
Probing the nature of charge carriers in one-dimensional conjugated polymers: a review of the theoretical models, experimental trends, and thermoelectric applications
Yang
Liu
 *a,
Shengtao
Gao
b,
Xinyu
Zhang
c,
John H.
Xin
d and
Chao
Zhang
a
*a,
Shengtao
Gao
b,
Xinyu
Zhang
c,
John H.
Xin
d and
Chao
Zhang
a
aDepartment of Biomedical Engineering, Sun Yat-sen University, Shenzhen, China. E-mail: liuyang56@mail.sysu.edu.cn
bSchool of Chemical Engineering, Anhui University of Science and Technology, Huainan, Anhui Province, China
cDepartment of Chemical Engineering, Auburn University, Auburn, Alabama, USA
dInstitute of Textiles and Clothing, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR
First published on 24th November 2022
Abstract
Since the first discovery of polyacetylene in the late 1970s, one-dimensional (1D) conjugated polymers have attracted immense research interest as a result of their high conductivity, wide and tunable band gap, and solution processability, making them extraordinary materials for the next-generation organic electronics, photovoltaics, and thermoelectrics. Recently, more research interest has been devoted to integrating the 1D conjugated polymers with organic tissues, e.g., implantable electrodes and electronic plants, as their excellent biofunctions and biocompatibilities are demonstrated. However, to achieve optimal material designs for the 1D conjugated polymers in practical applications, it is essential to understand their fundamental charge transport mechanisms. The current reviews are mainly focused on the functional properties and applications of the conjugated polymers while the theoretical models for charge transport and their correlations with the experimental observations are barely discussed systematically. In this review article, we try to elucidate the nature of charge carriers by taking the total footprints of the research and development in conjugated polymers for consideration, from molecular orbitals to thermally activated conductivity. The electronic structures and mathematical description of the charge carriers, i.e., solitons, polarons, and bipolarons, are discussed and analyzed based on the frontier molecular orbital theory, non-interacting model, Su–Schrieffer–Heeger model, density functional theory, and Holstein-based models. The predicted electronic structures for the charge carriers are model-dependent which can be either weakly or strongly correlated with the Coulomb interactions in the valence and conduction bands. Origins of the optical signatures of the charge carriers, e.g., polarons and bipolarons, in the visible, near infrared, and mid infrared ranges are described and discussed, which may provide semi-quantitative information on the conjugation length and delocalization of the charge carriers. Experimental observations of the spectroscopic characteristics of the charge carriers and their correlations with the molecular configuration, conductivity, crystallinity, and composition of the conjugated polymers are carefully reviewed and compared based on doping induced dopant–host interactions. The transport mechanism of the charge carriers can be described two-dimensionally in the microscale and probed by analyzing the thermoelectric properties of the conjugated polymers and modelling the thermally activated conductivity as a function of the thermopower at the device scale.
 Yang Liu | Dr Yang Liu graduated from Donghua University, Shanghai, China, in 2009 with a bachelor's degree in textile chemistry. He then joined Prof. Xinyu Zhang's lab in Auburn University as a master student and conducted research in the synthesis and carbonization of conducting polymer nanostructures for biosensing and energy-storage applications. He then joined Prof. John Xin's group in the Hong Kong Polytechnic University to study the interfacial physics of conducting polymer-derived nanostructured carbons and semiconductors as a PhD student. He has joined Sun Yat-sen University as a research associate professor since 2017 after graduation. |
 Shengtao Gao | Dr Shengtao Gao, male, born in June 1987. His research interest includes polymer-based nanocomposites and electromagnetic wave absorbing materials. He has published nearly 30 academic papers in peer-reviewed international journals. He has won one youth project from the National Natural Science Foundation of China (No. 52200139), one provincial humanities and social science research project for higher education (key project), one state key laboratory open fund, and participated in two projects endorsed by the national natural science foundation of China (No. 51173002 and 51477002). He won the second prize and the third prize of teaching achievement in Anhui Province. |
 Xinyu Zhang | Prof. Xinyu Zhang received his doctoral degree with Professors Alan G. MacDiarmid and Sanjeev K. Manohar in materials chemistry, from the University of Texas at Dallas in 2005. He is a professor in Chemical Engineering at Auburn University, and his research mainly focuses on microwave-initiated ultrafast nanomanufacturing for hierarchical, multifunctional nanocomposites, fabrication of polymer coatings for anti-corrosion and anti-microbial applications, and production of conducting polymer-based nanocomposites for catalysis, sensing and energy storage applications. |
 John H. Xin | Prof. John H. Xin is a Chair Professor of Textile Chemistry at the Hong Kong Polytechnic University. Prof. Xin's main research includes functional surface modification for textile fibres, biomimetic and bioinspired materials synthesis and applications, new membrane filtration technology for salt removal, green environmentally friendly coloration technologies, etc. He is a Fellow of the Society of Dyers and Colourists (SDC). Based on his outstanding performance, he was appointed as the Lee Family Endowed Professor. He has won many prestigious awards, including the prestigious Gold Award from SDC and China National Science and Technology Progress Award. |
 Chao Zhang | Prof. Chao Zhang received his BSc, MSc, and PhD degrees of Polymer Chemistry and Physics from the Wuhan University, China; during 2006–2010, he conducted postdoctoral research at the University of Wisconsin-Milwaukee and University of California, San Diego in biomaterials and tissue engineering, respectively. In 2010, Dr Zhang joined the Sun Yat-sen University as Associate Professor in Biomedical Engineering and was promoted to Full Professor in 2020. Dr. Zhang's research interests include polymer chemistry, biomaterial science, regenerative medicine, and additive manufacturing for implantable medical devices. |
1. Introduction
Conjugated polymers are a subcategory of polymers that have unique molecular backbone structures, which are composed of alternating single and double bonds. Conjugated polymers are typically formed by polymerizing free radicals of unsaturated alkenes or heterocyclic compounds, i.e., acetylene, pyrrole, aniline, and thiophene, leading to the formation of long-chain polyenes. The formation of conjugated polymers starts from a process called “dimerization”, which refers to the formation of a covalent bond between a pair of unsaturated carbon atoms in chemical terms. In physical terms, the process of dimerization may include the sharing of one unpaired π electron between two unsaturated carbon atoms, which may lead to the formation of an intermediate polyradical state, as shown in Fig. 1. In this state, the unpaired π electrons can be delocalized over the polymer backbone, resulting in a conductor with zero band gap. However, this state is thermodynamically unstable and will be subsequently transformed into alternating short and long bonds via Peierls transition, which are depicted as double and single bonds (Fig. 2), respectively.1 An electronic density gap is created during this transition, which subsequently transforms a polyene from a conductor to a semiconductor. The electronic density gap, which corresponds to the energy difference between the two band structures, i.e., the valence band and conduction band, arises from the molecular orbitals associated with the bonding and anti-bonding states of the main-chain-carbon π electrons.2 According to the frontier molecular orbital theory (FMOT), the bottom of the conduction band corresponds to the lowest unoccupied molecular orbital (LUMO) while the top of the valence band corresponds to the highest occupied molecular orbital (HOMO).3 In the ground state, the π electrons orbit in the HOMO; whenever the π electrons receive energy from external sources, i.e., light, heat, and radiation, that are larger than the electronic density gap between the conduction band and valence band, the π electrons jump from the HOMO to the LUMO, generating the electrical conductivity. For example, the band gap of polyacetylene is around 1.5 eV; for polythiophene, the value increases to around 2.0 eV, and for polypyrrole, the band gap is around 2.8 eV.4,5 Schematic illustrations of the band structure in polyacetylene are shown in Fig. 3.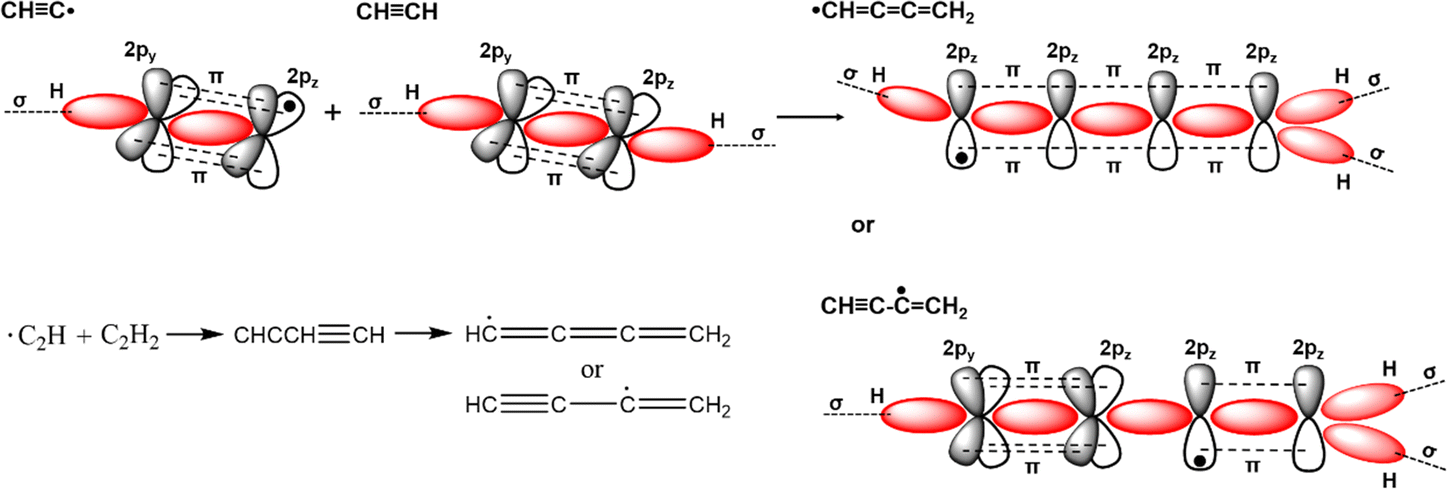 | ||
| Fig. 1 Schematic illustration of the dimerization process of acetylene. The unpaired π electron of the ethynyl radical may react with the unsaturated acetylene molecule, leading to the formation of butatrienyl radical dimers of different structures. | ||
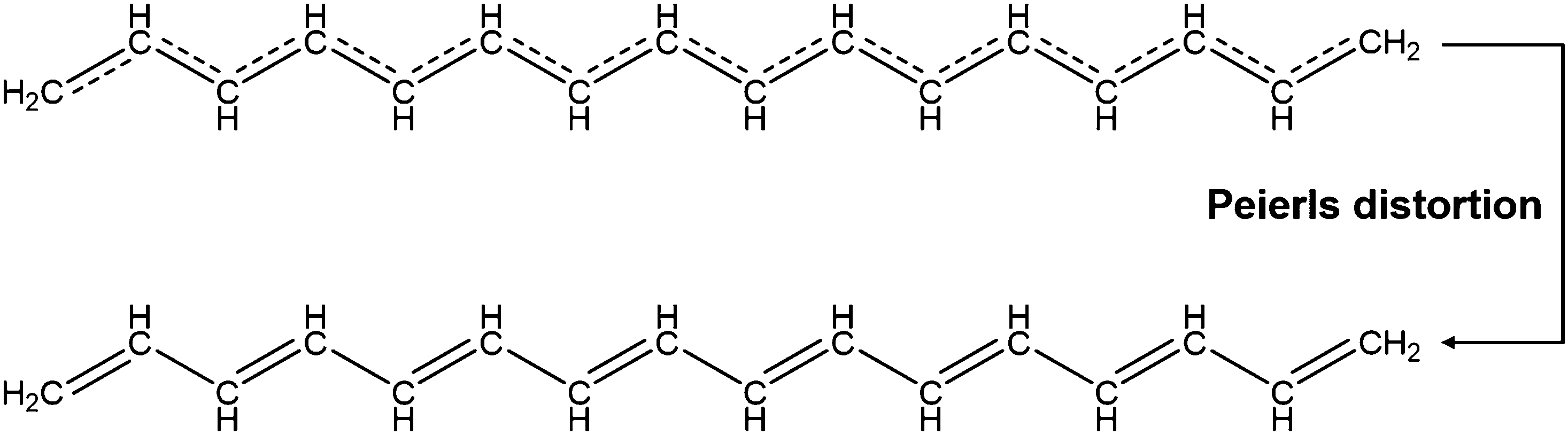 | ||
| Fig. 2 Schematic illustration of the Peierls distortion takes place on the polyacetylene molecule. Delocalization of the π electrons over the polymer backbone may be inhibited by the bond alternation caused by Peierls distortion. | ||
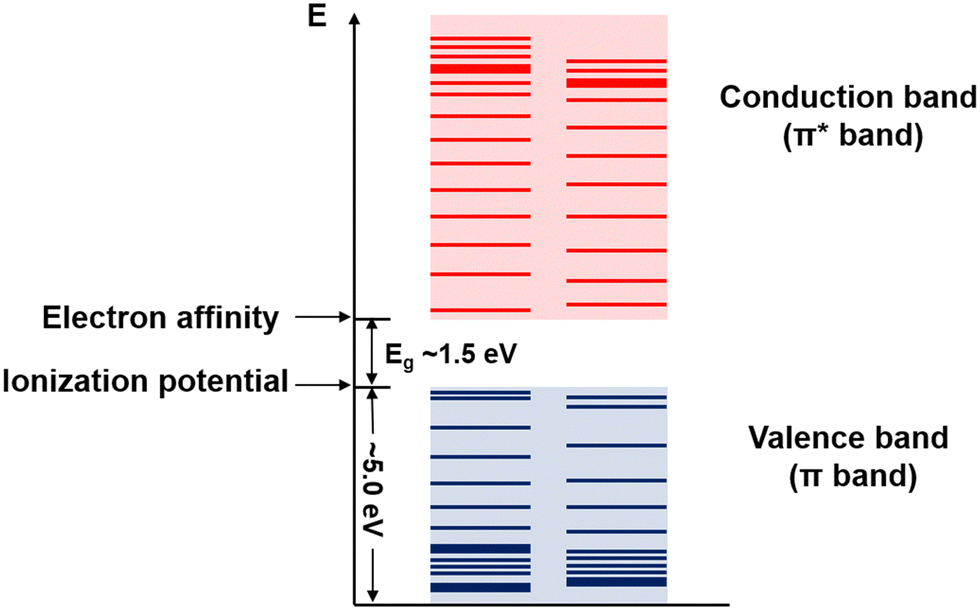 | ||
| Fig. 3 Schematic illustration of the band structure of polyacetylene. The values and margins of the band gap (Eg), ionization potential, and electron affinity are specified. | ||
The emergence of conductivity for conjugated polymers is fundamentally different from that for inorganic semiconductors, i.e., silicon, germanium, and titanium dioxide. At room temperature, the pristine inorganic semiconductors show conductivities typically in the range between 10−8 S cm−1 and 10−2 S cm−1.6 The conductivity of a few inorganic semiconductors, i.e., germanium, can reach a magnitude of 101 S cm−1 in their pristine state.7 The conductivities of other inorganic semiconductors, i.e., GaAs and CdS, are in the range between 10−7 S cm−1 and 100 S cm−1.8–10 For the pristine conjugated polymers, their conductivities are in the same range as those of most of the inorganic semiconductors. For example, the conductivity of pristine polyacetylene is around 10−9–10−8 S cm−1;11 the conductivity of undoped polythiophene is measured to be 1.48 × 10−12 S cm−1;12 and the conductivity of undoped polyaniline is around 10−10 S cm−1.13 The electrical and electronic properties of both organic and inorganic semiconductors, including conductivity, field-effect mobility, band gap, etc., can be engineered through a process called “doping”. Generally speaking, the doping process involves the incorporation of foreign atoms, molecules, and ions into the lattice/matrix of the semiconductor. Depending on the nature of charge carriers generated during the doping process, the doping process can be classified into n-type and p-type doping, which corresponds to electrons and holes as the major charge carriers, respectively. Upon doping, the electronic properties of the semiconductors can be significantly enhanced, and their conductivities are increased by several orders of magnitude. For example, the conductivity of polyacetylene may increase from 10−10 S cm−1 to 105 S cm−1 upon doping with iodine.14 The conductivity of Si single crystals may increase from 10−4 S cm−1 to 103 S cm−1 by heavy phosphorus doping.15
There are fundamental differences in the doping process for the inorganic semiconductors and organic conjugated polymers. Besides the differences in dopant types and doping techniques, the dopant concentration required to transform the semiconductor into a conductor is substantially different in the case of inorganic semiconductors and organic conjugated polymers. Generally speaking, it requires a dopant concentration ranging from 1014 dopant atoms per cm3 to 1020 dopant atoms per cm3 to achieve applicable electronic properties for inorganic semiconductors.16 Silicon crystals with dopant concentrations between 1018 atoms per cm3 and 1020 atoms per cm3 can be called heavily doped silicon.15,17 However, the absolute weight percentages of dopant concentration in inorganic semiconductors are extremely small based on the mass of the intrinsic semiconductor. For example, a dopant concentration of 4.5 × 1016 atoms per cm3 is equal only to 1 ppm for phosphorus doped silicon.18 On the other hand, the dopant concentration required to transform conjugated polymers into conductors is significantly higher than that required to transform into inorganic semiconductors. For example, the iodine doping levels of cis-polyacetylene can be tuned from 2.2 mol% to 18.7 mol% to obtain conductivities ranging from 102 S cm−1 to 10−1 S cm−1, respectively.19,20
The fundamental differences between inorganic semiconductors and conjugated polymers in the doping process arise from their intrinsic atomic, lattice, crystalline, and defect structures. On the other hand, the conductivities of the intrinsic conjugated polymers are found to be in the insulator range, while the conductivities of the intrinsic inorganic semiconductors are generally in the semiconductor range. It is reasonable to expect that the intrinsic conjugated polymers may require much higher dopant concentrations to modify their electronic structures, generating sufficient charge carriers to conduct electricity. Unlike doped inorganic semiconductors, which allow the free movement of electrons and holes in their conduction and valence bands, the movements of electrons and holes in doped conjugated polymers are non-linear and non-resonant, resulting in unique electronic, photonic, and thermoelectric properties of doped conjugated polymers.
The thermoelectricity of a conjugated polymer can be evaluated by the thermoelectric figure of merit (ZT), which can be determined using the following equation:
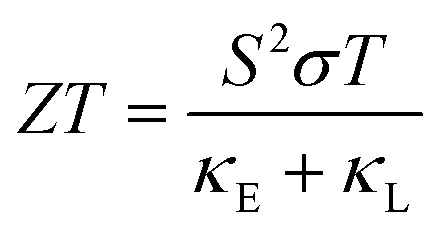 | (1) |
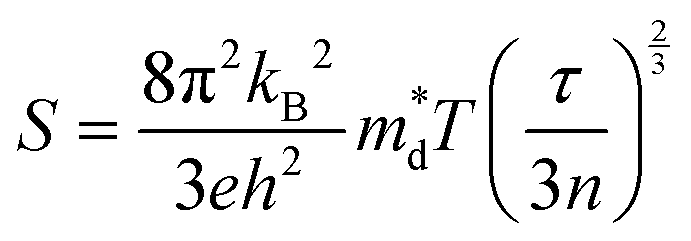 | (2) |
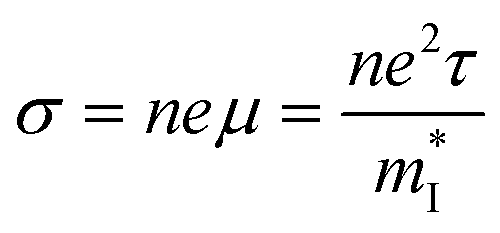 | (3) |
| κ = κE + κL = LσT + κL | (4) |
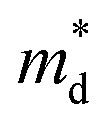 is the density of states effective mass, T is the absolute temperature, τ is the relaxation time, n is the charge carrier concentration, μ is the charge carrier mobility,
is the density of states effective mass, T is the absolute temperature, τ is the relaxation time, n is the charge carrier concentration, μ is the charge carrier mobility, 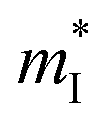 is the inertia band mass, κ is the total thermal conductivity and L is the Lorenz number.
is the inertia band mass, κ is the total thermal conductivity and L is the Lorenz number.
It can be seen from eqn (2) that S is negatively dependent on the charge carrier concentration (n), while σ is proportional to n. Moreover, S is proportional to the density of states of effective mass 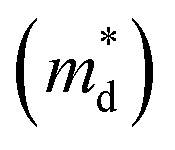 , while σ is inversely proportional to the inertial mass
, while σ is inversely proportional to the inertial mass 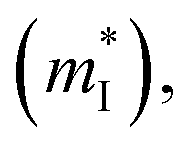 which is correlated with
which is correlated with 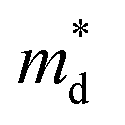 by
by 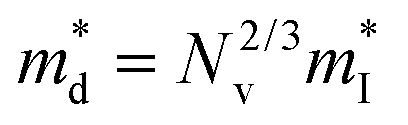 (Nv is the number of band valleys). Therefore, it is impossible to increase both S and σ simultaneously, while it is more rational to systematically engineer the parameters that are correlated with S and σ to achieve an optimized power factor (S2σ). The most effective way to engineer the power factor of the conjugated polymers is to tune their doping levels. The charge carrier concentration, n, can be significantly increased as the doping level of a conjugated polymer is increased from low to high. However, as the increase in charge carrier concentration may result in the nonequilibrium increase in σ and decrease in S, there exists an optimal doping level where the highest power factor can be reached. On the other hand, intercalation of dopant ions into the polymer lamellar structure may loosen the crystalline region and reduce the crystallinity, which may mitigate the effective phonon scattering by grain boundaries and interfaces, resulting in increased κLκ, and decreased τ. However, the increase in the effective structural disorder with elevated doping levels may in turn enhance the electron–phonon coupling strength in conjugated polymers, resulting in a higher charge carrier mobility by lowering the activation energy Ea based on the Arrhenius temperature dependence:
(Nv is the number of band valleys). Therefore, it is impossible to increase both S and σ simultaneously, while it is more rational to systematically engineer the parameters that are correlated with S and σ to achieve an optimized power factor (S2σ). The most effective way to engineer the power factor of the conjugated polymers is to tune their doping levels. The charge carrier concentration, n, can be significantly increased as the doping level of a conjugated polymer is increased from low to high. However, as the increase in charge carrier concentration may result in the nonequilibrium increase in σ and decrease in S, there exists an optimal doping level where the highest power factor can be reached. On the other hand, intercalation of dopant ions into the polymer lamellar structure may loosen the crystalline region and reduce the crystallinity, which may mitigate the effective phonon scattering by grain boundaries and interfaces, resulting in increased κLκ, and decreased τ. However, the increase in the effective structural disorder with elevated doping levels may in turn enhance the electron–phonon coupling strength in conjugated polymers, resulting in a higher charge carrier mobility by lowering the activation energy Ea based on the Arrhenius temperature dependence: 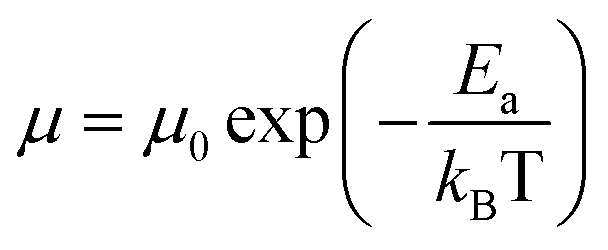 ,25 where μ0 is the infinite temperature mobility for all materials. Compared to the inorganic semiconductors, the conjugated polymers may have intrinsically low electron density, and a weak electron–electron interaction. Therefore, the electrons or holes generated during the doping process would tend to dielectrically polarize the polymer lattice, falling into one of the discrete levels, and localize.26 When an external electric field is applied, the polarized states may move like negative or positive charges, acting as the current carriers. Compared to electrons or holes, the effective mass of the polarized states is much larger,27 which may confer the conjugated polymers with intrinsically high Seebeck coefficients. Compared to the inorganic semiconductors, which may require careful engineering of the band structure, elemental composition, and dopants to achieve high ZT values, and tuning of the doping level, charge carrier density, mobility, and microstructure of the conjugated polymers can be completed simultaneously in a simple doping process based on the redox mechanism. Moreover, the conjugated polymer-based thermoelectrics are more solution processable and can be used to create a large-area patterned surface.
,25 where μ0 is the infinite temperature mobility for all materials. Compared to the inorganic semiconductors, the conjugated polymers may have intrinsically low electron density, and a weak electron–electron interaction. Therefore, the electrons or holes generated during the doping process would tend to dielectrically polarize the polymer lattice, falling into one of the discrete levels, and localize.26 When an external electric field is applied, the polarized states may move like negative or positive charges, acting as the current carriers. Compared to electrons or holes, the effective mass of the polarized states is much larger,27 which may confer the conjugated polymers with intrinsically high Seebeck coefficients. Compared to the inorganic semiconductors, which may require careful engineering of the band structure, elemental composition, and dopants to achieve high ZT values, and tuning of the doping level, charge carrier density, mobility, and microstructure of the conjugated polymers can be completed simultaneously in a simple doping process based on the redox mechanism. Moreover, the conjugated polymer-based thermoelectrics are more solution processable and can be used to create a large-area patterned surface.
In general, charge carriers in conjugated polymers can be generated by the formation of localized mid-gap states between the valence band and the conduction band. At first, holes or electrons are generated in the valence band and the conduction band of the conjugated polymer by redox doping, photo-excitation, or charge injection, respectively.28–30 Subsequently, the holes or electrons would distort the nearby lattice structures to lower their energy levels, forming localized topological excitons, e.g., soliton, polaron, bipolaron, as the charge carriers. Based on the reduction of energy by lattice distortion, the energy levels of the charge carriers from high to low can be written as soliton > polaron > ½ bipolaron. Taking one of the most studied conjugated polymers, poly(3-hexylthiophene) (P3HT), as an example, polarons can be generated in the lattice structure of P3HT typically by a p-doping process. As an electron is transferred to the dopant through a redox mechanism, a hole can be generated at a redox active site of the polymer chain, which subsequently distorts the surrounding lattice structures and forms a polaron with a width of ten monomer units, as shown in Scheme 1.
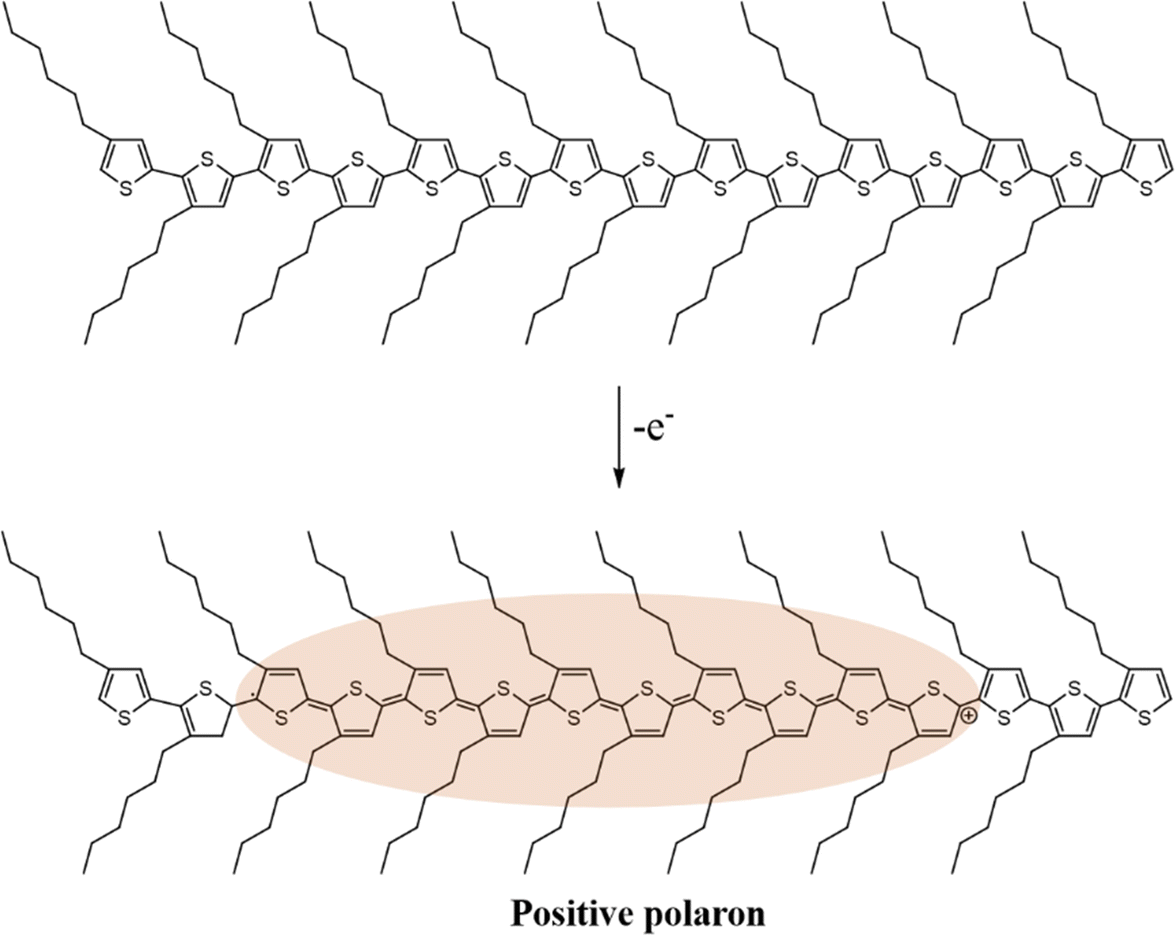 | ||
| Scheme 1 Formation of a positive polaron in the chain of P3HT. | ||
In conjugated polymers, polarons can be transported along the polymer matrix to conduct electricity in an external electric field. However, the transport of polarons is temperature-dependent and considered to be thermally activated. Different from the band transport of electrons and holes that is observed in metals, which shows a negative temperature dependence on mobility, the conjugated polymers generally show a positive temperature dependence on mobility, which can be explained by hopping transport mechanisms. In a hopping transport model, the polaron can be transported along the chain and between the chain. During an intrachain hopping process, the polaron hops from one site to the neighboring site along the chain through electronic coupling. On the other hand, the polaron hops from one chain to another through the proximate thiophene rings in the chains during an interchain hopping process. The activation energies for the hopping process differ significantly on the transport modes, crystallinity, and distances. For the hopping transport along the chain, the activation energy is negligible compared to the thermal energy at 300 K, indicating a quasiparticle-like transport of polarons along the chain.31 Therefore, the rate-limiting step of the polaron transport in conjugated polymers is the interchain hopping process. It is believed that an ordered and aligned lamellar structure can reduce the activation energy of the interchain hopping transport, while a disordered and amorphous structure can increase the activation energy, inducing a low mobility.32
However, a more precise control over the thermoelectric properties of conjugated polymers may require a full understanding of the quantum mechanism of the charge carriers in conjugated polymers, and a brief summary of the existing theories is given in the next section.
2. Band structures and models
The theoretical models regarding the band structures of conjugated polymers are inspired by the observations of low-energy, long-wavelength excitations in the mid-gap.33 This is because the characteristic correlation length (ξ) is significantly larger than the lattice constant (a) in many existing conjugated polymers, i.e., polyacetylene, polythiophene, and polypyrrole.34–36 Basically, a simple model, namely the noninteracting model, was constructed to describe the electronic structures of conjugated polymers derived from the Landau theory of normal Fermi liquids.37 The noninteracting model can provide an accurate prediction of the electronic structures of polyacetylene with low energy features (Fig. 4). However, the predicted properties by the model corresponding to the states near the Fermi surface are model-independent, whilst the properties corresponding to the states near band edges are model-dependent. Therefore, the noninteracting model may fail in predicting the properties of conjugated polymers corresponding to the states near band edges; however, to validate the noninteracting model under such circumstances, a few prerequisites must be satisfied (Heeger et al., 1988):38| Eg/Wc ≃ a/ξ ≪ 1 | (5) |
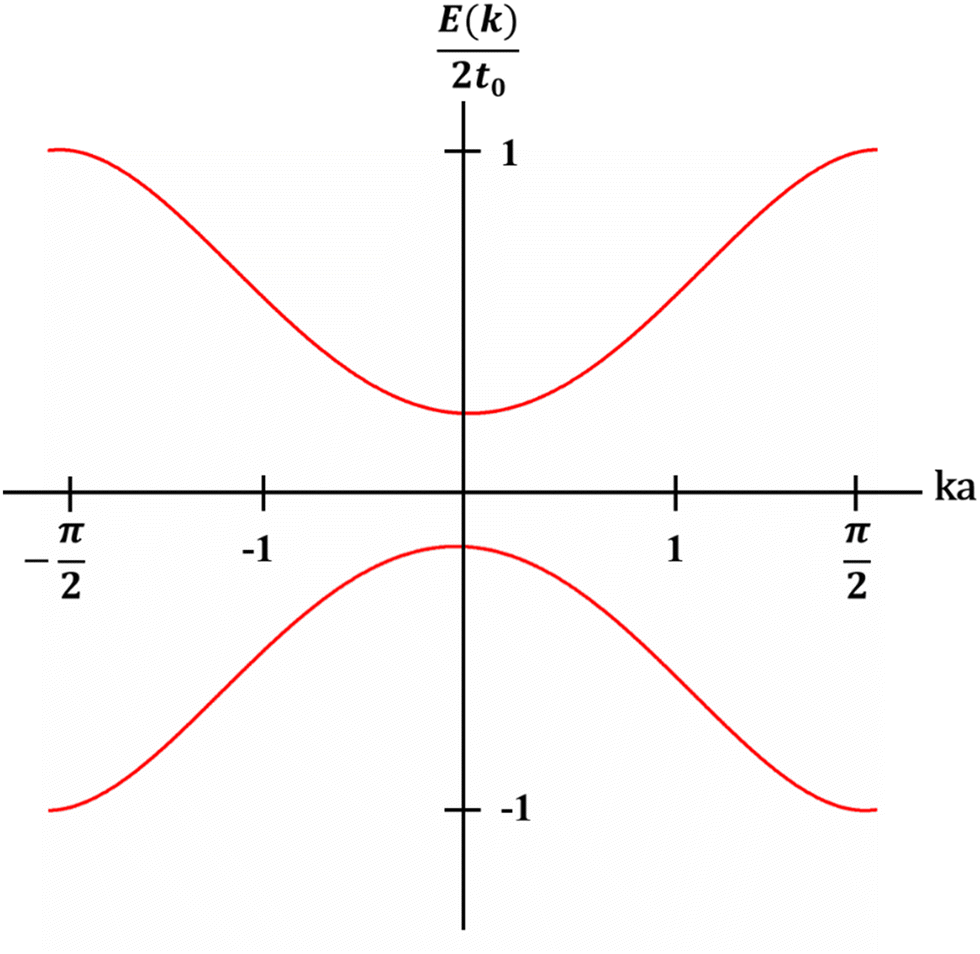 | ||
| Fig. 4 E–k plot of trans-polyacetylene obtained from the noninteracting model, where the energy of the electron (E) is plotted against the wavenumber k, and the corresponding band structure is revealed. | ||
Also (Kivelson et al., 1985)40
| U/Wc ≪ 1 | (6) |
| |Ec − EF| ≤ U | (7) |
The noninteracting model can predict the band structures of many conjugated polymers (e.g., polyacetylene) with satisfactory accuracy, which can be partially attributed to the weak electron–electron interactions in those systems. However, the noninteracting model did not correlate the low-energy, long-wavelength excitations with the characteristic lattice structures of the material; therefore, it cannot fully explain the physical origin of the conductivity for the doped conjugated polymers or predict the electronic properties of the conjugated polymers based on their lattice structures. To address this issue, the Su–Schrieffer–Heeger (SSH) model was proposed to explain the structural changes associated with low-energy excitations, which generated quasiparticle solutions such as soliton and anti-soliton to correlate the lattice distortion associated with the low-energy excitation.42 The simplified Hamiltonian of the SSH model can be written as (Su, W. P., Schrieffer, J. R. and Heeger, A. J., 1980)43
| HSSH = Hπ + Hπ–ph + Hph | (8) |
Provided n is the location of the carbon site residing in the center of low-energy excitation, for odd n, the shape of the low-energy excitation can be interpreted as44
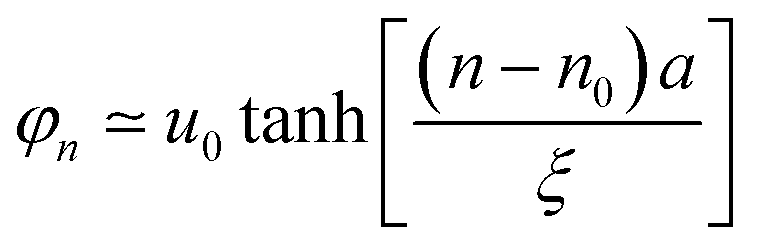 | (9) |
As a result of the two-fold ground-state degeneracy of the SSH model, the low-energy excitation can have two different phases, i.e., φn = u0 (odd n) and φn = −u0 (even n). For even n, the low-energy excitation can be interpreted as44
| φn ≃ −u0tanh(na/ξ) | (10) |
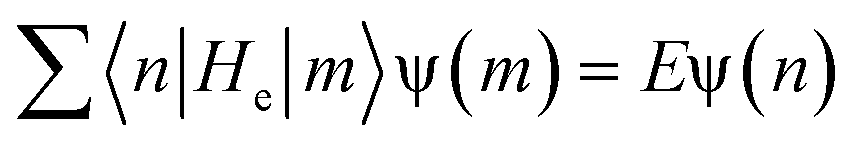 | (11) |
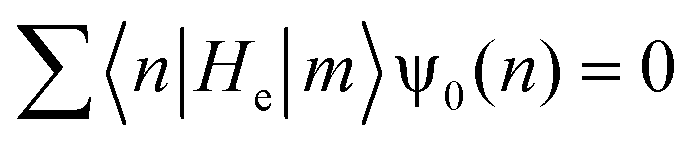 | (12) |
| ψ0(n + 1) = −Rψ0(n − 1) | (13) |
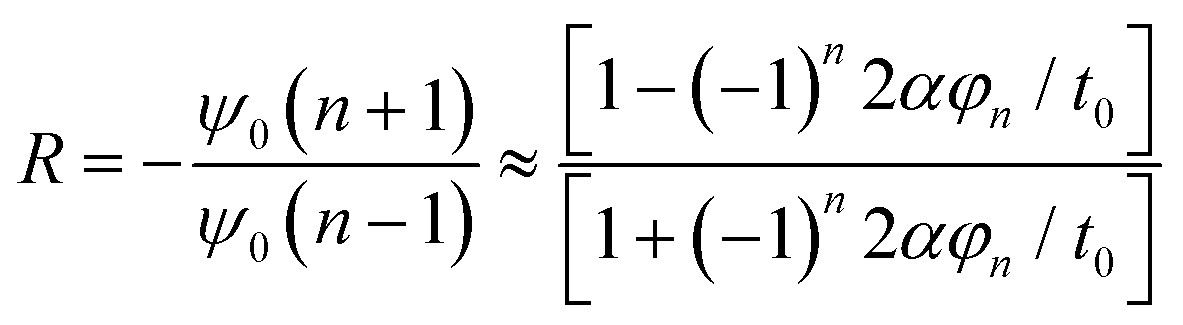 | (14) |
However, quasiparticles such as solitons and anti-solitons can only exist in conjugated polymers with a degenerate ground state, e.g., polyacetylene, for the geometric alternation of single and double bonds in the polymer backbone results in the ground states with the same energy.46 For the conjugated polymers with non-degenerate ground states, i.e., polythiophene, polypyrrole, and polyaniline, the coupling modes of charges to the underlying lattice structures may behave differently.47–49 For example, a spinless charged soliton is created when an electron is removed from the chain of polyacetylene, which can serve as the charge carrier.50 On the other hand, a radical ion with a spin of ½ is created when an electron is removed from the chain of polythiophene, which can serve as the charge carrier.51 For the non-degenerate conjugated polymers, the creation of a radical ion in the polymer chain may result in a localized charge associated with a local lattice distortion, which simultaneously generates a localized electronic structure between the LUMO and the HOMO. This electronic structure is called a polaron,52 and its corresponding band structure is shown in Fig. 5.
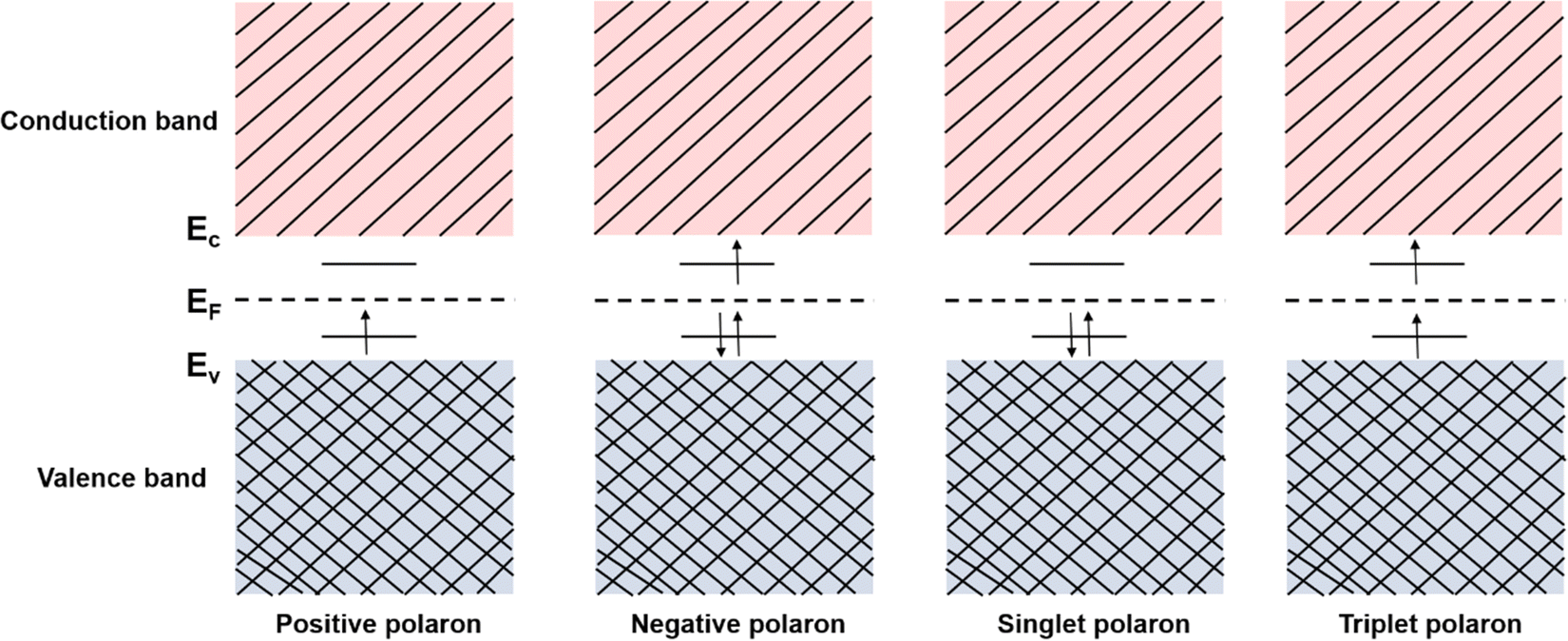 | ||
| Fig. 5 Schematic illustrations of the electronic structures of polaron. During the injections of holes and electrons into the conjugated polymer, the polarons would spontaneously form. Depending on the energy states and exciton interaction, there exist positive polarons (s = ½,⊕), negative polarons (s = ½, e−), singlet polarons (s = 0, j = 1), and triplet polarons (s = 1, j = 3). Ec, EF, and Ev mark the energy levels of the bottom of the conduction band, Fermi surface, and the top of the valence band, respectively. | ||
In inorganic semiconductors, the generation of electrons or holes in the lattice structure by doping may result in an extra donor level or acceptor level appearing close to the conduction band and the valence band, respectively.53 These emerged energy levels are close to the Fermi levels, which makes the electrons easily excited, generating free electrons in the conduction band (p-doped) and free holes in the valence band (n-doped), as shown in Fig. 6. On the other hand, in organic semiconductors, the doping process may lower the vertical ionization potential Eip or increase the electron affinity of the conjugated polymer to an extent of ΔE.54 The vertical ionization potential, Eip, corresponds to the difference in energy between the ground state and the ionized state of the conjugated polymers, which is constituted of the lattice distortion energy Ed, ionization energy of the distorted lattice Eid, and relaxation energy Ere, as shown in Fig. 7.
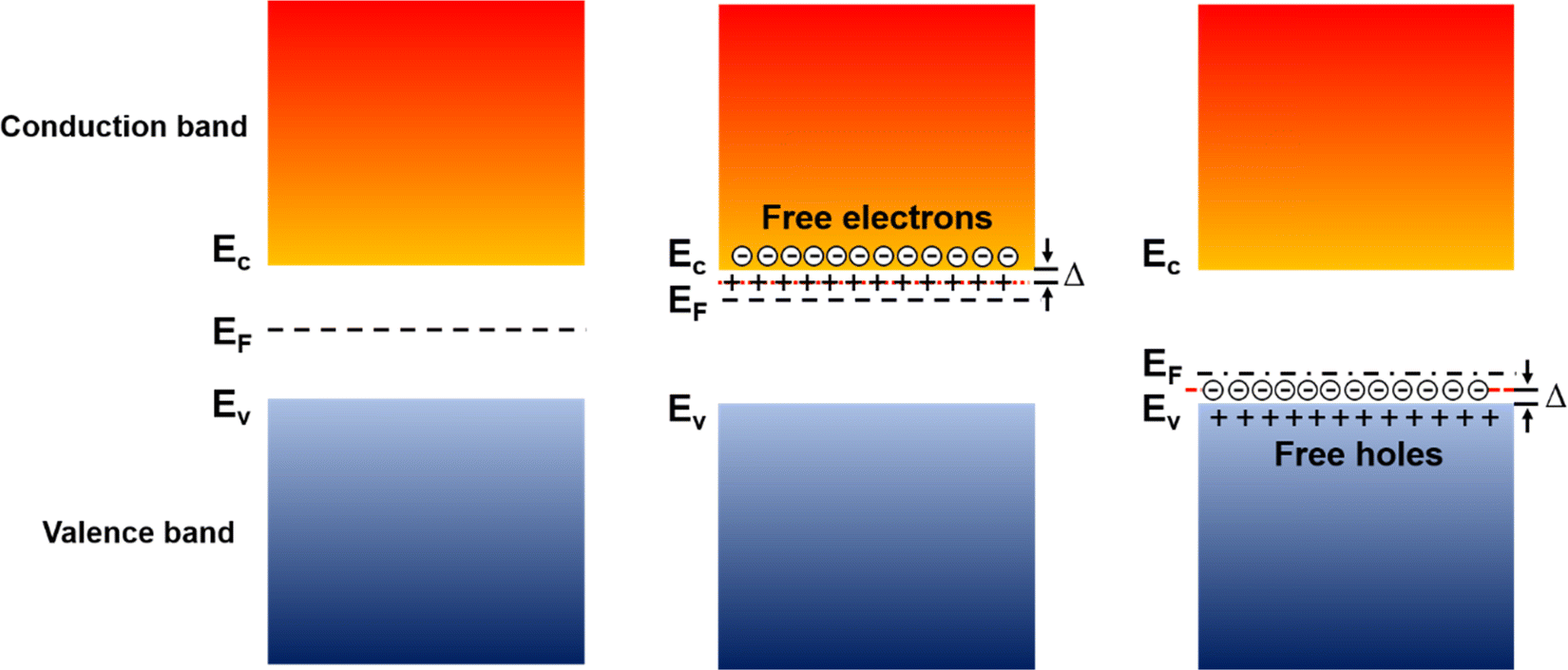 | ||
| Fig. 6 Schematic illustrations of the band structures of a neutral (left), n-doped (middle), and p-doped (right) inorganic semiconductor. Free electrons and free holes are created in the LUMO and HOMO of the conduction band and valence band, respectively, by overcoming the activation energy Δ required to dissociate the electrostatic interactions between the charge carriers and the dopant counterions. | ||
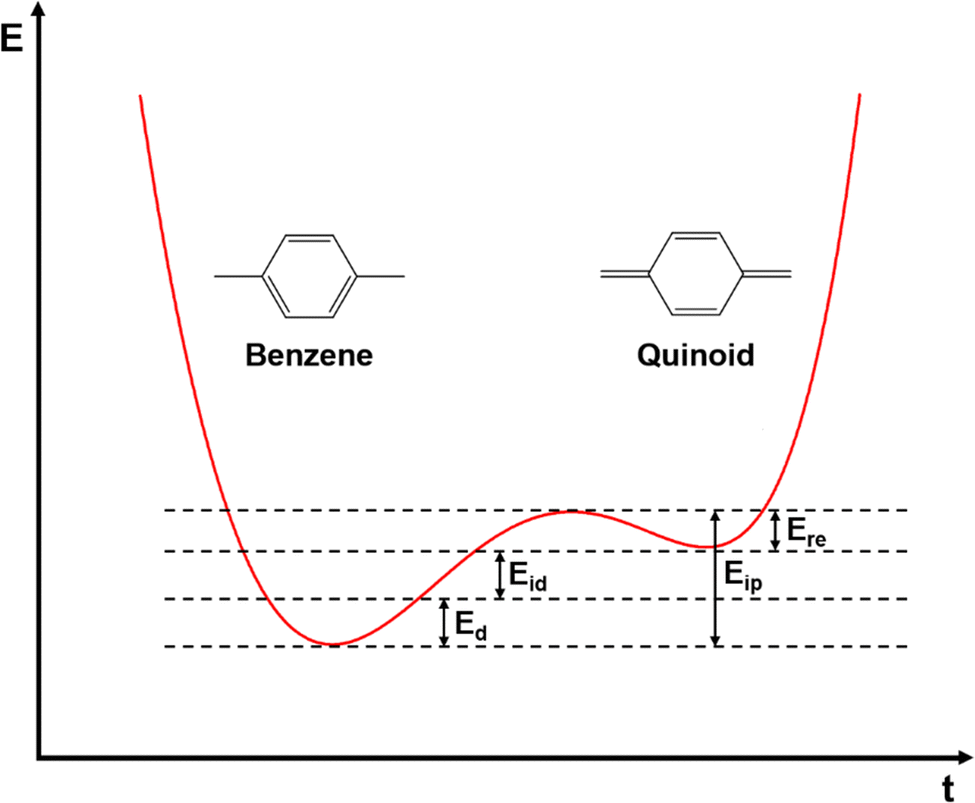 | ||
| Fig. 7 Schematic illustration of the changes in the states of energy during the ionization process of a conjugated polymer with nondegenerate ground states. The geometric structure of the molecules changed from benzene-like to quinoid-like as the energy of the molecules was elevated from the ground state by Eip. The components of Eip, i.e., Ed, Eid, and Ere, are depicted in the plot. | ||
Consider the case of taking an electron away from the backbone of a conjugated polymer through doping, which would lower the ionization energy by an amount of E0. If E0 > Ed, the molecular lattice may distort and couple to the as-created free radical ion, forming topological excitations. This thermodynamic process may result in the transition of the electronic structure, specifically, a downward movement of the LUMO and an upward movement of the HOMO, generating localized electronic states such as polarons. Due to the existence of an unpaired electron, a polaron has a spin of ½. If one more electron is removed from the backbone of conjugated polymer, the second polaron or a bipolaron can be formed, depending on the energy and location of the excitation. A bipolaron is a localized dication confined by strong lattice distortions. Since Ed for the generation of a bipolaron is higher than that for a polaron, the creation of a bipolaron may result in larger downward shifts of the LUMO, and larger upward shifts of the HOMO, respectively; as no unpaired electrons exist in bipolarons, they are spinless (Fig. 8). Whether one bipolaron or two polarons are formed depends on the binding energy Eb. In Hückel theory, Eb can be defined as the difference between ΔE and Ed.55 In several conjugated polymers, such as PPy and PPP, Eb for the generation of a bipolaron is larger than the Eb for the generation of two polarons, implying that the generation of a bipolaron is thermodynamically more favorable in these conjugated polymer systems.56,57
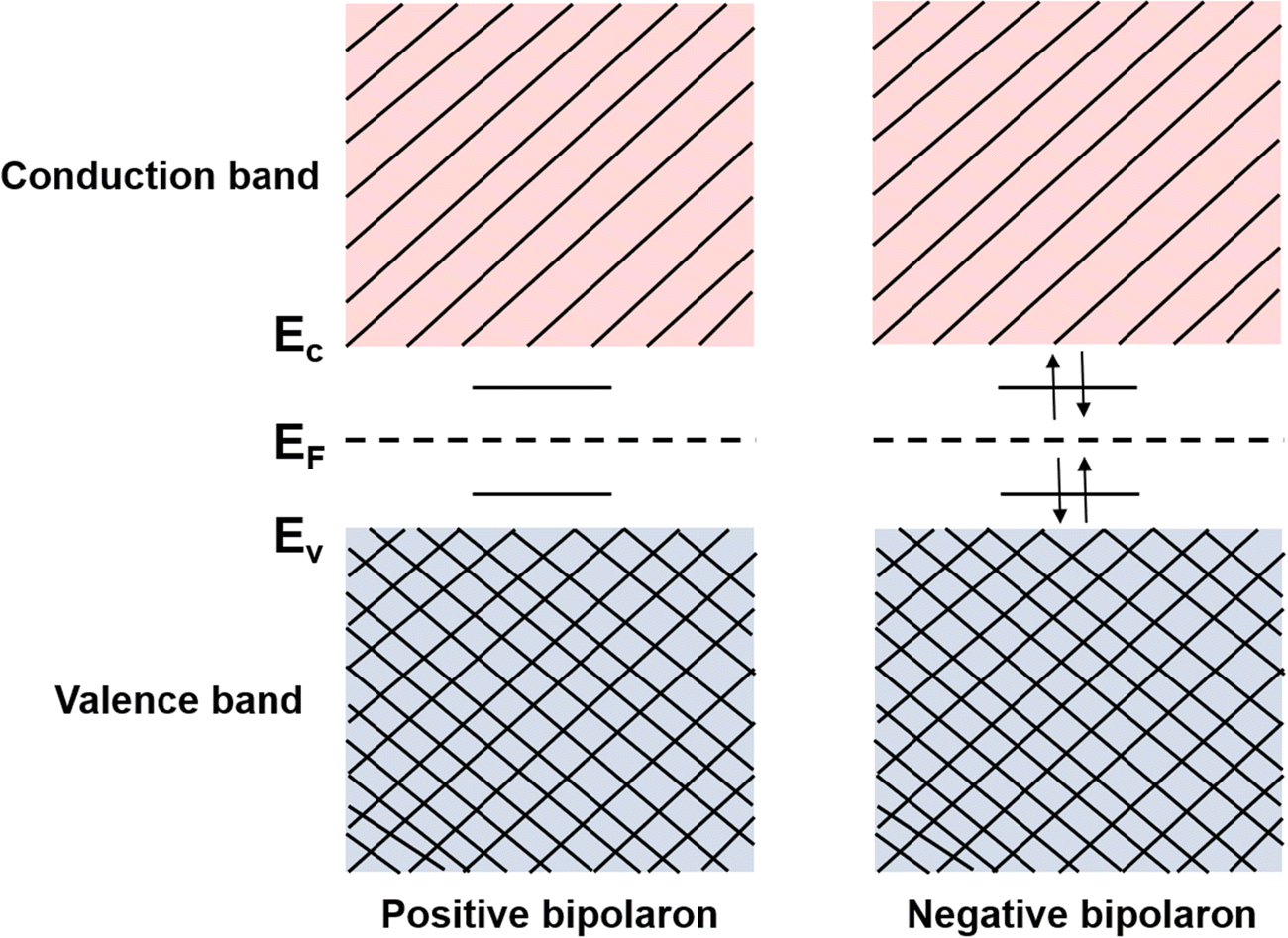 | ||
| Fig. 8 Schematic illustration of the electronic structures of a bipolaron. A bipolaron is formed by the combination of two free polarons. Depending on the nature of the polarons, there exist positive bipolarons (s = 0, 2+), and negative bipolarons (s = 0, 2e−). | ||
The theory for the generation of hole- and electron-polarons based on mid-gap states has been widely accepted and used to describe the band structures and transport phenomena in semiconductors. However, inconsistencies can be found by considering the generation of a second hole or the addition of a second electron, in the hole polaron and the electron polaron, respectively. Due to the formation of the singly occupied energy level above the valence band edge, the ionization energy for the generation of a second hole, IE+, should be smaller than the ionization energy for the generation of the first hole, IE0; on the other hand, due to the existence of the singly occupied energy level below the conduction band edge, the electron affinity for the addition of a second electron, EA+, should be larger than the electron affinity for the addition of the first electron, EA0, based on the mid-gap state theory. Recently, Koch N. et al. have investigated the electronic structure of polarons in C60 by using a combination of experimental techniques and density functional theory (DFT).58 The contradiction between the results of the inverse photoelectron spectroscopy (IPES) and ultraviolet photoelectron spectroscopy (UPS) in the presence of excess charges indicated that the frontier molecular orbitals can be split due to the Coulomb interactions (Hubbard interactions) between the local electrons. In this regard, the HOMO of the hole polaron would split into an upper, unoccupied sub-level which shifts into the band gap by the reorganization energy (λ); and a lower occupied sub-level below the neural HOMO by U-λ, as shown in Fig. 9a. On the other hand, the neutral LUMO of electron polaron would split into a lower, singly occupied sub-level shifts downwards by λ and an upper, empty sub-level shifts upwards by U-λ, as shown in Fig. 9b. Therefore, the correlations of IE0 < IE+ and EA0 > EA+ are more plausible for the polarons by considering the on-site Coulomb interactions.
