
Halogen-free photosensitizers based on meso-enamine-BODIPYs for bioimaging and photodynamic therapy†
Ruth
Prieto-Montero‡
 a,
Aitor
Díaz Andres‡
b,
Alejandro
Prieto-Castañeda‡
c,
Andrea
Tabero
d,
Asier
Longarte
e,
Antonia R.
Agarrabeitia
cf,
Angeles
Villanueva
d,
María J.
Ortiz
c,
Raúl
Montero
*g,
David
Casanova
*bh and
Virginia
Martínez-Martínez
*a
a,
Aitor
Díaz Andres‡
b,
Alejandro
Prieto-Castañeda‡
c,
Andrea
Tabero
d,
Asier
Longarte
e,
Antonia R.
Agarrabeitia
cf,
Angeles
Villanueva
d,
María J.
Ortiz
c,
Raúl
Montero
*g,
David
Casanova
*bh and
Virginia
Martínez-Martínez
*a
aDepartamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 48080 Bilbao, Spain. E-mail: virginia.martinez@ehu.eus; Tel: +34-946-015-969
bDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain
cDepartamento de Química Orgánica, Facultad de CC. Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain
dDepartamento de Biología, Universidad Autónoma de Madrid, Darwin 2, 28049 Madrid, Spain
eSpectroscopy Laboratory, Departamento Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), Apartado 644, 48080 Bilbao, Spain
fSección Departamental de Química Orgánica, Facultad de Óptica y Optometría, Universidad Complutense de Madrid, Arcos de Jalón 118, 28037 Madrid, Spain
gSGiker Laser Facility, Universidad del País Vasco (UPV/EHU), Sarriena s/n, 48940 Leioa, Spain
hIKERBASQUE, Basque Foundation for Science, 48009 Bilbao, Euskadi, Spain
First published on 23rd November 2022
Abstract
The search for efficient heavy atom free photosensitizers (PSs) for photodynamic therapy (PDT) is a very active field. We describe herein a simple and easily accessible molecular design based on the attachment of an enamine group as an electron-donor moiety at the meso position of the BODIPY core with different alkylation patterns. The effect of the alkylation degree and solvent polarity on the photophysical properties in terms of splitting absorption bands, fluorescence efficiencies and singlet oxygen production is analyzed in depth experimentally using spectroscopic techniques, including femtosecond and nanosecond transient absorption (fs- and ns-TA) and using computational simulations based on time-dependent density functional theory. The correlation between the theoretical/experimental results permits the rationalization of the observed photophysical behavior exhibited by meso-enamine-BODIPY compounds and the determination of mechanistic details, which rule the population of the triplet state manifold. The potential applicability as a theragnostic agent for the most promising compound is demonstrated through in vitro assays in HeLa cells by analyzing the internalization, localization and phototoxic action.
Introduction
The search for single systems able to diagnose and treat diseases, mainly cancer, has attracted great interest in modern medicine. Theragnosis (diagnosis and therapy) permits the in situ visualization of cancer tissues and cells, enhancing its treatment.1–4 Nevertheless, searching for a theragnostic agent is not a trivial task.5–9 A promising route could be the combination of fluorescence bioimaging for diagnosis and photodynamic therapy (PDT) as treatment, by the use of organic dyes.4,10–16 PDT requires the presence of molecular oxygen (O2), an organic dye (photosensitizer) and a specific light source. In PDT, the photosensitizer (PS) is activated under light, generating reactive oxygen species (ROS), mainly singlet oxygen (1O2), Fig. 1. This species of oxygen are cytotoxic and able to destroy cancer cells by apoptosis or necrosis.17–19 However, fluorescence and ROS generation are competing photophysical processes and a suitable balance between these antagonistic features should be achieved.20–23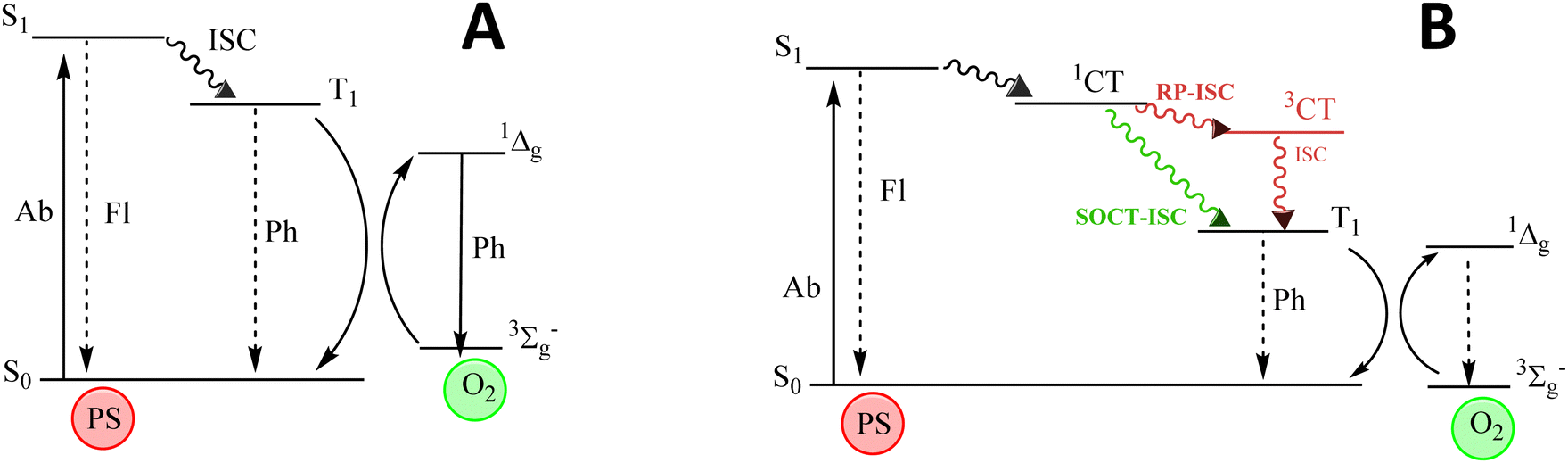 | ||
| Fig. 1 Jablonski diagram showing the population of triplet states by the conventional intersystem crossing (ISC) mechanism from the first singlet excited state (A) and by radical-pair intersystem crossing (RP-ISC) or spin–orbit charge transfer intersystem crossing (SOCT-ISC) mechanisms (B). | ||
Recently, one type of new lab-made PS has been based on the BODIPY chromophore (boron dipyrromethene), considered a chemically versatile small chromophore with excellent photophysical properties (intense absorption and emission bands and high photoresistance), stable under physiological conditions and insensitive to environmental changes.24–27 One strategy to design a theragnostic agent is the reduction of fluorescence by adding heavy atoms such as transition metals (Ru, Pd or Pt) or halogen atoms (Br or I) into the fluorophore structure, enhancing the intersystem crossing (ISC) driven by spin–orbit coupling (SOC) and as a consequence, increasing the triplet state population,19,20,22,28–32Fig. 1A. For instance, upon addition of heavy atoms to the BODIPY core, especially iodinated atoms at 2 and 6 positions, its fluorescence drastically decreases in favor of the triplet state population by ISC (S1 → Tn), and high singlet oxygen generation is achieved.18,20,24,31,33–39 Nevertheless, the efficiency of the photosensitizing action depends significantly on the lifetime of the lowest triplet state (T1) and the presence of a heavy atom increases also the ISC process back to the ground state (T1 → S0), considerably reducing the triplet state lifetime. Besides, halo-BODIPYs suffer from poor photostability and undesirable toxicity, diminishing their applicability and biocompatibility for biomedical uses.18,19,37
To address these drawbacks, the design of heavy atom-free photosensitizers is currently a very active research field.40–53 One of the strategies relies on the combination of electron donor (D) and acceptor (A) moieties to induce charge transfer (CT) states, which can act as mediators between singlet and triplet excited states by two different pathways: radical pair intersystem crossing (RP-ISC)54,55 or spin–orbit charge transfer intersystem crossing (SOCT-ISC),56–59Fig. 1B. Roughly, RP-ISC usually occurs when the electronic coupling between D and A units is weak, e.g., in spatially separated dyads by a linker, whereas the SOCT-ISC efficiency rapidly decays with the distance and it often takes place in directly linked D–A structures with a nearly orthogonal disposition. Generally, by these types of mechanisms, singlet oxygen generation (triplet population) and fluorescence efficiency (singlet population) of PSs can be efficiently modulated by the relative electron ability of A and D units, their chemical connection and relative geometrical disposition, as well as the polarity of the media.8,46,47,57,60–63
Some examples of halogen-free BODIPYs, in which different electron-donor groups have been incorporated into the chromophore core,39,45,47,48,53,63–67 have been already reported as potential theragnostic agents. In most cases, the electron-donating unit is added to the meso position, considering the most sensitive one in terms of the photophysical impact because of the marked change in the electronic density that occurs upon excitation (Fig. 2-Top).
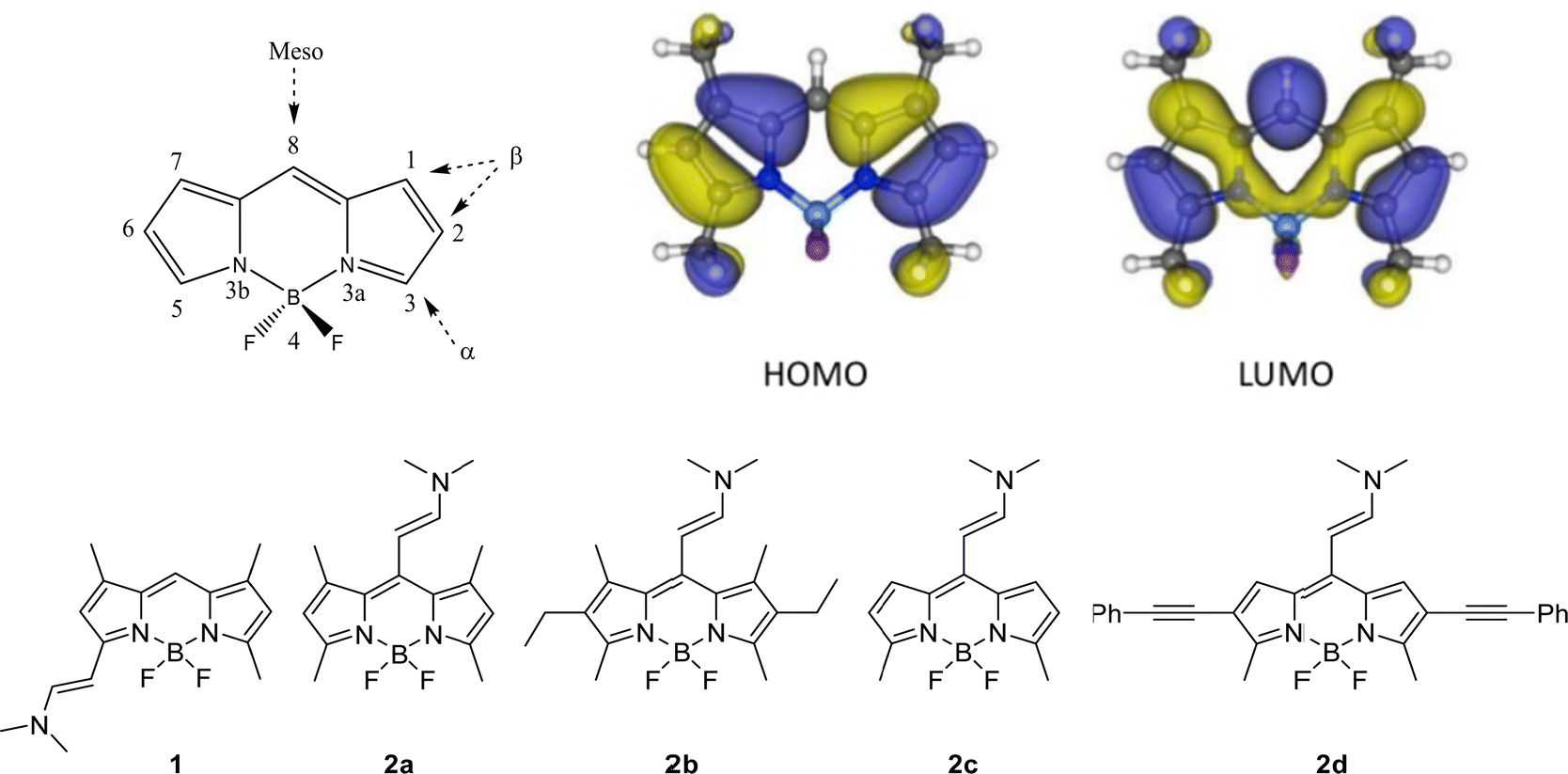 | ||
| Fig. 2 (Top) General BODIPY structure. The different positions in the core are indicated and numbered according to the IUPAC system.68 Frontier orbitals involved in the lowest electronic transitions (HOMO and LUMO) of a representative BODIPY; Bottom) Molecular structures of the different enamine substituted BODIPY derivatives. | ||
In this work, unlike other previous studies, a small group, an enamine, is selected as electron donor substituent in the set of studied BODIPYs shown in Fig. 2.39,45,48,53,63,65–67,69 All these compounds were synthesized using the Vilsmeier–Haack reaction, showing a new family of structurally simple and easily accessible compounds with push–pull character.70 First, the effect exerted on photophysics by the substitution of this group at the α- vs. meso-positions of the BODIPY core is compared (1vs.2a in Fig. 2-Bottom). Then, in the search for the best fluorescent-PS agent for PDT, several meso-enamine-BODIPY derivatives varying the alkylation pattern of the BODIPY core are analyzed (2a–2c, Fig. 2-Bottom). The effects of the alkylation degree and solvent polarity on the resultant photophysical features are deeply analyzed by theoretical simulations that allow characterizing the electronic states involved in the excitation/relaxation processes. To further understand the observed photophysical behavior, the relaxation pathway and the energy of the involved states were explored by femtosecond and nanosecond transient absorption (TA) and triplet energy and lifetime characterization. Finally, the collected evidence allows us to postulate a new BODIPY, with ethynyl phenyl groups at 2- and 6-positions to extend the delocalization of the π-system (2d in Fig. 2), as a suitable candidate with a red-shifted absorption band for theragnostic applications. The internalization, localization and phototoxic action of this compound are tested in vitro in HeLa cells with very promising results.
Results and discussion
Enamine substitution of BODIPY
First, the impact of the position of the donor enamine group on the photophysical properties was analyzed by comparing the enamine-BODIPYs substituted at the 3- or meso-position (1vs.2a). As expected, the enamine substitution at the α-position extends the π-conjugation of the BODIPY core, as shown by the π-electron distribution of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) (Fig. S1, ESI†), which induces a bathochromic shift with respect to the unsubstituted green BODIPYs, placing their respective absorption and emission bands in the orange region of the visible spectra (575 nm and 593 nm, respectively, Table 1). The relative shift in the absorption bands of 1 and 2a is well recovered by the computed vertical excitations at the Franck–Condon region (Table S1, ESI†). Compound 1 keeps a relatively high fluorescence quantum yield (60%) and negligible singlet oxygen production (Fig. 3 and Table 1).| Compound | λ ab (nm) | ε max 10−4 (M−1 cm−1) | λ fl (nm) | Φ fl | τ fl (ns) | Φ Δ |
|---|---|---|---|---|---|---|
| a λ ex = 530 nm, λem = 585 nm (for 1); λex = 480 nm, λem = 530 nm (for 2c); λex = 490 nm, λem = 600 nm (for 2d). | ||||||
| 1 | 575 | 4.3 | 593 | 0.60 | 0.69 (10%) | 0 |
| 4.04 (90%) | ||||||
| 2a | 494/451 | 5.7 | 518/640 | <0.01 | — | 0.04 |
| 2b | 516/449 | 3.3 | 540/670 | <0.01 | — | 0 |
| 2c | 496/460 | 6.6 | 529 | 0.08 | 0.92 (97%) | 0.20 |
| 5.29 (3.0%) | ||||||
| 2d | 551/490 | 5.7 | 599 | 0.42 | 3.23 | 0.31 |
 | ||
| Fig. 3 Height-normalized absorption (solid curves) and emission spectra (dash curves) in chloroform for 1 (blue) vs.2a (black). | ||
Conversely, by simply attaching the enamine group at the meso position, the photophysical properties of the BODIPY are drastically changed. Compound 2a brings an important difference in the location of their respective spectroscopic bands, and it shifted to shorter wavelengths with respect to compound 1 (Table 1 and Fig. 3). This hypsochromic effect is typically found when electron donor groups are anchored in the meso position, especially for amine groups.71,72 Moreover, the absorption spectra, unlike common BODIPY dyes, show an important shoulder contribution at shorter wavelengths, placed at around 450 nm. The formation of H-type aggregates is discarded as the spectrum is registered in a diluted solution (∼2 × 10−6 M). The nature of this contribution will be discussed in detail below. Besides, the enamine substitution at the meso position induces drastic fluorescence emission quenching, shortening its lifetime (Table 1). Although the fluorescence is weak for 2a (Table 1), it was possible to record a dual emission (Fig. 3 and Table 1), showing a narrower emission band at 518 nm assigned to the locally excited (LE) state and a broader band centered at around 640 nm, characteristic of radiative deactivation of an intramolecular charge transfer (CT) state.73,74 Although the presence of an enamine unit at the meso position in compound 2a enhances the non-radiative deactivation pathways, the triplet state is not effectively populated, yielding relatively low singlet oxygen production (4% in chloroform, Table 1).
Role of alkylation in meso-enamine substituted BODIPYs
Further alkylation of the BODIPY core with two extra ethyl groups at 2,6-positions, compound 2b, does not ameliorate the photophysical properties (Table 1), rendering practically null fluorescence and singlet oxygen production in all solvents of any polarity (Table 1 and Table S3, ESI†). Intriguingly, the absorption spectrum of compound 2b clearly shows two independent bands, with the peak centered at shorter wavelengths slightly blue-shifted with respect to the previously denoted as a shoulder for compound 2a. Contrarily, the main absorption band is red-shifted with respect to that recorded for the homologous compound 2a (Table 1 and Fig. 4A). The splitting of the two absorption bands of 2b increases as the polarity of the solvent decreases (Fig. 4B). Decreasing the polarity of the solvent, acetonitrile > chloroform > toluene > c-hexane, has a double effect on the absorption peak at shorter wavelengths, that is, a gradual intensity decrease and a hypsochromic shift. On the other hand, the main absorption band (at higher wavelengths) suffers a bathochromic shift and becomes progressively more intense. Indeed, for apolar solvents, e.g., c-hexane, the absorption bands are nicely divided into two distinguished peaks. In contrast, in polar solvents, e.g., acetonitrile, both absorption contributions overlap as they become closer in energy and intensity.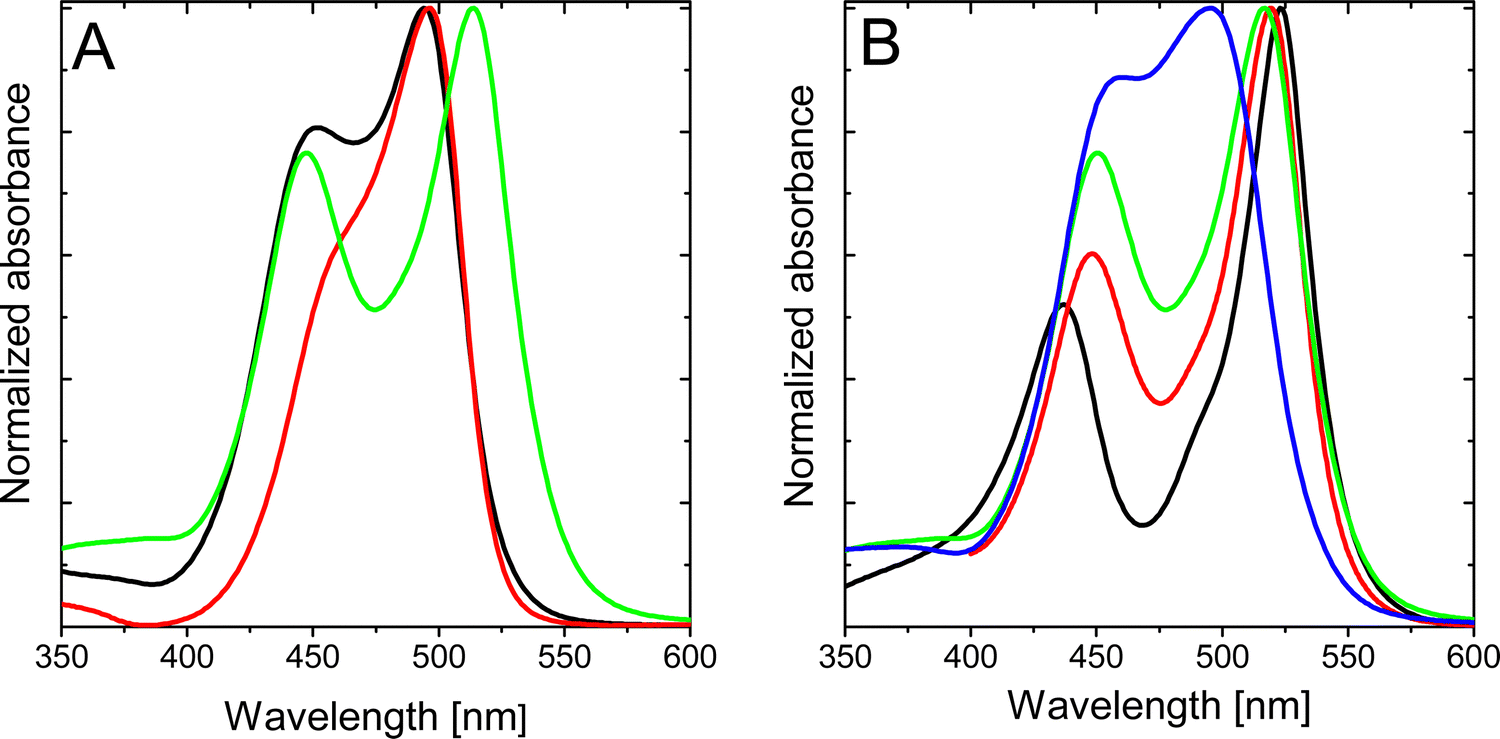 | ||
| Fig. 4 (A) Normalized absorption spectra of 2a (black), 2b (green) and 2c (red) in chloroform; and (B) normalized absorption spectra of 2b in different solvents: c-hexane (black), toluene (red), chloroform (green) and acetonitrile (blue). | ||
To gain insight into the nature of the two bands in the absorption spectra, rationalize the differences between the studied compounds and understand the changes induced by the polarity of the solvent, we have carried out electronic structure calculations for molecules 2a–c. The ground state energy minimum of molecule 2b exhibits a relative torsion of θ ∼ 30° between the group at the meso position and BODIPY's molecular plane. Nonplanarity is promoted by steric hindrance of the enamine with the methyl groups at positions 1 and 7. TDDFT calculations at the Franck–Condon geometry identify the two lowest singlet-to-singlet electronic transitions in 2b as optically active, i.e., with large oscillator strengths, in good agreement with the presence of two absorption bands registered in solution (Fig. 4). The lowest excited singlet state is mainly obtained as the excitation from the HOMO to the LUMO, whereas S2 can be described as the electronic promotion from the HOMO−1 to the LUMO (Fig. 5a). The HOMO of 2b is localized on the BODIPY unit, while HOMO−1 emerges from the coupling of the enamine and BODIPY π-systems. Finally, the LUMO is largely delocalized over both enamine and BODIPY moieties.
 | ||
| Fig. 5 (a) Frontier molecular orbitals of 2b computed with CAM-B3LYP/cc-pVDZ in chloroform. (b) Orbital energy diagram (in eV) of 2b in c-hexane, toluene, chloroform and acetonitrile. (c) Zoom of HOMO and HOMO−1 orbital energies. | ||
Interestingly, frontier orbital energies change with the polarity of the environment. Concretely, solvents with larger dielectric constants stabilize and destabilize the HOMO and HOMO−1, respectively (Fig. 5b and c). On the other hand, the energy of the LUMO remains rather constant. As a consequence, the transition energy to S1 (S2) increases (decreases) with the solvent polarity, and the S1 to S2 energy gap decreases. It is worth noting that the computed oscillator strengths (f) suggest a change in the relative intensity of the two bands, with the f(S2)/f(S1) ratio increasing with the polarity of the solvent (Table 2). These results explain the change in the absorption profiles upon the change in the solvent polarity, i.e., the frequency shift of the two bands and the change in relative intensities (Fig. 4B).
| Solvent | E(S1) | E(S2) | ΔE |
|---|---|---|---|
| c-Hexane | 2.86 (0.635) | 3.32 (0.588) | 0.46 |
| Toluene | 2.88 (0.628) | 3.30 (0.599) | 0.42 |
| Chloroform | 2.90 (0.624) | 3.22 (0.651) | 0.32 |
| Acetonitrile | 2.98 (0.569) | 3.21 (0.649) | 0.23 |
Dealkylation of positions 2 and 6 in 2a with respect to 2b preserves the torsion of the enamine with respect to the BODIPY fragment. The main difference between the electronic structures of 2a and 2b is the orbital energy of the HOMO, since it is the frontier orbital presenting the largest electron density at the 2 and 6 carbon atoms. Substitution of the alkyl groups by a more electronegative hydrogen stabilizes the HOMO (Fig. S2, ESI†), shifting the S1 transition to shorter wavelengths and reducing the energy separation between the S1 and S2 states (Table 3), in agreement with the absorption spectra (Fig. 4A). Similarly, dealkylation of positions 1 and 7 in 2c mainly stabilizes the HOMO and LUMO, as both orbitals present sizeable electron densities on these atoms. As a consequence, the S1 energy barely changes with respect to 2a, while the S2 transition slightly redshifts (∼ 0.1 eV), resulting in a rather small separation between the S1 and S2 transitions. In fact, the absorption spectrum of 2c does not show a noticeable shoulder feature at shorter wavelengths and the main band resembles the one for pristine BODIPY (no substitution). It is worth noting that the no-alkylation in 1 and 7 atoms slightly increases the molecular planarity in 2c (θ ∼ 26°) with respect to 2b and 2a, allowing for larger electronic mixing of the BODIPY and enamine π-systems (Fig. S1, ESI†).
| Molecule | E(S1) | E(S2) | ΔE |
|---|---|---|---|
| 2b | 2.90 (0.624) | 3.22 (0.651) | 0.32 |
| 2a | 3.00 (0.585) | 3.25 (0.635) | 0.25 |
| 2c | 3.02 (0.559) | 3.12 (0.692) | 0.10 |
On the other hand, regarding the fluorescence and singlet oxygen quantum yield, our results clearly indicate that methylation at 1- and 7-positions in meso-enamine-BODIPYs noticeably influences the fluorescence efficiency, and more significantly, the singlet oxygen generation (Table 1 and Table S3, ESI†). Compound 2c, without methyl groups at 1- and 7-positions, shows the highest singlet oxygen and fluorescence quantum yields of the series, whereas compounds 2a and 2b show nearly no emission and singlet oxygen production, being internal conversion (IC) the main deactivation pathway (see next section). Moreover, the fluorescence quantum yield and also the singlet oxygen quantum yield of 2c increase as the polarity of the solvent decreases (Table S3, ESI†). The less efficient radiative deactivation of the singlet excited state, as well as the singlet to triplet transition in polar solvents, was also observed for other BODIPY dyads.47,57,60
Decay dynamics of photoactivated enamine-BODIPYs
In order to extract mechanistic information on the excited state relaxation of the present compounds, TA experiments have been carried out on 2b (Fig. 6) and 2c (Fig. 7), considering them as representative examples in terms of methylation degree, in solvents with different polarities (toluene, CHCl3 and ACN). Fig. 6a shows the TA spectra of 2b in toluene at selected pump-probe delays. At short delays (0.15 ps) the TA spectrum is composed mainly of 3 contributions: a positive band at 350–400 nm attributable to excited state absorption (ESA), the ground state bleach (GSB) at 425–525 nm and a lower negative feature extending from 525 to 700 nm and corresponding to stimulated emission (SE). The blue end of the latter contribution reflects the emission from the initially prepared excited state. It decays in 1 ps, denoting a short living LE state. Within the same time scale, a positive feature appears at 570 nm corresponding to the absorption from an excited species formed during the LE state decay. A comparison with previous studies on BODIPY donor and acceptor groups75 allows us to identify this feature as the absorption of a CT state, in which the enamine plays the role of the donor and the BODIPY core acts as the acceptor. The remarkable red shift of the emission during the first picoseconds seems to support this hypothesis. | ||
| Fig. 6 Transient spectra and DAS of 2b in toluene (a and b), CHCl3 (c and d) and ACN (e and f). | ||
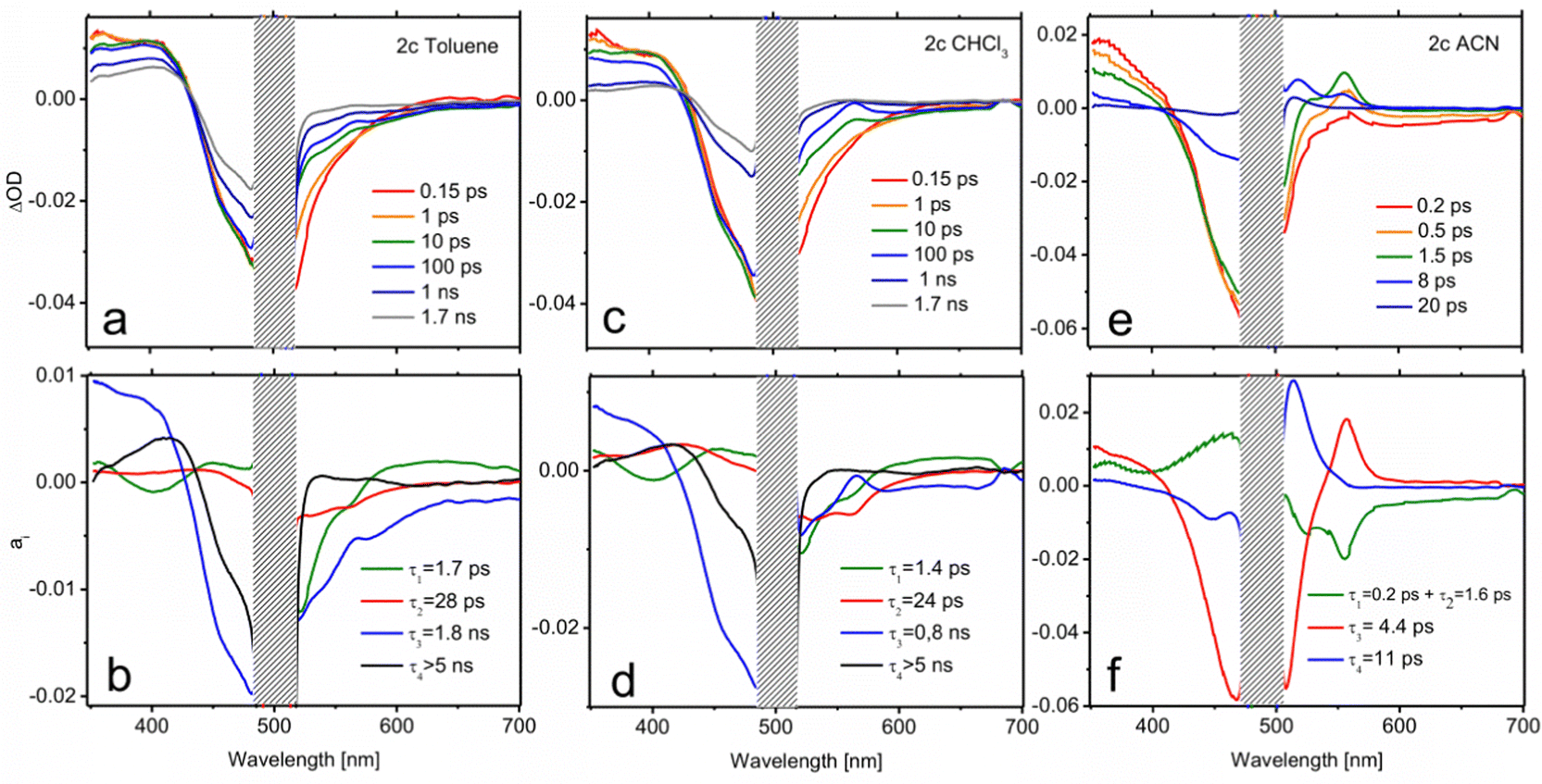 | ||
| Fig. 7 Transient spectra and DAS of 2c in toluene (a and b), CHCl3 (c and d) and ACN (e and f). | ||
A multiexponential fit with four components (τ1 = 1.1 ps; τ2 = 19 ps; τ3 = 118 ps; τ4 > 5 ns) yields a satisfactory description of the transient spectra. The distribution of preexponential factors resulting from this analysis is presented in the decay associated spectra (DAS) in Fig. 6b (see the ESI† for further details on data analysis). As is observed there, for 2b the amplitude of the slower decay (a4) is negligible, but it is included for the latter comparison with 2c. The distribution of a1 (τ1 = 1.1 ps) shows changes in the ESA band without affecting the evolution of the GSB, meaning that the nature of the excited state is modified in this time scale. Indeed, a1 describes simultaneously the LE emission decay (550 nm) and the onset of the CT features (positive band at 570 nm and emission at wavelengths > 650 nm). Therefore, a1 accounts for the population that is initially excited to S1 (LE), and reaches the CT state. Regarding τ3, as displayed by the GSB band, the excited state population decays to the ground state with 118 ps lifetime. The spectral characteristics of the CT state in the 500–700 nm region are also perceptible in the a3 distribution, which indicates that IC to the ground state mainly occurs from the CT state. Finally, the scrutiny of a2 reflects that certain CT formation takes place in a slower time scale (τ2 = 19 ps). In fact, a1 and a2 spectra are very similar except for the emission contribution in the red edge. Differently, a2 shows emission at intermediate wavelengths between the LE and CT bands, which could be related to the cooling (vibrational relaxation) on the CT potential well.
A higher polarity solvent such as CHCl3 (panels c and d in Fig. 6) does not change qualitatively the dynamics, as illustrated by the resemblance of the transient spectra and DAS with those in the previous solvent. Only a substantial acceleration of CT formation is noticeable. Thus, in CHCl3, τ1 and τ2 are respectively 0.6 and 3.5 ps, while IC remains almost unaltered (τ3 = 112 ps). Following the same trend, Fig. 6e and f show that further stabilization of the CT state with a higher-polarity solvent (ACN) gives rise to even faster dynamics (τ1 = 0.2 ps; τ2 = 1.3 ps; τ3 = 13 ps; τ4 = 78 ps). In this case, two components of tens of picoseconds, τ3 and τ4, account for the independent radiationless relaxation pathways of the CT state. The origin of these dual deactivation channels to the ground state is unclear.
Although notable differences in the kinetics are observed for 2c, its TA data (Fig. 7) can be understood by the same basic model as that employed for 2b. In toluene, the decay of the LE fluorescence and the onset of the red shifted emission of the CT formation take place in τ1 = 1.7 ps and τ2 = 28 ps, respectively (Fig. 7b). The decay of the CT state, described by the a3 spectrum, is much slower in this case (τ3 = 1.8 ns). It is also worth noting that the CT emission in 2c covers the 520–600 nm region, pointing to a less marked CT character. Indeed, the CT absorption band at 570 nm appears now as a small dip on top of the emission band. The most remarkable feature in the relaxation of 2c is the appearance of a very long-living species (τ4 > 5 ns) that can be identified as the T1 state on the basis of its TA spectrum (a4). This triplet state has to be formed from the CT relaxation, although its formation dynamics cannot be resolved from the recorded data. The triplet quantum yield can be determined on the grounds of the a4 component through the ratio between the GSB band maximum in the a4 spectrum and at t = 0 ns, yielding a value of ΦT = 0.37. This number is in total agreement with the singlet oxygen quantum yield obtained in this solvent by the direct registration of its photoluminescence signal (Table S3, ESI†). Moreover, the assignation of the TA spectrum at τ4 > 5 ns to the triplet state absorption features is confirmed by the registration of the TA spectra in the nanosecond range (Fig. S3, ESI†). Similarly, the ns-resolved TA (ns-TA) presents a positive band around 375–430 nm, attributed to the absorption of the T1 state, a negative contribution in the 425–525 nm range that corresponds to ground state bleaching (GSB) associated with S0 → S1 transition, and a weak and broad contribution at > 525 nm, also assigned to triplet absorption.47,60,74,76,77 The lifetimes registered at 405 nm and 540 nm measured under nitrogen-saturated conditions were 16 μs, long enough for efficient oxygen quenching (Fig. S3, ESI†). The spin-triplet multiplicity nature of this state is confirmed by quenching registered within the triplet lifetime range under aerated conditions (Table S4, ESI†).
The CT dynamics of 2c in CHCl3 is very similar. However, its lifetime is noticeably shorter (τ3 = 0.8 ns) and the triplet quantum yield is lower (ΦT = 0.24), which is once again in agreement with the singlet oxygen production obtained in this solvent. This fact suggests that the formation of a long-lived CT state is required in order to efficiently couple to the triplet state. The ns-TA spectrum is comparable to that obtained in toluene (Fig. S3, ESI†), but a much longer triplet lifetime is registered (113 μs, Table S4, ESI†). Finally, in ACN, extremely fast relaxation of the CT is observed (panels e and f in Fig. 7 and Fig. S5, ESI†). The formation of the CT state occurs at a similar scale (τ1 = 0.2 ps and τ2 = 1.6 ps), both of which, owing to their very similar spectra, have been plotted together in Fig. 7f for simplicity. However, the subsequent decay is much faster, draining the population to the ground state with τ3 = 4.4 ps. This channel precludes the formation of a triplet state. Accordingly, the distribution of a4 is compatible with the cooling of the excitation energy in the ground state (vibrational relaxation) that is now perceptible due to the very fast IC.
Further experiments were also carried out for 2b and 2c in CHCl3 using excitation energies resonant with the S2 state (445 nm). The observed dynamics was essentially the same recorded after pumping to S1, pointing to a very fast S2–S1 conversion in both species (Fig. S6, ESI†).
ISC mechanism
TA measurements of 2b and 2c indicate that the triplet state population occurs from a CT state, suggesting a SOCT mechanism. Electronic structure calculations of 2c identify two excited state minima on the S1 potential energy surface. At small torsion angles between BODIPY and the meso-enamine fragments, there is a local S1 minimum with the hole and electron largely localized on the BODIPY and with the participation of enamine's π-system, which can be related to the LE state identified by TA spectroscopy (Fig. 8). The LE geometry exhibits a small enamine-BODIPY dihedral angle (∼ 20°) and presents a sizeable transition dipole moment and oscillation strength to the ground state (Table S2, ESI†). Hence, we identify it as the state responsible for the fluorescence emission recorded in the 500–540 nm region. Additionally, the molecule presents a lower energy excited state minimum with an orthogonal disposition between the two fragments in which the hole is localized on the enamine unit and the electron on the BODIPY fragment, i.e., a CT state. Notably, the relative energy between LE and CT states strongly depends on the solvent polarity, with polar solvents stabilizing the orthogonal CT with respect to the LE state. The vertical energy gap to the ground state at the CT minimum severely diminishes upon increasing the polarity of the solvent (Table S2, ESI†), in line with the increase of non-radiative decay rates and the hindering of singlet oxygen generation. | ||
| Fig. 8 Energy profiles (in kcal mol−1) of the lowest excited singlet state (S1) of 2c computed in c-hexane (orange) and ACN (blue) along the molecular torsion between BODIPY and enamine moieties. The molecular torsion model has been obtained by constrained geometry optimization of the S1 state in ACN at different torsion angles. S1 energies are relative to the value in the planar arrangement. Inset: HOMO and LUMO for LE and CT states. | ||
At the LE minimum, although the S1 state presents non-vanishing SOCs to the two lowest triplet states, singlet–triplet energy gaps are rather large, preventing efficient ISC, as in pristine BODIPY (Fig. 9). On the other hand, molecular torsion towards the CT minimum largely reduces S1/Tn energy differences and modifies the electronic nature of low-lying states. Along the molecular orthogonalization, T1 strongly localizes on the BODIPY moiety, whereas T2 becomes a purely CT excitation, like the lowest excited singlet. As a consequence, in the perpendicular arrangement S1/T2 nearly degenerates and presents a small SOC, in accordance with El Sayed's rule. Simultaneously, the S1/T1 energy gap (SOC) decreases (increases) considerably. We note that despite the reduction of the singlet–triplet gap, it remains rather large (Fig. 9a). On the other hand, it is well-known that TDDFT functionals tend to underestimate and overestimate the energies of T1 and S1, respectively.78 Therefore, these results make us conclude that ISC in 2b (and 2c) is triggered by excited state relaxation via BODIPY-enamine torsion, and takes place through the SOCT-ISC mechanism between the CT excited singlet and low-lying triplets, in particular the BODIPY-localized T1 state, but also the second excited triplet T2.
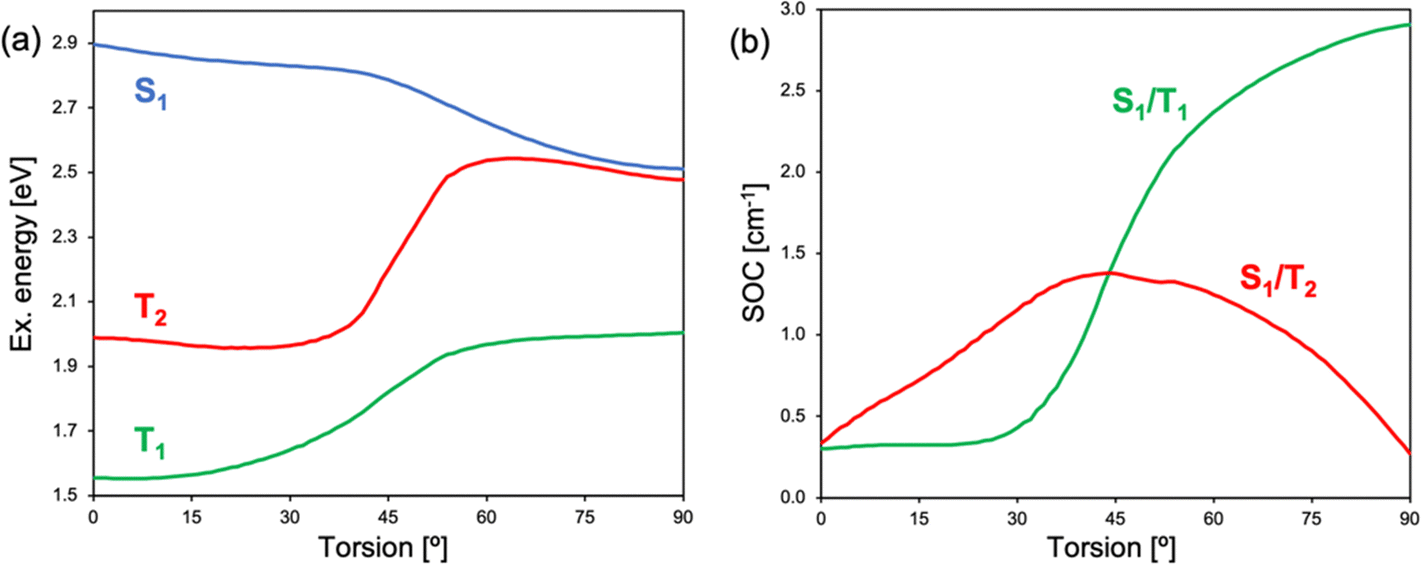 | ||
| Fig. 9 (a) Excitation energy (in eV with respect to the ground state at the planar geometry) of the lowest excited singlet (S1) and triplet (T1 and T2) states and (b) singlet/triplet SOCs computed in ACN of 2c along the molecular torsion between BODIPY and enamine moieties. | ||
To further characterize the energy of the lowest triplet state (T1), luminescence measurements are recorded at 78 K for 2c (Fig. S7, ESI†). The band placed at 655 nm was assigned to phosphorescence emission, with average lifetimes in the ms range. The energy of this low-lying triplet state (182.8 kJ mol−1) is higher than the energy gap between the triplet ground state of O2 (3Σg) and the first singlet excited state (1Δg) of O2 (94.2 kJ mol−1), fulfilling the necessary conditions to generate singlet oxygen by the type-II energy transfer mechanism (Fig. 1). With 2c being the best compound of the series in terms of singlet oxygen quantum production and fluorescence efficiency, a more π-conjugated meso-enamine BODIPY, Fig. 2, 2d, was formulated. By attaching ethynyl phenyl groups at 2 and 6 positions a notable shift takes place in both absorption and emission bands (Fig. S8, ESI†), resulting in an even better balance between fluorescence and singlet oxygen quantum yields (Table 1 and Table S1, ESI†), and showing a relatively long lived triplet state close to 100 μs (Fig. S4 and Table S4, ESI†). Hence, 2d could be considered as a purely organic agent with a suitable balance between fluorescence and singlet oxygen capacities with potential use in theragnostic applications.
In vitro tests
Compound 2d was selected as the most suitable enamine-BODIPY to perform assays in HeLa cells. Subcellular localization experiments confirm that 2d can be internalized inside cells, allowing sharp fluorescence imaging following a typical lipid droplet pattern, indicative of selective accumulation (Fig. 10A). Taking into account the broad fluorescence emission spectrum of compound 2d, it was not possible to carry out colocalization experiments with commercial organelle-targeted fluorescent probes, since the emissions of probes and compound 2d would overlap. However, Fig. S9 (ESI†) shows that the subcellular distribution pattern of compound 2d is absolutely similar to that of lipid droplets, and besides, quite different from mitochondria or lysosomes in HeLa cells. Interestingly, the fluorescence image obtained with 2d is much sharper and brighter than compound 1, besides its lower fluorescence efficiency (Table 1).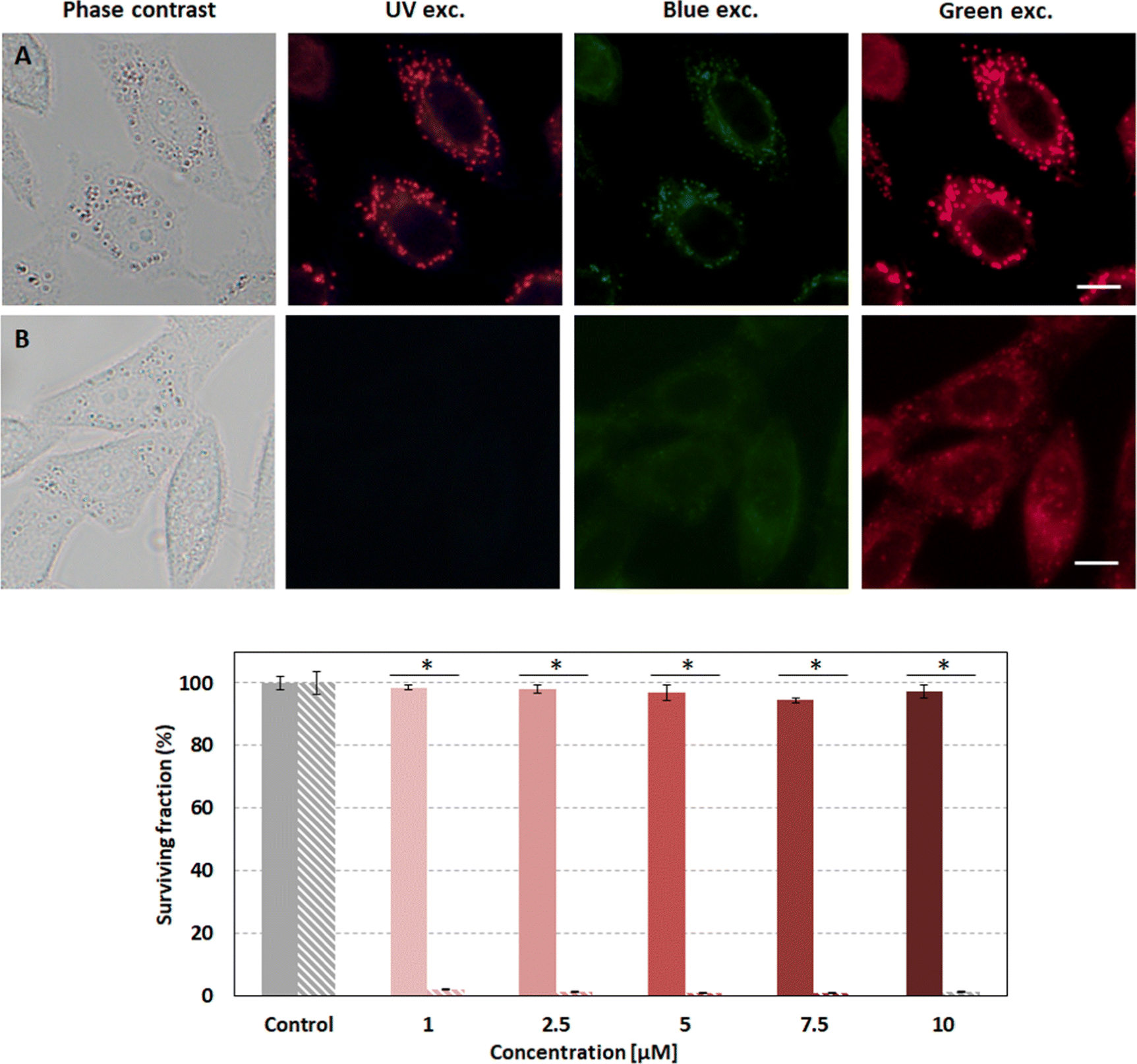 | ||
| Fig. 10 (Top) Fluorescence microscopy images of HeLa cells incubated with 2d (A) and 1 (B). Bottom) MTT assay results of 2d under dark conditions (striped bars) and green irradiation (plain bars; 10 J cm−2). Scale bar: 10 μm. Statistical significance for the surviving fraction was obtained by using one-way ANOVA. Statistically significant differences are labelled as * (p < 0.001). | ||
Given the better performance of 2d in cellular internalization, we selected this compound for further experiments in order to assess its phototoxicity. After 24 h of incubation with 2d and 10 J cm−2 irradiation with 518 nm light, HeLa cell viability was evaluated using an MTT assay. The results shown below (Fig. 10) demonstrate that 2d induces high phototoxicity after green irradiation at low light doses (10 J cm−2) even at the lowest 2d concentration assayed (98% of cell death at 1 μM). Null or very low dark toxicity was detected under our experimental conditions.
Conclusions
This study evidences that meso-enamine-BODIPYs are a new family of structurally simple and easily accessible BODIPYs with the capability to act as halogen-free 1O2 photosensitizers for PDT or even for theragnosis based on PDT and fluorescence. The attachment of an enamine group (electron-donor) at the meso position activates a charge transfer state, the key to populate the triplet state via SOCT-ISC. This mechanism is triggered by excited state relaxation via BODIPY-enamine torsion (towards orthogonalization), taking place between the CT excited singlet and low-lying triplets, in particular the BODIPY-localized T1 state, but also the second excited triplet T2.Theoretical simulations and experimental characterization of the excited state relaxation by femto-transient absorption demonstrated that the formation and energetic stabilization of a CT state induce a faster deactivation kinetics, promoted by the polarity of the solvent and/or alkylation of the BODIPY core (mainly at 1- and 7-positions), accelerate non-radiative decay rates hindering singlet oxygen generation and fluorescence ability. In fact, the CT state must be formed with a sufficient lifetime (timescale set at ≥ 1.8 ns) to allow the population of triplet states.
The rational design of a meso-enamine BODIPY without alkylation at C1 and C7, but with extended conjugation at C2 and C6, has demonstrated its viability as a biomarker of lipid droplets and a photosensitizer agent in HeLa cells being very photoefficient at low green light doses (10 J cm−2) and low concentrations (≤ 1 μM) and no cytotoxic under dark conditions.
Further studies to explore the possibility of the development of red-absorbing photosensitizers based on the present strategy (enamine group at the meso position of the BODIPY core) by the introduction of larger extended π-conjugated moieties at C2 and C6 and/or at C3 and C5 positions would be interesting to research.
Data availability
All of the additional information and experimental data are provided in the ESI.†Author contributions
A.P.-C., A. R. A. and M. J. O. synthesized and chemically characterized all compounds. R. P.-M. and V. M.-M. measured the photophysical properties. A. L. and R. M. carried out the femtosecond measurements. A. D. A. and D. C. performed the theoretical simulations. A. T. and A. V. conducted the in vitro assays. R. M., D. C. and V. M.-M. wrote the manuscript. V. M.-M. revised and edited the final manuscript. V. M.-M. conceived and supervised the study. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by MCIN/AEI/10.13039/501100011033 (projects no. PID2020-114347RB-C32, PID2020-114755GB-C31, PID2019-109555GB-I00 and RED2018-102815-T) and Gobierno Vasco (project no. IT1639-22 and PIBA19-0004). R. P.-M. and A. D. A. thank UPV/EHU, MIU and NGEU for their respective postdoctoral (MARSA21/71) and predoctoral (PRE2020-092036) fellowships. We also thank the SGIker Laser Facility of the UPV/EHU for technical support.References
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- A. Yordanova, E. Eppard, S. Kürpig, R. A. Bundschuh, S. Schönberger, M. Gonzalez-Carmona, G. Feldmann, H. Ahmadzadehfar and M. Essler, OncoTargets Ther., 2017, 10, 4821–4828 CrossRef PubMed.
- S. Navalkissoor, G. Gnanasegaran and R. Baum, Br. J. Radiol., 2018, 91, 20189004 CrossRef PubMed.
- D. van Straten, V. Mashayekhi, H. de Bruijn, S. Oliveira and D. Robinson, Cancers, 2017, 9, 19–73 CrossRef.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- W. Hu, H. Ma, B. Hou, H. Zhao, Y. Ji, R. Jiang, X. Hu, X. Lu, L. Zhang, Y. Tang, Q. Fan and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 12039–12047 CrossRef CAS.
- W. Hu, X. Miao, H. Tao, A. Baev, C. Ren, Q. Fan, T. He, W. Huang and P. N. Prasad, ACS Nano, 2019, 13, 12006–12014 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1901778 CrossRef.
- X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- B. del Rosal, B. Jia and D. Jaque, Adv. Funct. Mater., 2018, 28, 1803733 CrossRef.
- C. N. Ko, G. Li, C. H. Leung and D. L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS.
- M. DeRosa, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- C. Hopper, Lancet Oncol., 2000, 1, 212–219 CrossRef CAS PubMed.
- K. Moghissi, K. Dixon and S. Gibbins, Surg. J., 2015, 01, e1–e15 CrossRef CAS.
- H. T. Bui, D. K. Mai, B. Kim, K.-H. Choi, B. J. Park, H.-J. Kim and S. Cho, J. Phys. Chem. B, 2019, 123, 5601–5607 CrossRef CAS.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- B. Kim, B. Sui, X. Yue, S. Tang, M. G. Tichy and K. D. Belfield, Eur. J. Org. Chem., 2017, 25–28 CrossRef CAS.
- C. S. Kue, S. Y. Ng, S. H. Voon, A. Kamkaew, L. Y. Chung, L. V. Kiew and H. B. Lee, Photochem. Photobiol. Sci., 2018, 17, 1691–1708 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- G. Durán-Sampedro, N. Epelde-Elezcano, V. Martínez-Martínez, I. Esnal, J. Bañuelos, I. García-Moreno, A. R. Agarrabeitia, S. de la Moya, A. Tabero, A. Lazaro-Carrillo, A. Villanueva, M. J. Ortiz and I. López-Arbeloa, Dyes Pigm., 2017, 142, 77–87 CrossRef.
- S. Zhen, S. Wang, S. Li, W. Luo, M. Gao, L. G. Ng, C. C. Goh, A. Qin, Z. Zhao, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1706945 CrossRef.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024–213109 CrossRef CAS.
- A. Treibs and F.-H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- F. López Arbeloa, J. Bañuelos, V. Martínez, T. Arbeloa and I. López Arbeloa, Int. Rev. Phys. Chem., 2005, 24, 339–374 Search PubMed.
- X. F. Zhang and J. Zhu, J. Lumin., 2019, 205, 148–157 CrossRef CAS.
- R. Prieto-Montero, A. Prieto-Castañeda, R. Sola-Llano, A. R. Agarrabeitia, D. García-Fresnadillo, I. López-Arbeloa, A. Villanueva, M. J. Ortiz, S. Moya and V. Martínez-Martínez, Photochem. Photobiol., 2020, 96, 458–477 CrossRef CAS PubMed.
- R. Padrutt, V. Babu, S. Klingler, M. Kalt, F. Schumer, M. I. Anania, L. Schneider and B. Spingler, ChemMedChem, 2020, 694–701 Search PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- I. S. Turan, G. Gunaydin, S. Ayan and E. U. Akkaya, Nat. Commun., 2018, 9, 805 CrossRef.
- W. Wu, Y. Geng, W. Fan, Z. Li, L. Zhan, X. Wu, J. Zheng, J. Zhao and M. Wu, RSC Adv., 2014, 4, 51349–51352 RSC.
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC.
- J. H. Gibbs, Z. Zhou, D. Kessel, F. R. Fronczek, S. Pakhomova and M. G. H. Vicente, J. Photochem. Photobiol., B, 2015, 145, 35–47 CrossRef CAS PubMed.
- R. Lincoln, A. M. Durantini, L. E. Greene, S. R. Martínez, R. Knox, M. C. Becerra and G. Cosa, Photochem. Photobiol. Sci., 2017, 16, 178–184 CrossRef CAS.
- J. Zou, Z. Yin, K. Ding, Q. Tang, J. Li, W. Si, J. Shao, Q. Zhang, W. Huang and X. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 32475–32481 CrossRef CAS PubMed.
- A. J. Sánchez-Arroyo, E. Palao, A. R. Agarrabeitia, M. J. Ortiz and D. García-Fresnadillo, Phys. Chem. Chem. Phys., 2017, 19, 69–72 RSC.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- V. N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- Y. Dong, A. Elmali, J. Zhao, B. Dick and A. Karatay, Chem. Phys. Chem., 2020, 21, 1388–1401 CrossRef CAS PubMed.
- J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS.
- Z. J. Jakubek, M. Chen, M. Couillard, T. Leng, L. Liu, S. Zou, U. Baxa, J. D. Clogston, W. Y. Hamad and L. J. Johnston, J. Nanopart. Res., 2018, 20, 98 CrossRef.
- Y. Zhao, R. Duan, J. Zhao and C. Li, Chem. Commun., 2018, 54, 12329–12332 RSC.
- X. F. Zhang and N. Feng, Chem. – Asian J., 2017, 12, 2447–2456 CrossRef CAS PubMed.
- Z. Wang, M. Ivanov, Y. Gao, L. Bussotti, P. Foggi, H. Zhang, N. Russo, B. Dick, J. Zhao, M. Di Donato, G. Mazzone, L. Luo and M. Fedin, Chem. – Eur. J., 2020, 26, 1091–1102 CrossRef CAS PubMed.
- J. Jiménez, R. Prieto-Montero, B. L. Maroto, F. Moreno, M. J. Ortiz, A. Oliden-Sánchez, I. López-Arbeloa, V. Martínez-Martínez and S. de la Moya, Chem. – Eur. J., 2020, 26, 601–605 CrossRef PubMed.
- W. Hu, Y. Lin, X. F. Zhang, M. Feng, S. Zhao and J. Zhang, Dyes Pigm., 2019, 164, 139–147 CrossRef CAS.
- T. C. Pham, S. Heo, V. N. Nguyen, M. W. Lee, J. Yoon and S. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 13949–13957 CrossRef CAS PubMed.
- V. N. Nguyen, S. J. Park, S. Qi, J. Ha, S. Heo, Y. Yim, G. Baek, C. S. Lim, D. J. Lee, H. M. Kim and J. Yoon, Chem. Commun., 2020, 56, 11489–11492 RSC.
- L. A. Ortiz-Rodríguez and C. E. Crespo-Hernández, Chem. Sci., 2020, 11, 11113–11123 RSC.
- V. N. Nguyen, S. Heo, C. W. Koh, J. Ha, G. Kim, S. Park and J. Yoon, ACS Sens., 2021, 6, 3462–3467 CrossRef CAS.
- W. Hu, X.-F. Zhang and M. Liu, J. Phys. Chem. C, 2021, 125, 5233–5242 CrossRef CAS.
- X. Wang, Y. Song, G. Pan, W. Han, B. Wang, L. Cui, H. Ma, Z. An, Z. Xie, B. Xu and W. Tian, Chem. Sci., 2020, 11, 10921–10927 RSC.
- E. A. Weiss, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2003, 107, 3639–3647 CrossRef CAS.
- D. Liu, A. M. El-Zohry, M. Taddei, C. Matt, L. Bussotti, Z. Wang, J. Zhao, O. F. Mohammed, M. Di Donato and S. Weber, Angew. Chem., Int. Ed., 2020, 59, 11591–11599 CrossRef CAS.
- M. A. Filatov, Org. Biomol. Chem., 2019, 18, 10–27 RSC.
- D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136–158 CrossRef CAS.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Phys. Chem. C, 2019, 123, 22793–22811 CrossRef CAS.
- X. F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 9050–9055 CrossRef CAS PubMed.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- L. A. Ortiz-Rodríguez, S. J. Hoehn, A. Loredo, L. Wang, H. Xiao and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2021, 143, 2676–2681 CrossRef PubMed.
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem. – Eur. J., 2021, 27, 9934–9947 CrossRef CAS PubMed.
- S. Rihn, P. Retailleau, N. Bugsaliewicz, A. De Nicola and R. Ziessel, Tetrahedron Lett., 2009, 50, 7008–7013 CrossRef CAS.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606–615 CrossRef CAS.
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS PubMed.
- J. Zhao, K. Chen, Y. Hou, Y. Che, L. Liu and D. Jia, Org. Biomol. Chem., 2018, 16, 3692–3701 RSC.
- J. B. Prieto, F. L. Arbeloa, V. M. Martínez, T. A. López and I. L. Arbeloa, J. Phys. Chem. A, 2004, 108, 5503–5508 CrossRef.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- E. Palao-Utiel, L. Montalvillo-Jiménez, I. Esnal, R. Prieto-Montero, A. R. Agarrabeitia, I. García-Moreno, J. Bañuelos, I. López-Arbeloa, S. de la Moya and M. J. Ortiz, Dyes Pigm., 2017, 141, 286–298 CrossRef CAS.
- I. Esnal, I. Valois-Escamilla, C. F. A. Gómez-Durán, A. Urías-Benavides, M. L. Betancourt-Mendiola, I. López-Arbeloa, J. Bañuelos, I. García-Moreno, A. Costela and E. Peña-Cabrera, Chem. Phys. Chem., 2013, 14, 4134–4142 CrossRef CAS PubMed.
- V. P. Yakubovskyi, M. P. Shandura and Y. P. Kovtun, Dyes Pigm., 2010, 87, 17–21 CrossRef CAS.
- N. Epelde-Elezcano, E. Palao, H. Manzano, A. Prieto-Castañeda, A. R. Agarrabeitia, A. Tabero, A. Villanueva, S. de la Moya, Í. López-Arbeloa, V. Martínez-Martínez and M. J. Ortiz, Chem. – Eur. J., 2017, 23, 4837–4848 CrossRef CAS.
- Y. Liu, J. Zhao, A. Iagatti, L. Bussotti, P. Foggi, E. Castellucci, M. Di Donato and K. L. Han, J. Phys. Chem. C, 2018, 122, 2502–2511 CrossRef CAS.
- R. Montero, V. Martínez-Martínez, A. Longarte, N. Epelde-Elezcano, I. Lamas and I. L. Arbeloa, J. Phys. Chem. Lett., 2021, 12, 7439–7441 CrossRef CAS PubMed.
- Y. Lee, R. M. Malamakal, D. M. Chenoweth and J. M. Anna, J. Phys. Chem. Lett., 2020, 11, 877–884 CrossRef CAS PubMed.
- Q. Zhou, M. Zhou, Y. Wei, X. Zhou, S. Liu, S. Zhang and B. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 1516–1525 RSC.
- V. Postils, F. Ruipérez and D. Casanova, J. Chem. Theory Comput., 2021, 17, 5825–5838 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01515c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |