
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymorph- and molecular alignment-dependent lasing behaviors of a cyano-substituted thiophene/phenylene co-oligomer†
Tomomi
Jinjyo
*a,
Hitoshi
Mizuno
 *a,
Fumio
Sasaki
b and
Hisao
Yanagi
a
*a,
Fumio
Sasaki
b and
Hisao
Yanagi
a
aGraduate School of Science and Technology, Nara Institute of Science and Technology (NAIST), 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. E-mail: jinjo.tomomi.jn0@ms.naist.jp; hitoshi352-17@ms.naist.jp
bResearch Institute for Advanced Electronics and Photonics, National Institute of Advanced Industrial Science and Technology, Ibaraki 305-8568, Japan
First published on 17th January 2023
Abstract
Four types of crystals with different molecular orientations and crystal morphologies (green platelet, yellow platelet, yellow fiber, and orange disk) consisting of 5,5′-bis(4′-cyanobiphenyl-4-yl)-2,2′-bithiophene (BP2T-CN), a type of thiophene/phenylene co-oligomer (TPCO), were obtained by changing the crystal preparation method. Each crystal exhibited high photoluminescence quantum efficiency values of 61–85%. For green- and yellow-emitting (platelet and fiber) crystals, lasing was observed from the crystal end facets acting as Fabry–Pérot (F–P) resonators. Strong exciton–photon coupling was demonstrated by energy-wavevector dispersion plots obtained from the energies of each interference peak in the mode structure of the F–P resonator. Moreover, a low lasing threshold of 17 μJ cm−2 was achieved in the case of yellow-emitting fiber-like crystals, owing to the high absorption efficiency resulting from the lying-down molecular orientation in the crystal. For the orange-emitting crystal, whispering-gallery mode lasing was observed above 156 μJ cm−2. This higher threshold resulted from the low absorption efficiency that is attributable to the 50° angle (slip angle) formed between the transition dipole moment and crystal basal plane ((100) plane). The crystal structure- and molecular alignment-dependent optical properties can be obtained by controlling the molecular packing and magnitude of intermolecular interactions.
1. Introduction
The solid-state fluorescence of organic molecules strongly depends on their molecular structure and intermolecular interactions.1–14 It is possible to achieve conformation-dependent optoelectronic properties and varied emission colors from the same fluorophore using crystal polymorphs, which can be controlled by preparation methods.1–14 The use of molecular alignment-control technology allows us to readily change the crystal morphology from one to three dimensions and depending on the structure of the freely constructed molecular array, different optoelectronic properties are manifested (conformation-dependent optoelectronic properties). Since low-dimensional organic crystals have well-defined crystal planes wherein the molecules are aligned in a regular fashion with specific orientations, the molecular arrangement in a particular crystal plane affects the overall photoluminescence (PL) properties and photoluminescence quantum yield (PLQY).15,16 In pentacene crystals, crystal-plane-dependent PL behavior has been observed. The PL properties have been attributed to the arrangement of pentacene molecules in the (010) and (001) planes and the main crystal planes in one-dimensional (wire) and two-dimensional (disk) crystals.15,16 The difference in the PL spectra of one-dimensional wires and two-dimensional disks has been ascribed to the alignment of pentacene molecules on each crystal plane relative to the direction of incident light.15,16 Moreover, the molecular alignment is generally governed by the interaction between molecules and substrates, base layer (buffer layer) material, and molecular modification. In organic light-emitting field-effect transistors,18–23 organic light-emitting diodes,18,22,24,25 organic photovoltaic cells,26–28 and organic solid-state lasers,18,29–37 achieving a lying or upright molecular orientation with respect to the substrate is important for improving the device performance particularly in terms of carrier transport properties. The use of crystal polymorphs is an efficient molecular alignment-control approach, including the interaction between the molecules and substrates and the use of a buffer layer.1–14 Using the crystal polymorphs, the optoelectronic properties can be varied without considering the substrate or buffer layer.Crystal polymorphs have frequently been used to tune the singlet emissions of organic dyes. However, crystal polymorph-based organic solid-state lasers are under-developed owing to the lack of investigation on the relationship between molecular conformations/structures and optical properties. In a study investigating the impact of polymorphism on the optoelectronic properties of a low-bandgap polymer, two different semi-crystalline polymorphs, β1 and β2, were explored. β2 has been reported to have a lower optical bandgap, reduced π-stacking distance, higher hole mobility in field-effect transistors, and improved photocurrent generation in polymer solar cells.28 In a study on the effect of crystal polymorphs (consisting of thienothiophene–thiazolothiazole molecules) on the solid-state and transistor properties, Schneider et al. reported that among the two crystal polymorphs of 1-Red and 1-Yellow, only 1-Yellow displayed semiconductor properties (p-type, hole mobility (μh): 3.3 × 10−3 cm2 V−1 s−1).10
For the organic polymorphs of blue- or green-emissive microribbons (MRs) consisting of difluoroboron avobenzone (BF2AVB), it has been reported that the blue- and green-emissive MRs exhibit monoclinic (J-aggregates, PL efficiency: 68%) and orthorhombic phases (H-aggregates, PL efficiency: 24%), respectively.6 The laser oscillation thresholds for the blue- and green-emissive MRs (observed with femtosecond laser pumping) were as high as 530 and 1126 μJ cm−2. Fluorene trimer 2,2′:7′,2′′-ter(9,9-dimethylfluorene) (TDMeF) has four polymorphs with different molecular conformations.7 However, all the crystalline polymorphs of TDMeF exhibited blue emissions, and no color tuning was observed.7 For the lasing behavior of TDMeF crystals, the quality factor (Q) estimated from the lasing spectra was as low as 786 or 1144.
Meanwhile, the occurrence of aggregation-induced emission and aggregation quenching (concentration quenching) has been reported in many organic crystals and aggregates.14 Among various organic crystals, single crystals of thiophene/phenylene co-oligomers (TPCOs)21,34–48 exhibit high PLQYs ranging from 40 to 63% even in the solid state. To develop high-performance organic lasers and organic light-emitting devices, materials that exhibit high PLQYs, irrespective of conformational and molecular packing changes, are required. The formation and optical properties of crystal polymorphs of TPCO crystals have not been reported yet. Herein, we report the optical properties of crystal polymorphs with controlled crystal structures, molecular orientations, and crystal growth planes using 5,5′-bis(4′-cyanobiphenyl-4-yl)-2,2′-bithiophene (BP2T-CN) (n-type),37,38,48 a type of TPCO. We also demonstrate the optically pumped lasing properties of four different BP2T-CN crystals with controlled crystal morphologies and molecular alignments.
2. Experimental section
BP2T-CN was purchased from Sumitomo Seika Chemicals Co. and was used as such, without further purification. Tetrahydrofuran (THF) and o-dichlorobenzene (o-DCB) were purchased from Sigma-Aldrich Co. Ltd.BP2T-CN single crystals were prepared using physical vapor transport (PVT) and solution growth methods. In the PVT method, the BP2T-CN powder, placed in a nitrogen-filled glass tube was heated at 280 °C for 24 h using an electric furnace with a temperature gradient, and green emitting BP2T-CN crystals were obtained. The flow rate of nitrogen gas was adjusted to 0.01 L min−1 using a flow meter. After cooling to 40 °C, the single crystals were selected and mounted onto a quartz substrate using a tungsten needle. When preparing the platelet- or fiber-like BP2T-CN crystals exhibiting yellow emission, solution growth method was used. To grow the platelet-like crystals, 2 mg of BP2T-CN powder was dissolved in 20 mL of THF, and the solution was heated to 60 °C. The single crystals were precipitated by gradual cooling to 27 °C for 24 h using a temperature controller. After cooling, the solution was filtered and allowed to dry naturally. Single crystals on the filter were then selected and mounted onto a quartz substrate using a tungsten needle. To prepare the fiber-like crystals, 3 mg of BP2T-CN powder was dissolved in 20 mL of o-DCB, and the solution was heated to 150 °C. The BP2T-CN fiber-like crystals were precipitated after slow cooling to 27 °C for 20 h using a temperature controller. BP2T-CN crystals with orange emission were prepared using the melting method. BP2T-CN powder was placed on a quartz substrate and melted using a gas burner. The heating was stopped immediately after the BP2T-CN powder was melted over a gas burner. The temperature of the quartz substrate surface was determined to be approximately 360 °C by using a thermometer. The prepared BP2T-CN crystals were allowed to cool naturally.
Optical absorption spectra were measured using a UV-visible spectrometer (JASCO Japan Spectroscopy V-730). The fluorescence micrographs of the BP2T-CN crystals were obtained using a fluorescence microscope (Olympus, BX-51). The PL spectra were measured using a spectrometer (Princeton Instruments, HRS-300) equipped with a thermoelectrically cooled CCD (charge-coupled device) detector (Princeton Instruments, PIXIS-256) and a neodymium (Nd):yttrium aluminum garnet (YAG) laser (λex = 355 nm, pulse duration: <1.1 ns, repetition rate: 1.2 kHz). θ/2θ measurements of the single crystal were performed at 103 K using an X-ray diffractometer (Rigaku ValiMax Rapid) with a Mo Kα line (λ = 0.71075 Å). For time-resolved PL measurements, the second-harmonic light of a Ti:sapphire pulsed laser (Coherent Mira 900F, time duration: 100–150 fs, repetition rate: 76 MHz) was used as an excitation source (λex = 400 nm). The luminescence of the single crystals was measured and focused on the entrance slit of an imaging polychromator (Hamamatsu Photonics, C5094). The emission decay profiles of the BP2T-CN crystals were measured using a streak camera (Hamamatsu, C4334) (time resolution: 15 ps).
Differential scanning calorimetry (DSC) purity analysis and thermogravimetry-differential thermal analysis (TG-DTA) were performed on a Hitachi DSC7000X/STA7200 under nitrogen gas flow at a heating rate of 1 °C min−1 and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C min−1, respectively.
°C min−1, respectively.
3. Results and discussion
The molecular structure of BP2T-CN is shown in Fig. 1(a). Fig. 1(b) and (c) show the fluorescence micrographs of the BP2T-CN single crystals prepared using the THF solvent by the PVT and liquid-phase growth methods, respectively. The BP2T-CN single crystals prepared by the PVT method showed green emission, and bright emission was observed at the crystal edges. On the other hand, BP2T-CN single crystals grown using the liquid-phase growth method showed yellow emission from the entire crystal body. The PL and absorption spectra of the green- and yellow-emitting crystals, measured at room temperature (300 K) are shown in Fig. 1(d) and (e), respectively. For the green-emitting crystals prepared by the PVT method (Fig. 1(d)), the 0-1, 0-2, and 0-3 PL bands were observed at 2.34, 2.17, and 2.00 eV, respectively. The 0-0, 0-1, and 0-2 absorption bands were observed at 2.56, 2.73, and 2.90 eV, respectively. In the yellow-emitting crystals prepared using the liquid-phase growth method (Fig. 1(e)), the 0-1, 0-2, and 0-3 transitions in the PL spectra appear at 2.26, 2.09, and 1.92 eV, respectively. The 0-0, 0-1, and 0-2 absorption bands appear at 2.48, 2.65, and 2.82 eV. Although the lowest 0-0 transition is forbidden, weak 0-0 transitions in the absorption spectra of BP2T-CN crystals (similar to other TPCO crystals) were observed which can be attributed to the antiparallel exciton coupling between adjacent molecules aligned in the crystal. Compared to the energy of the 0-1 PL bands of the green-emitting single crystal, the yellow-emitting crystal was red-shifted by 0.07 eV. | ||
| Fig. 1 Molecular structure of BP2T-CN (a). Fluorescence micrographs of green- (b) and yellow-emitting (c) BP2T-CN crystals. Optical absorption and photoluminescence (PL) spectra of the green- (d) and yellow-emitting (e) crystals. Gray circle, triangles, diamonds, and squares in the absorption and PL spectra indicate the 0-0, 0-1, 0-2, and 0-3 bands, respectively. | ||
X-ray structural analysis of the BP2T-CN crystals was performed to investigate the factors responsible for the energy difference in the PL spectra of the green- and yellow-emitting crystals. The crystal structures of the green- and yellow-emitting crystals are shown in Fig. 2(a)–(d). The crystal structure of the green-emitting crystal prepared by the PVT method was reproduced using the data reported by Hatano et al. (monoclinic, a = 18.34 Å, b = 7.24 Å, c = 18.446 Å, β = 100.5°).37 The transition dipole moment of the BP2T-CN molecule is parallel to the long molecular axis, and the dipole moments (molecules) align parallel to the ac plane of the crystal, as shown in Fig. 2(a). The crystal plane of the green-emitting crystal is (201), and the long axis of the molecule forms an angle of 25° against the crystal plane, as shown in Fig. 2(a). Because of the lying orientation (with respect to the crystal plane) of the molecules, both surface and edge emissions were obtained from the platelet-like crystals (Fig. 1(b)). As shown in Fig. 2(b), the shortest distance between two adjacent molecules was 6.41 Å. Single-crystal X-ray structure analysis of the yellow-emitting crystals shown in Fig. 1(c) revealed that the BP2T-CN molecules formed a triclinic crystal with a = 7.124 Å, b = 12.255 Å, c = 14.512 Å, α = 98.39°, β = 102.08°, and γ = 97.67°. Thus, BP2T-CN forms two different molecular arrangements and crystal structures, green- and yellow-emitting crystals as crystalline polymorphs. The differences in molecular packing change the magnitude of intermolecular interactions, thereby changing the optical transition energy. In case of the yellow-emitting crystal, the BP2T-CN molecules align along the ac plane of the crystal because the crystal plane of the platelet-like crystal structure (Fig. 1(c)) corresponds to the (01−1) plane. The molecular long axis forms an angle of 34° against the crystal basal plane ((01−1) plane), and the molecules result in an inclined orientations to the basal plane. The closest distance between the two neighboring molecules is 3.68 Å (Fig. 2(d)), which is approximately 0.57 times lesser than that in the case of green-emitting crystals. The mechanism responsible for the energy change in response to the change in molecular arrangement and crystal structure is explained as follows: With the aggregation of organic molecules, the excited energy levels split into two levels (high and low energy levels), as shown in Fig. 2(e).1 For a majority of TPCO crystals reported so far, the lowest excited energy level is forbidden due to the antiparallel exciton coupling in the excited state, as discussed above. However, the misaligned molecular arrangements, as in the case of BP2T-CN (Fig. 2(e)) result in a partially allowed transition at the lowest excited energy level.31,32,36,45 As the intermolecular distance between the two neighboring molecules shortens, the width of energy splitting increases, and the lowest excited energy level becomes more stable. Therefore, it is inferred that the emission color changed from green to yellow because the distance between the two closest neighboring molecules was shorter and the lowest excited energy level was more stabilized in the yellow polymorphs prepared by the liquid-phase growth method. The dimer model can also explain this change in emission energy. In the dimer model reported by Kasha,49 excitonic interaction energy between two transition dipole moments is given by Eexciton = |μ|2(1 − 3cos2θ)/r3, where Eexciton, μ, θ, and r are excitonic interaction energy, transition dipole moment, angle, and distance between two transition dipole moments, respectively. In the crystal polymorphs of BP2T-CN, the angle and distance between two transition dipole moments affect the excitonic interaction energy. Using the transition dipole moment (16.3 D),48 slip angle (angle between two adjacent molecules forming a diagonal pair and crystal basal plane) (40° and 1°) (see Fig. 2(a) and (c)), the distance between the two dipoles (6.41 Å and 3.68 Å) (see Fig. 2(a) and (c)), and the magnitude of Eexciton are larger in the yellow-emitting crystal than that in the green-emitting crystal. Hence, since the splitting energy in the dimer is larger in the yellow-emitting crystal than that in the green-emitting crystal, the red-shifted emission is obtained in the yellow-emitting crystal.
 | ||
| Fig. 2 Crystal structures of the green- (a and b) and yellow-emitting (c and d) BP2T-CN crystals. The crystal structures of the green-emitting crystal were reproduced using the reported data.37 Schematic diagram for the splitting energy in molecular aggregates increasing with decreasing intermolecular distance and slip angle (e). Slip angles are also shown in (e). | ||
To investigate the emission lifetimes and PLQYs of the green and yellow crystal polymorphs, we measured the temperature dependence of their time-resolved PL spectra (10–300 K). Fig. 3(a) and (b) show the decay curves of the 0-1 emission bands measured at 300 K for the green and yellow crystal polymorphs, respectively. Using convolution fitting (sky blue solid line) with the incident laser pulse (black dashed line), the emission decay profiles were characterized by two time constants: 530 ps (89%) and 1.3 ns (11%) for the green-emitting crystal, and 720 ps (99%) and 2.5 ns (1%) in the case of the yellow-emitting crystal. In the green crystal, BP2T-CN platelet crystal polymorphs, the fast and slow components are attributed to the two different types of singlet exciton states: free and self-trapped excitons (shallow self-trapped excitons), respectively, as reported earlier.48 It can be inferred from this mechanism that the main components with 530 ps time constant in the green crystal polymorph and 720 ps time constant in the yellow crystal polymorph are due to the free excitons. To determine the PLQY values for crystal polymorphs, the temperature dependence of emission lifetimes of 0-1 band (dominant component) was analyzed using the following equation, which has been used in earlier works.43,48
| τ(T) = 1/(kf + knrexp(−Ea/(kBT))) | (1) |
| Φ = kf/(kf + knr). | (2) |
 | ||
| Fig. 3 Emission decay profiles of 0-1 emission bands measured at 300 K for the green (a) and yellow (b) crystal polymorphs. Temperature dependence of the emission lifetimes (dominant component) of the 0-1 bands for the green (green color) and yellow (yellow color) polymorphs (c). | ||
| Green platelet crystals | Yellow platelet crystals | Yellow fiber crystals | Orange disk crystals | |
|---|---|---|---|---|
| Lifetime (τ) at 300 K | 0.53 ns (89%) | 0.72 ns (99%) | 0.74 ns | 1.1 ns |
| 1.3 ns (11%) | 2.5 ns (1%) | |||
| k f | 14.3 × 108 s−1 | 13.2 × 108 s−1 | 11.0 × 108 s−1 | 7.6 × 108 s−1 |
| k nr | 9.1 × 108 s−1 | 2.3 × 108 s−1 | 5.2 × 108 s−1 | 4.2 × 108 s−1 |
| E a | 18 meV | 35 meV | 20 meV | 25 meV |
| PLQY | 0.61 | 0.85 | 0.68 | 0.64 |
To investigate the optically pumped lasing behavior of the green and yellow crystal polymorphs exhibiting high PLQY values, we measured the PL spectra under high-density photoexcitation using a Nd:YAG laser. A spot-shaped excitation beam (0.12 cm2, p-polarization) was focused on the crystal polymorphs at an incidence angle of 45° against the substrate, and the light emitted from the crystal edges was detected using a CCD spectrometer (Hamamatsu PMA-50). The BP2T-CN green crystal polymorph shown in Fig. 1(b) had a length (L) of 100 μm and a thickness (t) of 3.7 μm, and BP2T-CN yellow crystal polymorph shown in Fig. 1(c) had L of 120 μm and t of 14.8 μm. Fig. 4(a) and (b) show the excitation density dependence of the PL spectra and integrated PL intensity of the 0-1 band (2.31–2.33 eV) for the green-emitting crystal. For the green-emitting crystals, when the excitation density increased above 69 μJ cm−2, emission amplification was observed in the 0-1 band (2.32 eV). As shown in Fig. 4(b), the integrated 0-1 band intensity increased linearly in the low excitation density range, and super linearly above the threshold of 69 μJ cm−2. In the yellow-emitting single crystal shown in Fig. 1(c), a broad spontaneous emission spectrum was observed at a low excitation density of 46 μJ cm−2 (Fig. 4(c)). The integrated 0-2 band PL intensity (2.10–2.14 eV) increased super linearly at excitation densities above 214 μJ cm−2, as shown in Fig. 4(c) and (d). The emission amplification in the 0-2 band in the yellow-emitting crystal can be ascribed to the reabsorption effect due to the thicker crystal (14.8 μm). The lasing thresholds for green- and yellow-emitting crystals are affected by the absorption efficiency of the excitation light owing to their specific molecular orientations with respect to the crystal basal planes. The angles between the electric field plane of the excitation light and the molecular transition dipole moment, against the crystal basal planes ((201) or (01−1) planes) in the green- and yellow-emitting crystals were 110° and 101°, respectively. The yellow-emitting crystal can absorb the excitation light more efficiently than the green-emitting crystal. However, a lower lasing threshold was obtained for the BP2T-CN green-emitting crystal. The one-dimensional optical confinement effect is responsible for a lower threshold lasing in the BP2T-CN green-emitting crystal. Fig. 5(a) and (b) show high-resolution PL spectra showing lasing for the green- and yellow-emitting crystals measured at a spectral resolution of 0.2–0.3 nm. When the excitation density increased above the threshold, longitudinal multimode lasing was observed in the 0-1 and 0-2 emission bands for the green- and yellow-emitting crystals, respectively. With a mode spacing of Δν = 11 cm−1 in the lasing spectrum (Fig. 5(a)) and the crystal length L of 100 μm (Fig. 1(b)), the group refractive index, ng of the BP2T-CN green-emitting crystal was estimated to be ng = 1/(2 LΔν) = 4.6. This ng value is almost consistent with the values reported for other TPCO crystals.34–36 Thus, in the green-emitting crystal shown in Fig. 5(a), the lasing has been attributed to Fabry–Pérot (F–P) resonance effect caused by the longitudinal end faces in the crystal acting as a mirror. The Q value estimated from the lasing spectrum was 4250. For the lasing spectrum in the yellow-emitting crystal shown in Fig. 5(b), ng was calculated to be 3.2 with a mode spacing of Δν = 13 cm−1 in the lasing spectrum and a crystal length L of 120 μm in the longitudinal direction. Longitudinal multimode lasing was observed in the yellow-emitting crystal, originating from the F–P resonance in the BP2T-CN platelet-like crystal resonator with mirror surfaces at both ends. The Q value for the yellow-emitting crystal, estimated from the lasing spectrum (Fig. 5(b)) was 2360. The lower Q value in the yellow-emitting crystal in comparison to that in the green-emitting crystal can be ascribed to emission leakage from the crystal surface, as shown in Fig. 1(c). The high ng and Q values obtained in the green-emitting crystal can be attributed to the one-dimensional optical confinement resulting from the well-aligned molecules in the longitudinal direction coupled with the excellent optical waveguide quality.
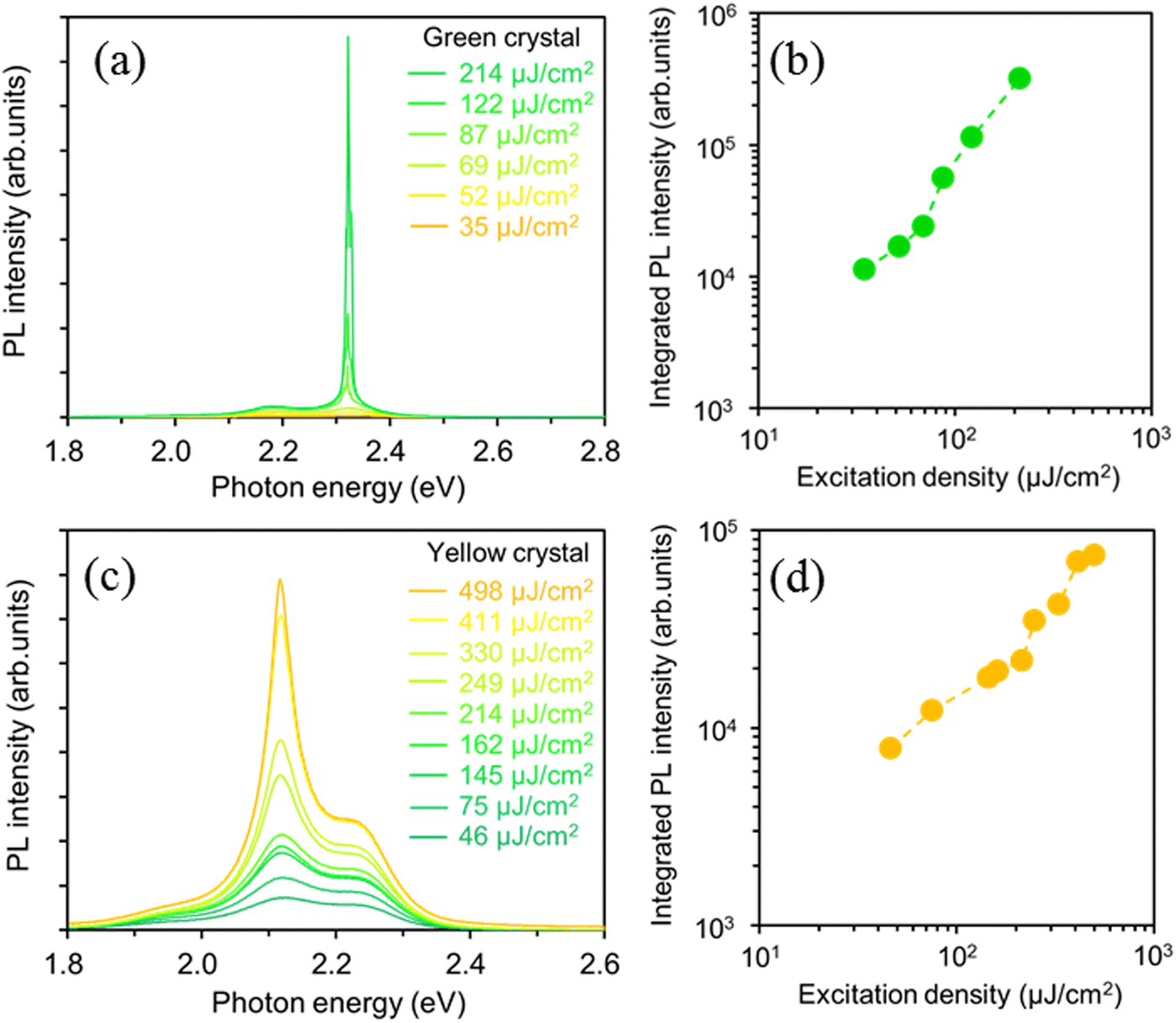 | ||
| Fig. 4 Excitation density dependence of PL spectra (a) and integrated PL intensity (b) of 0-1 band (2.31–2.33 eV) measured for the green-emitting crystal. Excitation fluence dependence of PL spectra (c) and integrated PL intensity (d) of 0-2 band (2.10–2.14 eV) for the yellow-emitting crystal. | ||
 | ||
| Fig. 5 High-resolution PL spectra (spectral resolution: 0.2–0.3 nm) showing lasing for the green- (a) and yellow-emitting (b) crystals. | ||
Next, we show the optical properties of BP2T-CN fiber-like crystals for different growth planes obtained by changing the solvent used in the liquid-phase growth method from THF to o-DCB. Fig. 6(a) and (b) show the fluorescence micrograph and PL spectrum of the yellow-emitting fiber-like crystals. In Fig. 6(b), the 0-1, 0-2, and 0-3 transitions in the PL spectrum were observed at 2.30, 2.13, and 1.96 eV, respectively. To examine the molecular orientation of these fiber-like crystals, X-ray diffraction (XRD) θ/2θ measurements were performed by mounting the crystals on the glass substrate. The XRD patterns of the fiber-like crystals are shown in Fig. 6(c). Diffraction peaks were observed at 7.36°, 20.37°, and 21.34°, corresponding to the (010), (10−3), and (022) planes, respectively. The crystal structures displaying the (010), (10−3), and (022) planes corresponding to each crystal basal plane are shown in Fig. 6(d)–(f), respectively. The highest diffraction intensity in the (022) plane suggests that the (022) plane is parallel to the substrate in most fiber-like crystals. The simulated powder XRD pattern (Fig. 6(c)) using the crystallographic data for the yellow-emitting crystal shown in Fig. 1(c) is in good agreement with the diffraction pattern of the fiber-like crystals. This result indicates that the yellow-emitting fiber-like crystal has the same crystal structure as the yellow-emitting platelet-like crystal. In the yellow-emitting fiber-like crystals, the transition dipole moments of the BP2T-CN molecules were tilted by 6° with respect to the (022) plane, and fiber-like crystals were obtained due to π-stacking along the a-axis.
 | ||
| Fig. 6 Fluorescence image (a) and fluorescence spectrum (b) of BP2T-CN fiber-like crystal. X-ray diffraction (XRD) pattern (green) measured using the fiber-like crystals mounted on the glass substrate (c). The simulated powder XRD pattern using the crystallographic data for the yellow-emitting crystal was also shown by a black line in (c). Crystal structures displaying (010) (d), (10−3) (e), and (022) (f) planes corresponding to each crystal basal plane. (g) The temperature dependence of the 0-1 band emission lifetimes for the fiber-like crystal. | ||
We also obtained the PLQY value by measuring the temperature dependence of emission lifetimes for the BP2T-CN fiber-like crystal using eqn (1). Fig. 6(g) shows the temperature dependence of the 0-1 band emission lifetimes for the fiber-like crystal. The emission decay profile exhibited a single exponential decay with a lifetime of 0.74 ns at 300 K. The parameters were listed in Table 1.
We examined the effects of the molecular orientation and crystal shape on the lasing properties. The fiber-like crystal shown in Fig. 6(a) had L of 40 μm, crystal width of approximately 1.6 μm, and t of 100 nm. As shown in Fig. 7(a) and (b), as the excitation density increased above 17 μJ cm−2, the PL intensity increased significantly with a decrease in the emission spectral width. Fig. 7(c) shows the high-resolution PL spectrum (spectral resolution: 0.2–0.3 nm) of the fiber-like crystal at 85 μJ cm−2. As shown in Fig. 7(b), when the excitation density increased above the threshold excitation density of 17 μJ cm−2, longitudinal multimode lasing was observed in the 0-2 emission band. The lower lasing threshold of 17 μJ cm−2 can be attributed to the two-dimensional optical confinement along the fiber direction and the higher absorption efficiency to the lying molecular orientation (molecules were tilted by 6° against the (022) plane). Despite the thin thickness of 100 nm, lasing was observed in the 0-2 emission band probably due to the reabsorption effect. The reabsorption effect is ascribed to the increased absorption efficiency resulting from more lying-down molecular orientations in comparison to the yellow-emitting platelet-like crystal (Fig. 1(c)). The ng value of the fiber-like crystal was determined to be 7.8 with mode spacing, Δν of 16 cm−1 and L of 40 μm as shown in the lasing spectrum in Fig. 7(c). The Q value estimated from the lasing spectrum was 3700. The higher ng value in the fiber-like crystal compared to the green-emitting platelet-like and the yellow-emitting platelet-like crystal may be due to the fact that the emission propagates as exciton polariton along the fiber-like crystal.48,51 We have already reported that the lower polariton branch propagates along the BP2T-CN microrod crystal in our previous work.48 The exciton-polariton property in the BP2T-CN fiber-like crystal is described below. Fig. 8(a) shows a schematic representation of exciton-polariton formation in BP2T-CN fiber-like crystal. Fig. 8(b) represents the PL spectrum of BP2T-CN fiber-like crystal detected from the crystal edge (blue line). In Fig. 8(b), the blue solid, red solid, and black dashed lines indicate the PL spectrum, reproduced spectrum using the sum of Lorentzian functions, and Lorentz functions corresponding to each resonance energy, respectively. Using the PL spectrum in Fig. 8(b), the corresponding energy–wavevector (E–kz) dispersion of propagating modes was obtained by plotting the energies of each interference peak (energy of each Lorentzian function) with respect to kz. The E–kz dispersion plots are shown in Fig. 8(c). To demonstrate exciton–polariton formation in the BP2T-CN fiber-like crystal, the exciton–polariton dispersion was analyzed using a coupled oscillator model for exciton–photon coupling:48
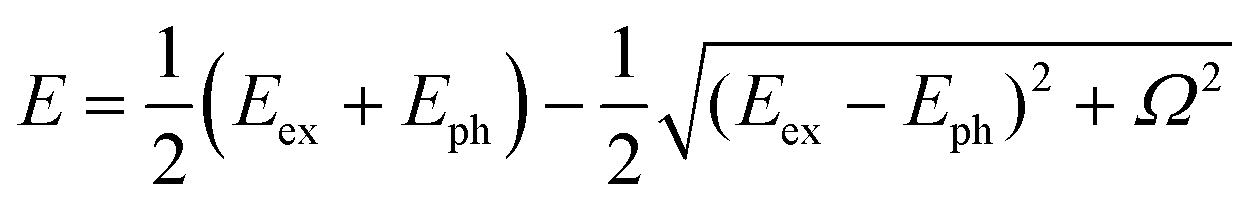 | (3) |
 | (4) |
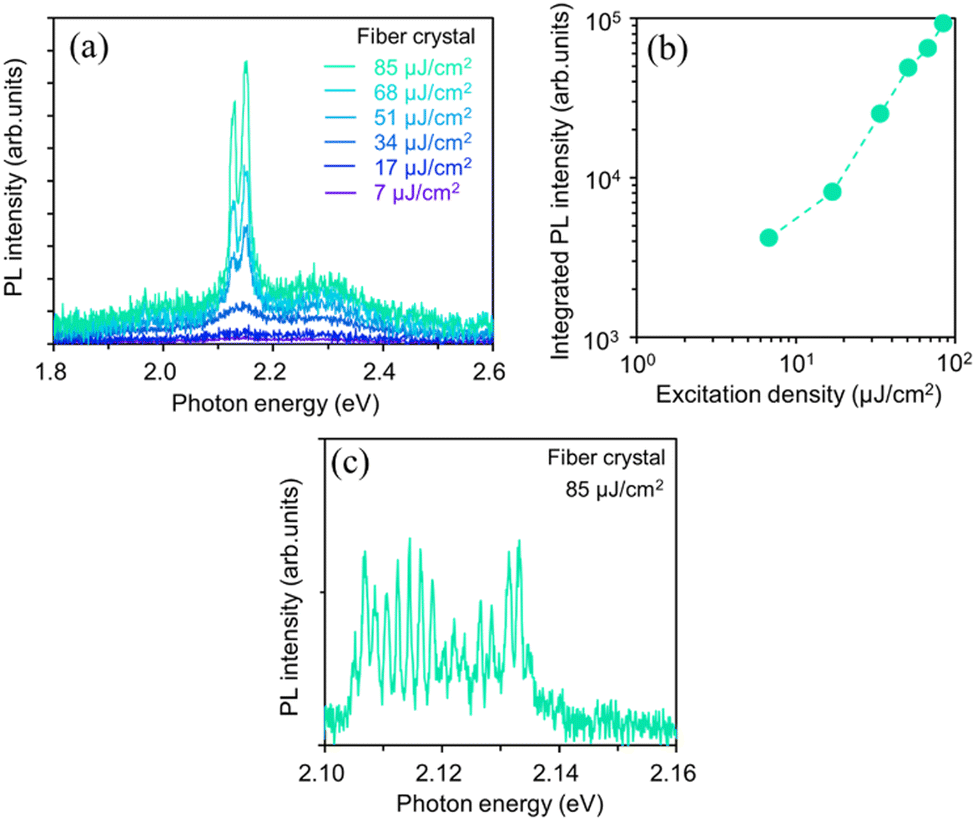 | ||
| Fig. 7 PL spectra (a) and integrated 0-2 emission band intensity (b) of BP2T-CN fiber-like crystal measured as a function of excitation density. High-resolution PL spectrum (spectral resolution: 0.2–0.3 nm) measured at 85 μJ cm−2 in the fiber-like crystal (c). | ||
 | ||
| Fig. 8 (a) Schematic representation of exciton-polariton formation in BP2T-CN fiber-like crystal. (b) PL spectrum of BP2T-CN fiber-like crystal detected from the crystal edge (blue line). The red line shows reproduced spectrum using the sum of Lorentzian functions corresponding to each resonance energy (black dashed lines). (c) Energy–wavevector (E–kz) dispersion plots obtained from the energies of each interference peak (energy of each Lorentzian function) in (b). The blue lines in (c) show upper and lower polariton branches (UPB, LPB) analyzed using a coupled oscillator model. | ||
Next, we discuss the emission properties of orange-emitting crystals with molecular orientations different from the three BP2T-CN crystals described above. Fig. 9(a) and (b) show the fluorescence image, absorption, and PL spectra of the BP2T-CN orange-emitting crystal prepared using the melting method. Polarized light microscopy observations confirmed that orange-emitting samples were single crystals (see the ESI,† Fig. S1). The 0-0, 0-1, 0-2, and 0-3 absorption bands appeared at 2.41, 2.59, 2.77, and 2.95 eV, respectively. The 0-1, 0-2, and 0-3 PL bands are observed at 2.22, 2.04, and 1.86 eV, respectively. XRD measurements were performed to investigate the molecular orientation of the BP2T-CN orange-emitting crystals. The results demonstrated that the crystal structure of the orange-emitting BP2T-CN crystals was the same as that of the green-emitting crystal (Fig. 1(b)) (Fig. S2, ESI†). The orange luminescence was not caused by impurities or the mixture of amorphous and monoclinic phase crystal. To investigate that the orange emission was not due to thermal decomposition or impurities, the DSC purity analysis and TG-DTA were performed. The DSC purity analysis resulted in the purity of 99.64% for the BP2T-CN orange-emitting crystals. From the TG-DTA spectra, it was found that no thermal decomposition did not occur under the crystal fabrication condition at approximately 360 °C using a gas burner (Fig. S3, ESI†). The difference in the peak energy in the PL spectra between the orange- and green-emitting crystals is on account of the crystal plane-dependance of the PL properties. Few studies have been conducted on such crystal plane-dependent PL properties.15,16 It is conceivable that the emission energy was red-shifted in the orange-emitting crystal because of the appearance of the (100) plane (basal plane), not observed in the case of the green-emitting crystal. Meanwhile, in 9,10-bis(phenylethynyl)anthracene (BPEA) microtubes/microrods, it has been reported that the optical loss in the BPEA microtubes is ascribed to reabsorption, while scattering and the coupling between the BPEA microrod crystal and the substrate that causes optical loss (waveguide loss) in light propagation are responsible for the change in emission color.16,17 Because the orange-emitting crystal retains its emission color even after being peeled from the substrate, it implies that there is negligible coupling between the crystal and the substrate. In addition, in the BP2T-CN orange-emitting crystal, wherein luminescence is confined within the disk-shaped crystal, it is unlikely that optical loss may be the cause of the red-shifted PL spectrum. Therefore, the red-shifting of approximately 0.12 eV in the PL spectrum of the orange-emitting crystal in comparison to the green-emitting crystal is attributed to the crystal plane change.
 | ||
| Fig. 9 Fluorescence image (a), absorption (black), and PL spectra (orange) (b) of the BP2T-CN orange-emitting crystal. Gray circle, triangles, diamonds, and squares in the absorption and PL spectra indicate the 0-0, 0-1, 0-2, and 0-3 bands, respectively. XRD patterns measured for the orange-emitting crystals (c). Crystal structure of the orange-emitting crystal displaying a (100) plane (d). (e) The temperature dependence of the emission lifetimes of the 0-1 band for the orange crystal. | ||
Fig. 9(c) shows the XRD patterns of the orange-emitting crystals. The diffraction pattern was indexed using the crystallographic data for the green-emitting crystal shown in Fig. 1(b). A diffraction peak, observed at 4.65° (Fig. 9(c)) corresponds to the (100) plane, as shown in Fig. 9(d). For the molecular orientation in the orange-emitting crystal, the angle between the transition dipole moment of the BP2T-CN molecule and the (100) plane is 50°, as shown in Fig. 9(d). In the orange-emitting crystal as shown in Fig. 9(d), the slip angle, which is the angle between two adjacent molecules forming a diagonal pair and the (100) plane, was 12°. This smaller slip angle compared to that in the green-emitting crystal is the cause of the red-shifted spectra. Comparison of the parameters obtained from four types of BP2T-CN crystals is summarized in Table 2.
| Green platelet (Fig. 1(b)) | Yellow platelet (Fig. 1(c)) | Yellow fiber (Fig. 6(a)) | Orange disk (Fig. 9(a)) | |
|---|---|---|---|---|
| Crystal structure | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Crystal basal plane | (201) | (01−1) | (022) | (100) |
| Angle between the long axis of molecule and the crystal basal plane | 25° | 34° | 6° | 50° |
| Slip angle | 40° | 1° | 12° | |
| Lasing threshold | 69 μJ cm−2 | 214 μJ cm−2 | 17 μJ cm−2 | 156 μJ cm−2 |
| Cavity length L | 100 μm | 120 μm | 40 μm | 92 μm |
| n g | 4.6 | 3.2 | 7.8 | 3.9 |
| Q factor | 4250 | 2360 | 3700 | 3140 |
Fig. 9(e) shows the temperature dependence of the emission lifetimes of the 0-1 band for the orange crystal. The PLQY value was determined for the orange-emitting crystal using eqn (1). The emission decay curve showed a single exponential decay with a lifetime of 1.1 ns at 300 K. The obtained parameters are summarized in Table 1.
To investigate the effects of the molecular orientation and crystal geometry on the light amplification properties, we examined the lasing properties of the orange-emitting crystal. The orange-emitting crystal shown in Fig. 9(a) is a disk-shaped crystal with a diameter, R of 29 μm and a thickness, t of 600 nm. Fig. 10(a) and (b) show the excitation-density dependence of the PL spectra and the integrated PL intensity of the orange-emitting crystal. As the excitation density increased above 156 μJ cm−2, the PL intensity in the 0-2 emission band (2.06 eV) increased super linearly with the increase in excitation density. Fig. 10(c) shows the high-resolution PL spectrum (spectral resolution: 0.2–0.3 nm) measured at 405 μJ cm−2 for the orange-emitting crystal. At 405 μJ cm−2, sharp lasing peaks appeared at the 0-2 emission band with a linewidth, Δν of 14 cm−1. The ng value was calculated to be 3.9 from the Δν value and the length of the circumference (92 μm) of the disk-shaped crystal. This ng value is equivalent to the values obtained for the BP2T-CN crystals described above. This indicates that the observed lasing in the orange-emitting crystal is attributed to the whispering-gallery mode (WGM). The Q value was also estimated to be 3140 from the lasing spectrum, indicating that the orange-emitting crystal had a strong light confinement effect, similar to the other BP2T-CN crystals described above.
 | ||
| Fig. 10 Excitation density dependence of PL spectra (a) and integrated 0-2 PL band intensity (2.01–2.13 eV) (b) measured for the orange-emitting crystal. Lasing spectrum of the orange-emitting crystal measured at 405 μJ cm−2 (c). | ||
4. Conclusions
We prepared two types of BP2T-CN crystal polymorphs using the PVT and liquid-phase growth techniques. XRD measurements revealed that the green-emitting platelet-like and yellow-emitting platelet-like crystals had monoclinic and triclinic forms, respectively. The changes in the molecular arrangements and intermolecular interactions resulted in the formation of green and yellow crystal polymorphs. By changing the crystal preparation method, the green platelet, yellow platelet, yellow fiber, and orange disk crystals were prepared. These four types of crystals displayed high PLQY values of 61–85%. In the green- and orange-emitting crystals that have the monoclinic form, the red-shifted emission in the orange-emitting crystal is attributed to the smaller slip angle. Multimode lasing was observed from the crystal end facets acting as F–P resonators in the green- and yellow-emitting (platelet, fiber) crystals. For the fiber-like crystal, lasing, at a lower threshold of 17 μJ cm−2 was obtained, owing to two-dimensional optical confinement effect and a higher absorption efficiency. In the fiber-like crystal, exciton polariton formation was also demonstrated by E–kz dispersion plots obtained from the energies of each interference peak in the mode structure of the F–P resonator. In the orange-emitting crystal, high Q-factor (3140) WGM lasing was observed above the threshold excitation density of 156 μJ cm−2. The results obtained herein show that the optical properties dependent on the crystal structure and molecular alignment can be obtained by controlling the molecular packing and magnitude of intermolecular interactions via a change in the crystal growth conditions.Author contributions
H. Mizuno designed this study. H. Mizuno and T. Jinjyo constructed an optical setup for a laser-spectroscopy system. T. Jinjyo performed all experimental work and analyzed data with the aid of H. Mizuno. T. Jinjyo wrote the manuscript with contributions from H. Mizuno, F. Sasaki and H. Yanagi.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 21H01370 (Grant-in-Aid for Scientific Research(B)). This article is partially based on the results obtained from a project, JPNP20004, commissioned by the New Energy and Industrial Technology Development Organization (NEDO). The work was also supported by JST CREST (JPMJCR02T4). The authors acknowledge Mr Y. Okajima, Nara Institute of Science and Technology, for the support in time-resolved PL measurements, Mr S. Katao, Nara Institute of Science and Technology, for XRD measurements, and Mr M. Fujihara, Nara Institute of Science and Technology, for the support of TG-DTA. We would like to thank Editage (www.editage.com) for the English language editing.Notes and references
- C. Kitamura, Chem. Rec., 2012, 12, 506–514 CrossRef CAS PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- Z. Xie, H. Wang, F. Li, W. Xie, L. Liu, B. Yang, L. Ye and Y. Ma, Cryst. Growth Des., 2007, 7, 2512–2516 CrossRef CAS.
- K. Jo, S. Lee, A. Yi, T.-Y. Jeon, H. H. Lee, D. Moon, D. M. Lee, J. Bae, S.-T. Hong, J. Gene, S. G. Lee and H. J. Kim, ACS Omega, 2019, 4, 19705–19709 CrossRef CAS PubMed.
- A. O. F. Jones, B. Chattopadhyay, Y. H. Geerts and R. Resel, Adv. Funct. Mater., 2016, 26, 2233–2255 CrossRef CAS.
- Q. Liao, X. G. Wang, S. Lv, Z. Xu, Y. Zhang and H. Fu, ACS Nano, 2018, 12, 5359–5367 CrossRef CAS PubMed.
- C.-J. Ou, X.-H. Ding, Y.-X. Li, C. Zhu, M.-N. Yu, L.-H. Xie, J.-Y. Lin, C.-X. Xu and W. Huang, J. Phys. Chem. C, 2017, 121, 14803–14810 CrossRef CAS.
- L. Pithan, C. Cocchi, H. Zschiesche, C. Weber, A. Zykov, S. Bommel, S. J. Leake, P. Schäfer, C. Draxl and S. Kowarik, Cryst. Growth Des., 2015, 15, 1319–1324 CrossRef CAS.
- Q. Qi, J. Zhang, B. Xu, B. Li, S. X.-A. Zhang and W. Tian, J. Phys. Chem. C, 2013, 117, 24997–25003 CrossRef CAS.
- J. A. Schneider, H. Black, H. P. Lin and D. F. Perepichka, ChemPhysChem, 2015, 16, 1173–1178 CrossRef CAS PubMed.
- S. Shen, G. Xia, Z. Jiang, Q. Shao, W. Shan and H. Wang, Cryst. Growth Des., 2018, 19, 320–327 CrossRef.
- S. Varghese, S. K. Park, S. Casado, R. Resel, R. Wannemacher, L. Lüer, S. Y. Park and J. Gierschner, Adv. Funct. Mater., 2016, 26, 2349–2356 CrossRef CAS.
- X. Wang, Z.-Z. Li, S.-F. Li, H. Li, J. Chen, Y. Wu and H. Fu, Adv. Opt. Mater., 2017, 5, 1700027 CrossRef.
- Z. Zhang, X. Song, S. Wang, F. Li, H. Zhang, K. Ye and Y. Wang, J. Phys. Chem. Lett., 2016, 7, 1697–1702 CrossRef CAS PubMed.
- J. E. Park, M. Son, M. Hong, G. Lee and H. C. Choi, Angew. Chem., 2012, 124, 6489–6494 CrossRef.
- W. Yao and Y. S. Zhao, Nanoscale, 2014, 6, 3467–3473 RSC.
- Y. S. Zhao, J. Xu, A. Peng, H. Fu, Y. Ma, L. Jiang and J. Yao, Angew. Chem., Int. Ed., 2008, 47, 7301–7305 CrossRef CAS PubMed.
- R. Ding, M. H. An, J. Feng and H. B. Sun, Laser Photonics Rev., 2019, 13, 1900009 CrossRef CAS.
- T. Hasegawa and J. Takeya, Sci. Technol. Adv. Mater., 2009, 10, 024314 CrossRef PubMed.
- S. Mondal, W.-H. Lin, Y.-C. Chen, S.-H. Huang, R. Yang, B.-H. Chen, T.-F. Yang, S.-W. Mao and M.-Y. Kuo, Org. Electron., 2015, 23, 64–69 CrossRef CAS.
- K. Sugahara, T. Nakagawa, R. Hirase, T. Katagiri, Y. Inada, T. Yamao and S. Hotta, Jpn. J. Appl. Phys., 2018, 57, 04FL02 CrossRef.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef PubMed.
- S. K. Park, J. H. Kim and S. Y. Park, Adv. Mater., 2018, 30, e1704759 CrossRef PubMed.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef PubMed.
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao, K. Xian, H. Y. Woo, Z. Ge and J. Hou, Adv. Mater., 2019, 31, e1903441 CrossRef PubMed.
- M. Li, A. H. Balawi, P. J. Leenaers, L. Ning, G. H. L. Heintges, T. Marszalek, W. Pisula, M. M. Wienk, S. C. J. Meskers, Y. Yi, F. Laquai and R. A. J. Janssen, Nat. Commun., 2019, 10, 2867 CrossRef PubMed.
- Z. Chen, C. Dai, W. Xiong, Y. Che and C. Zhang, Commun. Chem., 2021, 4, 97 CrossRef CAS.
- M. Chu, B. Qiu, W. Zhang, Z. Zhou, X. Yang, Y. Yan, J. Yao, Y. J. Li and Y. S. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 42740–42746 CrossRef CAS PubMed.
- H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 RSC.
- J.-J. Wu, X.-D. Wang and L.-S. Liao, ACS Photonics, 2019, 6, 2590–2599 CrossRef CAS.
- S. Zhao, H. Yamagishi, O. Oki, Y. Ihara, N. Ichiji, A. Kubo, S. Hayashi and Y. Yamamoto, Adv. Opt. Mater., 2021, 10, 2101808 CrossRef.
- H. Mizuno, I. Ohnishi, H. Yanagi, F. Sasaki and S. Hotta, Adv. Mater., 2012, 24, 2404–2408 CrossRef CAS PubMed.
- H. Mizuno, U. Haku, Y. Marutani, A. Ishizumi, H. Yanagi, F. Sasaki and S. Hotta, Adv. Mater., 2012, 24, 5744–5749 CrossRef CAS PubMed.
- Y. Tanaka, K. Goto, K. Yamashita, T. Yamao, S. Hotta, F. Sasaki and H. Yanagi, Appl. Phys. Lett., 2015, 107, 163303 CrossRef.
- R. Hatano, K. Goto, K. Yamashita, F. Sasaki and H. Yanagi, Jpn. J. Appl. Phys., 2017, 56, 04CL02 CrossRef.
- S. Dokiya, F. Sasaki and H. Yanagi, J. Cryst. Growth, 2017, 468, 792–795 CrossRef.
- H. Mizuno, T. Jinjyo, C. M. Laurio, H. Katsuki, I. Hiromitsu, F. Sasaki and H. Yanagi, Jpn. J. Appl. Phys., 2020, 59, SDDA14 CrossRef CAS.
- V. A. Postnikov, Y. I. Odarchenko, A. V. Iovlev, V. V. Bruevich, A. Y. Pereverzev, L. G. Kudryashova, V. V. Sobornov, L. Vidal, D. Chernyshov, Y. N. Luponosov, O. V. Borshchev, N. M. Surin, S. A. Ponomarenko, D. A. Ivanov and D. Y. Paraschuk, Cryst. Growth Des., 2014, 14, 1726–1737 CrossRef CAS.
- T. Katagiri, S. Ota, T. Ohira, T. Yamao and S. Hotta, J. Heterocycl. Chem., 2007, 40, 853–862 CrossRef.
- S. Hotta, M. Goto, R. Azumi, M. Inoue, M. Ichikawa and Y. Taniguchi, Chem. Mater., 2004, 16, 237–241 CrossRef CAS.
- S. Kanazawa, M. Ichikawa, T. Koyama and Y. Taniguchi, ChemPhysChem, 2006, 7, 1881–1884 CrossRef CAS PubMed.
- S. Dokiya, F. Sasaki, S. Hotta and H. Yanagi, Jpn. J. Appl. Phys., 2016, 55, 03DC13 CrossRef.
- S. Dokiya, H. Ishigami, T. Akazawa, F. Sasaki and H. Yanagi, Jpn. J. Appl. Phys., 2020, 59, 041004 CrossRef CAS.
- C.-F. Liu, X. Liu, W.-Y. Lai and W. Huang, Adv. Mater., 2018, 30, 1802466 CrossRef PubMed.
- Q. Ou, Q. Peng and Z. Shuai, Nat. Commun., 2020, 11, 4485 CrossRef CAS PubMed.
- H. Mizuno, T. Jinjyo, K. Bando, F. Sasaki, K. Yamashita and H. Yanagi, J. Mater. Chem. C, 2021, 9, 11189–11197 RSC.
- M. Kasha, Molecular Excitons in Small Aggregates, in Spectroscopy of the Excited State, ed. B. Di Bartolo, D. Pacheco and V. Goldberg, 1976, pp. 337–363 Search PubMed.
- K. Bando, T. Nakamura, S. Fujiwara, Y. Masumoto, F. Sasaki, S. Kobayashi, Y. Shimoi and S. Hotta, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 045205 CrossRef.
- K. Takazawa, J. Inoue, K. Mitsuishi and T. Takamasu, Phys. Rev. Lett., 2010, 105, 067401 CrossRef PubMed.
- Q. Liao, Z. Xu, X. Zhong, W. Dang, Q. Shi, C. Zhang, Y. Weng, Z. Li and H. Fu, J. Mater. Chem. C, 2014, 2, 2773–2778 RSC.
- X. Wang, Q. Liao, Z. Xu, Y. Wu, L. Wei, X. Lu and H. Fu, ACS Photonics, 2014, 1, 413–420 CrossRef.
- Q. Shang, S. Zhang, Z. Liu, J. Chen, P. Yang, C. Li, W. Li, Y. Zhang, Q. Xiong, X. Liu and Q. Zhang, Nano Lett., 2018, 18, 3335–3343 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04151k |
| This journal is © The Royal Society of Chemistry 2023 |