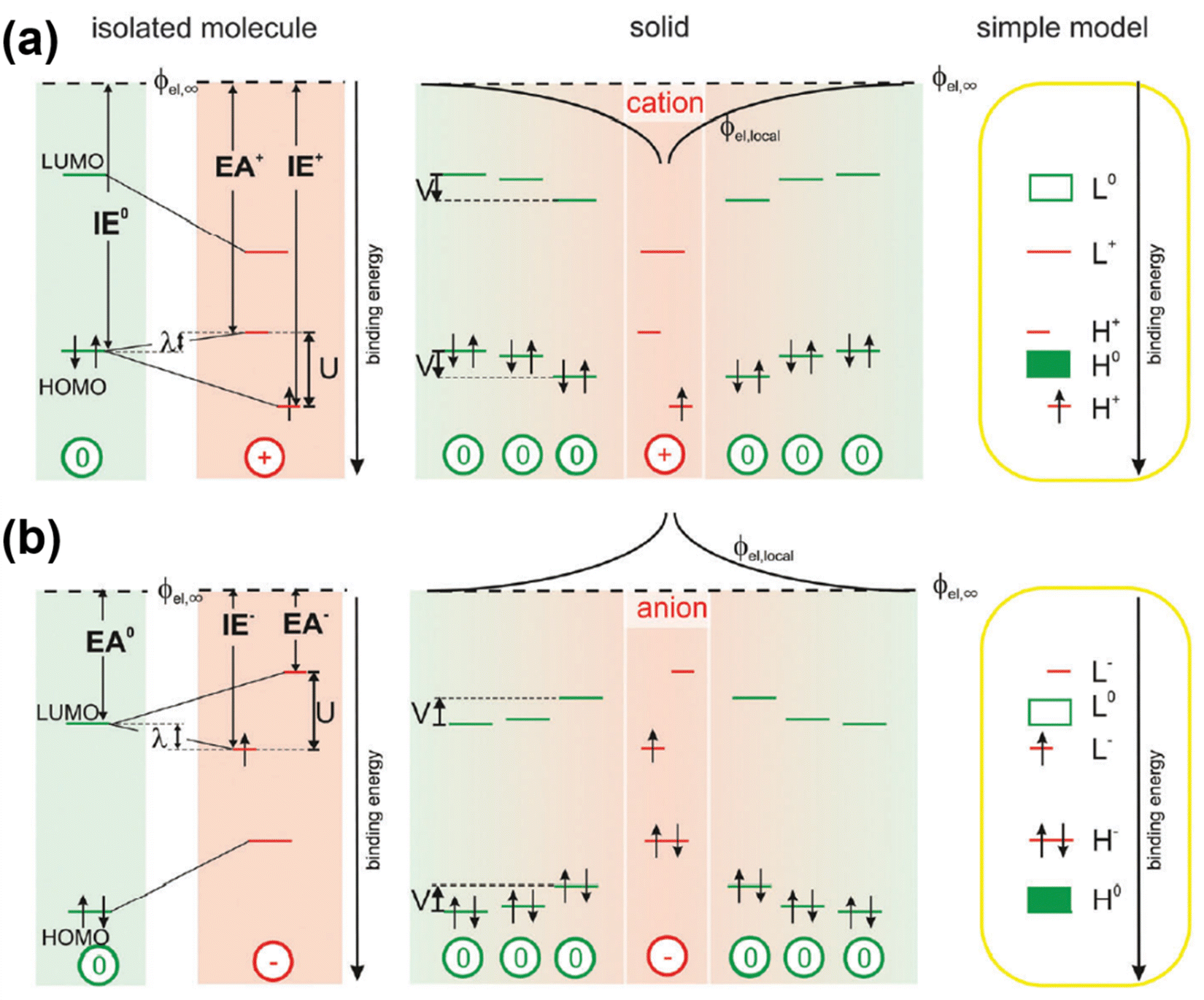 | ||
| Fig. 9 Schematic illustration of the band structures of a hole polaron (a) and an electron polaron (b) by considering the Coulomb interactions in the valence and conduction bands. Reproduced from ref. 58, under the license of CC-BY 3.0. | ||
The modified mid-gap theory which included the Coulomb interactions was supported by the findings of Heimel G., who used DFT calculations to compute the band structure of a conjugated polymer, poly(para-phenylene).59 By integrating the effects of adiabatic lattice relaxation, Coulomb interaction, and delocalization of excess charge on the conjugated polymers, Heimel has shown that for the electron polaron, the general optical transition of conjugated polymers in the low energy region (P1) can be ascribed to the excitations from the upper, singly occupied sub-level to the conduction band; and the optical transition in the high energy region (P2) can be ascribed to the excitations from the lower, doubly occupied sub-level bulged upwards to the conduction band, as shown in Fig. 10a. On the other hand, the P1 transition can be attributed to the excitations from the valence band to the lower, unoccupied sub-level for the hole polaron, and its P2 transition can be attributed to the excitations from the valence band to the upper, empty sub-level bulged downwards to the valence band, as shown in Fig. 10b. The modified model also satisfied IE0 < IE+ and EA0 > EA+.
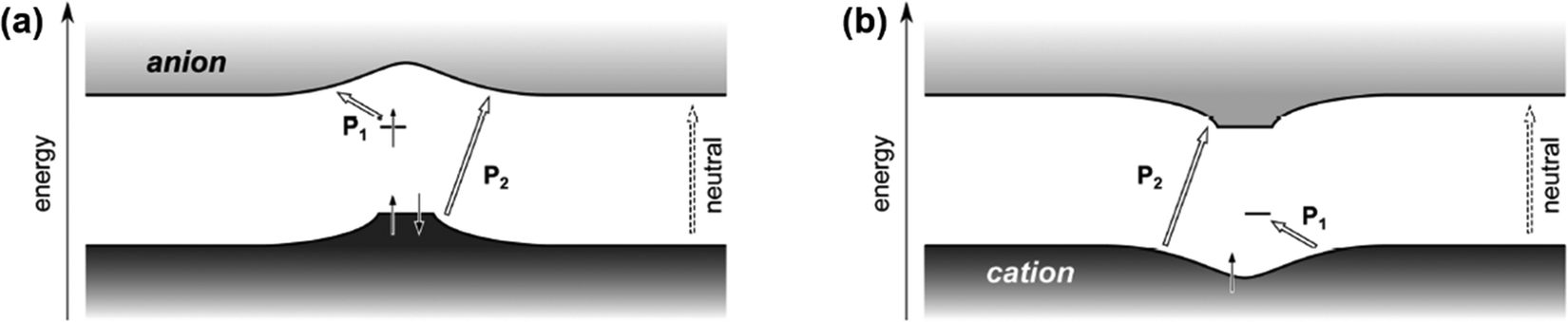 | ||
| Fig. 10 Schematic illustration of the band structures and P1, P2 transitions of an electron polaron (a) and a hole polaron (b) predicted by the nonempirical DFT calculation based on the exchange–correlation functional. Reproduced from ref. 59, with permission from American Chemical Society, 2016. | ||
By using a series of hole-doped triarylamine-fluorene (TAF) copolymers as the model system, Png R. et al. investigated the effects of the short-range (Hubbard) and long-range (Madelung) Coulomb interactions on the band structure of polarons.60 Significant broadening of the UPS spectra of the TAFs with increasing doping levels was observed, which was further interpreted by DFT calculations. The calculation results revealed a reversal of orbital ordering after adding a hole to the HOMO, as the HOMO may split into a new singly occupied molecular orbital (SOMO) which shifted downwards beneath the valence band edge, and a new empty SOMO* counterpart located above the valence band edge, due to the Hubbard interaction. This band model showed good consistency with the doping level- and counterion size-dependent work functions in organic semiconductors. Moreover, Zozoulenko I. et al. have reported the use of DFT, tight-binding DFT, and TD-DFT to simulate the band structures and UV-Vis-NIR absorption spectra of doped P3HT and doped P(g42T-T).61 In the DFT-based calculations, the conjugated polymers were primarily treated as oligomers of thiophene comprised of restricted repeating units, and the effects of π–π stacking, side chains, and dopants were also optimized and simulated. The UV-Vis-NIR spectra of the polythiophene oligomers predicted by DFT were in good agreement with the experimentally obtained spectra, indicating that the DFT-based model was feasible for quantitative applications. The band structure of polarons consisted of a single, unoccupied and empty level in the gap based on the DFT calculations, the P1 transition could be attributed to the excitation from the valence band to the polaron level, and the P2 transition could be attributed to the excitations from the valence band to the conduction band.
The emergence of P1 and P2 excitations in the mid- and near infrared regions of the optical absorption spectra of conjugated polymers is a signature formation of polarons localized in the polymer lattice. Since the polarons are primarily centered at the main chain carbon sites and the electronic coupling between the neighboring sites is strong, as a result of the one-dimensional electronic structure of the conjugated polymer, the transport of polarons along the chain (intrachain) is thermodynamically favorable. However, for the semicrystalline conjugated polymers, where the polymer chains are folded and layered to form an order lamellar structure with small interchain distances, the transport of polarons along the π-stacking direction (interchain) may become significant. Therefore, the polarons in the conjugated polymers are intrinsically two-dimensional.62 The effects of molecular structure and conformation, crystallinity, and molecular weight on the intrachain and interchain polaron transports can be used to predict and describe the differences in the electronic structure, property, and functions for different types of conjugated polymers. Intriguing perspectives have been provided by Spano F. C. et al., who used a Holstein-based Hamiltonian to describe 2D polaron transport in conjugated polymers nonadiabatically.63 By taking the disorder in the lamellar structure into account, Spano et al. were able to reproduce the mid-infrared absorptions of a semicrystalline P3HT experimentally obtained via transient pump–probe spectroscopy and charge modulation spectroscopy, with fine vibronic structures in correct positions and relative intensities. The fine reproduction of the experimental results by the Holstein-based model is achieved by the inclusion of spatially correlated diagonal (short-range) and off-diagonal (long-range) disorders. Specifically speaking, the disorder along the chain direction may attenuate the x-polarized spectrum, which may significantly affect the P1 transition originated from the intrachain transport in the higher energy region of the polaron spectrum; on the other hand, the disorder along the π-stacking direction may attenuate the y-polarized spectrum, which may significantly affect the DP1 transition originated from the interchain transport in the lower energy region of the polaron spectrum, as shown in Fig. 11.
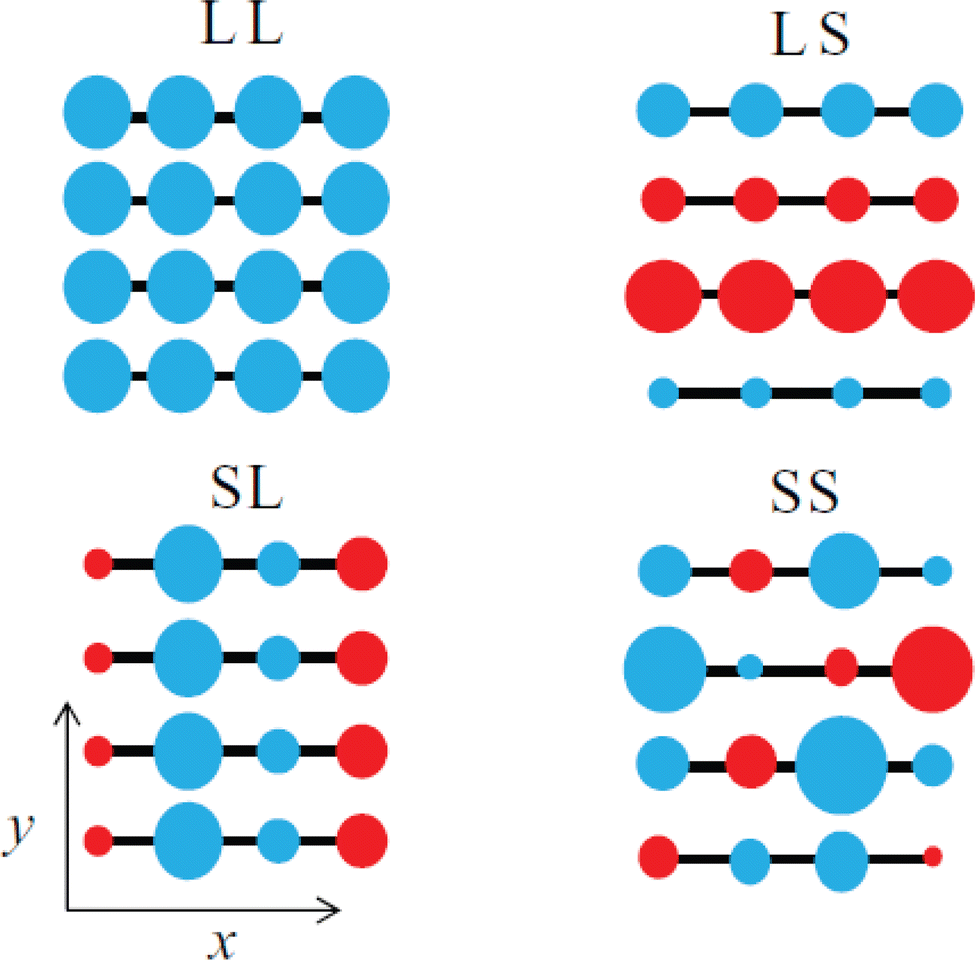 | ||
| Fig. 11 Schematic illustration of the spatial correlations of sited disorders in a 4 × 4 – π-stack polymer under ideal conditions, including LL, isotropic long-range broadening; LS, long-range broadening in the x-axis and short-range broadening in the y-axis; SL, short-range broadening in the x-axis and long-range broadening in the y-axis; SS, isotropic short-range broadening. The y-axis is the π-stacking direction, and the radius and color of the circle correspond to the energy and sign for deviations from planarity. Reproduced from ref. 63, with permission from AIP Publishing, 2014. | ||
By considering the correlations of the disorder effect and hole–dopant Coulombic bindings, Ghosh R. et al. further developed a modified model for predicting the mid-IR absorption of semicrystalline conjugated polymers using a Holstein-based Hamiltonian.64 Different from the Born–Oppenheimer descriptions, the low-energy excitation in the mid-IR absorption spectra (peak A) was derived from the Herzberg–Teller coupling as the oscillator strength was borrowed from the high energy excitation (peak B), in the strong polaron coupling domain. Therefore, the formation mechanism of peak A is intrinsically complicated, which can be attributed to the overlap of intra- and interchain spectral components mixed by disorder. Changes in peak A were closely correlated with the polaron coherence number, as the intensity of A would increase as the delocalization of polarons was enhanced, accompanied by a red shift and broadening of peak B. This new theoretical model can be used to describe the enhanced polaron delocalization with increased molecular weights observed in the mid-IR spectrum of regioregular (RR) F4TCNQ doped P3HT. Afterwards, Ghosh and Spano et al. further confirmed the consistency of the Holstein Hamiltonian-based model in predicting the mid-IR absorption spectra of oriented RR-P3HT films, and evidenced the adaptability of the new model to predict the mid-IR absorption spectrum of a diketopyrrolopyrrole (DPP) based donor–acceptor copolymer.65 Careful control over the disorder and dopants in the microstructures of the conjugated polymers may result in a significant enhancement in conductivity and thermoelectric properties. For example, Brinkmann M. et al. have recently reported the preferential doping of oriented P3HT films by tris(4-bromophenyl)ammoniumyl hexachloroantimonate (magic blue) in the amorphous region.66 Oriented by a high-temperature rubbing process,67 the P3HT film doped by magic blue can reach a conductivity of 3000 S cm−1 and a power factor of 170 ± 30 μW mK−2 parallel to the rubbing direction. Investigation of the polarized UV-Vis-NIR spectra perpendicular to the rubbing direction of the oriented P3HT films doped by different dopants showed that magic blue was able to induce the formation of the polaron bands, while other dopants, i.e., F4TCNQ and F6TCNNQ could not. No significant change was observed for the d-spacing of the (100) facet of P3HT crystallites after doping by magic blue. It thus indicated that magic blue was preferentially intercalated into the amorphous region of the oriented P3HT films, resulting in enhanced polaron delocalization in the crystalline region; hence significant improvements in the conductivity and power factor can be achieved, which coincides with the prediction from the Holstein-based model. The UV-Vis-NIR spectra of the magic blue-doped, oriented P3HT films showed an increased ratio between the A and B bands with increased dopant concentrations up to 2.0 g L−1, which was ascribed to enhanced polaron delocalization and the formation of bipolarons.
Since the spatial correlations of the intrachain and interchain polaron transport, and the short- and long-range Coulomb interactions may essentially formulate the bulk scale conductivity and thermoelectric properties of the conjugated polymers, it is important to investigate the effects of the molecular- and microstructure of the host polymer, dopant distribution, and dopant size on the polaron transport. Recent progress has been demonstrated by Chabinyc et al., who used a counter-ion exchange approach to investigate the effect of dopants with different sizes and functional groups on the conductivity and thermal stability of a conjugated polymer, poly[2,5-bis(3-tetradecylthiophen-2-yl)thieno[3,2-b]thiophene]-C14 (PBTTT).68 Dopants with different ionic diameters and functional groups, including (BF4)− (d = 5.2 Å), (PF6)− (5.8 Å), TCB− (6.6 Å) and PCF− (8.2 Å), were used to doped PBTTT; the corresponding mid-IR absorption spectra of PBTTT doped by different dopants manifested increased ratios between the intensities of peak A and peak B, and red shifts and broadening of peak B as the diameters of the dopants increased, which agreed with the enhancements in polaron delocalization depicted in the Holstein-based model, as shown in Fig. 12a. Moreover, the lamellar stacking distance (dL) was found to increase with the increasing dopant diameters, indicating that the lamellar structure would gently expand to incorporate the dopant counter ions, whilst the π-stacking distant (dπ) would slightly reduce, as shown in Fig. 12b. However, no significant enhancements in conductivity were observed along with the increases in dopant diameters, and the long-range orientational order and coherence length of the PBTTT crystallites were maintained after the dopant exchange process based on the results of resonant soft X-ray scattering and Williamson–Hall analysis. Eventually the authors came to a conclusion that the distance between the polaron and the dopant counter ion, rather than the identity of the dopant ion, may govern the electronic mobility of the polaron. This point of view was further supported by Müller C. et al., who investigated the chemical doping of a series of conjugated polymers with high ionization energies (IE > 5.3 eV) by magic blue (EA = 5.8 eV).69 The results showed that magic blue was capable of doping all of the sample conjugated polymers with 5.3 eV < IE < 5.8 eV. The effect of doping on the charge carriers of PDPP-3T was investigated in detail via potentiodynamic spectroelectrochemistry and chronoamperometry. The as-obtained UV-vis spectra of PDPP-3T doped by (PF6)− showed a consistent isosbestic point as the applied potentials changed from −0.45 V to +0.73 V, indicating that polarons were the only type of charge carrier formed from low to high doping levels. Compared to the PDPP-3T electrochemically doped by (PF6)−, the B peak of the UV-vis spectrum of PDPP-3T chemically doped by magic blue was slightly red-shifted for 40 nm, which was attributed to the larger thermochemical radius (r) of the counterions from magic blue [(SbCl6)−, r = 3.3 Å] compared to (PF6)− (r = 2.4 Å).
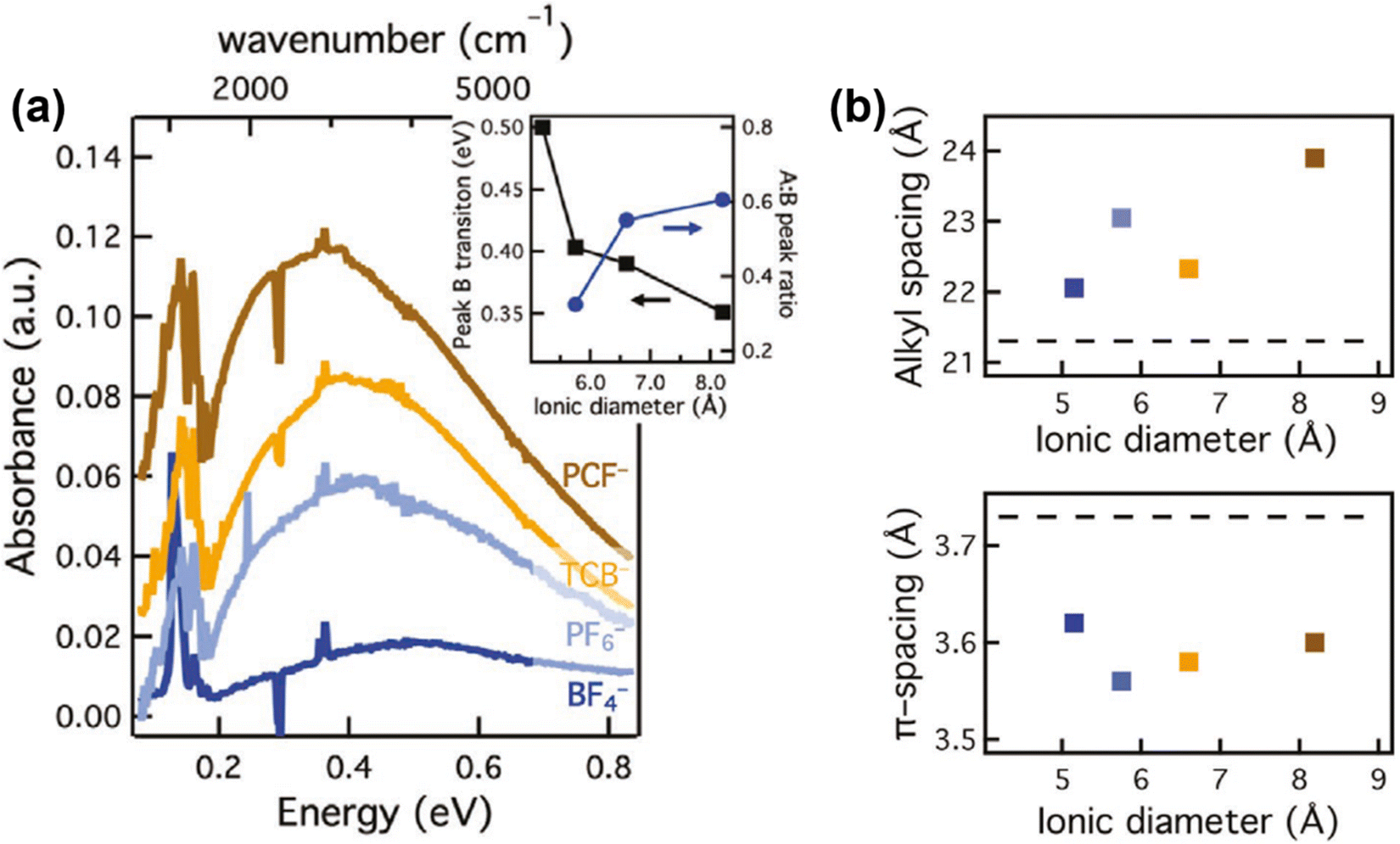 | ||
| Fig. 12 (a) FTIR spectra of PBTTT films containing PCF−, TCB−, (PF6)−, (BF4)−, acquired by the ion-exchange process. Inset shows the changes in the peak B positions and the ratios of the peak A and peak B intensities with respect to the ionic diameters of the counterions. (b) Alkyl and π-stacking spacing of the doped PBTTT films in (a) obtained from GIWAXS. The dashed lines correspond to the alkyl and π-stacking spacing of the pristine PBTTT. Reproduced from ref. 68, with permission from John Wiley and Sons, 2020. | ||
Chemically doped P3HT films with outstanding conductivity and power factor have been obtained by a combination of high-temperature rubbing process and utilization of molecular dopants with high EA and large size, as proposed by Brinkmann et al.70 In their report, P3HT thin films were firstly aligned uniaxially by the rubbing process and then doped by molybdenum dithiolene complex [Mo(tfd-COCF3)3] with both large size (d ∼ 11–14 Å) and high EA (∼5.6 eV). Due to the enhanced delocalization of the polarons, the charge carrier mobility of the oriented P3HT films can reach ∼9.3 cm2 V−1 s−1, and high conductivity (∼509 S cm−1) and power factor (160 μW m−1 K−2) can be simultaneously obtained. Scheunemann D. et al. also pointed out that large dopants with high EA can be very effective in generating polarons with high mobility.71 The distance between the dopant anion and the polaron, and the interaction between the dopant anion and the polaron, can also be effectively tuned by modifying the molecular structure of the conjugated polymers. For example, Nielsen C. B. et al. have reported the effect of polar side chains on the structure, charge transport, and thermal stability of a series of random copolymers of P3HT and poly(3-ethylene glycol methylthiophene) (P3EGMT) containing the ethylene glycol motif connected to the thiophene rings separated by a methyl group.72 It was found that as the content of P3EGMT in the copolymer increased from 5% to 30%, an increase in the coherence length from 160 Å to 240 Å was observed for the neutral P3HT and P3HT containing 20% P3EGMT, and increases in disorder accompanied by an expansion of lamellar spacing was observed for all the F4TCNQ doped P3HT-P3EGMT copolymers. The IR absorption spectra of the copolymers showed decreased intensities of the bands corresponding to the ag and b3g stretching modes of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, reduced ratios between the A and B peaks, and blue shifts of the B peaks, as the content of P3EGMT increased, which can be possibly attributed to the reduced planarity of the polymer chain, reduced polaron delocalization, and reduced distance between polarons and dopant anions. On the other hand, no significant changes in the dopant density and efficiency have been observed for the copolymers containing P3EGMT with polar side chains, indicating the observed deteriorated conductivity can be essentially ascribed to the reduced polaron mobility and interchain electronic coupling. The correlations between the molecular conformation, dopant size, EA and the doping mechanism were investigated in detail by Neher D. et al. by measuring the effect of different dopants on the formation of charge carriers in RR-P3HT and RRa-P3HT dissolved in chloroform, respectively.73 F4TCNQ, Mo(tfd-CO2Me)3 and tris(pentafluorophenyl)borane (BCF) were used as the dopants for comparison. Doping of RR-P3HT by these dopants showed identical absorption bands in the UV-Vis-NIR spectra, which was attributed to the integer charge transfer between the polymer and the dopant and the formation of polarons. Different polaron formation mechanisms for the dopants have been implied by the electron paramagnetic resonance (EPR). For F4TCNQ, the EPR spectrum indicated that the radical cations of RR-P3HT and F4TCNQ anions were closely coupled, forming relatively localized polarons. For BCF, the EPR spectrum was significantly broadened, which was attributed to the presence of protonated P3HT cations and protonated P3HT radicals. For Mo(tfd-CO2Me)3, the EPR spectrum showed a significantly weaker intensity compared to the case of F4TCNQ, and Mo(tfd-CO2Me)3 band independent from the polaron band, indicating a much weaker interaction between Mo(tfd-CO2Me)3 and RR-P3HT. However, F4TCNQ only formed a charge transfer complex with RRa-P3HT, and Mo(tfd-CO2Me)3 cannot induce the polaron formation in RRa-P3HT, which showed that the molecular conformation and planarity of the conjugated polymer may have more significant impact on the doping mechanism compared to the shape and size of the dopant molecule. Recently, Sirringhaus et al. also demonstrated the irrelevance between the ionic size of the dopant counterions to the electrical conductivity of the conjugated polymer by using the ion-exchange technique, whilst good correlations between the paracrystallinity and the electrical conductivity were observed.74
N, reduced ratios between the A and B peaks, and blue shifts of the B peaks, as the content of P3EGMT increased, which can be possibly attributed to the reduced planarity of the polymer chain, reduced polaron delocalization, and reduced distance between polarons and dopant anions. On the other hand, no significant changes in the dopant density and efficiency have been observed for the copolymers containing P3EGMT with polar side chains, indicating the observed deteriorated conductivity can be essentially ascribed to the reduced polaron mobility and interchain electronic coupling. The correlations between the molecular conformation, dopant size, EA and the doping mechanism were investigated in detail by Neher D. et al. by measuring the effect of different dopants on the formation of charge carriers in RR-P3HT and RRa-P3HT dissolved in chloroform, respectively.73 F4TCNQ, Mo(tfd-CO2Me)3 and tris(pentafluorophenyl)borane (BCF) were used as the dopants for comparison. Doping of RR-P3HT by these dopants showed identical absorption bands in the UV-Vis-NIR spectra, which was attributed to the integer charge transfer between the polymer and the dopant and the formation of polarons. Different polaron formation mechanisms for the dopants have been implied by the electron paramagnetic resonance (EPR). For F4TCNQ, the EPR spectrum indicated that the radical cations of RR-P3HT and F4TCNQ anions were closely coupled, forming relatively localized polarons. For BCF, the EPR spectrum was significantly broadened, which was attributed to the presence of protonated P3HT cations and protonated P3HT radicals. For Mo(tfd-CO2Me)3, the EPR spectrum showed a significantly weaker intensity compared to the case of F4TCNQ, and Mo(tfd-CO2Me)3 band independent from the polaron band, indicating a much weaker interaction between Mo(tfd-CO2Me)3 and RR-P3HT. However, F4TCNQ only formed a charge transfer complex with RRa-P3HT, and Mo(tfd-CO2Me)3 cannot induce the polaron formation in RRa-P3HT, which showed that the molecular conformation and planarity of the conjugated polymer may have more significant impact on the doping mechanism compared to the shape and size of the dopant molecule. Recently, Sirringhaus et al. also demonstrated the irrelevance between the ionic size of the dopant counterions to the electrical conductivity of the conjugated polymer by using the ion-exchange technique, whilst good correlations between the paracrystallinity and the electrical conductivity were observed.74
At high doping levels, bipolarons, other than polarons, may form preferentially in the conjugated polymers in the nondegenerate ground state, e.g., polythiophene, polypyrrole, P3HT. However, the removal of one electron from the polymer lattice may result in lifting of the degeneracy via a one-electron crystal potential:75
| Δ = Δi +Δe | (15) |
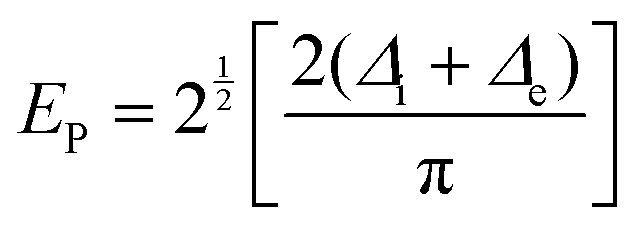 | (16) |
The minimum energy required to create a bipolaron is given by
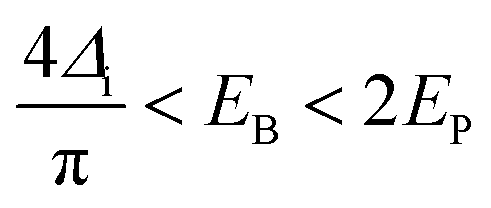 | (17) |
Compared to polarons, the mid-gap states of bipolarons are either fully occupied or empty, which means bipolarons are spinless, doubly charged carriers. It has been previously reported by Nowak et al. that the charge carrier configuration of single P3HT chains doped with NO+(PF6)− in dilute solution matched the existence of bipolarons, based on the EPR and spectroscopic measurements.77 On the other hand, the formation of bipolarons as the charge carrier at a low doping level may reflect a weak electron–electron Coulomb interaction in the band structures. However, the EPR measurements of polypyrrole showed an initial increase in the spin density at low doping levels, indicating the formation of polarons; this trend was reversed at high doping levels, indicating the formation of bipolarons.78 The co-existence of polarons and bipolarons in polypyrrole upon doping indicated that their energies were nearly degenerate, which can be attributed to strong electron–electron Coulomb interactions. By using the in situ spectroelectrochemistry technique, Enengl C. et al. systematically investigated the doping induced UV-Vis-NIR and mid-IR absorption bands of RR-P3HT thin films electrochemically, while acetonitrile and tetrabutylammonium hexafluorophosphate (TBAPF6) were used as the solvent and electrolyte, respectively.79 The RR-P3HT thin films showed polaron–bipolaron degeneracy analogous to polypyrrole, and as the increase of the absorption band of polarons (∼740 nm) dominated in the oxidation potential between +0.4 V and +0.9 V, the absorption band of bipolarons (in the mid-IR range) gradually increased and dominated in the potential range between +0.9 V and +1.3 V, accompanied by the decrease in intensity of the polaron band. Interestingly, the mid-IR absorption band peak around 4200 cm−1 (0.52 eV), which is generally recognized as the P1 band, broadened and red-shifted to lower energies (0.35 eV) as the oxidation potential increased from +0.9 V to +1.3 V, as shown in Fig. 13a. The broadening and red-shift of the P1 band can be possibly attributed to the stabilization of the bipolarons by enhanced electronic correlations, as a result of the enhanced crystallinity as the doping level increased. In contrast, a blue shift in the P1 band was observed for RR-P3HT doped by a Lewis acid–base pair, BCF-benzoyl peroxide (BPO), as recently reported by Chabinyc et al.80 As shown in Fig. 13b, continuous increases in the intensities of P1 and P2 bands of RR-P3HT have been observed as the dopant concentrations increased from BCF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BPO = (0.05:0.025) to BCF:BPO = (0.3:0.15). The sub-absorption bands of P1, i.e., P1a (lower energy sharp peak) and P1b (higher energy broad peak), also redshifted as the dopant concentration was increased, indicating enhanced delocalization of the doping induced polarons. However, the P1 band abruptly changed as the dopant concentration increased to BCF:BPO = (1:0.5), where the P1b band significantly strengthened, broadened and blue-shifted, accompanied by the decreases in the intensities of the P1a and P2 bands. The incoherence between the mid-IR signatures at low and high doping concentrations indicating the formation of a charge carrier different from polarons at high doping levels. Due to the strong Coulomb interactions between the dopant counterions and polarons at high doping levels, the hole–hole repulsion between polarons may become screened, and the polarons may interact to form localized bipolarons, resulting in the blue-shifts of the P1 band.
:BPO = (0.05:0.025) to BCF:BPO = (0.3:0.15). The sub-absorption bands of P1, i.e., P1a (lower energy sharp peak) and P1b (higher energy broad peak), also redshifted as the dopant concentration was increased, indicating enhanced delocalization of the doping induced polarons. However, the P1 band abruptly changed as the dopant concentration increased to BCF:BPO = (1:0.5), where the P1b band significantly strengthened, broadened and blue-shifted, accompanied by the decreases in the intensities of the P1a and P2 bands. The incoherence between the mid-IR signatures at low and high doping concentrations indicating the formation of a charge carrier different from polarons at high doping levels. Due to the strong Coulomb interactions between the dopant counterions and polarons at high doping levels, the hole–hole repulsion between polarons may become screened, and the polarons may interact to form localized bipolarons, resulting in the blue-shifts of the P1 band.
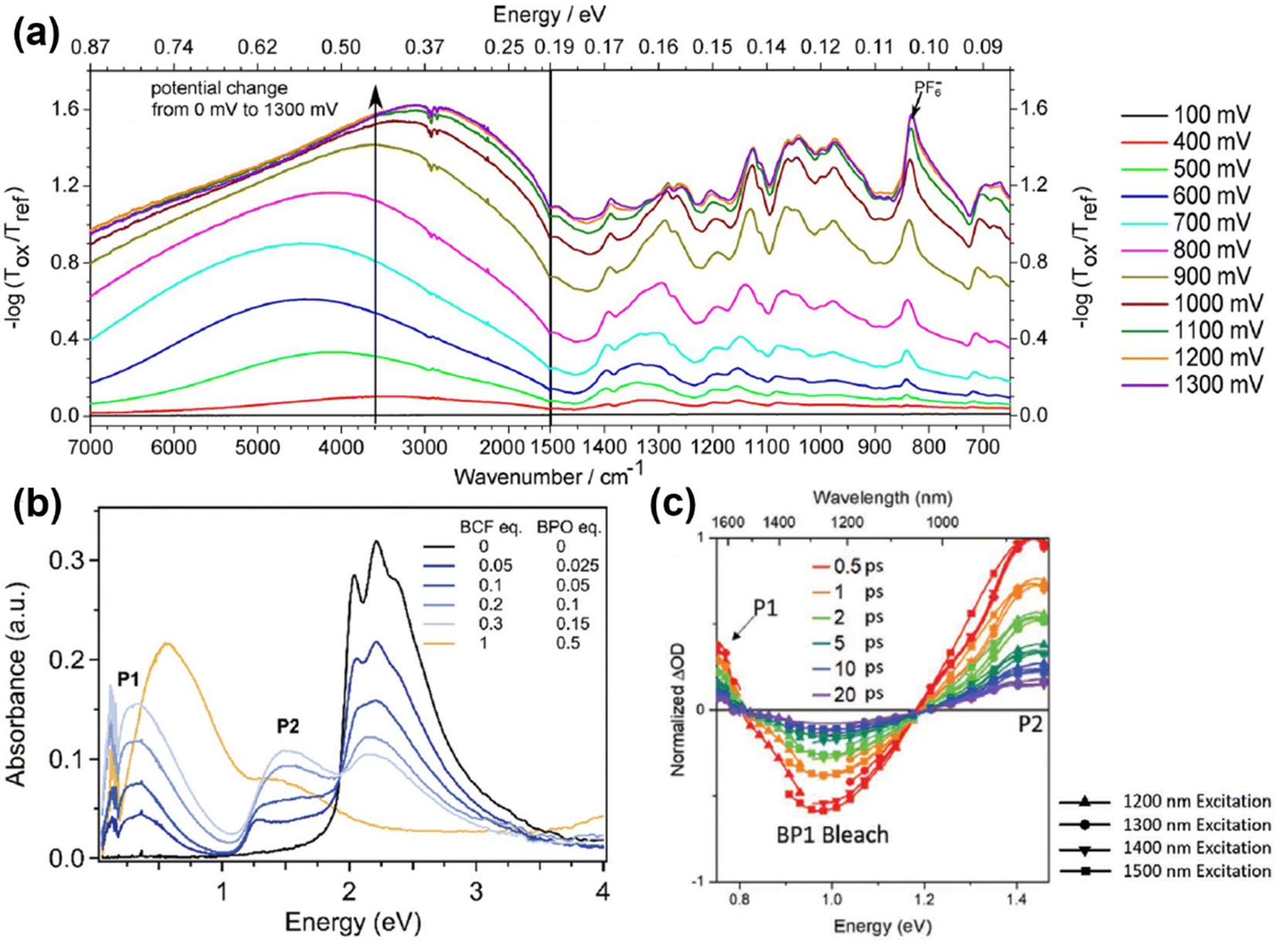 | ||
| Fig. 13 (a) In situ IR absorption spectra of the RR-P3HT thin film during electrochemical oxidation in the applied potential range from 400 mV to 1300 mV, taking 100 mV as the potential step. All spectra were calibrated by taking the spectrum obtained at an applied potential of 100 mV as the reference. Reproduced from ref. 79, with permission from John Wiley and Sons, 2016. (b) Combined FTIR and UV-vis spectra of RR-P3HT thin films doped by increasing BCF-BPO concentrations. Reproduced from ref. 80, with permission from Royal Society of Chemistry, 2022. (c) Ultrafast transient absorption spectra of PBTDTP doped by F4TCNQ excited at different delay times and wavelengths. Reproduced from ref. 81, with permission from John Wiley and Sons, 2020. | ||
Moreover, a blueshift of the P1 band and the absence of the P2 band have been observed for a donor–acceptor conjugated polymer, poly[(4-(2-hexyldecyl)-4H-dithieno[3,2-b:2′,3′-d′]pyrrole)-2,6-diyl-alt-(2,5-bis(3-dodecylthiophen-2-yl)benzo[1,2-d;4,5-[4]d′]bisthiazole)] (PBTDTP), at low doping levels, according to Schwartz et al.81 Compared to the other donor–acceptor conjugated polymers which generally contain no more than 4 rings in the donor segment,82 PBTDTP has a larger donor size of 5 rings, which may significantly weaken the electronic correlations between the donor and the acceptor units. The IR absorption spectra of F4TCNQ doped PBTDTP showed a single absorption band peak at ∼0.83 eV, which was in between the energies of P1 (∼0.5 eV) and P2 (∼1.5 eV) excitations. The intensity of the absorption band increased with elevated dopant concentrations, while no additional IR bands were observed to emerge, indicating the existence of a single type of charge carrier, as shown in Fig. 13c. However, by splitting the single absorption band using ultrafast transient absorption spectroscopy, the P1 and P2 bands of polaron emerged and decayed slowly (>20 picoseconds), indicating that the single IR absorption band was caused by bipolarons. The sole existence of bipolarons as the charge carriers in PBTDTP can be attributed to the lifted ground-state degeneracy by the energetic difference between the bulky donor and the acceptor segments, and the blue shift of bipolaron band can be ascribed to the screened hole–hole repulsion in the main chain by the proximate dopant counterions.83
The nature of charge carriers may have a significant impact on the electronic, electrical, and thermoelectric properties of the conjugated polymers. However, the allowed types of charge carriers are intrinsically encoded in the electronic structure of the conjugated polymer, which can be induced, transformed, and reversed by the application of suitable doping processes. For conjugated polymers with lifted ground-state degeneracy upon doping, a transition from the polarons to bipolarons as the doping-induced charge carriers can be observed. During the transition, the mid-IR signature of polaron (P1) can be either red-shifted or blue-shifted, depending on the degeneracy between polarons and bipolarons. Moreover, the degeneracy can be effectively tuned by the short- and long-range Coulomb interactions in the polymer lattice, e.g., molecular structure, conformation, dopant–polaron distance, paracrystallinity, as weak Coulomb interactions may favor the formation bipolarons at high doping levels.
3. Unveil the electronic structures of conjugated polymer: experimental studies and theoretical models
Over the decades, intensive research effort has been devoted to understanding the electrical, electronic, optical, thermal, and interfacial properties of various kinds of conjugated polymers, while their structural–property correlations have been carefully characterized. However, the theoretical nature of the electronic properties of conjugated polymer are still under debate, for the nomenclature of “polaron” and “bipolaron” may correspond to different charged species in conjugated polymers of different molecular and micro-structures. For example, similar UV-vis-NIR spectra can be obtained for doped conjugated polymers with different molecular structures; in general, three excitations corresponding to the neutral state, polaron, and bipolaron may appear in the spectrum.84 However, the position, full width at half maximum, and intensity of the excitations may differ, depending on the doping level, charge carrier density, charge carrier mobility, and microstructure of the conjugated polymers (Fig. 14).85–88 In general, the UV-Vis-NIR spectrum of the conjugated polymers in the neutral state may show an essential absorption band in the high energy region, typically between 4.00 eV (∼310 nm) and 1.77 eV (∼700 nm), which corresponds to the band gap transition.85,89 For example, the UV-Vis-NIR spectrum of P3HT showed a distinct broad band absorption from 400 nm to 650 nm, which showed multiple absorption peaks (Fig. 14a). Upon doping the P3HT thin films by using the chronoamperometry technique, it was observed that the intensity of the absorption band corresponding to the band gap transition gradually decreased as the applied oxidation voltage gradually increased. On the other hand, a new absorption band corresponding to the polaron excitation centered at ∼800 nm emerged and enhanced with elevated oxidation potential, indicating the increasing doping levels, as shown in Fig. 14a. The doping level of the conjugated polymer can also be controlled by changing the dopant concentration. For example, the doping level of a series of linear copolymers consisting of diketopyrrolopyrrole (DPP) and thiophene (T) as the repeating units, can be increased by raising the concentration of the dopant solution, as evidenced by the emergence and enhancement of the polaron band centered at ∼1500 nm (Fig. 14b). Moreover, the doping process may create analogous electronic structures in the conjugated polymer, regardless the kinds of dopant used. For example, the UV-Vis-NIR spectra of polyphenylene vinylene (PPV) doped by different dopants, i.e., FeCl3, H2SO4, I2, can be generally deconvoluted into four components, while the component bands labelled as 1, 2, 3 can be attributed to the formation of polarons and bipolarons (Fig. 14c). For PPV doped with different dopants, the shapes and intensities of the bands 1, 2, and 3 were analogous to each other while their positions differed, indicating the differences in strength for the polymer–dopant interaction. The microstructure of the conjugated polymer would also affect its UV-Vis-NIR spectrum, as a result of a complex effect involved the synthesis process, morphology, and doping level. For example, Chen, Y. F. et al. reported the use of etched ion-track polycarbonate (PC) templates to synthesize PPy nanowires and tubules with different diameters using the potentiometry technique.88 Diameter-dependent UV-Vis-NIR absorption characteristics were observed for the as-obtained Ppy nanostructures, as the polaronic bands may shift to longer wavelengths with increasing diameters (Fig. 14d). Moreover, higher doping levels were found in the as-obtained Ppy nanowires and nanotubules with larger diameters, indicated by the red shift of the polaron bands and the emergence of bipolaron bands in the UV-Vis-NIR spectra.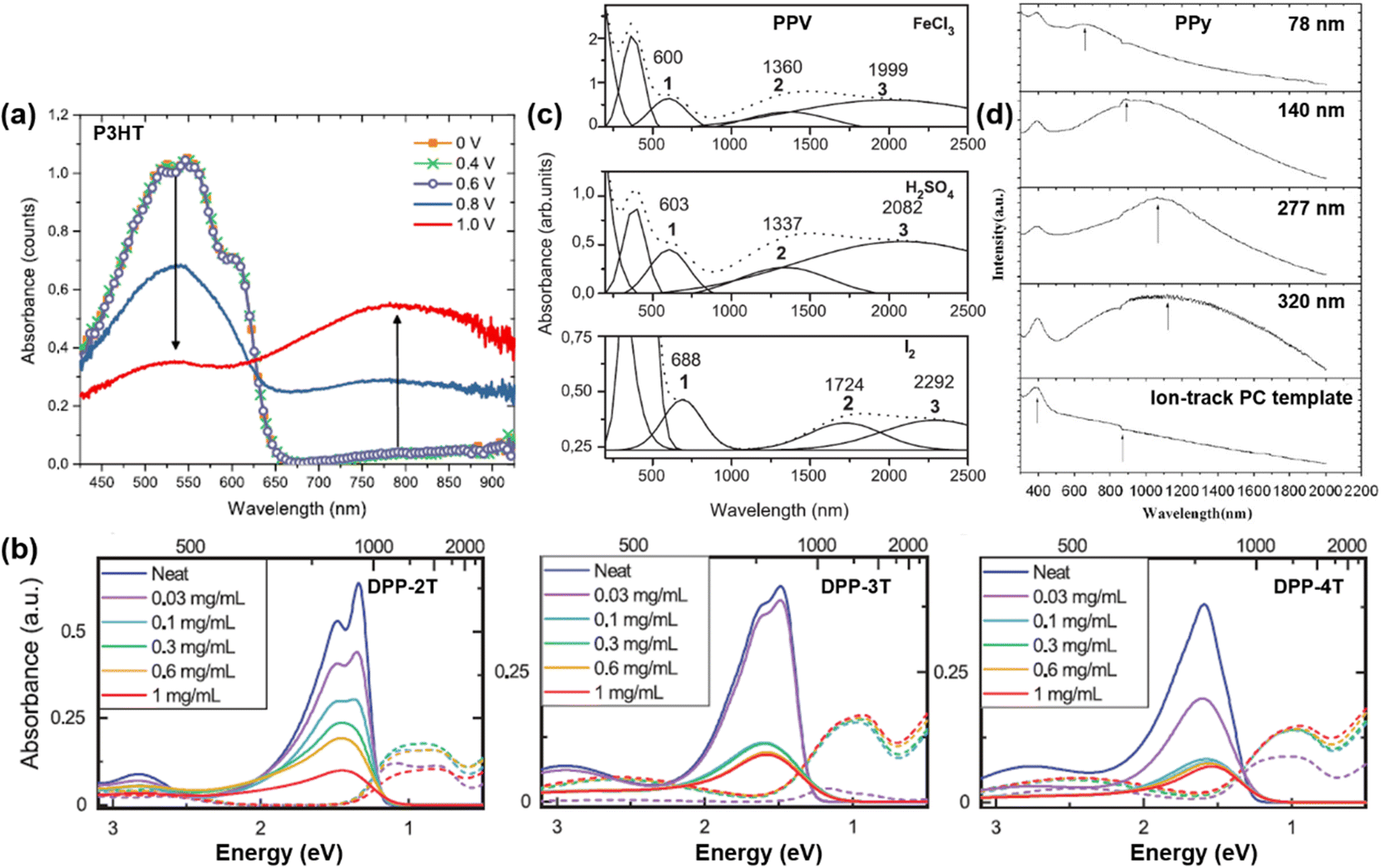 | ||
| Fig. 14 (a) UV-Vis-NIR spectra of P3HT:TBAPF6 thin films prepared by using different electrochemical oxidation potentials. Reproduced from ref. 85, with permission from American Chemical Society, 2018. (b) UV-Vis-NIR spectra of the periodic copolymers of DPP and thiophene with different arrangements of repeating sequence [-T-DPP-(T)n-DPP-T-], where n = 2, 3, 4 and corresponds to the number of the thiophene unit, respectively. Reproduced from ref. 86, with permission from John Wiley and Sons, 2022. (c) UV-Vis-NIR spectra (dot lines) of PPV thin films doped by FeCl3, H2SO4, and I2, respectively. The sum spectra can be deconvoluted into four component bands and the polaronic bands were marked with the numbers 1, 2, and 3. Reproduced from ref. 87, with permission from Elsevier, 2005. (d) UV-Vis-NIR spectra of PPy nanowires and nanotubules possessing different diameters synthesized by using the ion-track PC templates. Reproduced from ref. 88, with permission from Elsevier, 2010. | ||
For conjugated polymers with the same chemical formula, even the UV-vis-NIR spectra may merely reflect the same appearance of a different nature (Fig. 15).90,91 In other words, the phase condition, microstructure, and additive may have significant impacts on the energy states and the UV-Vis-NIR spectra of the conjugated polymers. For example, the UV-Vis-NIR spectrum of a regioregular P3HT (RR-P3HT) in solution was significantly different from the spectrum of the same RR-P3HT thin film (Fig. 15a: i). Compared to the RR-P3HT solution, the UV-Vis-NIR spectrum of the RR-P3HT thin film was broadened and red-shifted to longer wavelengths as a result of the formation of planar, highly conjugated and aggregated chains, as evidenced by the emergence of fine vibronic structures (0-2, 0-1, 0-0 transitions) in the UV-Vis-NIR and the photoluminescence (PL) spectra originated from the energy transfer from the coil chain segments to the aggregate chains(Fig. 15a: ii).90 The UV-Vis-NIR spectrum of the RR-P3HT can be further tuned by adding silicon nanocrystals (Si-ncs) or FeCl3 as the dopant. Bulk heterojunctions (BJHs) of RR-P3HT:Si-ncs can be formed by adding Si-ncs in the P3HT thin film, which further enhanced the optical density of the UV-Vis-NIR spectrum of P3HT by integrating the absorption of the Si-ncs (Fig. 15a: ii). On the other hand, the emergence of the polaron bands can be observed both in the UV-Vis-NIR spectrum and the PL spectrum of the RR-P3HT film after FeCl3 doping (Fig. 15a: iii). Different absorption and vibronic characteristics were observed between the regiorandom P3HT (Rra-P3HT) thin film and the regioregular P3HT thin film with enhanced chain aggregation by ethyl acetate (aggRR-P3HT), as the UV-Vis-NIR spectrum of the aggRR-P3HT thin film showed a more red-shifted absorption with fine vibronic excitations, induced by the aggregated polymer chains (Fig. 15b: i). When the aggRR-P3HT or Rra-P3HT thin film layer was combined with a polysilicon layer to form a planar heterojunction (PHJ), the UV-Vis-NIR spectrum of the PHJ was found to be the superposition of the absorption spectra of the individual functional layers (Fig. 15b: ii). The orientation of thin films may also have a significant impact on the UV-vis-NIR spectrum of the conjugated polymer. For example, the orientation of a regioregular P3HT thin film prepared by doctor-blading the solution of P3HT (ortho-dichlorobenzene as the solvent) at 160 °C can be controlled by rubbing the P3HT thin film using a rotating cylinder covered with a microfiber cloth, and the temperature of the thin film during rubbing can be controlled using a hot plate.91 As the rubbing temperature (TR) increased, the UV-Vis-NIR spectra of the P3HT thin films doped with F4TCNQ− showed significantly enhanced polarization for the polaronic band (P2) and the vibronic bands of F4TCNQ− (768 nm, 875 nm) in the high-temperature (TR ≥ 166 °C), as shown in Fig. 15c: i. The absorptions corresponding to the neutral (N) and polaronic bands (P1, P2) shifted to longer wavelengths as a result of the structural changes induced by the rubbing process; the ratio between the intensities of N and P1 bands also significantly increased when TR ≥ 148 °C (Fig. 15c: ii). To unveil the physical meaning of “polaron”, “bipolaron”, “trion” in conjugated polymers with different dopants, molecular structures, and microstructures, it is necessary to use a combination of spectroscopic, electrical, and electronic characterization techniques to acquire a full picture of the characteristics of the charged carriers.
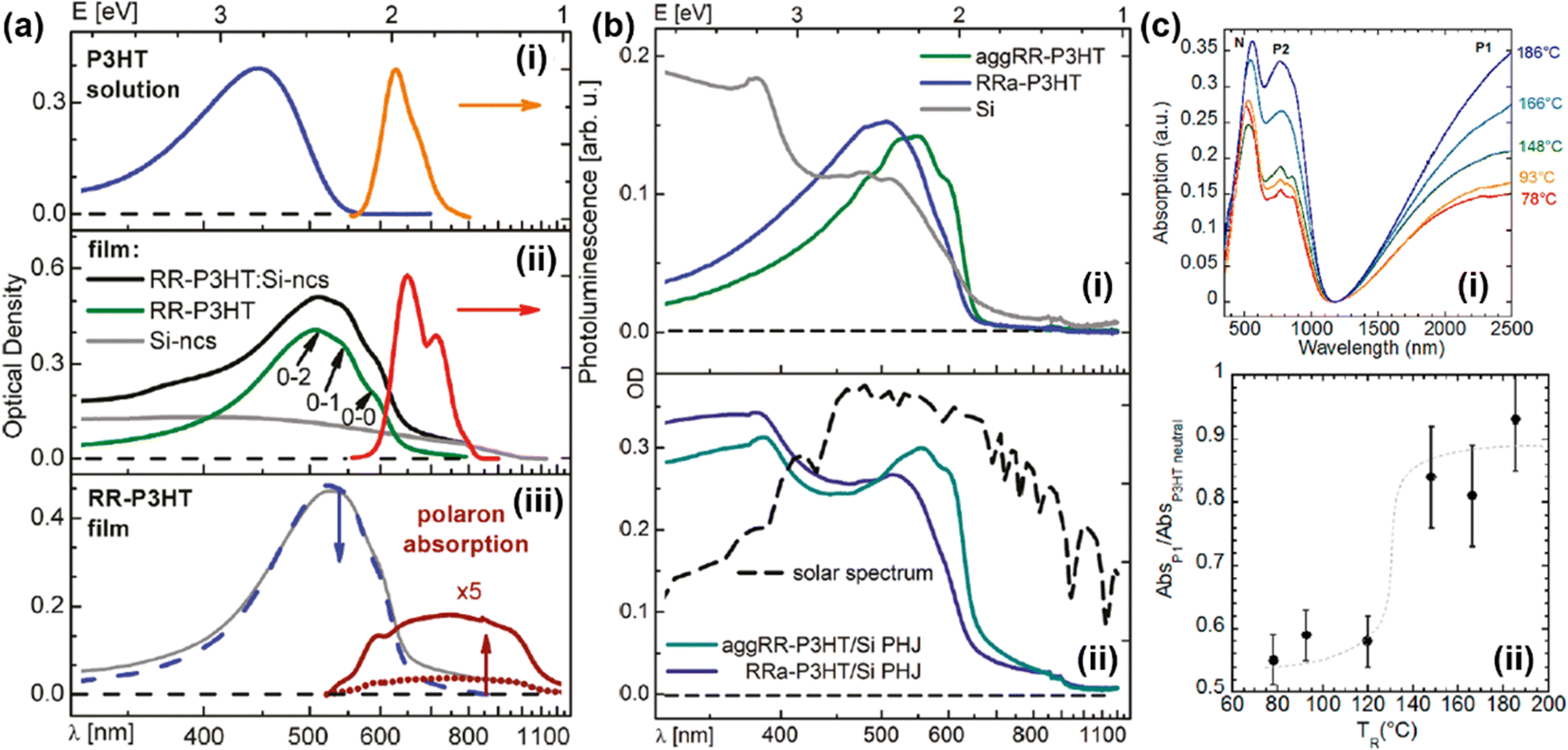 | ||
| Fig. 15 (a) UV-Vis-NIR and photoluminescence spectra of (i) P3HT solution (dissolved in diluted chloroform), (ii) RR-P3HT thin film, Si-ncs, and RR-P3HT:Si-ncs BHJ. 0-2, 0-1, and 0-0 marks the locations of the fine vibronic structures. Reproduced from ref. 90, with permission from American Chemical Society, 2011. (b) UV-Vis-NIR spectra of (i) aggRR-P3HT, RRa-P3HT, and polycrystalline silicon (Si), the spectra of the PHJs composed of aggRR-P3HT:Si and RRa-P3HT:Si are shown in panel (ii). The solar spectrum was also shown for comparison. Reproduced from ref. 90, with permission from American Chemical Society, 2011. (c: i) UV-Vis-NIR spectra of the P3HT:F4TCNQ thin films oriented by rubbing at different temperatures. The spectra were obtained by setting the light polarization parallel to the rubbing direction. (c: ii) Plot of the ratio between the intensities of the P1 and N bands of the P3HT:F4TCNQ thin films as a function of the rubbing temperatures TR. Reproduced from ref. 91, with permission from American Chemical Society, 2020. | ||
3.1 The effect of dopants
It is well-known that the electrical conductivity of the conjugated polymers can be effectively tuned by the doping process. Dopants, which are introduced into the macromolecular structure of the conjugated polymers during doping, generate polarons and bipolarons through an electron/charge transfer process in which the dopant and the polymer are involved. Due to the metallic characteristics of the conjugated polymers, their conductivities can be extended to 105 S cm−1 at room temperature upon doping,92 while similar thermoelectric and thermoelectromagnetic properties can also be found for the doped conjugated polymers and metals.93,94 Therefore, the doped conjugated polymers with metal-like conductivities are referred as “synthetic metals” by A. G. McDiarmid et al.95 It is speculated that the doped conjugated polymers may show high-temperature superconductivity surpassing that of metals, provided they follow different structure–property correlations in conducting electricity.96–98 However, the state-of-the-art research on conjugated polymers lies in investigating the effects of dopant on the electrical, electronic, and thermoelectric properties of the conjugated polymers, which can be rationally categorized into three major ones, i.e., the dopant concentration, size of dopant, and type of dopant.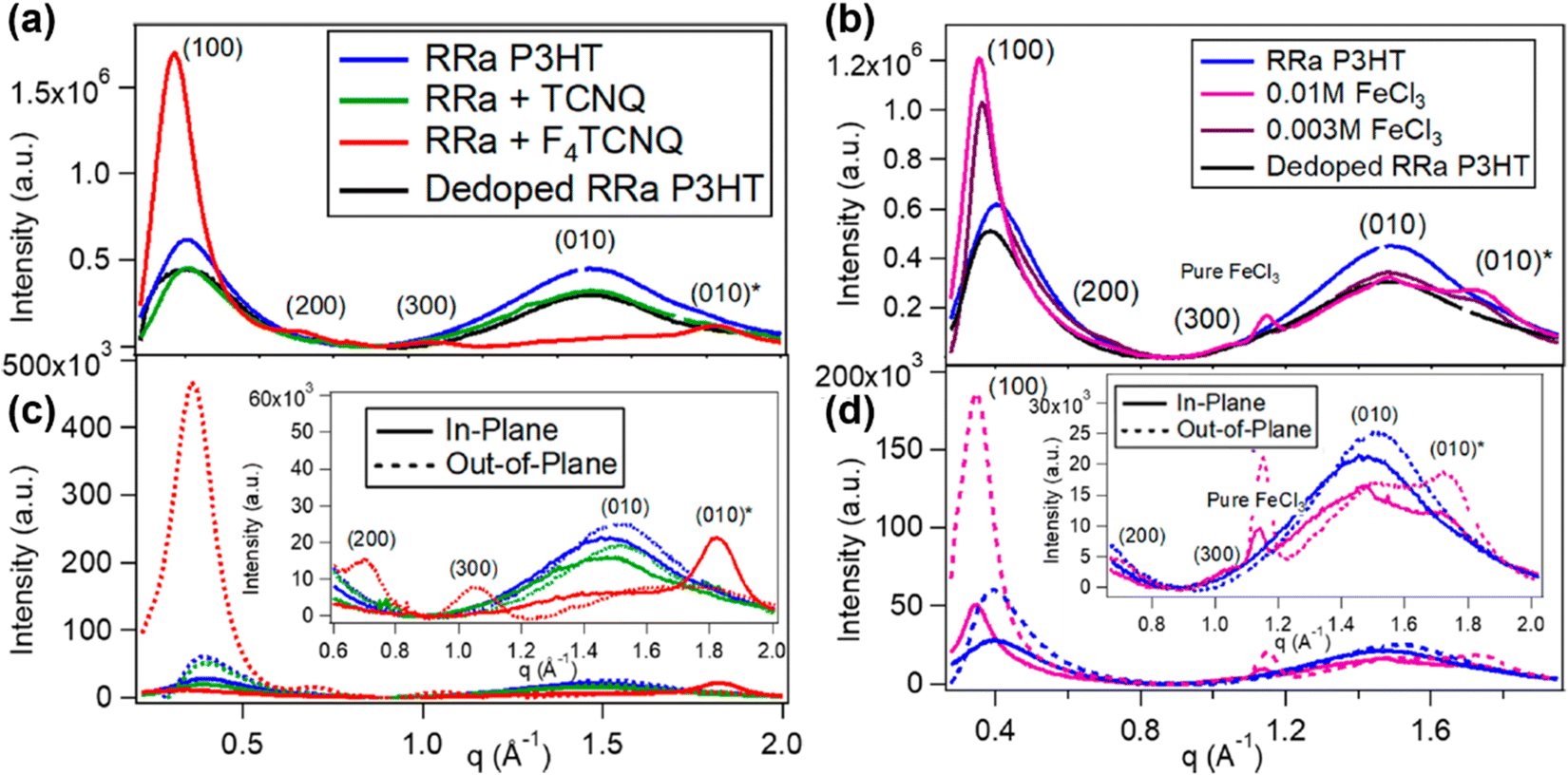 | ||
| Fig. 16 Radically integrated X-ray diffraction spectra of the RRa-P3HT films from the corresponding GIWAXS diffractograms. The spectra of RRa-P3HT doped by 0.003 M F4TCNQ and 0.01/0.003 M FeCl3 are shown in (a) and (b), respectively. The diffraction spectra of the pristine Rra-P3HT, TCNQ treated RRa-P3HT, and thermally dedoped RRa-P3HT are also shown for comparison. (c) and (d) show the selective in-plane and out-of-plane integration of the spectra in (a) and (b), respectively. The insets show the zoomed-in view of the lamellar overtones and π-stacking region. Reproduced from ref. 101, with permission from American Chemical Society, 2019. | ||
| Label | Dopant | σ RT (S cm−1) | T 0 (103 K) | T A (K) | T B (K) | Method of synthesis |
|---|---|---|---|---|---|---|
| PF6(-40a) | PF6 | 340 | 662 | 369 | Current, 0.3 mA cm−2; temp, −40 °C | |
| PF6(-40b) | PF6 | ∼340 | 471 | 298 | Current, 0.15 mA cm−2; temp, −40 °C | |
| PF6(-40c) | PF6 | 180 | 426 | 203 | Current, 0.07 mA cm−2; temp, −40 °C | |
| PF6(0) | PF6 | 150 | 582 | 256 | Current, 0.3 mA cm−2; temp, 0 °C | |
| PF6(20) | PF6 | 80 | 39 | Current, 0.3 mA cm−2; temp, 20 °C | ||
| pTS | pTS | 50 | 6.7 | Current, 12 mA cm−2 | ||
| CHEM | DBSA | 25 | Chemical | |||
| PMMA | pTS | 4 | 31 | Deposited on PMMA | ||
| W-pTS(3m) | pTS | 3 | 284 | Wrinkled, deposited on ITO | ||
| W-pTS(5m) | pTS | 2 | 179 | Wrinkled, deposited on ITO | ||
| W-DDS | DDS | 4 | 188 | Wrinkled, deposited on ITO | ||
| W-DDBS | DDBS | 835 | Wrinkled, deposited on ITO | |||
| W-PF6 | PF6 | Wrinkled, deposited on ITO | ||||
| W-ClO4 | ClO4 | Wrinkled, deposited on ITO | ||||
| S-pTS | pTS | ∼18 | 620 | Sensor, electrochemical deposition | ||
| S-OS | OS | ∼1.2 | 1760 | Sensor, electrochemical deposition | ||
| S-DHBS | Tiron® | ∼1.1 | 7680 | Sensor, electrochemical deposition |
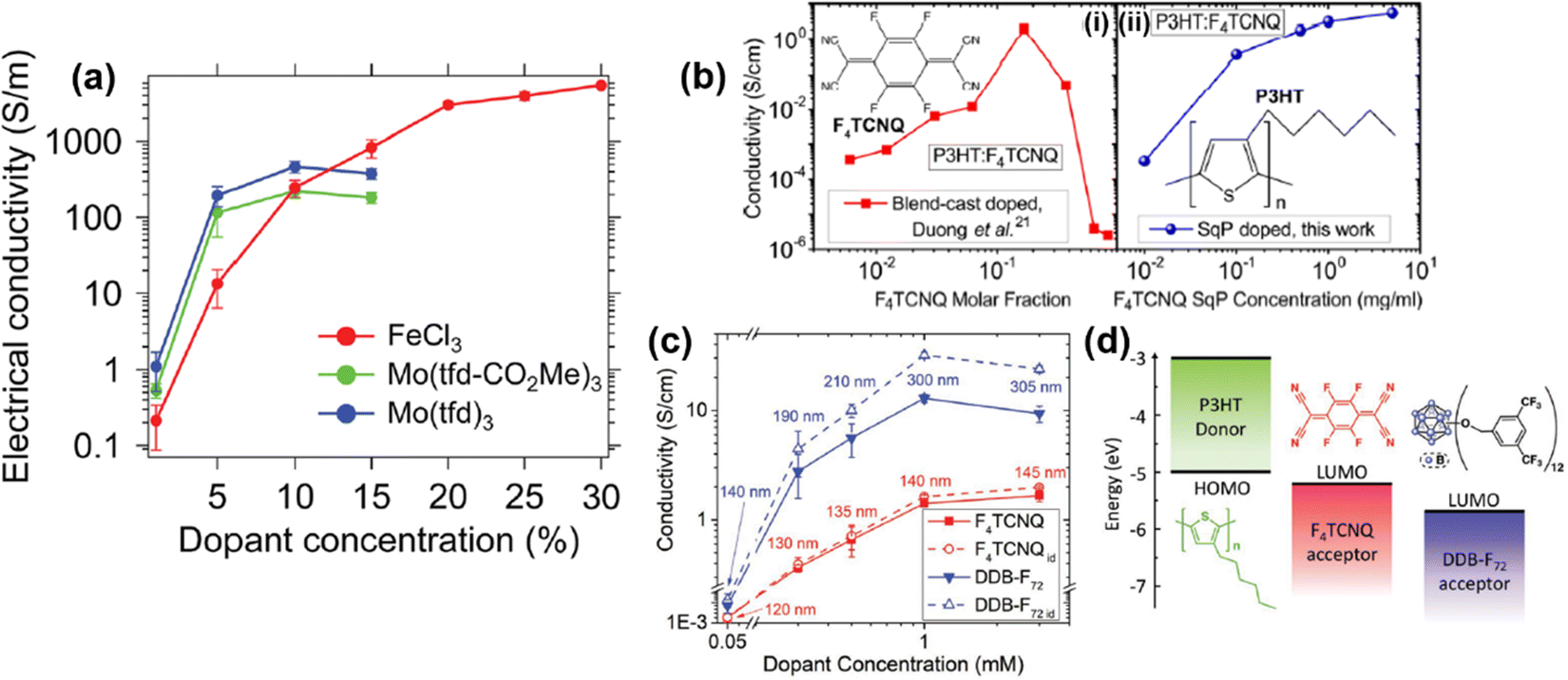 | ||
| Fig. 17 (a) Plot showing the correlations of conductivity vs. dopant concentration for RR-P3HT doped by FeCl3, Mo(tfd-CO2Me)3, and Mo(tfd)3. The P3HT films were prepared by single-solution processing. Reproduced from ref. 104, with permission from Royal Society of Chemistry, 2018. (b) Plots of conductivity vs. dopant concentration for F4TCNQ doped P3HT thin films prepared by blend-cast doping (i) and sequential doping (ii). Reproduced from ref. 105, with permission from American Chemical Society, 2015. (c) Plot of conductivity vs. dopant concentration for P3HT thin films sequentially doped by F4TCNQ or DDB-F72. Data points in the solid lines were calculated from the measured film thickness, and the data points in the dot lines were calculated by using the original film thickness (120 nm). Reproduced from ref. 106, with permission from John Wiley and Sons, 2019. (d) Schematic illustrations of the energy diagram and chemical structures of F4TCNQ and DDB-F72. Reproduced from ref. 106, with permission from John Wiley and Sons, 2019. | ||
The structural change of the conjugated polymer thin film induced by the doping process may significantly change the correlation between conductivity and dopant concentration. For example, it has been reported that the conductivity of P3HT films doped with different dopants, i.e., F4TCNQ, DDB-F72, increased with elevated dopant concentrations in the concentration range between 0.05 mM and 1 mM.106 Beyond this range, the conductivity will reach a plateau and gradually decrease (Fig. 17c). For the measured dopant concentrations, the conductivity of the P3HT thin films doped by DDB-F72 was an order of magnitude higher than the P3HT films doped by F4TCNQ. However, the thickness of the films would significantly increase after being doped by DDB-F72, as compared to F4TCNQ (Fig. 17c, solid lines). Swelling of the film can be ascribed to the intercalation of the DDB-F72 dopants with large molecular size (Fig. 17d) into the film, and the accumulation of DDB-F72 on the film surface. In view of the plot of conductivity vs. dopant concentration, it is rational to speculate that the effect of dopant (size, charge distribution, etc.) may critically impact the electrical conductivity of the conjugated polymer and its microstructure. Similar conductivity vs. dopant correlations have also been observed for other conducting polymers, such as polyaniline and PEDOT. Lenin R. et al. have reported that the conductivity of polyaniline doped by chloride ions increased from 1.98 S cm−1 to 10.2 S cm−1 as the concentration of chloride increased from 0.275 M to 1.0 M.107 Moreover, Shuai Z. et al. have reported that the conductivity of heavily doped PEDOT:Tos (Tos, p-toluenesulfonate) can reach 9.26 × 104 S cm−1, much higher than the conductivity of 1.95 × 103 S cm−1 of the lightly doped PEDOT:Tos.108
The doping process involves the charge-transfer interaction between the conjugated polymers and the dopants, forming new polaron and bipolaron states in the band gap, and the polarons and bipolarons can serve as the charge carriers. To realize fast and efficient charge transportation, the doped conjugated polymer should have high charge carrier concentration, high charge carrier mobility, and high inter-chain hopping efficiency. By increasing the dopant concentration, the charge carrier concentration of the conjugated polymer can be simultaneously increased, resulting in improved conductivity. The optimal conductivity can be achieved by consolidation of the mixed valence state consisting of both polarons and bipolarons within the optimal dopant concentration range. On the other hand, when the dopant concentrations are elevated to a substantially high level, the charge carriers become trapped by the excess electrostatic interactions with the dopants, resulting in decreased mobility and electrical conductivity. For example, Schwartz B. J. et al. have reported that the ratio between free and trapped polarons may decrease at high dopant concentrations, depending on the distance between the dopant molecules and the polarons. They further defined the polarons located ∼0.4 nm away from the dopants as trapped, and the ones located 0.7–0.9 nm away as free.106 On the other hand, polarons combine to form bipolarons with respect to the elevated dopant concentrations since bipolarons are more thermodynamically stable. However, the mobility of the bipolarons is significantly lower than the polarons due to their strong interactions with the defects. Bondarenko N. et al. have shown that the bipolarons were largely immobilized by the defects as a result of the electronic configuration transition, while the polarons can be mobile.109 Furukawa Y. et al. have studied the electronic structures of regioregular P3HT films doped by FeCl3 of different levels.110 It was shown that the P3HT films were transferred from polaron dominated to bipolaron dominated as the doping level of FeCl3 was substantially increased. The measured conductivity of the polaron-dominated P3HT films were two-orders-of-magnitude higher than the bipolaron-dominated films, indicating that the mobility of polarons was 100 times higher than that of the bipolarons.
As mentioned previously, the doping process may generate new electronic structures, e.g., polarons and bipolarons, in the band gap of conjugated polymer and change the band gap. For the conjugated polymers with non-degenerate ground states, bipolarons are more thermodynamically favorable charge carriers. In this regard, dopants with higher electron affinities may be more efficient in generating bipolarons. On the other hand, the size, polarity, and diffusivity of the dopant molecules may have a significant impact on the dopant–host interactions, which may determine the concentration, distribution, and microstructure of the dopant-intercalated host. The charge distribution of dopants may also remarkably affect the strength of the electrostatic interactions between the dopant and the host molecule. Therefore, these characteristics of dopant may have great impacts on the charge carrier concentration, transport energy, and charge carrier mobility of the doped conjugated polymer. To realize conjugated polymers with outstanding thermoelectric properties, it is vital to utilize the molecular design of the dopant, and advanced doping techniques to achieve high doping efficiencies, i.e., low dopant concentration, minimum host structural change, and high conductivity.
The doping efficiency is highly dependent on the interaction between the host and the dopant to generate free charge carriers in the macromolecular structure. In inorganic semiconductors, the host must have a low ionization potential and high electron affinity in order to activate the dopants and achieve a higher doping efficiency. This concept also holds partially true for p-doping organic semiconductors, e.g., the conjugated polymers. For example, in the conjugated polymers which have non-degenerated ground states, e.g., polypyrrole, polythiophene, PEDOT, the polymer backbones may take up a quinoid geometry near the location of the charges, which is shown to have low ionization potential and high electron affinity.56,111,112 However, controversies have been observed in n-doping organic semiconductors such as C60. Ortman F. et al. have reported the utilization of the benzimidazoline radical (2-Cyc-DMBI), which has a low ionization potential and high ionization efficiency, to dope the C60 host which has a high electron affinity.113 As the C60 host was doped to the ionized state, its electron affinity was decreased, which was substantially different from the case of conjugated polymers. Furthermore, the total conductivity of C60 doped by different dopants correlated strongly with the decrease in electron affinity before and after doping. The smaller the absolute value of the decrease (Δ1), the higher the conductivity, as shown in Fig. 18a. Fig. 18a: i shows the plot of conductivity vs. the molar concentration of the dopant (nD) for the C60 thin films doped by different dopants, and it is clearly observed that the thin films doped by di-metal complexes of 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidine (hpp), i.e., Cr2(hpp)4 and W2(hpp)4, show higher conductivities with respect to the increasing nD. These trends can be described by the small |Δ1| of C60 doped by Cr2(hpp)4 and W2(hpp)4 (Fig. 18a: ii), indicating a low energy barrier for the generated charge carriers to become mobile. This phenomenon can be attributed to the different molecular structures of conjugated polymers and C60. For the conjugated polymers, their polymer chains are able to relax locally around the charges upon doping, e.g., from aromatic to quinoid, due to the difference in the ionization energy and distortion energy. The ability of the conjugated polymer chains to change their geometries around the charge can lower the ionization potential and increase the electron affinity upon doping. On the other hand, the polyhedral structure of C60 is highly stable and the intermolecular interactions of C60 are weak, making it hard to change the polymer geometry upon doping, and a higher ionization potential and lower electron affinity can be expected after doping.114 Therefore, compared to C60, the conjugated polymers, especially the one-dimensional ones, can be doped to a greater extent due to the increase of electron affinity upon doping.
 | ||
| Fig. 18 (a) i: Plot of conductivity vs. dopant concentration for C60 thin films doped by different dopants, i.e., Cr2(hpp)4, W2(hpp)4, RuCp*(mes), 2-Cyc-DMBI. Ii: Plot of conductivity vs. Δ1 for the C60 thin films doped by different dopants. Reproduced from ref. 113, with permission from Springer Nature, 2018. (b) EPR spectra of ETE-S thin films treated by different electrochemical potentials, the correlations between (i) the densities of the ½ spin, (ii) the line widths derived from the EPR spectra, and the applied electrochemical potentials are shown in the right panel. Reproduced from ref. 116, with permission from John Wiley and Sons, 2017. (c) Ultrafast transient absorption (TA) and photoinduced absorption (PIA) spectra of the PFO thin films composed solely of the α phase (0% β phase), and 23% β phase. (d) Correlations between the polaron yield and β phase fraction calculated from the TA spectra obtained at different excitation wavelengths (λexc). Reproduced from ref. 123, with permission from American Chemical Society, 2019. | ||
The charge stabilized by the local distortion may reflect the true physical meaning of polarons. In conjugated polymers, polarons are defined as radical ions with a spin of ½; however, when the second charge is introduced locally at the polarons, a pair of charge stabilized by larger distortions will form, and so the bipolaron. Compared to polarons, the bipolarons are spinless, which are considered similar to Cooper pairs in the BCS theory.57 Bipolarons are thermodynamically much more stable than polarons and have significantly higher binding energies. It has been proved both theoretically and experimentally that the bipolarons are significantly immobile at the charge pair locations by both the geometry distortion and dopant counter ions.115 The existence of polarons and bipolarons in the matrix of conjugated polymers can be qualitatively characterized by electron paramagnetic resonance (EPR). High-intensity EPR signals can be detected at relatively low dopant concentrations, indicating the existence of polarons with a spin of ½. However, the EPR signals may decrease as the dopant concentration is increased, and no signals can be detected at high dopant concentrations, indicating spinless charge carriers, i.e., bipolarons, are dominant. For example, Volkov A. V. et al. have studied the nature of the charge carriers in a self-doped oligomer sodium salt of bis[3,4-ethylenedioxythiophene]-3-thiophene butyric acid ((ETE-S)).116 It was observed that the spin densities and line widths of the EPR spectra of the ETE-S thin films increased with the increasing electrochemical oxidation potentials in the ranges from −1.0 V to 0 V and 0 V to +0.7 V, respectively (Fig. 18b). In the max reduced state measured (−1.0 V), the charge carriers in ETE-S showed a g factor of 2.0030, which was different from the g factor (2.0027) of the remaining measured potentials, indicating that the carriers may be traps rather than polarons. However, the intensity of polarons increased with the electrochemical oxidation potentials up to 0.0 V, and drastically decreased to the minimum as the oxidation potential reached +0.7 V; this phenomenon can be attributed to the combination of the polarons to form the spinless bipolarons, accompanied by the abrupt increase in line widths from 0.0 V to +0.7 V. Moreover, bipolarons can be transformed into quasi-particles of different functions in the lattice of non-degenerate conjugated polymers, e.g., a coupled confined spinless soliton–antisoliton pair upon scattering.117
State-of-the-art strategies for improving the doping efficiency include tuning the ionization potential of dopants or polymers, manipulating the distance between dopants and charge carriers, and designing the molecular and crystallization structures of the host.118–120 For example, mutual electrical doping has been achieved by using a p-doped conjugated polymer with low ionization potential, bithiophene-co-thiophene [P(g42T-T)], and a n-doped conjugated polymer with high electron affinity, poly(benzimidazophenanthroline) (BBL), to dope each other.121 Blends of the mutual doped polymers show a conductivity of 0.2 S cm−1 and high thermal stability at 200 °C. This new way of doping offers alternatives to the existing doping techniques and may provide new insight for designing conjugated polymer–dopant systems with high electrical conductivity and thermal stability. On the other hand, dodecaborane (DDB)-based dopants with high redox potential and localized electron density with steric protection have been developed.122 High-intensity free charge carriers can be produced by doping P3HT films with DDB-based dopants, as a result of the redox potential driven intercalation of DDB into the lamellar structure of P3HT, and the shielded electrostatic interactions between DDB and polarons. The DDB doped P3HT films showed conductivities approximately an order of magnitude higher than that of F4TCNQ doped P3HT films. Cheetham N. J. et al. have reported the effect of the interplay between the highly ordered “β-phase” and amorphous “α-phase” (glassy phase) on the electronic properties of poly(9,9-dioctylfluorene) (PFO).123 It has been shown that the ordered β-phase was dispersed in the major α-phase, forming heterojunctions which were beneficial for electron–hole separation. The ultrafast transient absorption (TA) spectrum of the glassy-phase sample showed characteristic absorption corresponding to the triplet excitation (α-phase, ∼1.55 eV) and the polarons (∼2.1 eV), while the TA spectrum of the sample containing 23% β-phase showed characteristic absorption composed of the superpositions of the triplet and polaron excitations of both the α and the β phase (Fig. 18c). However, the presence of the β-phase can significantly enhance the longevity and lifetime of the photo-generated polarons; as the absorption corresponding to the polarons may completely disappear in the photo-induced absorption (PIA) spectrum of the α-phase only sample, it can be still observed in the PIA spectrum of the sample containing 23% β-phase (∼1.85 eV). It was speculated that the β phase can function as a highly efficient energy trap to localize the triplet and polaron excitations generated by the α phase. By tuning the fraction of the β-phase, the polaron yield at 385 nm excitation wavelength increased significantly from 1.6% to 4.4% as the fraction of the β-phase increased from 0 to 8%. However, a decrease in the polaron yield at 437 nm was observed as the fraction of the β-phase further increased to 23% (Fig. 18d).
Schwartz B. J. et al. have investigated the effect of crystallinity on the conductivity of a statistical copolymer of poly(3-heptylselenophene) (P37S) and P3HT (P37S-stat-P3HT).124 It showed that the crystallinity of P37S-stat-P3HT significantly increased as the percentage of P37S was increased from 25% to 75%. However, the P37S (75%)-stat-P3HT (25%) sample with the highest crystallinity showed the lowest conductivity after doping with F4TCNQ in dichloromethane (DCM). By comparing the grazing incidence wide angle X-ray scattering (GIWAXS) diffractograms of P37S (75%)-stat-P3HT (25%) before (Fig. 19a: i) and after (Fig. 19a: ii) the doping process, it can be observed that its crystallinity was largely reduced by doping. On the other hand, the P37S (25%)-stat-P3HT (75%) showed much lower crystallinity compared to P37S (75%)-stat-P3HT (25%) before doping, but enhanced crystallinity was observed after the doping process, and a higher conductivity could be achieved (Fig. 19a: iii). The measured charge carrier densities for the doped statistical copolymers coincided with the changes in their conductivities (Fig. 19a: iv), except for the P37S (25%)-stat-P3HT (75%) and the P37S (100%) samples, which may show a higher conductivity at a lower level of charge carrier density. These phenomena can be attributed to the different swelling, intercalation and recrystallization behaviors of the P37S-P3HT copolymers in the DCM solution containing F4TCNQ. The effect of solvent is also significant for the semicrystalline PEDOT. As reported by Simonato J. P. et al., the addition of N-methyl pyrrolidone (NMP) as a cosolvent into the polymerization solution can slow down the polymerization rate of EDOT, promoting the formation of larger crystallites and higher structural orders in the PEDOT matrix.125 According to the GIWAXS diffractograms shown in Fig. 19b, the addition of NMP can promote both the out-of-plane stacking and in-plane packing orders of the PEDOT thin film, indicating by the enhanced diffraction intensities of the (020) orthorhombic PEDOT (q = 1.82 Å−1) and (h00) peaks (q ≈ 0.88 Å−1). By replacing PSS by trifluoromethanesulfonate (Otf), the out-of-plane stacking of the PEDOT molecules can be significantly enhanced, indicating by the emergence of the diffraction peaks at q = 0.46 Å−1. The conductivity of the as-obtained PEDOT can reach 3600 S cm−1 while the PEDOT synthesized without the addition of NMP only showed a conductivity of 1200 S cm−1. By further treating the as-obtained PEDOT film with dilute sulfuric acid, the film can be further oxidized and the trifluoromethanesulfonate anions was replaced by the sulfate anion, resulting in the enhancement in conductivity from 3600 S cm−1 to 5400 S cm−1.
 | ||
| Fig. 19 (a) Radially integrated GIWAXS diffractograms of the thin films of statistical copolymers of P3HT and P37S before (i) and after (ii) being sequentially doped by 1 mg mL−1 F4TCNQ. The measured conductivities and charge carrier densities extracted from the AC Hall effect measurements of the statistical copolymer samples are shown in (iii) and (iv), respectively. Reproduced from ref. 124, with permission from American Chemical Society, 2019. (b) Synchrotron GIWAXS in-plane and out-of-plane diffractograms of PEDOT thin films prepared by adding different dopants and cosolvents, i.e., PSS, Otf, Otf plus NMP, and sulfuric acid plus NMP. Reproduced from ref. 125, with permission from American Chemical Society, 2016. (c) Two-dimensional GIWAXS diffractograms of the pristine (i, ii) and TDAE doped (iii, iv) RR-P(NDI2OD-T2) and RI-P(NDI2OD-T2), respectively. Reproduced from ref. 126, under the license of CC-BY-NC-ND. (d) EPR spectra of the RR-P(NDI2OD-T2) and RI-P(NDI2OD-T2) thin films doped by exposing to the TADE vapor for 15 hours. The corresponding line widths of the EPR spectra are shown in the right panel. Reproduced from ref. 126, under the license of CC-BY-NC-ND. | ||
The effect of regiochemistry on the electronic properties of P(NDI2OD-T2) was investigated by Fabiano S. et al.126 The regioregular (RR) P(NDI2OD-T2) showed much higher crystallinity than the regioirregular (RI) P(NDI2OD-T2), as indicated by the GIWAXS diffractograms shown in Fig. 19c. The diffractogram of the pristine RR-P(NDI2OD-T2) showed a strong diffraction spot at q ≈ 0.30 Å−1, while there was no spot in the same location of the diffractogram of the pristine RI-P(NDI2OD-T2), indicating that the RR-P(NDI2OD-T2) may obtain a higher degree of lamellar orientation. Upon n-doping with tetrakis(dimethylamino)ethylene (TADE), the crystallinity of the RR-P(NDI2OD-T2) increased at a low doping level and significantly decreased at a high doping level, indicated by the conversion of the diffraction spot into a diffraction ring after exposing to TADE vapor for 15 hours (Fig. 19c: left-bottom panel). On the other hand, the crystallinity of RI-P(NDI2OD-T2) was merely affected by the doping process, as no obvious changes could be found in its diffractogram (Fig. 19c: right-bottom panel). Interestingly, the TADE doped RI-P(NDI2OD-T2) showed a higher conductivity and significantly higher maximum carrier concentration than the TADE doped RR-P(NDI2OD-T2), and the measured conductivities for both samples were of the same magnitude. Similar correlations in conductivity could be observed for the RR-P(NDI2OD-T2) and RI-P(NDI2OD-T2) doped by dimethylbenzimidazoline (N-DMBI). However, the RR-P(NDI2OD-T2) showed a higher charge carrier mobility (μ) than its regioirregular counterpart, evidenced by the small ratio (≈6) between μregular and μirregular, while this value could exceed 1000 for P3HT. The EPR spectrum of the TDAE doped RI-P(NDI2OD-T2) showed a much higher peak intensity than the regioregular one, indicating that the polaron density in the doped regioirregular sample was significantly higher (Fig. 19d). On the other hand, the RI-P(NDI2OD-T2) showed a significant larger line width compared to the RR-P(NDI2OD-T2), indicating that its doping level was much higher.127 Therefore, compared to P3HT, P(NDI2OD-T2) is insensitive to regiochemistry and shows much higher tolerance to the structural disorder with respect to the doping efficiency, which can be attributed to its high chain stiffness induced limited conformation. In other words, the doping efficiency is higher for the RI-P(NDI2OD-T2) compared to RR-P(NDI2OD-T2).
3.2. Doping processes and techniques
Doping processes may have significant impacts on the free charge carrier density, conductivity, and thermoelectric properties of the conjugated polymers. Different doping processes have been developed for the conjugated polymers, with the utilization of different techniques. A good doping process may typically have characteristics such as ease of processing, fine control over the doping concentration, high stability, reproducibility and simple instrumentation. Based on the electron/charge transfer mechanisms, current doping processes can be generally grouped into three categories, i.e., molecular doping, electrochemical doping, and mutual doping.In the molecular doping process, the electron/charge transfer takes place between the dopant molecules and the conjugated polymers.128 To achieve high doping concentration, the solvent for dissolving the dopant molecules must be carefully selected: for example, it should have good solubility with respect to the dopant molecules, and high solution stability at high processing temperatures and under high shear forces; it should be inert to redox reactions and have a high dielectric constant, as it would not participate in the doping process and can sufficiently dissociate the dopant counterions; moreover, the solvent is good to have good swellability with respect to the host and a low boiling point, making it easier for the dopant ions to intercalate into the crystalline region of the host and for the quick removal of the solvent molecules.124,125,129,130 The sequential doping process (SqP) is one of the most frequently used techniques for the molecular doping process. It has relatively simple instrumentation and the major devices include a spin coater, a hot plate and a glove box. A typical SqP process may involve four steps, including (i) spin-coating of the conjugated polymer thin film, (ii) thermal annealing of the thin film to remove the residue solvent, (iii) spin-coating of the dopant solution, and (iv) thermal annealing of the doped film at different temperatures to control the doping levels.131,132 The thickness of the doped conjugated polymer thin films prepared by SqP is typically ranged from 20 nm to 60 nm, depending on the concentration of the polymer solution.131–133 Thin films prepared by SqP may have uniform sizes and good crystallinity, making it suitable for processing the semicrystalline conjugated polymers such as PEDOT:PSS, P3HT, and PBTTT.134 However, for high-molecular-weight conjugated polymers with low solubility, e.g., Ppy, PANI, PT, the doping process can be completed simultaneously in the polymerization solution with the addition of both the dopants and the initiators.135–137
In the molecular doping process, the dopant molecules may serve as both the electron/charge acceptor and the counterion, which may decrease the doping efficiency as the counterions in the dopant oxidants may not have optimal electrostatic interaction with the host molecules and pose new challenges for optimization. For example, the closely packed molecular chains of poly(2,2′-bithiazolothienyl-4,4′,10,10′-tetracarboxydiimide) (PDTzTI) with a high degree of planarity can effectively keep the counterions distant from the polymer backbone, resulting in a significantly enhanced charge mobility;138 the small counterions dissociated from the oxidizing cations may easily diffuse out from the polymer matrix, resulting in de-doping of the polymer.139 Another unfavorable effect associated with the dopant oxidants is the formation of charge transfer complexes (CTX).140 It is found that CTX may strongly localize the charge carriers, resulting in a significant decrease of conductivity over time. Pemberton J. E. et al. reported that there were two possible reaction pathways for P3HT doped with F4TCNQ: one was the formation of free charge carriers through integer charge transfer (ICT), and the other was the generation of trapped charge carriers through the formation of CTX.141 It was found that ICT was kinetically more favorable and CTX was thermodynamically more favorable, and the conversion from ICT to CTX may responsible for the drastic decrease in conductivity for the P3HT:F4TCNQ thin films under ambient conditions. According to the FTIR spectra of the P3HT:F4TCNQ thin films with a relatively low F4TCNQ molar fraction (χ = 0.05), the v(CN) b1u band (2194 cm−1) corresponding to the ICT state decreased with the prolonged storage time, while the absorption band corresponding to the CPX state (v = 2201 cm−1) increased remarkably with the prolonged storage time (Fig. 20a). The same trend can be observed for the P3HT:F4TCNQ thin films with a high F4TCNQ molar fraction (χ = 0.11), but the enhancement in absorption for the CTX band was not as significant as the low χ sample (Fig. 20b). Based on the results shown in Fig. 20c, it can be inferred that the transformation of ICT to CTX is more pronounced in the range of χ < 0.096. For χ > 0.096, the enhancement of the CTX absorption as a function of the storage time is insignificant with respect to the ICT absorption (Fig. 20d). An effort to circumvent the effect of CTX has been proposed by Sirringhaus H. et al., who used a high concentration ionic liquid (BMP-TFSI) and an oxidizing agent (FeCl3) to formulate the dopant solution, and subsequently applied the dopant solution to the P3HT thin film through SqP.142 During the doping process, the TFSI− ion will replace the FeCl4− ion at the charge sites of the polymer backbone, resulting in the high-efficient intercalation of the TFSI− ions into the lamellar structures of P3HT, and a high ion exchange efficiency of >99% can be achieved in a short doping time of 100 s. As a result of the favorable dopant–host interaction, the electrical conductivity of the P3HT:TFSI can reach the value of 1000 S cm−1.
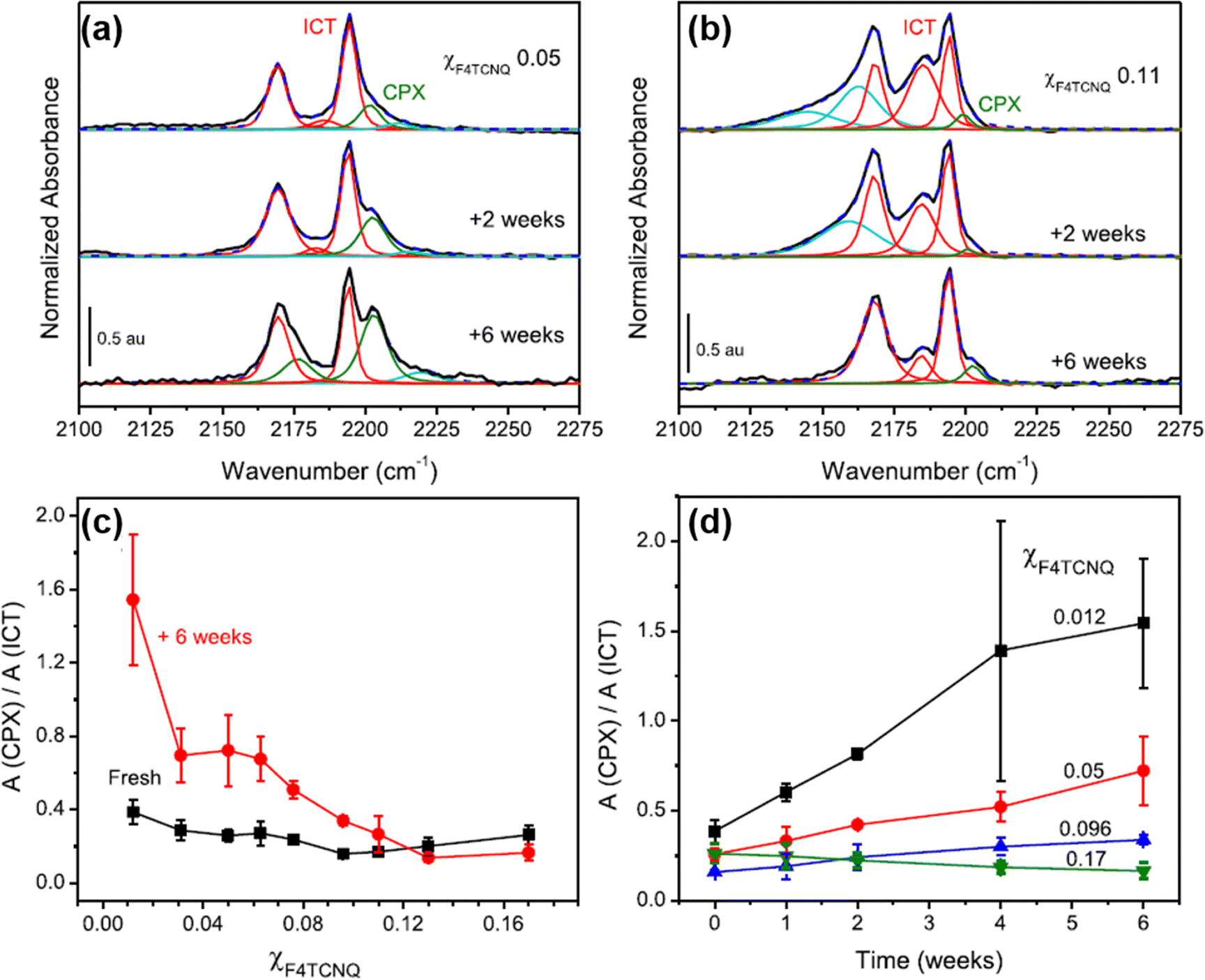 | ||
| Fig. 20 (a) FTIR spectra of a freshly prepared P3HT:F4TCNQ thin film with a F4TCNQ molar fraction χ = 0.05, and the same film after being stored under dark (glovebox) and inert (Ar atmosphere, <15 ppm O2) conditions for 2 or 6 weeks, respectively. (b) FTIR spectra of a P3HT:F4TCNQ thin film with χ = 0.11, the experimental conditions were the same for (a). (c) Ratio of the CPX and ICT absorption intensities as a function of χ, and (d) plot of the CPX/ICT ratio as a function of the prolonged storage time. Reproduced from ref. 141, with permission from American Chemical Society, 2019. | ||
Different from the molecular doping process, the charge/electron transfer takes place between the conjugated polymer and the electrode in the electrochemical doping process.143 To realize the electrochemical doping process, the first step is to coat the conjugated polymer onto the electrode surface. Both electrodeless and electrochemical techniques can be applied to finish this step, i.e., spin-coating is one of the most widely used electrodeless technique,144 while cyclic voltammetry (CV) and potentiostatic techniques are frequently used among the electrochemical techniques.145,146 However, for the spin-coating process, the conjugated polymer must be dissolved in a suitable solvent prior to use, while for the CV and potentiostatic processes, monomers of the conjugated polymer are generally used for the in situ polymerization of the conjugated polymer layer on the electrode surface. By adding electrolytes containing dopant counterions, the doping process can be completed simultaneously during the in situ electrochemical polymerization. The doping level and charge carrier density can be easily controlled by changing the electrochemical parameters, i.e., potential windows, scan rates, CV cycles, and potentiostat deposition time.147–149 Notably high doping efficiency can be achieved by the electrochemical doping process as unfavorable side reactions can be eliminated, provided a suitable oxidation potential is used.150 Ludwigs S. et al. have reported the electrochemical doping of RR-P3HT thin films using the potentiostatic technique.151 By changing the oxidation potential, the in situ UV-Vis-NIR spectra of the RR-P3HT thin films showed characteristic absorptions of the neutral (525 nm), polaron (805 nm), and bipolaron (1600 nm) bands, as shown in Fig. 21a. The RR-P3HT thin film can be easily doped to ∼40% at an applied potential of +0.8 V, corresponding to a hole density of 1021 cm−3, as shown in Fig. 21b and c. A maximum conductivity as high as 224 S cm−1 can be achieved at a doping level of ∼32.5% and an applied potential of +0.7 V. At the same time, the doping level can be facilely tuned from 15% to 40% in the potential range from +0.4 V to +0.8 V, while the conductivity shifted in the range between ∼5 S cm−1 and 224 S cm−1 (Fig. 21d), respectively. Compared to the molecular doping methods, i.e., SqP, the electrochemical doping methods may be less effective in controlling the microstructures of the as-prepared thin film, due to the random nucleation and polymerization processes on the electrode surface. Moreover, the conductivity of the RR-P3HT film can reach 224 S cm−1 after doping, but the conductivity of the doped RI-P3HT film was only 10 S cm−1, indicating the significant impact of the molecular conformation on the charge carrier mobility.
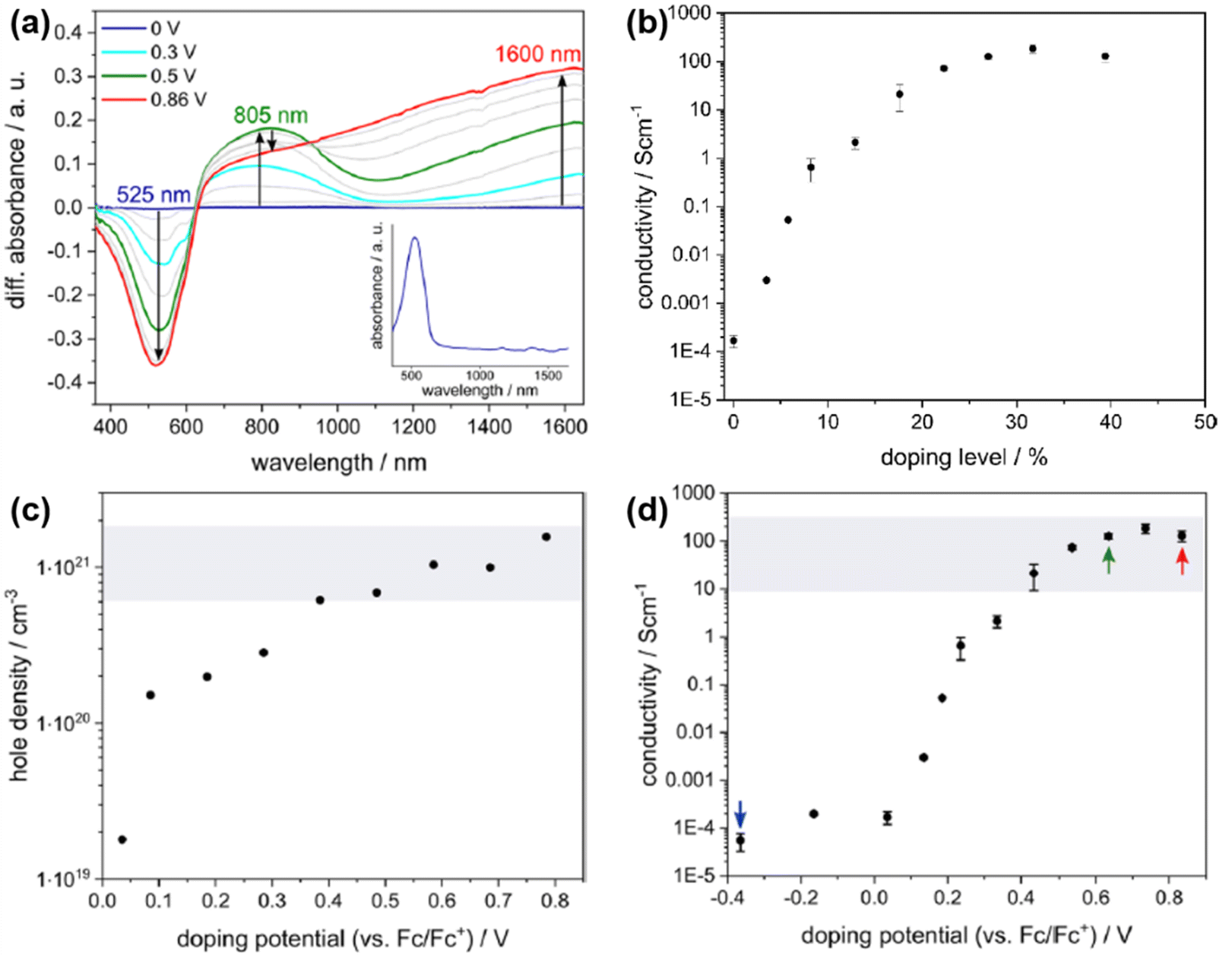 | ||
| Fig. 21 (a) In situ UV-Vis-NIR spectra of the RR-P3HT thin films doped via the potentiostatic technique. Different oxidation potentials were used for the doping process, including 0.3 V, 0.5 V, and 0.86 V. The absorption spectrum of the pristine RR-P3HT thin film is shown in the small panel and is used as the baseline. (b) Plot of conductivity vs. doping level, and (c) plot of hole density vs. doping potential, and (d) plot of conductivity vs. doping potential for the RR-P3HT thin films doped via the potentiostatic technique. Reproduced from ref. 151, with permission from American Chemical Society, 2020. | ||
The electronic properties of the doped conjugated polymers are generally obtained in the experimental environment with inert gas protection, and may undergo serious degradation under ambient conditions, which significantly limits the practical applications of the conjugated polymers.152 To address this issue, the concept of ground-state electron transfer (GSET) has been recently proposed by Fabiano S. et al.153 To achieve GSET, a p-type conjugated polymer, poly(benzimidazobenzo-phenanthroline) (BBL), was used to blend with an n-type conjugated polymer, poly(bithiophene-co-thiophene) [P(g42T-T)], and the resulting polymer blends showed five to six orders of magnitude enhancement in conductivity compared to each of the components in their pristine state. The EPR measurements showed that the charge carrier density of the P(g42T-T)-BBL GSET bilayer film reached 4–5 × 1019 cm−3, while the charge carrier density of the non-GSET P3HT-BBL bilayer film was only 3.5 × 1018 cm−3 (Fig. 22a). The GSET is effective in generating both n-type and p-type polarons, resulting in enhanced UV-Vis-NIR absorption compared to the pristine conjugated polymers (Fig. 22b). The UV-Vis-NIR spectrum of the P(g42T-T)-BBL GSET bilayer film showed characteristic absorption bands of both n-type polaron (800 nm) and p-type polaron (1030 nm), as shown in Fig. 22c. The positions of the measured polaronic bands were also consistent with the band positions modelled by the DFT/TDDFT simulations (Fig. 22d), which confirmed that the polaronic absorption of the subtracted UV-Vis-NIR spectrum of the bilayer film was the superposition of the spectra of the doped films of the pristine polymers. Superior thermal stability was shown by the GSET polymer blends, which can maintain a conductivity ∼2.3 × 10−2 S cm−1 after thermal annealing at 200 °C for 20 h in a glove box.
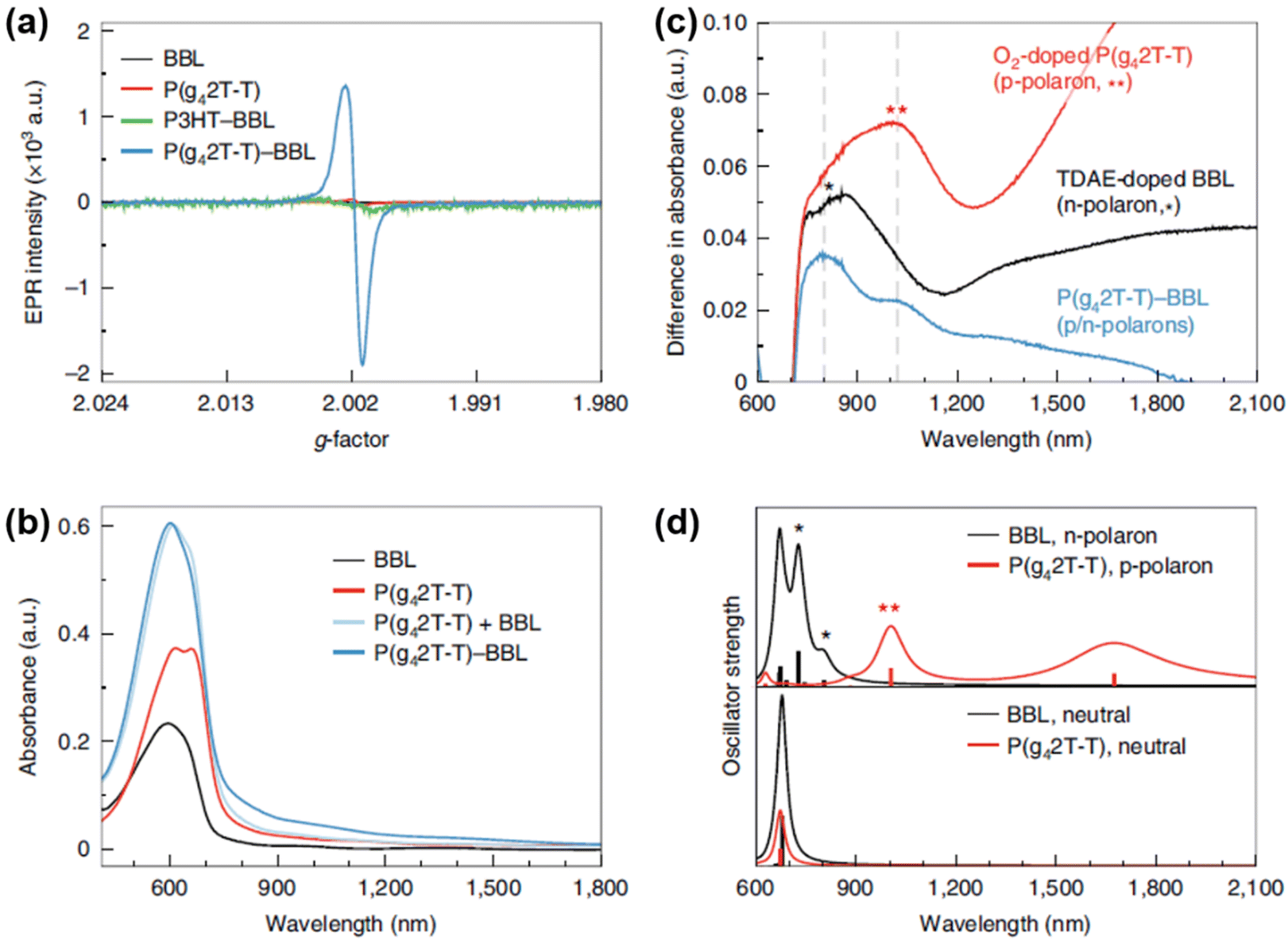 | ||
| Fig. 22 (a) EPR spectra of the pristine BBL and P(g42T-T) thin films, and the P3HT-BBL and P(g42T-T)-BBL bilayer thin films. (b) UV-Vis-NIR spectra of the BBL, P(g42T-T), P(g42T-T) + BBL mixed, P(g42T-T)-BBL bilayer thin films. (c) Subtracted absorption spectra of the O2 doped P(g42T-T), TDAE doped BBL, and P(g42T-T)-BBL bilayer thin films. (d) DFT and TDDFT simulated spectra of doped and neutral BBL and P(g42T-T), respectively. Reproduced from ref. 153, with permission from Springer Nature, 2020. | ||
3.3. Thermoelectric models and applications
Due to the wide-range tunability of the Seebeck coefficient, conductivity, and low thermal conductivity, the conjugated polymers are considered good candidate materials for high-efficiency thermoelectrics.154 By optimizing the interplay between Seebeck coefficient and conductivity, a figure of merit (ZT) of 0.25 at room temperature can be achieved for PEDOT:Tos films by carefully controlling the degree of oxidation, through a reduction process using TDAE as the reducing agent.155 This value of ZT is considered practically applicable for efficient thermoelectric devices. By dispersing PEDOT nanowires into the composite of PEDOT:PSS and PEDOT:Tos, and adding the electron scavenger dimethyl sulfoxide, the power factor of the nanocomposite can be enhanced 9-fold to 446.6 μW m−1 K−2, and a ZT as high as 0.44 can be obtained.156 By using 1,3,5-trichlorobenzene as the epitaxial template, highly anisotropic P3HT thin films which grew along the [100] direction can be obtained. After doping the anisotropic P3HT thin films with iron trifluoromethanesulfonimide [Fe(TFSI)3], outstanding thermoelectric properties, including high power factor (62.4 μW mK−2) and high ZT (0.1), can be achieved at 365 K.157 Compared to the commercial thermoelectric materials, e.g., the bismuth-telluride (Bi2Te3) based alloys, which can reach ZT values close to 1.0,158 the ZT values currently reported for the conjugated polymers are relatively low. However, for constructing efficient thermoelectric devices of practical significance, the ideal thermoelectric materials should have characteristics such as low cost, solution processible, and tunable film-forming properties, which are considered intrinsically present in the conjugated polymers.For obtaining conjugated polymer-based thermoelectrics, a major challenge exists in the interplay between the Seebeck coefficient (S) and the conductivity (σ), for ZT = S2σT/κ, where T is the absolute temperature and κ is the thermal conductivity. However, it has been widely observed that there is a tradeoff between S and σ for the conjugated polymers, and the general trend is found to be 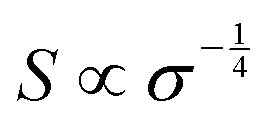 .159 Moreover, the conjugated polymers are generally observed to show an Arrhenius-type temperature dependency, as their conductivities increase with elevated temperatures, indicating that the charge transport in the electronic structures of the conjugated polymers is thermally activated.160 However, S and σ are analogous to the two faces in one coin, and the interplay between S and σ is essential for monitoring the thermoelectric properties of the conjugated polymer while it can serve as the starting point in the understanding of the charge transport mechanisms in conjugated polymers. To address the charge transport mechanisms in organic semiconductors, the mobility edge model and the polaron hopping model were proposed by Mott N. F. et al.161 Both these models used a generalized Boltzmann transport equation to describe conductivity, which can be written as
.159 Moreover, the conjugated polymers are generally observed to show an Arrhenius-type temperature dependency, as their conductivities increase with elevated temperatures, indicating that the charge transport in the electronic structures of the conjugated polymers is thermally activated.160 However, S and σ are analogous to the two faces in one coin, and the interplay between S and σ is essential for monitoring the thermoelectric properties of the conjugated polymer while it can serve as the starting point in the understanding of the charge transport mechanisms in conjugated polymers. To address the charge transport mechanisms in organic semiconductors, the mobility edge model and the polaron hopping model were proposed by Mott N. F. et al.161 Both these models used a generalized Boltzmann transport equation to describe conductivity, which can be written as
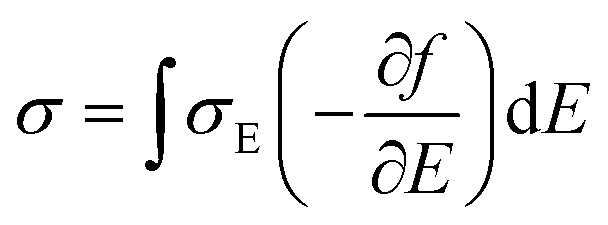 | (18) |
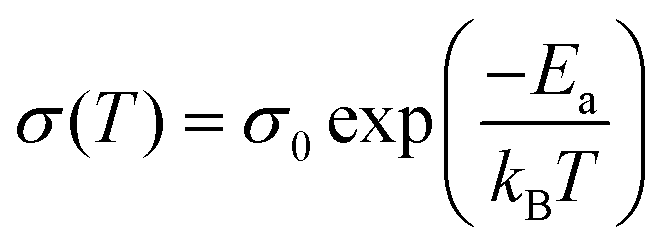 | (19) |
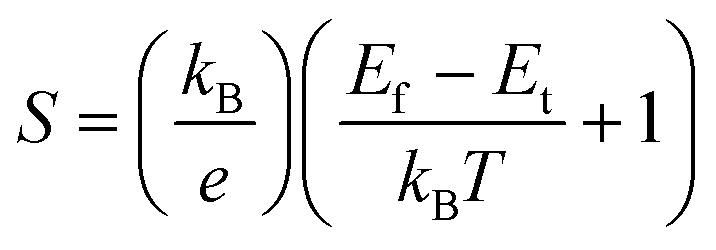 | (20) |
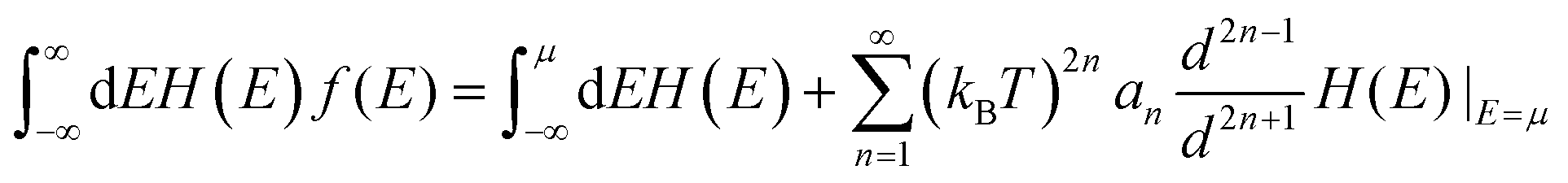 | (21) |
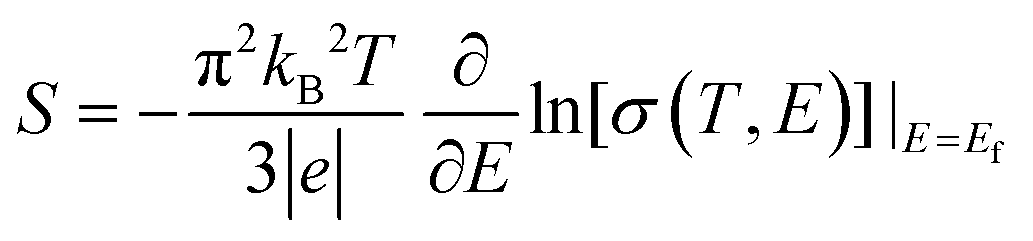 | (22) |
| σ(T, E) = e2g(E)D(E) | (23) |
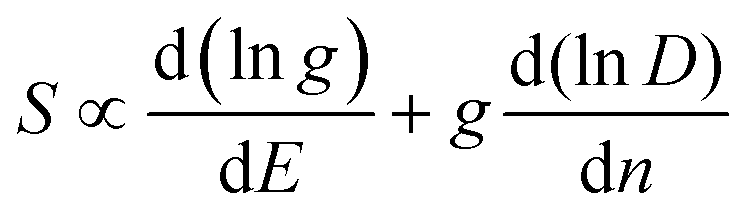 | (24) |
The modified Mott's model was used to simulate the thermoelectric properties of an electrochemical transistor based on PEDOT:PSS. The simulated correlations for S and σ against the charge carrier concentrations showed good qualitative agreement with the experimentally measured results, at a temperature difference of 2 K. Based on the results of simulation, it was found that the concentrations of free charge carrier follows a different correlation with the oxidation level in the range of n/N0 < 0.2, while N0 corresponds to the total concentration of the charge carriers. A dimensionless parameter b with the order of unity should be introduced to rescale the value of n, in order to achieve good agreement between the experimental results and the simulated models. However, the modified Mott's model cannot accurately predict the temperature dependency of the conductivity for conjugated polymers, as more general solutions should be applied to the transport function. To address this issue, Snyder G. J. et al. have reported the introduction of the concept of “transport edge” (Et) to take place on the “mobility edge” in Mott's model.164 Different from the mobility edge, which is derived from the activation energy of the free charge carriers, the transport edge is directly related to the thermopower, leaving out the effort to distinguish the real numbers of free charge carrier. Moreover, the transport edge is defined as the energy above which the charge carriers can conduct electricity; compared to the mobility edge, a thermal dimension (the activation energy) has been associated with the transport edge. The transport function in Snyder's model can be written as
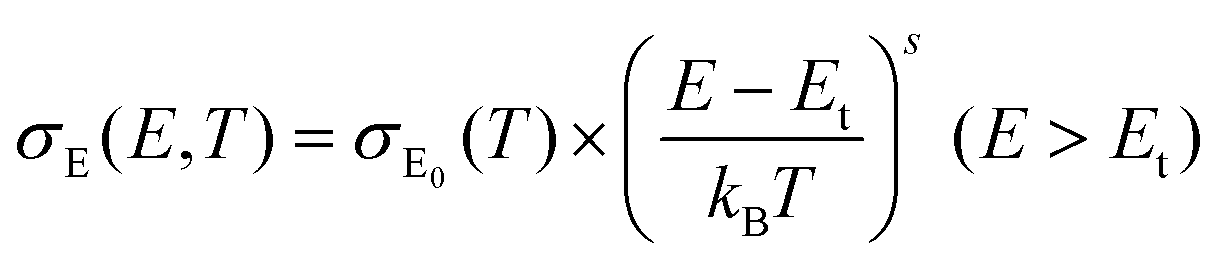 | (25) |
With the expression of σE(E,T) in eqn (25), the functions of σ and S can be simplified to
| σ = σE0(T) × sFs−1(η) | (26) |
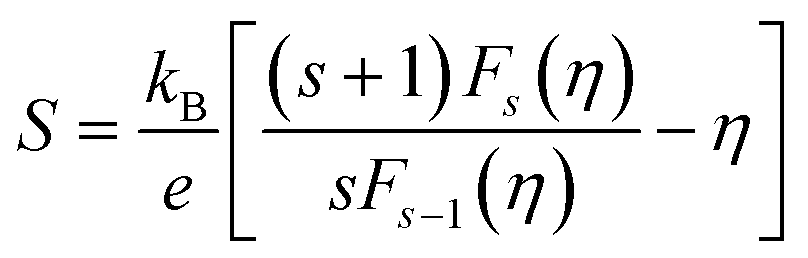 | (27) |
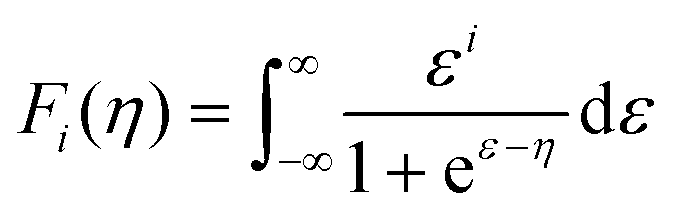 | (28) |
By measuring S, the reduced chemical potential η can be obtained from eqn (27), and σE0(T) can be subsequently obtained from eqn (26). In Snyder's model, the transport of charge carriers in the conjugated polymer is interpreted using a percolation transport mechanism, as the electrical conductivity is limited by the density of the percolation paths (Fig. 23a), and the range of conductivity is defined by the transport parameter s. When the energy of the charge carriers is smaller than Et (E < Et), the density of states is small and the percolation paths are limited, resulting in localized states; there exists a general activation energy W to excite the charge carriers from the localized states to the conductive states (Fig. 23b). Using eqn (20)–(23), the temperature-dependent transport coefficient σE0 of polyacetylene has been successfully simulated (Fig. 23c). By using equation (23) to fit the σE0vs. T−1/2 plots of polyacetylene with different doping concentrations, the as-obtained curve fits shared a same value of slope, corresponding to a same activation energy of W. However, the results obtained by analyzing the same data set using the variable range hopping (VRH) model showed different hopping energies for the polyacetylene samples, depending on the doping concentration.
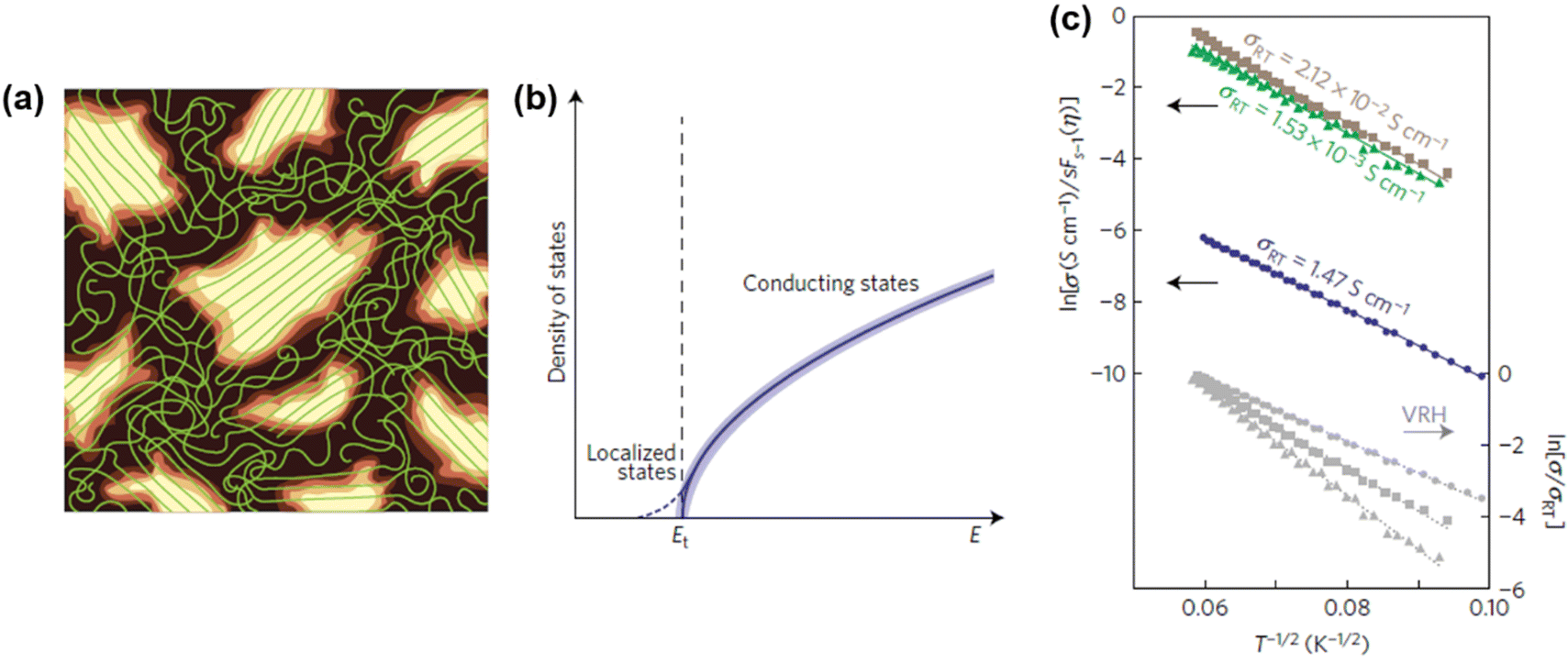 | ||
| Fig. 23 (a) Schematic illustration of the percolation transport in conjugated polymers, the transport coefficient is limited by the percolation paths that connect the conducting, ordered domains. (b) The density of states with an energy below Et is considered localized, and a thermal activation energy is required to transfer the localized states into the conductive states. (c) The correlations between the transport coefficient and the temperature of polyacetylene with different doping concentrations, the data points are obtained by modelling the thermopower and conductivity of the polyacetylene samples using eqn (10)–(13). The solid lines are obtained by fitting the data points using eqn (13). The room temperature conductivity (σRT) of each sample is shown along with the line. The modelling results using the VRH model are shown as the gray points and dot lines, respectively. Reproduced from ref. 164, with permission from Springer Nature, 2017. | ||
The transport edge model can effectively predict the thermally activated charge transport properties of the conjugated polymers with transport mechanisms in between band transport and hopping transport, based on the concepts of transport edge and temperature-dependent distribution of chemical potential. However, a few disordered conjugated polymers may show transitions of the transport properties with respect to temperatures. For example, Takeya J. et al. have investigated the transport properties of an organic field-effect transistor (FET) based on a uniaxially-oriented and disordered conjugated polymer, polycyclopentadithiophene-benzothiadiazole (CDT-BTZ).165 Surprisingly, it was observed that the CDT-BTZ based FET showed a negative temperature dependence of mobility, as its mobility increased from 5 cm2 V−1 s−1 to 6.5 cm2 V−1 s−1 as the temperature decreased from 300 K to 240 K. The negative temperature dependence of mobility was commonly found in highly ordered single crystals of small molecule semiconductors, and were considered evidence for band transport. Results of the Hall measurement indicated that the Hall factor for the FET based on CDT-BTZ was close to unity as the temperature was above 300 K, but it decreased to ∼0.5 as the temperature lowered to 280 K. Therefore, it can be speculated that CDT-BTZ may undergo a temperature dependent transition from hopping to band transport, which is unique for disordered organic semiconductors. Moreover, it is evident that the disorder induced decoherence can be mitigate by lowering the temperature, resulting in enhanced charge carrier mobility comparable to ordered crystals.
4. Summary and outlook
The unique electrical and electronic properties of conjugated polymers make them promising candidates in the metal-free, organic electronics, such as solar cells, photodiodes, field-effect transistors, and thermoelectrics. The organic nature of the conjugated polymers also makes them intrinsically compatible with biological tissues; if their electronic properties can be engineered properly, it may further unveil their important applications in tissue–electronic interfaces and electronic plants. The key to achieving precise engineering of the electronic structure of conjugated polymers is unraveling the mystery of their conductivity. By reviewing the existing literature on the charge transport models of conjugated polymers, it is clear that their fundamental assumptions are built on the fact that the charge carriers in conjugated polymers are low energy, long wavelength excitations. The non-interacting model assumes that the electron–electron interactions in conjugated polymers are weak, which can appropriately describe the behavior of charge carriers near the Fermi surface (E > EF). To better describe the charge transport properties near the band edge, the SSH model was proposed to depict the topological shapes of the charge carriers. In this regard, the charge carriers can be depicted as spinless topological solitons that propagate along the lattice of conjugated polymers without changing their shape and energy. For conjugated polymers with degenerate ground states, e.g., polyacetylene, the soliton and anti-soliton may stand for the solutions of the low-energy excitations for the conjugated systems with an ionization energy of 0. However, for conjugated polymers with non-degenerate ground states, e.g., P3HT, PANI, Ppy, the ionization energy is required to transfer the conjugated system from the insulating neutral state to the ionized conducting state. To accomplish this task, dopants with high electron affinity are generally used for the p-doping process, while dopants with low ionization potential are used for the n-doping process. During the doping process, the dopant molecules may deeply integrate with the conjugated polymer through the host–guest interactions. The charge transfer between the dopant molecules and the conjugated polymer may generate polarons, which are radical ions with a spin of 1/2, bound by the lattice distortion in the polymer matrix. Two polarons can combine to form one spinless bipolaron, depending on the binding energy. Both polarons and bipolarons can act as the charge carrier in the conjugated polymers with non-degenerate ground state. Generally speaking, as a result of the lattice distortion induced by the formation of polarons and bipolarons, the crystalline structure of conjugated polymers can be remarkably deteriorated, which may have negative impacts on the conductivity, thermopower, and work function. However, doping induced crystallinity can also be observed in a few types of conjugate polymers, i.e., P3HT. Therefore, a high doping efficiency is desired for achieving high conductivity without significantly degrading the crystallinity.However, the doping efficiency can be described by the correlation between conductivity and dopant concentration. In general, the conductivity of conjugated polymers may increase with increasing dopant concentrations until a “plateau” or an inflection point is reached, indicating saturation. Therefore, high doping efficiency can be realized by reaching the conductivity plateau at low dopant concentrations. To achieve this goal, the electron affinity, molecular structure, and size of the dopant should be carefully designed to achieve optimal Coulomb interactions in the polymer lattice. In other words, when selecting a dopant for a conjugated polymer host, the key parameters that should be taken into account may include the electron affinity, charge distribution, side chains, functional groups, and diffusion coefficients, rather than the type of dopant. The key to achieving high doping efficiencies is the establishment of favorable dopant–host interactions; existing strategies may include the usage of organometallic complex with high electron affinity or localized electron density, the selection of good solvents to swell the polymer, and side-chain engineering to tune the dopant redox potential, or thermally align the polymer lattice by high-temperature rubbing. Generally speaking, dopants with high electron affinity, high electrostatic shielding, high diffusion coefficient, and high efficiency of intercalation are favorable to generate a high intensity of delocalized charge carriers that may contribute to the total conductivity. On the other hand, for the conjugated system composed of given conjugated polymers and dopants, the plateau of conductivity can be reached when mixed electronic states composed of both polarons and bipolarons are created in the band gap. To precisely control the dopant concentration and improve the doping stability, different doping techniques have been developed, such as SqP, ion exchange, and ground state electron transfer. The selection of a proper doping technique can effectively enhance the dopant–host interactions, hence increasing the doping efficiency. However, current doping techniques, such as SqP, ion exchange, potentiostatic, apply the dopant–host interactions mainly by solid-state or solution diffusion of the dopant ions, in which the doping efficiency may be limited by the diffusion coefficient and the diffusion pathways. The GSET can impose the doping process in the interface between the n-type and p-type conjugated polymers, which omits the needs for ion diffusion. Moreover, a sufficient dopant concentration is also important for maintaining stable conductivity and impeding the transition from integer charge transfer to the charge transfer complex.
To achieve both high conductivity and high thermopower in conjugated polymers, a high intensity of free charge carriers and a low transport edge are highly required, according to Mott's mobility model and Snyder's transport model, respectively. In Snyder's model, the description of free charge carrier density is replaced by the transport edge, above which the charge carriers start to conduct electricity. A percolation transport mechanism similar to that for amorphous oxide semiconductors can be derived from the Snyder's model, indicating that the charge carriers are transported through the percolation paths connecting the crystalline, highly conductive regions under thermal activation. For the future design and development in the high-performance electronic or thermoelectric devices based on conjugated polymers, it is important to increase the percolation pathways and regularize the distribution of the micro crystallites in the polymer microstructure. The existing approaches may include alignment of the polymer microstructure by stretching, high-temperature rubbing, and thermal annealing; the addition of cosolvents and phase-transfer reagents during the polymerization and film formation steps can remarkably enhance the crystallite formation. Stability is another critical issue for the practical applications of the conjugated polymer-based organic electronics. Methods increasing the intercalation efficiency of dopants, the usage of sufficient dopant concentrations, the synthesis of self-doped conjugated polymers, etc. can be used to overcome the property degradation upon aging.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (81801851) and Sun Yat-sen University.References
- J. Adjizian, P. Briddon, B. Humbert, J. Duvail, P. Wagner, C. Adda and C. Ewels, Dirac cones in two-dimensional conjugated polymer networks, Nat. Commun., 2014, 5, 5842 CrossRef CAS PubMed.
- G. Galeotti, F. Marchi, E. Hamzehpoor, O. Maclean, M. R. Rao, Y. Chen, L. V. Besteiro, D. Dettmann, L. Ferrari, F. Frezza, P. M. Sheverdyaeva, R. Liu, A. K. Kundu, P. Moras, M. Ebrahimi, M. C. Gallagher, F. Rosei, D. F. Perepichka and G. Contini, Synthesis of mesoscale ordered two-dimensional π-conjugated polymers with semiconducting properties, Nat. Mater., 2020, 19, 874–880 CrossRef CAS PubMed.
- C. P. Yu, S. Kumagai, T. Kushida, M. Mitani, C. Mitsui, H. Ishii, J. Takeya and T. Okamoto, Mixed-orbital charge transport in N-shaped benzene- and pyrazine-fused organic semiconductors, J. Am. Chem. Soc., 2022, 144, 11159–11167 CrossRef CAS PubMed.
- M. C. Scharber and N. S. Sariciftci, Low band gap conjugated semiconducting polymers, Adv. Mater. Technol., 2021, 6, 2000857 CrossRef CAS.
- T. P. Kaloni, G. Schreckenbach and M. S. Freund, Band gap modulation in polythiophene and polypyrrole-based systems, Sci. Rep., 2016, 6, 36554 CrossRef CAS PubMed.
- M. Yoon, K. Suh, S. Natarajan and K. Kim, Proton conduction in metal-organic frameworks and related modularly built porous solids, Angew. Chem., Int. Ed., 2013, 52, 2688–2700 CrossRef CAS PubMed.
- O. I. Podkopaev, A. F. Shimanskii, N. O. Molotkovskaya and T. V. Kulakovskaya, Effect of the microstructure on electrical properties of high-purity germanium, Phys. Solid State, 2013, 55, 949–951 CrossRef CAS.
- P. Hsieh, S. Wu, T. Liang, L. Chen and M. H. Huang, GaAs wafers possessing facet-dependent electrical conductivity properties, J. Mater. Chem. C, 2020, 8, 5456–5460 RSC.
- S. Tiwari and S. Tiwari, Electrical and optical properties of CdS nanocrystalline semiconductors, Cryst. Res. Technol., 2006, 41, 78–82 CrossRef CAS.
- Y. Long, Z. Chen, W. Wang, F. Bai, A. Jin and C. Gu, Electrical conductivity of single CdS nanowire synthesized by aqueous chemical growth, Appl. Phys. Lett., 2005, 86, 153102 CrossRef.
- S. C. Rasmussen, Acetylene and its polymers – 150+ years of history, Springer, Switzerland, 2018 Search PubMed.
- A. B. Chourasia and D. S. D. C. Kelkar, electrical conductivity and magnetic susceptibility of polythiophene doped with iodine, AIP Conf. Proc., 2013, 911, 1536 Search PubMed.
- V. I. Krinichnyi, S. D. Chemerisov and Y. S. Lebedev, EPR and charge-transport studies of polyaniline, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 16233–16243 CrossRef CAS.
- J. Tsukamoto, A. Takahashi and K. Kawasaki, Structure and electrical properties of polyacetylene yielding a conductivity of 105 S cm−1, Jpn. J. Appl. Phys., 1990, 29, 125–130 CrossRef CAS.
- P. W. Chapman, O. N. Tufte, J. D. Zook and D. Long, Electrical properties of heavily doped silicon, J. Appl. Phys., 1963, 34, 3291 CrossRef CAS.
- E. Brinciotti, G. Gramse, S. Hommel, T. Schweimboeck, A. Altes, M. A. Fenner, J. Smoliner, M. Kasper, G. Badino, S. Tuca and F. Kienberger, Probing resistivity and doping concentration of semiconductors at the nanoscale using scanning microwave microscopy, Nanoscale, 2015, 7, 14715–14722 RSC.
- H. T. Nguyen, Z. Li, Y. Han, R. Basnet, M. Tebyetekerwa, T. N. Truong, H. Wu, D. Yan and D. Macdonald, Contactless, nondestructive determination of dopant profiles of localized boron-diffused regions in silicon wafers at room temperature, Sci. Rep., 2019, 9, 10423 CrossRef PubMed.
- P. F. Fewster and A. F. W. Willoughby, The effect of silicon doping on the lattice parameter of gallium arsenide grown by liquid-phase epitaxy, vapour-phase epitaxy and gradient-freeze techniques, J. Cryst. Growth, 1980, 50, 648–653 CrossRef CAS.
- G. Kaindl, G. Wortmann, S. Roth and K. Menke, 129I-Mössbauer study of iodine-doped polyacetylene, Solid State Commun., 1982, 41, 75–78 CrossRef CAS.
- W. Röss, A. Philipp, K. Seeger, K. Ehinger, K. Menke and S. Roth, Magnetoresistance and corbino resistance of iodine doped polyacetylene, Solid State Commun., 1983, 45, 933–935 CrossRef.
- Y. Zhang, W. Wang, F. Zhang, K. Dai, C. Li, Y. Fan, G. Chen and Q. Zheng, Soft organic thermoelectric materials: Principles, current state of the art and applications, Small, 2022, 18, 2104922 CrossRef CAS PubMed.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Optimization of the thermoelectric figure of merit in the conducting polymer poly(3,4-ethylenedioxythiophene), Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed.
- X. Zhang and L. Zhao, Thermoelectric materials: Energy conversion between heat and electricity, J. Materiomics, 2015, 1, 92–105 CrossRef.
- Y. Pei, H. Wang and G. J. Snyder, Band engineering of thermoelectric materials, Adv. Mater., 2012, 24, 6125–6135 CrossRef CAS PubMed.
- R. P. Fornari, P. W. M. Blom and A. Troisi, How many parameters actually affect the mobility of conjugated polymers?, Phys. Rev. Lett., 2017, 118, 086601 CrossRef PubMed.
- E. E. Moore and D. Yaron, An explicit-solvent dynamic-dielectric screening model of electron–hole interactions in conjugated polymers, J. Chem. Phys., 1998, 109, 6147 CrossRef CAS.
- D. T. Haar, Collected Papers of L.D. Landau, 67 – The effective mass of the polaron, Pergamon, 1965 Search PubMed.
- M. Comin, V. Lemaur, A. Giunchi, D. Beljonne, X. Blase and G. D’Avino, Doping of semicrystalline conjugated polymers: dopants within alkyl chains do better, J. Mater. Chem. C, 2022, 10, 13815–13825 RSC.
- Y. Liu, H. Yu, C. Shi and Z. Xiang, Localized electron density modulation in conjugated polymer nanosheets for boosting photocatalytic H2 evolution, J. Mater. Chem. A, 2021, 9, 19625–19630 RSC.
- H. Yan, J. Wade, L. Wan, S. Kwon, M. J. Fuchter, A. J. Campbell and J. Kim, Enhancing hole carrier injection via low electrochemical doping on circularly polarized polymer light-emitting diodes, J. Mater. Chem. C, 2022, 10, 9512–9520 RSC.
- J. H. Bombile, S. Shetty, M. J. Janik and S. T. Milner, Polaron hopping barriers and rates in semiconducting polymers, Phys. Chem. Chem. Phys., 2020, 22, 4032–4042 RSC.
- J. Chang, H. Sirringhaus, M. Giles, M. Heeney and I. McCulloch, Relative importance of polaron activation and disorder on charge transport in high-mobility conjugated polymer field-effect transistors, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 205204 CrossRef.
- A. L. Botelho, Y. Shin, M. Li, L. Jiang and X. Lin, Unified Hamiltonian for conducting polymers, J. Phys.: Condens. Matter, 2011, 23, 455501 CrossRef PubMed.
- J. Ma, D. Djurado and J. E. Fischer, X-ray structure of Cs-doped polyacetylene, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 2971–2975 CrossRef CAS PubMed.
- J. Y. Na, B. Kang, D. H. Sin, K. Cho and Y. D. Park, Understanding solidification of polythiophene thin films during spin-coating: effects of spin-coating time and processing additives, Sci. Rep., 2015, 5, 13288 CrossRef PubMed.
- M. S. Kiani, N. V. Bhat, F. J. Davis and G. R. Mitchell, Highly anisotropic electrically conducting films based on polypyrrole, Polymer, 1992, 33, 4113–4120 CrossRef CAS.
- W. Wu and S. Kivelson, Theory of conducting polymers with weak electron–electron interactions, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8546 CrossRef PubMed.
- A. J. Heeger, S. Kivelson, J. R. Schrieffer and W. P. Su, Solitons in conducting polymers, Rev. Mod. Phys., 1988, 60, 781–850 CrossRef CAS.
- Y. Cao, Y. Wang and B. Chen, Calculation of band structure of doped polyacetylene, Int. J. Quantum Chem., 1990, 37, 679–683 CrossRef CAS.
- S. Kivelson and W. Wu, Effect of electron–electron interactions in conducting polymers, Mol. Cryst. Liq. Cryst., 1985, 118, 9–18 CrossRef CAS.
- S. Kivelson and W. Wu, Photoproduction of neutral soliton pairs in trans-(CH)x, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5423 CrossRef CAS.
- E. J. Meier, F. A. An and B. Gadway, Observation of the topological soliton state in the Su–Schrieffer–Heeger model, Nat. Commun., 2016, 7, 13986 CrossRef CAS PubMed.
- W. P. Su, J. R. Schrieffer and A. J. Heeger, Soliton excitations in polyacetylene, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 2099 CrossRef CAS.
- W. P. Su, J. R. Schrieffer and A. J. Heeger, Solitons in polyacetylene, Phys. Rev. Lett., 1979, 42, 1698 CrossRef CAS.
- S. H. Han, S. G. Jeong, S. W. Kim, T. H. Kim and S. Cheon, Topological features of ground states and topological solitons in generalized Su–Schrieffer–Heeger models using generalized time-reversal, particle-hole, and chiral symmetries, Phys. Rev. B, 2020, 102, 235411 CrossRef CAS.
- S. Roth and D. Carroll, One-dimensional metals: Conjugated polymers, organic crystals, carbon nanotubes and graphene, Wiley, Weinheim, 2015 Search PubMed.
- M. B. Qarai, R. Ghosh and F. C. Spano, Understanding bipolarons in conjugated polymers using a multiparticle Holstein approach, J. Phys. Chem. C, 2021, 125, 24487–24497 CrossRef CAS.
- M. Taunk, A. Kapil and S. Chand, Hopping and tunneling transport over a wide temperature range in chemically synthesized doped and undoped polypyrrole, Solid State Commun., 2010, 150, 1766–1769 CrossRef CAS.
- C. Lin, R. L. Li, S. Robbennolt, M. T. Yeung, G. Akopov and R. B. Kaner, Furthering our understanding of the doping mechanism in conjugated polymers using tetraaniline, Macromolecules, 2017, 50, 5892–5897 CrossRef CAS.
- K. H. Kim, S. Lara-Avila, H. He, H. Kang, S. J. Hong, M. Park, J. Eklöf, K. Moth-Poulsen, S. Matsushita, K. Akagi, S. Kubatkin and Y. W. Park, Probing variable range hopping lengths by magneto conductance in carbonized polymer nanofibers, Sci. Rep., 2018, 8, 4948 CrossRef PubMed.
- J. Y. Fu, D. S. Liu and S. J. Xie, Polarons and bipolarons in polythiophene in the presence of magnetic field, Phys. E, 2008, 40, 915–919 CrossRef CAS.
- W. F. Cunha, L. A. Ribeiro, A. L. Almeida-Fonseca, R. Gargano and G. M. Silva, Reactive scattering between excitons and charge carriers in conjugated polymers, J. Phys. Chem. C, 2014, 118, 23451–23458 CrossRef.
- J. Ristein, Surface transfer doping of semiconductors, Science, 2006, 313, 1057–1058 CrossRef CAS PubMed.
- Z. Liang, H. H. Choi, X. Luo, T. Liu, A. Abtahi, U. S. Ramasamy, J. A. Hitron, K. N. Baustert, J. L. Hempel, A. M. Boehm, A. Ansary, D. R. Strachan, J. Mei, C. Risko, V. Podzorov and K. R. Graham, N-type charge transport in heavily p-doped polymers, Nat. Mater., 2021, 20, 518–524 CrossRef CAS PubMed.
- K. Jug, P. C. Hiberty and S. Shaik, σ-π energy separation in modern electronic theory for ground states of conjugated systems, Chem. Rev., 2001, 101, 1477–1500 CrossRef CAS PubMed.
- J. L. Bredas, J. C. Scott, K. Yakushi and G. B. Street, Polarons and bipolarons in polypyrrole: Evolution of the band structure and optical spectrum upon doping, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 1023 CrossRef CAS.
- J. L. Bredas and G. B. Street, Polarons, bipolarons, and solitons in conducting polymers, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- S. Winkler, P. Amsalem, J. Frisch, M. Oehzelt, G. Heimel and M. Koch, Probing the energy levels in hole-doped molecular semiconductors, Mater. Horiz., 2015, 2, 427 RSC.
- G. Heimel, The optical signature of charges in conjugated polymers, ACS Cent. Sci., 2016, 2, 309–315 CrossRef CAS PubMed.
- R. Png, M. C. Y. Ang, M. Teo, K. Choo, C. G. Tang, D. Belaineh, L. Chua and P. K. H. Ho, Madelung and Hubbard interactions in polaron band model of doped organic semiconductors, Nat. Commun., 2016, 7, 11948 CrossRef CAS PubMed.
- I. Sahalianov, J. Hynynen, S. Barlow, S. R. Marder, C. Müller and I. Zozoulenko, UV-to-IR absorption of molecularly p-doped polythiophenes with alkyl and oligoether side chains: Experiment and interpretation based on density functional theory, J. Phys. Chem. B, 2020, 124, 11280–11293 CrossRef CAS PubMed.
- R. Österbacka, C. P. An, X. M. Jiang and Z. V. Vardeny, Two-dimensional electronic excitations in self-assembled conjugated polymer nanocrystals, Science, 2000, 287, 839–842 CrossRef PubMed.
- C. M. Pochas and F. C. Spano, New insights on the nature of two-dimensional polarons in semiconducting polymers: Infrared absorption in poly(3-hexylthiophene), J. Chem. Phys., 2014, 140, 244902 CrossRef PubMed.
- R. Ghosh, A. R. Chew, J. Onorato, V. Pakhnyuk, C. K. Luscombe, A. Salleo and F. C. Spano, Spectral signatures and spatial coherence of bound and unbound polarons in P3HT films: Theory versus experiment, J. Phys. Chem. C, 2018, 122, 18048–18060 CrossRef CAS.
- R. Ghosh, C. K. Luscombe, M. Hambsch, S. C. B. Mannsfeld, A. Salleo and F. C. Spano, Anisotropic polaron delocalization in conjugated homopolymers and donor-acceptor copolymers, Chem. Mater., 2019, 31, 7033–7045 CrossRef CAS.
- Y. Zhang, V. Untilova, D. Muller, S. Guchait, C. Kiefer, L. Herrmann, N. Zimmermann, M. Brosset, T. Heiser and M. Brinkmann, Preferential location of dopants in the amorphous phase of oriented regioregular poly(3-hexylthiophene-2,5-diyl) films helps reach charge conductivities of 3000 S cm−1, Adv. Funct. Mater., 2022, 32, 2202075 CrossRef.
- V. Vijayakumar, E. Zaborova, L. Biniek, H. Zeng, L. Herrmann, A. Carvalho, O. Boyron, N. Leclerc and M. Brinkmann, Effect of alkyl side chain length on doping kinetics, thermopower, and charge transport properties in highly oriented F4TCNQ-doped PBTTT films, ACS Appl. Mater. Interfaces, 2019, 11, 4942–4953 CrossRef CAS PubMed.
- E. M. Thomas, K. A. Peterson, A. H. Balzer, D. Rawlings, N. Stingelin, R. A. Segalman and M. L. Chabinyc, Effects of counter-ion size on delocalization of carriers and stability of doped semiconducting polymers, Adv. Electron. Mater., 2020, 6, 2000595 CrossRef CAS.
- A. I. Hofmann, R. Kroon, S. Zokaei, E. Järsvall, C. Malacrida, S. Ludwigs, T. Biskup and C. Müller, Chemical doping of conjugated polymers with the strong oxidant magic blue, Adv. Electron. Mater., 2020, 6, 2000249 CrossRef CAS.
- V. Untilova, J. Hynynen, A. I. Hofmann, D. Scheunemann, Y. Zhang, S. Barlow, M. Kemerink, S. R. Marder, L. Biniek and C. Müller, High thermoelectric power factor of poly(3-hexylthiophene) through in-plane alignment and doping with a molybdenum dithiolene complex, Macromolecules, 2020, 53, 6314–6321 CrossRef CAS PubMed.
- D. Scheunemann, E. Järsvall, J. Liu, D. Beretta, S. Fabiano, M. Caironi, M. Kemerink and C. Müller, Charge transport in doped conjugated polymers for organic thermoelectrics, Chem. Phys. Rev., 2022, 3, 021309 CrossRef CAS.
- P. A. Finn, I. E. Jacobs, J. Armitage, R. Wu, B. D. Paulsen, M. Freeley, M. Palma, J. Rivnay, H. Sirringhaus and C. B. Nielsen, Effect of polar side chains on neutral and p-doped polythiophene, J. Mater. Chem. C, 2020, 8, 16216 RSC.
- M. Arvind, C. E. Tait, M. Guerrini, J. Krumland, A. M. Valencia, C. Cocchi, A. E. Mansour, N. Koch, S. Barlow, S. R. Marder, J. Behrends and D. Neher, Quantitative analysis of doping-induced polarons and charge transfer complexes of poly(3-hexylthiophene) in solution, J. Phys. Chem. B, 2020, 124, 7694–7708 CrossRef CAS PubMed.
- I. E. Jacobs, G. D’Avino, V. Lemaur, Y. Lin, Y. Huang, C. Chen, T. F. Harrelson, W. Wood, L. J. Spalek, T. Mustafa, C. A. O’Keefe, X. Ren, D. Simatos, D. Tjhe, M. Statz, J. W. Strzalka, J. Lee, I. McCulloch, S. Fratini, D. Beljonne and H. Sirringhaus, Structural and dynamic disorder, not ionic trapping, controls charge transport in highly doped conducting polymers, J. Am. Chem. Soc., 2022, 144, 3005–3019 CrossRef CAS PubMed.
- L. Lauchlan, S. Etemad, T. C. Chung, A. J. Heeger and A. G. MacDiarmid, Photoexcitations in polyacetylene, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 3701 CrossRef CAS.
- S. A. Brazovskii and N. N. Kirova, Excitons, polarons, and bipolarons in conducting polymers, JETP Lett., 1981, 33, 6 CAS.
- M. Nowak, S. D. D. V. Rughooputh, S. Hotta and A. J. Heeger, Polarons and bipolarons on a conducting polymer in solution, Macromolecules, 1987, 20, 965–968 CrossRef CAS.
- F. Genoud, M. Guglielmi, M. Nechtschein, E. Genies and M. Salmon, ESR study of electrochemical doping in the conducting polymer polypyrrole, Phys. Rev. Lett., 1985, 55, 118 CrossRef CAS PubMed.
- C. Enengl, S. Enengl, S. Pluczyk, M. Havlicek, M. Lapkowski, H. Neugebauer and E. Ehrenfreund, Doping-induced absorption bands in P3HT: Polarons and bipolarons, ChemPhysChem, 2016, 17, 3836–3844 CrossRef CAS PubMed.
- K. A. Peterson and M. L. Chabinyc, Lewis acid-base pair doping of p–type organic semiconductors, J. Mater. Chem. C, 2022, 10, 6287 RSC.
- M. G. Voss, J. R. Challa, D. T. Scholes, P. Y. Yee, E. C. Wu, X. Liu, S. J. Park, O. L. Ruiz, S. Subramaniyan, M. Chen, S. A. Jenekhe, X. Wang, S. H. Tolbert and B. J. Schwartz, Adv. Mater., 2020, 2000228 Search PubMed.
- H. Jia, Z. Huang, P. Li, S. Zhang, Y. Wang, J. Wang, X. Gu and T. Lei, Engineering donor-acceptor conjugated polymers for high-performance and fast-response organic electrochemical transistors, J. Mater. Chem. C, 2021, 9, 4927–4934 RSC.
- M. B. Qarai, R. Ghosh and F. C. Spano, Understanding bipolarons in conjugated polymers using a multiparticle Holstein approach, J. Phys. Chem. C, 2021, 125, 24487–24497 CrossRef CAS.
- R. Ghosh and F. C. Spano, Excitons and polarons in organic materials, Acc. Chem. Res., 2020, 53, 2201–2211 CrossRef CAS PubMed.
- J. Nightingale, J. Wade, D. Moia, J. Nelson and J. Kim, Impact of molecular order on polaron formation in conjugated polymers, J. Phys. Chem. C, 2018, 122, 29129–29140 CrossRef CAS.
- A. J. Moule, G. Gonel, T. L. Murrey, R. Ghosh, J. Saska, N. E. Shevchenko, I. Denti, A. S. Fergerson, R. M. Talbot, N. L. Yacoub, M. Mascal, A. Salleo and F. C. Spano, Quantifying polaron mole fractions and interpreting spectral changes in molecularly doped conjugated polymers, Adv. Electron. Mater., 2022, 8, 2100888 CrossRef CAS.
- M. R. Fernandes, J. R. Garcia, M. S. Schultz and F. C. Nart, Polaron and bipolaron transitions in doped poly(p-phenylene vinylene) films, Thin Solid Films, 2005, 474, 279–284 CrossRef CAS.
- Y. F. Chen, J. Liu, H. J. Yao, D. Mo, J. L. Duan, M. D. Hou, Y. M. Sun, L. Zhang and K. Maaz, Electrochemical polymerization and characterization of polypyrrole nanowires and nanotubules, Phys. B, 2010, 405, 2461–2465 CrossRef CAS.
- J. Bakalis, A. R. Cook, S. Asaoka, M. Forster, U. Scherf and J. R. Miller, Polarons, compressed polarons, and bipolarons in conjugated polymers, J. Phys. Chem. C, 2014, 118, 114–125 CrossRef CAS.
- D. Herrmann, S. Niesar, C. Scharsich, A. Köhler, M. Stutzmann and E. Riedle, Role of structural order and excess energy on ultrafast free charge generation in hybrid polythiophene/Si photovoltaics probed in real time by near-infrared broadband transient absorption, J. Am. Chem. Soc., 2011, 133, 18220–18233 CrossRef CAS PubMed.
- V. Untilova, T. Biskup, L. Biniek, V. Vijayakumar and M. Brinkmann, Control of chain alignment and crystallization helps enhance charge conductivities and thermoelectric power factors in sequentially doped P3HT:F4TCNQ films, Macromolecules, 2020, 53, 2441–2453 CrossRef CAS.
- H. Naarmann and N. Theophilou, New process for the production of metal-like, stable polyacetylene, Synth. Met., 1987, 22, 1–8 CrossRef CAS.
- Z. U. Khan, J. Edberg, M. M. Hamedi, R. Gabrielsson, H. Granberg, L. Wagberg, I. Engquist, M. Berggren and X. Crispin, Thermoelectric polymers and their elastic aerogels, Adv. Mater., 2016, 28, 4556–4562 CrossRef PubMed.
- Y. Wang and X. Jing, Intrinsically conducting polymers for electromagnetic interference shielding, Polym. Adv. Technol., 2005, 16, 344–351 CrossRef CAS.
- N. Hall, Twenty-five years of conducting polymers, Chem. Commun., 2003, 1–4 Search PubMed.
- E. P. Goodings, Conductivity and superconductivity in polymers, Chem. Soc. Rev., 1976, 5, 95–123 RSC.
- J. Voit, Superconductivity in models of conducting polymers, Phys. Rev. Lett., 1990, 64, 323 CrossRef CAS PubMed.
- K. Zhang, R. Wang and X. Chen, Order–disorder transition in p-oligophenyls, Phys. Chem. Chem. Phys., 2019, 21, 13590 RSC.
- X. Guo and A. Facchetti, The journey of conducting polymers from discovery to application, Nat. Mater., 2020, 19, 922–928 CrossRef CAS PubMed.
- O. D. Parashchuk, T. V. Laptinskaya and D. Y. Paraschuk, Macromolecular dynamics of conjugated polymer in donor-acceptor blends with charge transfer complex, Phys. Chem. Chem. Phys., 2011, 13, 3775–3781 RSC.
- P. Y. Yee, D. T. Scholes, B. J. Schwartz and S. H. Tolbert, Dopant-induced ordering of amorphous regions in regiorandom P3HT, J. Phys. Chem. Lett., 2019, 10, 4929–4934 CrossRef CAS PubMed.
- G. Krauss, A. Hochgesang, J. Mohanraj and M. Thelakkat, Highly efficient doping of conjugated polymers using multielectron acceptor salts, Macromol. Rapid Commun., 2021, 42, 2100443 CrossRef CAS PubMed.
- N. T. Kemp, A. B. Kaiser, C. J. Liu, B. Chapman, O. Mercier, A. M. Carr, H. J. Trodahl, R. G. Buckley, A. C. Partridge, J. Y. Lee, C. Y. Kim, A. Bartl and L. Dunsch, Thermoelectric power and conductivity of different types of polypyrrole, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 953–960 CrossRef CAS.
- Z. Liang, Y. Zhang, M. Souri, X. Luo, A. M. Boehm, R. Li, Y. Zhang, T. Wang, D. Y. Kim, J. Mei, S. R. Marder and K. R. Graham, Influence of dopant size and electron affinity on the electrical conductivity and thermoelectric properties of a series of conjugated polymers, J. Mater. Chem. A, 2018, 6, 16495 RSC.
- D. T. Scholes, S. A. Hawks, P. Y. Yee, H. Wu, J. R. Lindemuth, S. H. Tolbert and B. J. Schwartz, Overcoming film quality issues for conjugated polymers doped with F4TCNQ by solution sequentially processing: Hall effect, structural, and optical measurements, J. Phys. Chem. Lett., 2015, 6, 4786–4793 CrossRef CAS PubMed.
- T. J. Aubry, J. C. Axtell, V. M. Basile, K. J. Winchell, J. R. Lindemuth, T. M. Porter, J. Liu, A. N. Alexandrova, C. P. Kubiak, S. H. Tolbert, A. M. Spokoyny and B. J. Schwartz, Dodecaborane-based dopants designed to shield anion electrostatics lead to increased carrier mobility in a doped conjugated polymer, Adv. Mater., 2019, 31, 1805647 CrossRef PubMed.
- R. Lenin, A. Singh and C. Bera, Effect of dopants and morphology on the electrical properties of polyaniline for various applications, J. Mater. Sci., 2021, 32, 24710–24725 CAS.
- W. Shi, T. Zhao, J. Xi, D. Wang and Z. Shuai, Unravelling doping effects on PEDOT at the molecular level: From geometry to thermoelectric transport properties, J. Am. Chem. Soc., 2015, 137, 12929–12938 CrossRef CAS PubMed.
- N. Bondarenko, O. Eriksson and N. V. Skorodumova, Polaron mobility in oxygen-deficient and lithium-doped tungsten trioxide, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 165119 CrossRef.
- J. Yamamoto and Y. Furukawa, Electronic and vibrational spectra of positive polarons and bipolarons in regioregular poly(3-hexylthiophene) doped with ferric chloride, J. Phys. Chem. B, 2015, 119, 4788–4794 CrossRef CAS PubMed.
- J. L. Bredas, F. Wudl and A. J. Heeger, Polarons and bipolarons in doped polythiophene: A theoretical investigation, Solid State Commun., 1987, 63, 577–580 CrossRef CAS.
- M. Zeng, X. Wang, R. Ma, W. Zhu, Y. Li, Z. Chen, J. Zhou, W. Li, T. Liu, Z. He, H. Yan, F. Huang and Y. Cao, Dopamine semiquinone radical doped PEDOT:PSS: Enhanced conductivity, work function and performance in organic solar cells, Adv. Energy Mater., 2020, 2000743 CrossRef CAS.
- C. Gaul, S. Hutsch, M. Schwarze, K. S. Schellhammer, F. Bussolotti, S. Kera, G. Cuniberti, K. Leo and F. Ortmann, Insight into doping efficiency of organic semiconductors from the analysis of the density of states in n-doped C60 and ZnPc, Nat. Mater., 2018, 17, 439–444 CrossRef CAS PubMed.
- B. Sundqvist, Mapping intermolecular bonding in C60, Sci. Rep., 2014, 4, 6171 CrossRef CAS PubMed.
- V. M. Geskin and J. L. Bredas, Polaron pair versus bipolaron on oligothiophene chains: A theoretical study of the singlet and triplet states, ChemPhysChem, 2003, 4, 498–505 CrossRef CAS PubMed.
- A. V. Volkov, S. K. Singh, E. Stavrinidou, R. Gabrielsson, J. F. Franco-Gonzalez, A. Cruce, W. M. Chen, D. T. Simon, M. Berggren and I. V. Zozoulenko, Spectroelectrochemistry and nature of charge carriers in self-doped conducting polymer, Adv. Electron. Mater., 2017, 1700096 CrossRef.
- L. A. Ribeiro, F. F. Monteiro, W. F. Cunha and G. M. Silva, Charge carrier scattering in polymers: a new neutral coupled soliton channel, Sci. Rep., 2018, 8, 6595 CrossRef PubMed.
- Q. Zhang, Y. Sun, Y. Qin, W. Xu and D. Zhu, Two soluble polymers with low ionization potentials: doping and thermoelectric properties, J. Mater. Chem. A, 2016, 4, 1432–1439 RSC.
- E. F. Aziz, A. Vollmer, S. Eisebitt, W. Eberhardt, P. Pingel, D. Neher and N. Koch, Localized charge transfer in a molecularly doped conducting polymer, Adv. Mater., 2007, 19, 3257–3260 CrossRef CAS.
- A. Abutaha, P. Kumar, E. Yildirim, W. Shi, S. Yang, G. Wu and K. Hippalgaonkar, Correlating charge and thermoelectric transport to paracrystallinity in conducting polymers, Nat. Commun., 2020, 11, 1737 CrossRef CAS PubMed.
- B. Kippelen, Mutual electrical doping in polymers, Nat. Mater., 2020, 19, 701–710 CrossRef PubMed.
- T. J. Aubry, K. J. Winchell, C. Z. Salamat, V. M. Basile, J. R. Lindemuth, J. M. Stauber, J. C. Axtell, R. M. Kubena, M. D. Phan, M. J. Bird, A. M. Spokoyny, S. H. Tolbert and B. J. Schwartz, Tunable dopants with intrinsic counterion separation reveal the effects of electron affinity on dopant intercalation and free carrier production in sequentially doped conjugated polymer films, Adv. Funct. Mater., 2020, 2001800 CrossRef CAS PubMed.
- N. J. Cheetham, M. Ortiz, A. Perevedentsev, L. Dion-Bertrand, G. M. Greetham, I. V. Sazanovich, M. Towrie, A. W. Parker, J. Nelson, C. Silva, D. D. C. Bradley, S. C. Hayes and P. N. Stavrinou, The importance of microstructure in determining polaron generation yield in poly(9,9-dioctylfluorene), Chem. Mater., 2019, 31, 6787–6797 CrossRef CAS.
- D. T. Scholes, P. Y. Yee, G. R. McKeown, S. Li, H. Kang, J. R. Lindemuth, X. Xia, S. C. King, D. S. Seferos, S. H. Tolbert and B. J. Schwartz, Designing conjugated polymers for molecular doping: The roles of crystallinity, swelling, and conductivity in sequentially-doped selenophene-based copolymers, Chem. Mater., 2019, 31, 73–82 CrossRef CAS.
- M. N. Gueye, A. Carella, N. Massonnet, E. Yvenou, S. Brenet, J. Faure-Vincent, S. Pouget, F. Rieutord, H. Okuno, A. Benayad, R. Demadrille and J. Simonato, Structure and dopant engineering in PEDOT thin films: Practical tools for a dramatic conductivity enhancement, Chem. Mater., 2016, 28, 3462–3468 CrossRef CAS.
- S. Wang, D. Fazzi, Y. Puttisong, M. J. Jafari, Z. Chen, T. Ederth, J. W. Andreasen, W. M. Chen, A. Facchetti and S. Fabiano, Effect of backbone regiochemistry on conductivity, charge density, and polaron structure of n-doped donor-acceptor polymers, Chem. Mater., 2019, 31, 3395–3406 CrossRef CAS PubMed.
- Y. Liu, L. Xiao, P. Yin and C. Zhang, One-step synthesis of polypyrrole nanofiber assemblies with enhanced electronic properties: Implications for next-generation organic electronics, ACS Appl. Nano Mater., 2022, 5, 8930–8941 CrossRef CAS.
- Y. Lu, J. Wang and J. Pei, Achieving efficient n-doping of conjugated polymers by molecular dopants, Acc. Chem. Res., 2021, 54, 2871–2883 CrossRef CAS PubMed.
- S. E. Yoon, J. M. Han, B. E. Seo, S. W. Kim, O. P. Kwon, B. G. Kim and J. H. Kim, Optimized selection of dopant solvents for improving the sequential doping efficiency of conjugated polymers, Org. Electron., 2021, 90, 106061 CrossRef CAS.
- G. E. J. Hicks, S. Li, N. K. Obhi, C. N. Jarrett-Wilkins and D. S. Seferos, Programmable assembly of π-conjugated polymers, Adv. Mater., 2021, 33, 2006287 CrossRef CAS PubMed.
- S. Wang, T. P. Ruoko, G. Wang, S. Riera-Galindo, S. Hultmark, Y. Puttisong, F. Moro, H. Yan, W. M. Chen, M. Berggren, C. Müller and S. Fabiano, Sequential doping of ladder-type conjugated polymers for thermally stable n-type organic conductors, ACS Appl. Mater. Interfaces, 2020, 12, 53003–53011 CrossRef CAS PubMed.
- P. Reiser, L. Müller, V. Sivanesan, R. Lovrincic, S. Barlow, S. R. Marder, A. Pucci, W. Jaegermann, E. Mankel and S. Beck, Dopant diffusion in sequentially doped poly(3-hexylthiophene) studied by infrared and photoelectron spectroscopy, J. Phys. Chem. C, 2018, 122, 14518–14527 CrossRef CAS.
- T. L. D. Tam, G. Wu, S. W. Chien, S. F. V. Lim, S. W. Yang and J. Xu, High spin pro-quinoid benzo[1,2-c;4,5-c′]bisthiadiazole conjugated polymers for high-performance solution-processable polymer thermoelectrics, ACS Mater. Lett., 2020, 2, 147–152 CrossRef CAS.
- V. Vijayakumar, E. Zaborova, L. Biniek, H. Zeng, L. Herrmann, A. Carvalho, O. Boyron, N. Leclerc and M. Brinkmann, Effect of alkyl side chain length on doping kinetics, thermopower, and charge transport properties in highly oriented F4TCNQ-doped PBTTT films, ACS Appl. Mater. Interfaces, 2019, 11, 4942–4953 CrossRef CAS PubMed.
- Y. Liu, N. Lu, S. Poyraz, X. Wang, Y. Yu, J. Scott, J. Smith, M. J. Kim and X. Zhang, One-pot formation of multifunctional Pt-conducting polymer intercalated nanostructures, Nanoscale, 2013, 5, 3872–3879 RSC.
- M. Khalid, J. J. S. Acuna, M. A. Tumelero, J. A. Fischer, V. C. Zoldan and A. A. Pasa, Sulfonated porphyrin doped polyaniline nanotubes and nanofibers: synthesis and characterization, J. Mater. Chem., 2012, 22, 11340–11346 RSC.
- B. Senthilkumar, P. Thenamirtham and R. K. Selvan, Structural and electrochemical properties of polythiophene, Appl. Surf. Sci., 2011, 257, 9063–9067 CrossRef CAS.
- J. Liu, Y. Shi, J. Dong, M. I. Nugraha, X. Qiu, M. Su, R. C. Chiechi, D. Baran, G. Portale, X. Guo and L. J. A. Koster, Overcoming coulomb interaction improves free-charge generation and thermoelectric properties for n-doped conjugated polymers, ACS Energy Lett., 2019, 4, 1556–1564 CrossRef CAS.
- R. Fujimoto, S. Watanabe, Y. Yamashita, J. Tsurumi, H. Matsui, T. Kushida, C. Mitsui, H. T. Yi, V. Podzorov and J. Takeya, Control of molecular doping in conjugated polymers by thermal annealing, Org. Electron., 2017, 47, 139–146 CrossRef CAS.
- I. E. Jacobs, C. Cendra, T. F. Harrelson, Z. I. B. Valdez, R. Faller, A. Salleo and A. J. Moule, Polymorphism controls the degree of charge transfer in a molecularly doped semiconducting polymer, Mater. Horiz., 2018, 5, 655–660 RSC.
- K. E. Watts, B. Neelamraju, E. L. Ratcliff and J. E. Pemberton, Stability of charge transfer states in F4TCNQ-doped P3HT, Chem. Mater., 2019, 31, 6986–6994 CrossRef CAS.
- I. E. Jacobs, Y. Lin, Y. Huang, X. Ren, D. Simatos, C. Chen, D. Tjhe, M. Statz, L. Lai, P. A. Finn, W. G. Neal, G. D’Avino, V. Lemaur, S. Fratini, D. Beljonne, J. Strzalka, C. B. Nielsen, S. Barlow, S. R. Marder, I. McCulloch and H. Sirringhaus, High-efficiency ion-exchange doping of conducting polymers, Adv. Mater., 2021, 2102988 Search PubMed.
- W. Yao, L. Shen, P. Liu, C. Liu, J. Xu, Q. Jiang, G. Liu, G. Nie and F. Jiang, Electrochemical doping engineering tuning of the thermoelectric performance of a π-conjugated free-standing poly(thiophene-furan) thin film, Mater. Chem. Front., 2020, 4, 597–604 RSC.
- Y. Wang, E. Zeglio, L. Wang, S. Cong, G. Zhu, H. Liao, J. Duan, Y. Zhou, Z. Li, D. Mawad, A. Herland, W. Yue and I. McCulloch, Green synthesis of lactone-based conjugated polymers for n-type organic electrochemical transistors, Adv. Funct. Mater., 2022, 32, 2111439 CrossRef CAS.
- J. S. Shayeh, A. Ehsani, M. R. Ganjali, P. Norouzi and B. Jaleh, Conductive polymer/reduced graphene oxide/Au nano particles as efficient composite materials in electrochemical supercapacitors, Appl. Surf. Sci., 2015, 353, 594–599 CrossRef.
- B. Gupta, B. Goudeau, P. Garrigue and A. Kuhn, Bipolar conducting polymer crawlers based on triple symmetry breaking, Adv. Funct. Mater., 2018, 28, 1705825 CrossRef.
- T. Lanz, E. M. Lindh and L. Edman, On the asymmetric evolution of the optical properties of a conjugated polymer during electrochemical p- and n-type doping, J. Mater. Chem. C, 2017, 5, 4706–4715 RSC.
- S. Inagi and T. Fuchigami, Electrochemical post-functionalization of conducting polymers, Macromol. Rapid Comm., 2014, 35, 854–867 CrossRef CAS PubMed.
- G. Salinas and B. A. Frontana-Uribe, Analysis of conjugated polymers conductivity by in situ electrochemical-conductance method, ChemElectroChem, 2019, 6, 4105–4117 CrossRef CAS.
- A. M. Österholm, J. F. Ponder, M. Keersmaecker, D. E. Shen and J. R. Reynolds, Disentangling redox properties and capacitance in solution-processed conjugated polymers, Chem. Mater., 2019, 31, 2971–2982 CrossRef.
- D. Neusser, C. Malacrida, M. Kern, Y. M. Gross and J. Slageren, High conductivities of disordered P3HT films by an electrochemical doping strategy, Chem. Mater., 2020, 32, 6003–6013 CrossRef CAS.
- M. Nikolka, I. Nasrallah, B. Rose, M. K. Ravva, K. Broch, A. Sadhanala, D. Harkin, J. Charmet, M. Hurhangee, A. Brown, S. Illig, P. Too, J. Jongman, I. McCulloch, J. L. Bredas and H. Sirringhaus, High operational and environmental stability of high-mobility conjugated polymer field-effect transistors through the use of molecular additives, Nat. Mater., 2017, 16, 356–362 CrossRef CAS PubMed.
- K. Xu, H. Sun, T. Ruoko, G. Wang, R. Kroon, N. B. Kolhe, Y. Puttisong, X. Liu, D. Fazzi, K. Shibata, C. Yang, N. Sun, G. Persson, A. B. Yankovich, E. Olsson, H. Yoshida, W. M. Chen, M. Fahlman, M. Kemerink, S. A. Jenekhe, C. Müller, M. Berggren and S. Fabiano, Ground-state electron transfer in all-polymer donor-acceptor heterojunctions, Nat. Mater., 2020, 19, 738–744 CrossRef CAS PubMed.
- L. Wang, C. Pan, A. Liang, X. Zhou, W. Zhou, T. Wan and L. Wang, The effect of the backbone structure on the thermoelectric properties of donor-acceptor conjugated polymers, Polym. Chem., 2017, 8, 4644–4650 RSC.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Optimization of the thermoelectric figure of merit in the conducting polymer poly(3,4-ethylenedioxythiophene), Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed.
- K. Zhang, J. Qiu and S. Wang, Thermoelectric properties of PEDOT nanowire/PEDOT hybrids, Nanoscale, 2016, 8, 8033 RSC.
- S. Qu, Q. Yao, L. Wang, Z. Chen, K. Xu, H. Zeng, W. Shi, T. Zhang, C. Uher and L. Chen, Highly anisotropic P3HT films with enhanced thermoelectric performance via organic small molecule epitaxy, NPG Asia Mater., 2016, 8, e292 CrossRef CAS.
- Y. Feng, X. Jiang, E. Ghafari, B. Kucukgok, C. Zhang, I. Ferguson and N. Lu, Metal oxides for thermoelectric power generation and beyond, Adv. Compos. Hybrid Mater., 2018, 1, 114–126 CrossRef CAS.
- V. Vijayakumar, Y. Zhong, V. Untilova, M. Bahri, L. Herrmann, L. Biniek, N. Leclerc and M. Brinkmann, Bringing conducting polymers to high order: Toward conductivities beyond 105 S cm−1 and thermoelectric power factors of 2 mW m−1 K−2, Adv. Energy Mater., 2019, 9, 1900266 CrossRef.
- R. P. Fornari, P. W. M. Blom and A. Troisi, How many parameters actually affect the mobility of conjugated polymers?, Phys. Rev. Lett., 2017, 118, 086601 CrossRef PubMed.
- N. F. Mott and E. A. Davis, Electronic processes in non-crystalline materials, Oxford University Press, Oxford, 2012 Search PubMed.
- J. Kübler, Theory of itinerant electron magnetism, Revised Edition, Oxford University Press, Oxford, 2017 Search PubMed.
- S. Ihnatsenka, X. Crispin and I. V. Zozoulenko, Understanding hopping transport and thermoelectric properties of conducting polymers, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 035201 CrossRef.
- S. D. Kang and G. J. Snyder, Charge-transport model for conducting polymers, Nat. Mater., 2017, 16, 252–257 CrossRef.
- Y. Yamashita, J. Tsurumi, F. Hinkel, Y. Okada, J. Soeda, W. Zajaczkowski, M. Baumgarten, W. Pisula, H. Matsui, K. Müllen and J. Takeya, Transition between band and hopping transport in polymer field-effect transistors, Adv. Mater., 2014, 26, 8169–8173 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |