps along with a longer one of ∼55 ± 6 ps for ZrMOF-630 at 360 nm excitation, and with two components of 3.17 ± 0.35 and 30 ± 4 ps, following 420 nm excitation (Table 4). We associate the fs-component that appears as a rise following excitation at 360 nm with the formation of the NIR band, which reflects an ultrafast LCCT process, generating CSSs in the MOFs. Similar time constant for the LCCT has been found for the NIR transients of Zr-NADC MOF in DCM.34 Note that due to the significantly lower transient signal following excitation at 420 nm, we observe a considerable contribution from the solvated electron signal in the first 200 fs, which does not allow us to adequately estimate the contribution of this component to the collected signals.
| λ exc (nm) | System | τ 1 (ps) | a 1 (%) | τ 2 (ps) | a 2 (%) | τ 3 (ps) | a 3 (%) | τ 4 (ps) | a 4 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 360 | ZrMOF-630 | 0.11 | (−)100 | 2.48 | 36 | 55 | 39 | >200 | 25 |
| ZrMOF-640 | 0.10 | (−)100 | 2.60 | 30 | 57 | 44 | >200 | 26 | |
| 420 | ZrMOF-630 | — | — | 3.17 | 59 | 30 | 20 | >200 | 21 |
| ZrMOF-640 | — | — | 3.05 | 56 | 32 | 25 | >200 | 19 |
The longer components of the TA signals are present as decays in the whole recorded spectral region. These two components have also been observed in the transients of the Zr-NADC MOF in DCM suspensions.34 Thus, we assign the 2.48 ps (3.17 ps) one to the vibrational relaxation (VR, cooling) of the photoexcited linkers, while the 55 ps (30 ps) component is attributed to a fast non-radiative relaxation of the LCCT state to trap states. A notable difference is evidenced in the value of the longer ps-component, which has a lower value (30 ps) following excitation at 420 nm. Since at this wavelength we predominantly excite the NACDC linker, we suggest that the presence of the electron withdrawing CN group can affect the electron orbital density around both the Zr node and the linker moieties, and thus their electronic coupling, which in turn can speed up the relaxation event to the trap states.
Nanosecond-transient absorption study of ZrMOF-630 and ZrMOF-640
To gather information on the slow relaxation processes of the two ZrMOFs in DCM suspensions, we carried out flash photolysis TA experiments in the ns–ms time regime. Fig. S16A (ESI†) shows TAS of ZrMOF-630 in DCM suspensions upon excitation at 355 nm recorded at different delay times (293 ns, 1 μs, and 3 μs). Fig. 5 displays selected decay traces of ZrMOF-630 when exciting at 355 nm, while the results for ZrMOF-640 is reported in Fig. S16B (ESI†).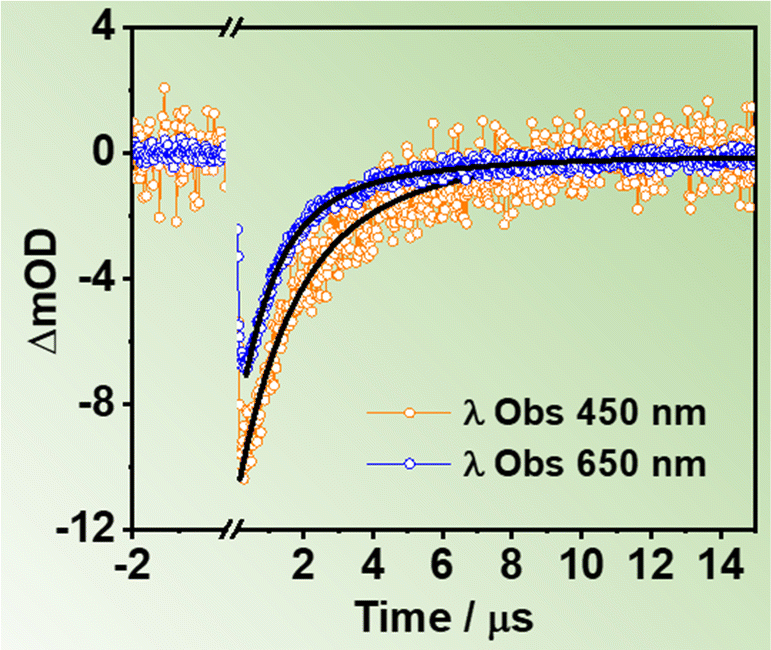 | ||
| Fig. 5 μs-transient decays of ZrMOF-630 in DCM suspensions. The excitation wavelength was 355 nm, and the observation ones are indicated in the inset. The solid lines are from the best multi-exponential fits. | ||
The TAS appear as a continuous negative band in the 400–700 nm region, with intensity maximum at the shortest wavelengths. Similar TAS have been observed earlier for Zr-NDC and Zr-NADC (65% NDC, 35% NADC) materials in DMF suspensions, and they have been ascribed to long-lived CSSs photoproduced after a LCCT process.34,44
No difference was observed for the decays registered under different atmospheric (air, O2, and N2) conditions, thus indicating that no triplet states are involved in the relaxation of the excited samples (Fig. S16C and D, ESI†). For ZrMOF-630 in DCM, the fits of the decays give two components (τ1 = 1.0 μs and τ2 = 3.7 μs) in the blue region, which become slightly longer (τ1 = 1.5 μs and τ2 = 5.4 μs) in the red region. On the other hand, for the ZrMOF-640 material the fits give only one component of 3.2 μs. Previous reports have suggested the existence of different trap states within the metal cluster part of the MOF.54–58 The origin of the observed multi-exponential decays has been associated to electron–hole (e−–h+) recombination from differently located trap states such as surface defects or linker vacancies. In addition, metal-based nanoparticles possessing d-orbitals have electron trap states, which can further contribute to the multi-exponential behavior.54,55,57,58 On the basis of these considerations, we suggest that the observed long-lived components of the investigated MOFs are due to e−–h+ recombination from two different trap states, in the case of ZrMOF-630, or just one trap state, in the case of ZrMOF-640. The different behaviour reflects the different nature/number of defects present in each MOF. It is worth noting that a recent report demonstrated that a more efficient LCCT will produce higher population of CSSs, and as a result, the photocatalysis activity of the MOFs is enhanced.45 Hence, these results indicate that ZrMOF-630 and ZrMOF-640 are potential candidates for photocatalysis applications.
To summarize, Scheme 2 illustrates the main findings and discussion of photoprocesses and deactivation pathways observed in ZrMOF-630 and ZrMOF-640 upon exciting predominantly at NDC (340–370 nm) and NACDC (400–430 nm) linkers.
Comparison of ZrMOF-630 and ZrMOF-640 with Zr-NDC and Zr-NACD MOFs
Finally, we discuss the differences between the spectral and photodynamical properties of ZrMOF-630 and ZrMOF-640 with those of Zr-NDC and Zr-NADC (10 and 35% of NADC linker). As we can see from Table 5, the QY values at 350 nm-excitation are drastically reduced by 96% going from Zr-NDC (QY = 0.3 ± 0.05) to Zr-NADC 35% (QY = 0.012 ± 0.008) due to a more efficient LCCT process enhanced by the amino group in the NADC linkers.44 On the other hand, the QY value of Zr-NADC (10%) is similar to those of ZrMOF-630 and ZrMOF-640, suggesting a comparable efficiency of the LCCT reaction. It occurs within the same temporal scale (∼100 fs) for all the investigated systems, the same as the ICT, which remains ultrafast. However, the most notable changes are observed in the time for the NDC excimer formation. For the pristine Zr-NDC MOF, the excimer formation occurs in 820 ps,36 whereas it decreases to 200 ps for the Zr-NADC (10%) because it is competing against the ET from the NDC linkers to the NADC ones.44 Notably, for the Zr-NADC (35%) no excimer formation was observed since the amount of NDC linkers decreased considerably. However, for the ZrMOF-630 and ZrMOF-640 (with only 10% of linker functionalization), no excimer formation was observed, due to a very efficient intra- and inter-particle ET reactions between the excited NDC linkers and the NACDC ones in times ranging from <10 to 160 ps.| System | QY | LCCT (fs) | ICT (fs) | ET (ps) | Excimer formation (ps) | ||
|---|---|---|---|---|---|---|---|
| Excit. @ 350 or 355 nm | Excit. @ 400 or 430 nm | Intra-particle | Inter-particle | ||||
| a From ref. 36. b From ref. 34. c From ref. 44. | |||||||
| Zr-NDC | 0.30 ± 0.05a | — | <100b | — | — | — | 840a |
| Zr-NADC (10%) | 0.07 ± 0.03 | 0.018 ± 0.008 | <100c | Ultrafastc | — | — | 200c |
| Zr-NADC (35%) | 0.012 ± 0.008c | 0.003 ± 0.002c | <100c | Ultrafastc | 1.2c | — | — |
| ZrMOF-630 | 0.04 ± 0.01 | 0.02 ± 0.01 | 110 | <100 | Ultrafast | <10–150 | — |
| ZrMOF-640 | 0.05 ± 0.01 | 0.03 ± 0.01 | 100 | <100 | Ultrafast | <10–160 | — |
Conclusions
In this work, we report on the effect of the addition of modulators on the photobehavior of two new Zr-based MOFs. The material, formed by Zr clusters and 90% of naphthalene-2,6-dicarboxylic acid (NDC) and 10% of 4-amino-8-cyanonaphthalene-2,6-dicarboxylic acid ligands (NACDC) was synthesized by the addition of acetic acid or benzoic acid, leading to two different MOFs, the ZrMOF-630 and ZrMOF-640, respectively. The two materials reveal efficient intra- and inter-particle ET reaction between the excited NDC linkers and the NACDC ones in <10 to 160 ps. The heterogeneity (in terms of different linkers) of the system generates two different NACDC species with CT character with emission lifetimes that depend on the local environment. Femtosecond spectroscopy shed light on the non-radiative ultrafast processes, which are independent of the modulators used for the MOFs’ synthesis. An ultrafast component of 110 fs is assigned to the LCCT process either from the excited NDC or NACDC linker. Interestingly, the slow e−–h+ recombination occurs mainly radiatively from two different trap states, in the case of ZrMOF-630, or just from a single trap state, in the case of ZrMOF-640. The different behaviours at the μs-time scale are ascribed to the different nature/number of defects present in the studied MOFs. Single crystal fluorescence microscopy results further showed that in the ensemble solids the NDC linkers in the MOFs undergo significant quenching due to the inter-particle ET interactions. We have demonstrated that simple modification in the structure of the secondary linker by introducing an electron-withdrawing CN group can dramatically change the photophysical behaviour of the single linker, Zr-NDC MOF, previously reported.36 These results may help in the design of new MOFs displaying efficient ET, considering defect engineering and improving of their applications in important fields of science and technology like photocatalysis.Author contributions
M. G. and M. R. N. performed the steady-state and time-resolved experiments, analysed the related data and co-wrote the manuscript. E. C. M. carried out the preliminary steady-state and picosecond experiments on MOFs in suspensions. F. S. synthesized and characterized the MOFs and wrote the related section in the paper. B. C. performed the femtosecond and fluorescence microscopy experiments, analysed and discussed the data and co-wrote the manuscript. A. D. planed the research and supervised the experiments, discussed the data, and co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants: PID2020-116519RB-I00, and PID2020-112590GB-C22 funded by MCIN/AEI/10.13039/501100011033 and by the European Union (EU); SBPLY/19/180501/000212 and SBPLY/21/180501/000108 funded by JCCM and by the EU through ‘‘Fondo Europeo de Desarollo Regional’’ (FEDER); and 2020-GRIN-28929 funded by UCLM (FEDER). We thank the Servicio de Instrumentación (IRICA) for the PXRD measurements and the Servicio Interdepartamental de Investigación (SIdI) of the Universidad Autónoma de Madrid (UAM) for the elemental analysis of the studied ZrMOFs.References
- J. Perego, C. X. Bezuidenhout, I. Villa, F. Cova, R. Crapanzano, I. Frank, F. Pagano, N. Kratochwill, E. Auffray, S. Bracco, A. Vedda, C. Dujardin, P. E. Sozzani, F. Meinardi, A. Comotti and A. Monguzzi, Nat. Commun., 2022, 13(1), 3504 CrossRef CAS.
- A. Monguzzi, M. Ballabio, N. Yanai, N. Kimizuka, D. Fazzi, M. Campione and F. Meinardi, Nano Lett., 2018, 18(1), 528–534 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112(2), 1105–1125 CrossRef CAS PubMed.
- J. F. Olorunyomi, S. T. Geh, R. A. Caruso and C. M. Doherty, Mater. Horiz., 2021, 8(9), 2387–2419 RSC.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47(13), 4710–4728 RSC.
- E. A. Dolgopolova and N. B. Shustova, MRS Bull., 2016, 41(11), 890–896 CrossRef CAS.
- H. Liu, Y. Wang, Z. Qin, D. Liu, H. Xu, H. Dong and W. Hu, J. Phys. Chem. Lett., 2021, 12(6), 1612–1630 CrossRef CAS PubMed.
- H.-Q. Yin and X.-B. Yin, Acc. Chem. Res., 2020, 53(2), 485–495 CrossRef CAS PubMed.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1(1), 100006 CrossRef.
- T. Jia, Y. Gu and F. Li, J. Environ. Chem. Eng., 2022, 10(5), 108300 CrossRef CAS.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9(3), 1779–1798 CrossRef CAS.
- K. Hemmer, M. Cokoja and R. A. Fischer, ChemCatChem, 2021, 13(7), 1683–1691 CrossRef CAS.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44(10), 957–968 CrossRef CAS PubMed.
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13(6), 7004–7020 CrossRef CAS PubMed.
- M. R. Saeb, N. Rabiee, M. Mozafari and E. Mostafavi, Materials, 2021, 14(13), 3652 CrossRef CAS PubMed.
- X. Liu, C.-F. Liu, S. Xu, T. Cheng, S. Wang, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2022, 51(8), 3181–3225 RSC.
- Y.-Z. Zhang, T. Cheng, Y. Wang, W.-Y. Lai, H. Pang and W. Huang, Adv. Mater., 2016, 28(26), 5242–5248 CrossRef CAS.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133(40), 15858–15861 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112(2), 1232–1268 CrossRef CAS.
- M. R. di Nunzio, V. Agostoni, B. Cohen, R. Gref and A. Douhal, J. Med. Chem., 2014, 57(2), 411–420 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43(16), 5815–5840 RSC.
- L. V. Meyer, F. Schönfeld and K. Müller-Buschbaum, Chem. Commun., 2014, 50(60), 8093–8108 RSC.
- Q. Gong, Z. Hu, B. J. Deibert, T. J. Emge, S. J. Teat, D. Banerjee, B. Mussman, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2014, 136(48), 16724–16727 CrossRef CAS.
- C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115(19), 11079–11108 CrossRef CAS PubMed.
- Z. Hu, G. Huang, W. P. Lustig, F. Wang, H. Wang, S. J. Teat, D. Banerjee, D. Zhang and J. Li, Chem. Commun., 2015, 51(15), 3045–3048 RSC.
- M. C. So, G. P. Wiederrecht, J. E. Mondloch, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51(17), 3501–3510 RSC.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46(11), 3185–3241 RSC.
- M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118(18), 8889–8935 CrossRef CAS.
- J. Yu, R. Anderson, X. Li, W. Xu, S. Goswami, S. S. Rajasree, K. Maindan, D. A. Gómez-Gualdrón and P. Deria, J. Am. Chem. Soc., 2020, 142(25), 11192–11202 CrossRef CAS PubMed.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32(20), 1905227 CrossRef CAS.
- S. Goswami, J. Yu, S. Patwardhan, P. Deria and J. T. Hupp, ACS Energy Lett., 2021, 6(3), 848–853 CrossRef CAS.
- M. Gutiérrez, Y. Zhang and J.-C. Tan, Chem. Rev., 2022, 122(11), 10438–10483 CrossRef.
- D. E. Williams and N. B. Shustova, Chem. – Eur. J., 2015, 21(44), 15474–15479 CrossRef CAS PubMed.
- M. Gutierrez, B. Cohen, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2016, 18(40), 27761–27774 RSC.
- M. Gutiérrez, F. Sánchez and A. Douhal, J. Mater. Chem. C, 2015, 3(43), 11300–11310 RSC.
- M. Gutiérrez, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2016, 18(7), 5112–5120 RSC.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28(11), 3749–3761 CrossRef CAS.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54(25), 7234–7254 CrossRef CAS PubMed.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30(37), 1704501 CrossRef PubMed.
- M. Gutiérrez, C. Martin, K. Kennes, J. Hofkens, M. Van der Auweraer, F. Sánchez and A. Douhal, Adv. Opt. Mater., 2018, 6(6), 1701060 CrossRef.
- M. Gutiérrez, C. Martín, B. E. Souza, M. Van der Auweraer, J. Hofkens and J.-C. Tan, Appl. Mater. Today, 2020, 21, 100817 CrossRef.
- M. Gutiérrez, C. Martín, M. Van der Auweraer, J. Hofkens and J.-C. Tan, Adv. Opt. Mater., 2020, 8, 2000670 CrossRef.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47(22), 8134–8172 RSC.
- M. Gutiérrez, F. Sánchez and A. Douhal, Chem. – Eur. J., 2016, 22(37), 13072–13082 CrossRef.
- A. Bhattacharyya, M. Gutiérrez, B. Cohen, A. Valverde-González, M. Iglesias and A. Douhal, Mater. Today Energy, 2022, 29, 101125 CrossRef CAS.
- J. G. Santaclara, F. Kapteijn, J. Gascon and M. A. van der Veen, CrystEngComm, 2017, 19(29), 4118–4125 RSC.
- M. Gutiérrez, L. Duplouy-Armani, L. Angiolini, M. Pintado-Sierra, F. Sánchez and A. Douhal, Int. J. Mol. Sci., 2020, 21(12), 4366 CrossRef PubMed.
- M. Ziółek, C. Martín, B. Cohen, H. Garcia and A. Douhal, J. Phys. Chem. C, 2011, 115(47), 23642–23650 CrossRef.
- V. Bon, I. Senkovska, M. S. Weiss and S. Kaskel, CrystEngComm, 2013, 15(45), 9572–9577 RSC.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20(2), 1347–1362 CrossRef CAS.
- L. Valenzuela, G. Amariei, C. I. Ezugwu, M. Faraldos, A. Bahamonde, M. E. G. Mosquera and R. Rosal, Sep. Purif. Technol., 2022, 285, 120351 CrossRef CAS.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5(1), 4176 CrossRef CAS PubMed.
- P. Chammingkwan, G. Y. Shangkum, L. T. T. Mai, P. Mohan, A. Thakur, T. Wada and T. Taniike, RSC Adv., 2020, 10(47), 28180–28185 RSC.
- J. Liang, Z. Deng, X. Jiang, F. Li and Y. Li, Inorg. Chem., 2002, 41(14), 3602–3604 CrossRef CAS.
- Z. Li, R. Wnetrzak, W. Kwapinski and J. J. Leahy, ACS Appl. Mater. Interfaces, 2012, 4(9), 4499–4505 CrossRef CAS PubMed.
- K. G. M. Laurier, E. Fron, P. Atienzar, K. Kennes, H. Garcia, M. Van der Auweraer, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, Phys. Chem. Chem. Phys., 2014, 16(11), 5044–5047 RSC.
- H. Sezen, M. Buchholz, A. Nefedov, C. Natzeck, S. Heissler, C. Di Valentin and C. Wöll, Sci. Rep., 2014, 4(1), 3808 CrossRef PubMed.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137(42), 13440–13443 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04460a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2023 |