
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomically dispersed platinum electrocatalysts supported on gadolinia-doped ceria nanoparticles for practical high-temperature solid oxide cells†
Jihong
Min‡
ab,
Haewon
Seo‡
 a,
Jisu
Shin
a,
Mi Young
Park
ac,
Sun-Young
Park
d,
Haneul
Choi
ae,
Soohyung
Park
f,
Sungeun
Yang
a,
Hye Jung
Chang
a,
Jongsup
Hong
*b and
Kyung Joong
Yoon
*a
a,
Jisu
Shin
a,
Mi Young
Park
ac,
Sun-Young
Park
d,
Haneul
Choi
ae,
Soohyung
Park
f,
Sungeun
Yang
a,
Hye Jung
Chang
a,
Jongsup
Hong
*b and
Kyung Joong
Yoon
*a
aEnergy Materials Research Center, Korea Institute of Science and Technology, Seoul, Republic of Korea. E-mail: kjyoon@kist.re.kr
bSchool of Mechanical Engineering, Yonsei University, Seoul, Republic of Korea. E-mail: jongsup.hong@yonsei.ac.kr
cDepartment of Materials Science and Engineering, Korea University, Seoul, Republic of Korea
dTechnological Convergence Center, Korea Institute of Science and Technology, Seoul, Republic of Korea
eDepartment of Materials Science and Engineering, Yonsei University, Seoul, 03722, Republic of Korea
fAdvanced Analysis Center, Korea Institute of Science and Technology, Seoul, Republic of Korea
First published on 30th October 2023
Abstract
Atomically dispersed catalysts provide excellent catalytic properties and atom utilization efficiency, but their high-temperature application has been limited by their low thermal stability. Herein, we report atomically dispersed Pt catalysts that are both highly active and thermally stable in fuel cells and electrolyzers operating above 600 °C. We developed a urea-based chemical synthetic method that strongly anchors atomic-scale Pt species on the surface of ceria nanoparticles and prevents their agglomeration at high temperatures. Doping the ceria with gadolinia further enhances their catalytic properties by increasing the oxygen vacancy concentration and promoting the oxygen exchange kinetics. This process enables in situ synthesis within the porous electrode of realistic solid oxide cells and significantly improves the power output and H2 production rate in fuel cell and electrolysis modes, respectively. Furthermore, this electrode stably operated without noticeable degradation during a long-term evaluation, thus proving the excellent thermal stability of atomically dispersed Pt/ceria catalysts.
Introduction
Solid oxide cells (SOCs), which jointly refers to solid oxide fuel cells and electrolysis cells, are expected to play a key role in making the future energy mix sustainable because they enable reversible conversion between chemical and electrical energy.1 SOCs operate at a high temperature, typically 600–900 °C, which affords unique advantages such as high efficiency and operational flexibility.2,3 However, such temperatures limit the choice of useable materials. For example, various electrocatalysts that exhibit excellent characteristics at low temperatures, such as atomically dispersed catalysts, have proven to be unsuitable for SOCs, mostly because they thermally degrade at high operating temperatures.Metal-based nanomaterials are an important category of heterogeneous catalysts. Their catalytic properties generally improve with decreasing particle size owing to favorable geometric and electronic alterations.4 In particular, atomically dispersed catalysts offer outstanding catalytic properties for various reactions and maximum atom utilization efficiency.5–8 The choice of support material also profoundly impacts the electrocatalytic performance, and significant research efforts have been devoted to designing the optimal support by engineering metal–support interactions.9–14 For atomic dispersion, the combination of Pt and ceria has been widely studied.4,15 Pt is well known for its excellent catalytic properties, and ceria is a reducible 4f oxide support that actively participates in catalytic reactions. Specifically, ceria is a mixed ionic and electronic conductor in a reducing atmosphere at high temperatures, and its surface can host electrocatalytic reactions by enabling access to both charge carriers.16,17 More importantly, Pt atoms are known to become preferentially trapped on the surface of ceria nanoparticles.15 Moreover, atomically dispersed Pt/ceria catalysts could be particularly effective in SOCs because high temperature activates the lattice oxygen of ceria.18 However, isolated Pt atoms are not thermally stable and readily agglomerate at high temperatures. Although many studies have improved the thermal stability of atomically dispersed catalysts, their feasible operating temperature remains below ∼300 °C.19–21 Thus, despite their promise, using atomically dispersed catalysts in SOCs at operating temperatures above 600 °C remains a major challenge.
Herein, we report atomically dispersed Pt/ceria electrocatalysts that are highly active and thermally stable under SOC operating conditions. These catalysts were synthesized by a urea-based chemical solution process, wherein urea functions as a complexing agent and offers two coordination sites: carbonyl O and amide N. Then, Pt and Ce ions favorably coordinate to N and O, respectively, to prevent phase separation in the solution until the cations precipitate.22,23 During the subsequent thermal treatment at ∼80 °C, the urea thermally decomposes and induces homogeneous precipitation by releasing ammonia and carbon dioxide into the solution, resulting in fine-scale precipitates with a homogenous composition.24–26 Finally, calcination yields nanostructured catalysts with atomic Pt species dispersed on the ceria surface.
Materials and methods
To synthesize atomically dispersed 4 at% Pt/Gd0.1Ce0.9O1.95 (GDC) powder samples, a precursor solution was prepared by mixing stoichiometric amounts of K2PtCl4, Ce(NO3)3·6H2O, and Gd(NO3)3·6H2O in distilled water, into which urea was dissolved at a urea-to-cation molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Then, ethanol was added with a distilled water/ethanol proportion of 1:3 and a total solution concentration of 0.125 mol L−1. After mixing, the solution was thermally treated (80 °C, 2 h) and then calcined in air (650 °C, 1 h). Ceria, GDC, and Pt/ceria powder samples were produced in the same manner for comparison.
:1. Then, ethanol was added with a distilled water/ethanol proportion of 1:3 and a total solution concentration of 0.125 mol L−1. After mixing, the solution was thermally treated (80 °C, 2 h) and then calcined in air (650 °C, 1 h). Ceria, GDC, and Pt/ceria powder samples were produced in the same manner for comparison.
The synthesized powder samples were analyzed by transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), X-ray absorption near edge structure (XANES), and Raman spectroscopy. Atomic-resolution high-angle annular dark-field-scanning tunneling electron microscopy (HAADF-STEM) images were recorded using a Cs-corrected microscope (FEI; Titan 80-300) with electron energy loss spectroscopy (EELS; Gatan, Quantum 966). Elemental maps were acquired using STEM equipped with super energy-dispersive X-ray spectroscopy (EDS; FEI, Talos F200X). XRD patterns were recorded using a D8 ADVANCE (Bruker, USA) with a nickel-filtered Cu Kα radiation source. XPS was performed using a PHI 5000 VersaProbe (Ulvac-PHI) system with a monochromator Al Kα X-ray source (1486.6 eV). XANES measurements were performed at the 1D XRS KIST-PAL beamline of the Pohang Accelerator Laboratory. The Raman spectrometer was a Horiba Jobin Yvon LabRAM ARAMIS with a He–Ne laser beam at an excitation radiation wavelength of 632.8 nm.
Symmetric cells were fabricated using ∼3 mm-thick 8 mol% yttria-stabilized zirconia (YSZ) discs as the electrolyte substrate. The pastes for the functional (66 wt% NiO + 34 wt% YSZ) and current-collecting (NiO) layers were prepared by mixing powders with desired amounts of solvent (α-terpineol), binder (ethyl cellulose), and dispersant (Hypermer KD15). The proportion of NiO and YSZ was previously found to maximize the length of the triple phase boundary (TPB) after the reduction of NiO into metallic Ni.27,28 The functional and current-collecting layers were sequentially screen-printed on both sides of the YSZ electrolyte substrate and sintered in air (1250 °C, 2 h). The precursor solution was injected into the sintered electrode using a microsyringe, followed by heat treatment (80 °C, 2 h) and calcination in air (650 °C, 1 h). The impedance spectra of the cells were collected using a Solartron 1260/1287 frequency response analyzer and potentiostat in an alumina tube reactor at 600–750 °C in a humidified H2 (3% H2O) atmosphere.
To fabricate full cells, a NiO–YSZ support layer was tape cast. The slurry was prepared by ball milling NiO (60 wt%), YSZ (40 wt%), and poly(methyl methacrylate) (PMMA) (10 wt% relative to the total oxides) powders in ethanol with Triton X-100 dispersant, polyvinyl butyral (PVB) binder, and polyethylene glycol (PEG) 400 plasticizer. To improve structural stability while maintaining the percolation of Ni, a slightly higher proportion of YSZ was used in the support layer than in the functional layer. After tape casting and drying, green tapes were cut into squares, laminated, and pre-sintered (1050 °C, 2 h). Then, a NiO–YSZ fuel electrode functional layer and a YSZ electrolyte were sequentially spin-coated. The inks were prepared by mixing powders into a vehicle made of α-terpineol and ethyl cellulose. We also added a carbon black pore former to the ink for the fuel electrode functional layer. After spin coating, the fuel electrode and electrolyte were co-sintered (1320 °C, 5 h), and a GDC interlayer was then spin-coated and sintered (1250 °C, 2 h). Next, a lanthanum strontium cobaltite (LSC, La0.6Sr0.4CoO3−δ) air electrode was screen printed and sintered (950 °C, 2 h). The active electrode area was 1.0 cm2. Finally, Pt/GDC was infiltrated into the fuel electrode in the same manner as for the symmetric cells. The fabricated cells were tested using metal interconnects, a glass-ceramic sealant, and Ni foam and Pt mesh current collectors. Humidified H2 (3% and 50% H2O for fuel cell and electrolysis modes, respectively) was supplied to the fuel electrode, and air was supplied to the air electrode. Prior to electrochemical testing, the cell was heat-treated at 800 °C for 2 h to form leak-tight seals. Electrochemical measurements were performed using a Solartron 1260A frequency response analyzer and the Solartron 1287A potentio/galvanostat electrochemical interface at 600–700 °C.
Results and discussion
Fig. 1(a) shows the TEM/EDS analysis of 4 at% Pt/ceria nanoparticles after calcination at 650 °C. The ceria nanoparticles are 20–30 nm in diameter, and Pt is finely dispersed over their entire surface with no indication of agglomeration. To further improve their catalytic properties, we doped ceria with 20 mol% gadolinia (GDC). Substituting Ce4+ ions with Gd3+ ions having a lower valence creates oxygen vacancies for charge neutrality and possibly promotes the reaction kinetics because oxygen ions are actively involved in SOC electrode reactions. Fig. 1(b) confirms that in GDC, ceria is homogeneously doped with gadolinia and that adding gadolinia does not alter the Pt distribution or particle morphology. Lower-magnification TEM images (Fig. S1†) corroborate the absence of major Pt agglomeration. By contrast, when the same composition is synthesized without urea, Pt agglomerates into large particles after calcination at 650 °C (Fig. S2†), highlighting the critical role of urea in dispersing and stabilizing Pt. Atomic-scale Pt species were more closely probed by STEM (Fig. 1(c)). In HAADF-STEM images, Pt appears bright owing to the strong z-contrast.29 Many isolated Pt atoms and sub-nanometer atomic clusters clearly appear, verifying the thermal stability of atomic-scale Pt at 650 °C. Fig. S3† shows additional HAADF-STEM images of atomically dispersed Pt.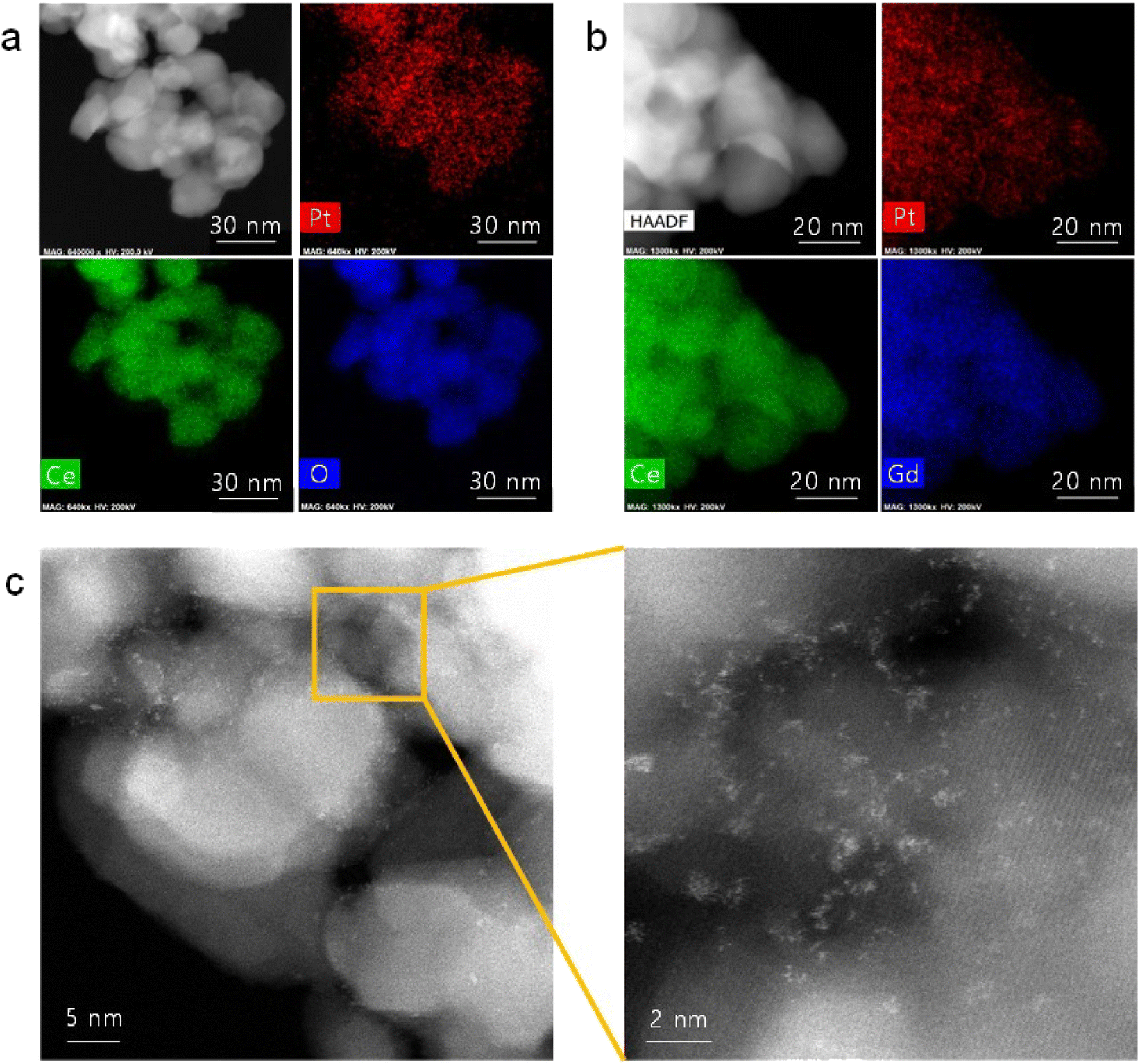 | ||
| Fig. 1 Atomic dispersion of Pt: (a and b) TEM/EDS elemental mapping of (a) Pt/ceria and (b) Pt/GDC nanoparticles. (c) HAADF-STEM images of Pt/GDC showing isolated Pt atoms and Pt atomic clusters. | ||
The intrinsic material properties of the Pt/GDC catalyst were also investigated (Fig. 2). In the XRD patterns (Fig. 2(a)), all major peaks of Pt/GDC synthesized using urea are indexed to the cubic fluorite structure of GDC. The Pt metal peak is very weak, indicating that most Pt species exist at an atomic scale and are not detected as a metal phase.30,31 In contrast, when the catalysts are synthesized without urea, Pt metal peaks are sharp because Pt metal particles aggregate, as previously observed (Fig. S2†). In the XPS analysis (Fig. 2(b)), the Pt 4f spectra of Pt/GDC were deconvoluted into two spin-splitting components, 4f7/2 and 4f5/2, which were assigned to the Pt(0) and Pt(II) ionic states, respectively.32–34 Pt is predominantly metallic in the sample synthesized without urea; by contrast, Pt2+ is the dominant species in that synthesized with urea, indicating that atomic-scale Pt species are mostly in the ionic state, as corroborated by XANES. Fig. 2(c) compares the normalized Pt L3-edges of the atomically dispersed Pt/GDC catalysts and those of model compounds (Pt metal (Pt(0)), Pt acetylacetonate (Pt acet) (Pt(II)), and PtO2 (Pt(IV))). The absorption intensity of Pt/GDC indicated by a white line, which reflects the electron transition from the 2p to the 5d orbital of the Pt atom,35 is considerably higher than that of the Pt metal, and the edge energy is closer to that of Pt acet rather than those of Pt metal and PtO2. These results indicate that the Pt(5d) orbital is unoccupied and that most of the Pt is in a Pt2+ oxidation state. The Raman spectra (Fig. 2(d)) show the synergetic effect of Pt and Gd in creating oxygen vacancies in ceria. The main ceria peak, which corresponds to the first-order Raman active F2g mode of the fluorite structure, appears at 460–470 cm−1, representing the symmetrical stretching vibration of the CeO8 unit.36 For GDC, an additional broad band appears at 500–700 cm−1, corresponding to dopant-induced oxygen vacancies.37,38 Previous studies also demonstrated the generation of oxygen vacancies with gadolinia doping in ceria.39–42 Adding atomic Pt to GDC substantially enlarges this band, indicating a sharply increased oxygen vacancy concentration. Considering Pt and Gd contents of 4 and 20 at%, respectively, the atomic dispersion of Pt effectively activates a large amount of lattice oxygen in ceria, imparting the SOC electrode with excellent redox properties.
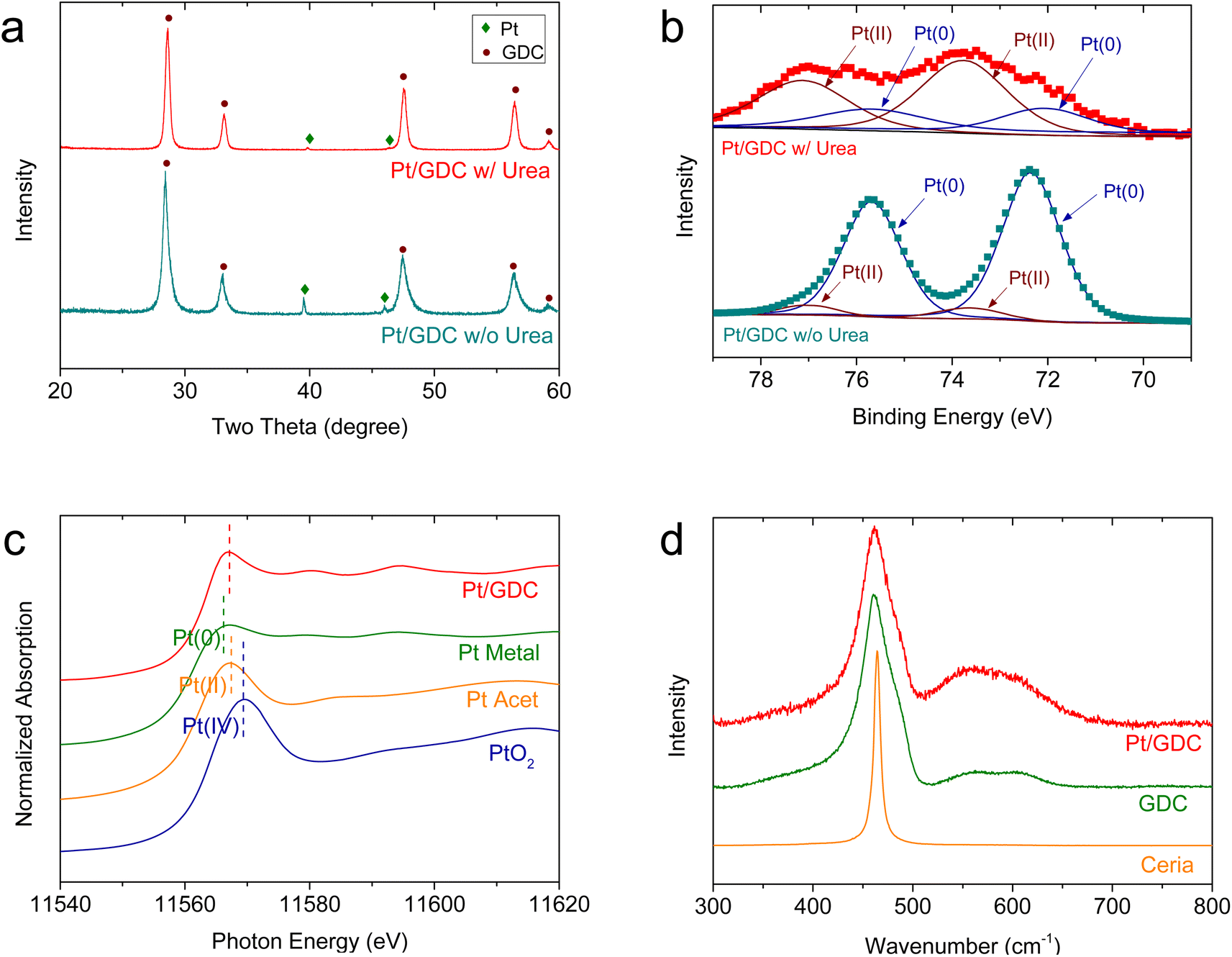 | ||
| Fig. 2 Intrinsic properties of atomically dispersed Pt on GDC: (a) XRD patterns and (b) XPS spectra of Pt/GDC synthesized with and without urea. (c) Normalized Pt L3-edge XANES spectra of atomically dispersed Pt/GDC and model compounds (Pt metal, Pt acet, and PtO2). (d) Raman spectra of atomically dispersed Pt/GDC, GDC, and undoped ceria. | ||
For device application, atomically dispersed Pt/GDC nanocatalysts were loaded into the pores of the SOC fuel electrode via an in situ synthetic process called infiltration. Fig. 3(a) shows the microstructure of the porous Ni–YSZ fuel electrode formed on a dense YSZ electrolyte. A typical SOC electrode comprises 30–40 vol% percolated pores, which are generally of sub-micrometer scale. To impregnate catalysts into such fine-scale electrodes, an in situ synthetic method is required because injecting and anchoring pre-synthesized particles is extremely difficult. During infiltration, the precursor solution is injected into the pores, and nanoparticles form on their inner surfaces via precipitation and calcination. In our urea-based chemical solution process, urea thermally decomposes at ∼80 °C and causes the homogeneous precipitation of cations. Then, fine precipitates strongly adsorb onto the pore walls by van der Waals attractions, and they remain immobile during subsequent heat treatment, thus uniformly covering the entire electrode with nanoparticles.43Fig. 3(b) shows the inner surface of the Ni–YSZ fuel electrode before and after infiltrating the Pt/GDC nanocatalysts. After infiltration, Pt/GDC nanocatalysts uniformly cover the entire surface of the Ni–YSZ fuel electrode, with no indication of agglomeration or excessive particle growth. Fig. S4† shows additional SEM images of infiltrated Pt/GDC nanocatalysts. In the TEM analysis (Fig. 3(c)), the particles of infiltrated Pt/GDC are estimated to be ∼20 nm, and the HAADF-STEM image confirms the presence of atomic-scale Pt species. During the infiltration process, Pt can possibly react with Ni rather than anchoring onto the surface of the GDC nanoparticles. We therefore performed a TEM-EDS analysis on the Pt/GDC nanocatalysts formed on Ni in the Ni–YSZ electrode, as shown in Fig. S5.† In the EDS mapping, Pt signals were mostly detected from the infiltrated particles, whereas they were very weak from the bulk Ni, confirming that Pt preferentially formed on the surface of GDC nanoparticles.
 | ||
| Fig. 3 In situ synthesize atomically dispersed Pt/GDC nanocatalysts within a fuel electrode: (a and b) SEM images of (a) the entire fuel electrode and (b) its inner surface before and after infiltration with Pt/GDC. (c) TEM and HAADF-STEM images of Pt/GDC nanocatalysts infiltrated into the fuel electrode. | ||
The electrochemical characteristics of atomically dispersed Pt/GDC nanocatalysts in the SOC fuel electrode were evaluated with symmetric cells, which comprised a ∼20 μm-thick Ni–YSZ functional layer and a ∼5 μm-thick Ni current collecting layer deposited on both sides of a ∼3 mm-thick YSZ electrolyte substrate (Fig. S6†). To systematically analyze the individual functions of constituent components, the impedance spectra of Ni–YSZ fuel electrodes infiltrated with ceria, GDC, and Pt/GDC were compared with those of a standard Ni–YSZ electrode. The catalyst loading was fixed at 9.51 × 10−6 mol cm−2, and the particle44 morphologies and surface coverages were similar for all infiltrated samples (Fig. S7†). Thus, the differences in the electrochemical performance could be attributed only to the catalytic properties rather than the microstructure. The Nyquist plot of the impedance spectra (Fig. 4(a)) displays the real and imaginary parts of the impedance on the x- and y-axis, respectively, and the width of the impedance arc represents the polarization resistance. The polarization resistance decreased in the order of the bare Ni–YSZ reference cell and the ceria- and GDC-infiltrated cells at 650 °C (0.94, 0.55, and 0.42 Ω cm2, respectively). However, it decreased dramatically to 0.28 Ω cm2 upon incorporating atomically dispersed Pt on the GDC surface. This polarization resistance is considerably lower than the majority of the reported values in the literature,45–49 suggesting the high electrocatalytic performance of the Pt/GDC catalysts. We also investigated the effect of the Pt content on the electrocatalytic activity. As shown in Fig. S8,† the polarization resistance gradually decreases with the increasing Pt loading up to 4 at% but plateaus with a further increase up to 6 at%. These results imply that the majority of Pt is retained in an atomically dispersed state up to 4 at%, beyond which Pt aggregates to form bulk particles. The impedance spectra of the fuel electrode comprise multiple overlapping arcs that represent individual rate-limiting processes. Fig. 4(b) shows the Bode plot with the imaginary part of the impedance spectra as a function of frequency, suggesting two major rate-limiting processes with characteristic frequencies at 103 to 104 and 101 to 102 Hz. For the standard Ni–YSZ fuel electrode, the high-frequency impedance in the kilohertz range is associated with surface reaction processes, and it contains the contributions of multiple elementary processes with similar time constants,50 including charge transfer, relaxation of electrical double layer, gas–solid interactions (adsorption, dissociation, desorption, etc.), surface diffusion, and hydrogen spillover.51–56 By contrast, the low-frequency arc below 102 Hz is generally attributed to gas-phase diffusion across the porous electrode.51–54 Clearly, nanocatalyst infiltration strongly impacts the high-frequency impedance but not the low-frequency one. For the quantitative analysis, the impedance spectra collected at 600–750 °C (Fig. S9†) were deconvoluted using an equivalent circuit model comprising two standard resistor (R)-constant phase element (CPE) units in series (Fig. S10†), and Table 1 summarizes the resulting high- and low-frequency resistances. At 650 °C, the overall impedance of the standard Ni–YSZ fuel electrode is dominated by the high-frequency arc, which is more than three times larger than the low-frequency arc. Upon Pt/GDC infiltration, the high-frequency arc substantially shrinks to less than half of the low-frequency impedance. The lower high-frequency impedance could be explained by the improved catalytic activity and extension of reaction sites. Specifically, for a standard Ni–YSZ electrode, the reaction zone is confined to the TPBs, and most of the high-frequency impedance is associated with the surface diffusion of adsorbed hydrogen to TPB and hydrogen spillover from Ni to YSZ. With infiltration, atomically dispersed Pt provides abundant adsorption sites and excellent catalytic properties. Furthermore, the reaction zone is extended from the TPBs to the entire surface of ceria because ceria provides a conduction path for both electrons and oxygen ions and catalyzes the hydrogen oxidation reaction.16,57,58 As the surface reaction is a thermally activated process, the lower high-frequency impedance significantly decreases the activation energy of the overall polarization resistance, as displayed in the Arrhenius plot of the polarization resistance (Fig. 4(c)).
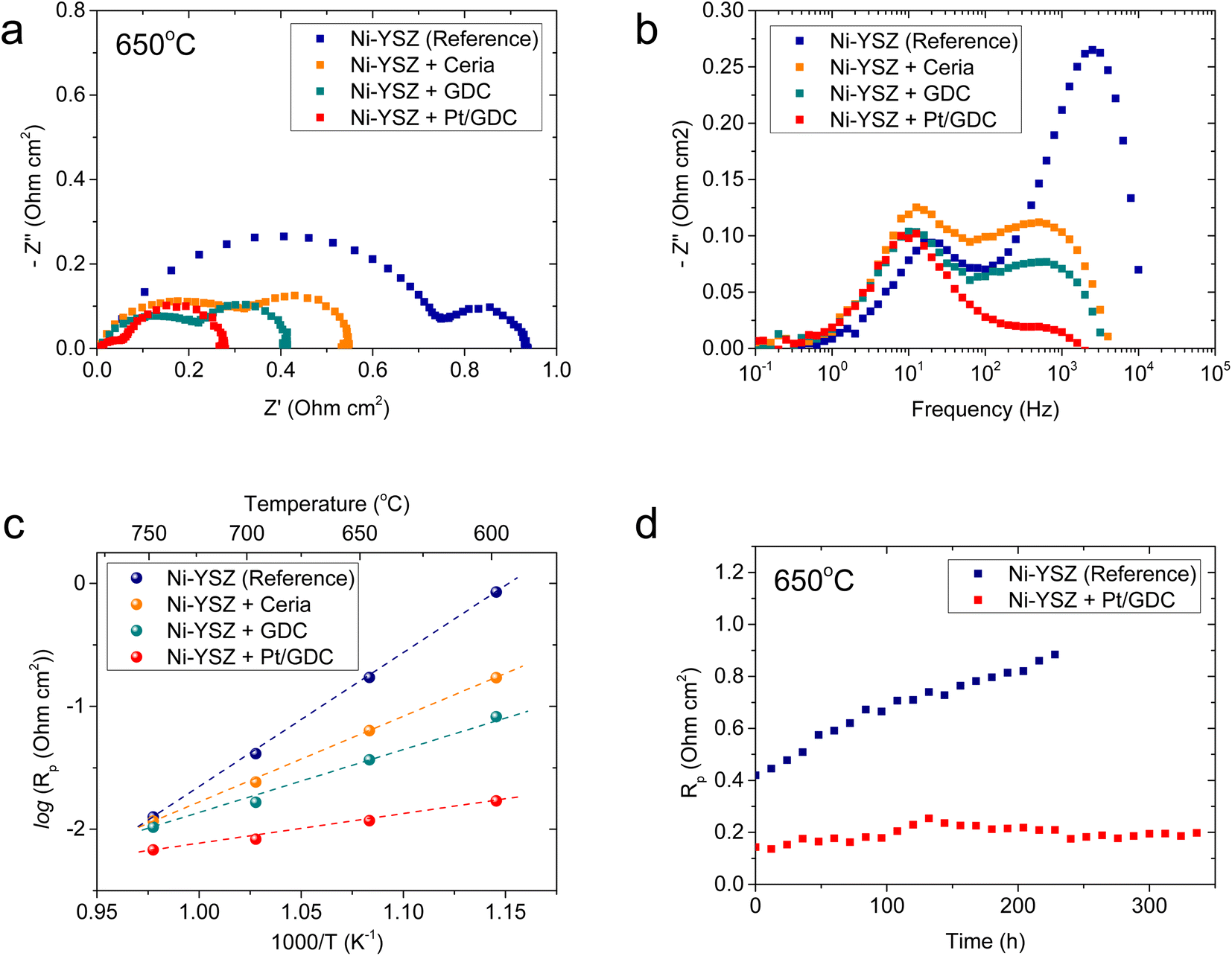 | ||
| Fig. 4 Electrochemical characteristics of fuel electrode loaded with atomically dispersed Pt/GDC: (a) Nyquist and (b) Bode plots of the impedance spectra, and (c) Arrhenius plot of the polarization resistance for Ni–YSZ fuel electrode loaded with ceria, GDC, and Pt/GDC. (d) Long-term test results of the Ni–YSZ reference cell and the Pt/GDC-infiltrated cell. | ||
| High-frequency resistance (Ω cm2) | Low-frequency resistance (Ω cm2) | |||||||
|---|---|---|---|---|---|---|---|---|
| 750 °C | 700 °C | 650 °C | 600 °C | 750 °C | 700 °C | 650 °C | 600 °C | |
| Ni–YSZ (reference) | 0.126 | 0.266 | 0.684 | 1.472 | 0.204 | 0.2 | 0.212 | 0.232 |
| Ni–YSZ + ceria | 0.09 | 0.156 | 0.348 | 0.658 | 0.232 | 0.234 | 0.25 | 0.264 |
| Ni–YSZ + GDC | 0.074 | 0.122 | 0.234 | 0.092 | 0.201 | 0.204 | 0.208 | 0.228 |
| Ni–YSZ + Pt/GDC | 0.028 | 0.052 | 0.092 | 0.168 | 0.202 | 0.198 | 0.192 | 0.194 |
The thermal stability of atomically dispersed Pt/GDC nanocatalysts was comparatively evaluated by periodically measuring the polarization resistance of Pt/GDC-infiltrated and standard Ni–YSZ symmetric cells at 650 °C for 340 and 230 h, respectively. Notably, the polarization resistance of the reference cell increased from 0.42 to 0.88 Ω cm2 in 230 h, whereas that of the Pt/GDC-infiltrated cell remained stable without noticeable degradation for 340 h (Fig. 4(d)). The performance of standard Ni–YSZ fuel electrodes degrades during initial operation for various reasons, such as changes in the TPB density, the loss of Ni percolation, and impurity poisoning.59–61 Long-term testing (Fig. 4(d)) indicated that incorporating atomically dispersed Pt/GDC prevents the intrinsic degradation of the Ni–YSZ fuel electrode because the electrochemical reaction mainly occurs at the Pt/GDC surface rather than the TPBs of Ni–YSZ. The stable performance of the Pt/GDC-infiltrated cell suggests that the atomic dispersion of Pt is retained during prolonged exposure to SOC operating environments. After long-term testing, the morphology of Pt/GDC nanoparticles did not observably change (Fig. S11†).
We then evaluated the practical applicability of atomically dispersed Pt/GDC catalysts by infiltration into realistic full cells. The major challenge in full-cell application is the lack of porosity in the functional layer of the fuel electrode in fuel-electrode-supported cells. In a typical fabrication process, the NiO–YSZ fuel electrode is co-sintered with the YSZ electrolyte at high temperatures to obtain a completely dense electrolyte film. During co-sintering, the fuel electrode functional layer also densifies (Fig. 5(a)), and pores form upon reducing NiO into metallic Ni during operation. Without sufficient porosity in the as-fabricated state, the precursor solution cannot penetrate into the functional layer where the electrochemical reaction occurs; thus, most nanocatalysts are synthesized in the outer support layer. Consequently, atomically dispersed Pt/GDC catalysts do not facilitate electrochemical reactions, and infiltration does not significantly affect the overall performance in standard cells (Fig. S12†). Therefore, the microstructure must be modified to maximize the effectiveness of the infiltration technique; we therefore added 1 wt% carbon black as a pore former, which formed fine pores with diameters of <500 nm uniformly throughout the functional layer (Fig. 5(b)). We fabricated full cells using this porous fuel electrode functional layer, and their microstructure is displayed in Fig. 5(c). The cells consisted of a ∼300 μm-thick Ni–YSZ support, a ∼12 μm-thick Ni–YSZ functional layer, a ∼2.5 μm-thick YSZ electrolyte, a ∼1.5 μm-thick GDC interlayer, and a ∼20 μm-thick LSC air electrode. After infiltration, all of the surfaces within the fuel electrode, including the support and functional layers, were uniformly covered with Pt/GDC nanoparticles, and the particle morphology in the full cell was similar to the one in the symmetric cell (Fig. 3(b)). Fig. 5(d) compares the current–voltage (I–V) curves of cells with and without Pt/GDC in fuel cell and electrolysis modes at 650 °C. The open-circuit voltages (OCVs) were very close to the theoretical values, which were calculated to be 1.128 V for fuel cell mode (97% H2–3% H2O and air) and 0.989 V for electrolysis mode (50% H2–50% H2O and air), indicating the leak-tightness of the cells and seals. Compared with the standard cell, the Pt/GDC-infiltrated cell performed substantially better with the maximum power density increasing from 0.58 to 0.83 W cm−2 in fuel cell mode. Fig. S13† compares the I–V curves and corresponding power densities of the two cells measured at 600–700 °C. The performance improvement by Pt/GDC infiltration was even more pronounced at lower temperatures. In electrolysis mode, Pt/GDC infiltration also significantly increased the current density at the thermo-neutral voltage (VTN, ∼1.28 V), which corresponds to the H2 production rate, from 0.24 to 0.43 A cm−2. Finally, the stability of the infiltrated cell was evaluated in electrolysis mode at 0.4 A cm−2 and 650 °C. The cell stably operated and maintained a consistent cell voltage at ∼1.25 V for 100 h (Fig. 5(e)). A TEM-EDS analysis was conducted after stability testing, as shown in Fig. S14(a)–(c),† revealing that fine Pt species remained homogeneously distributed over the entire surface of the GDC nanoparticles. The presence of atomic Pt is further confirmed in the HAADF-STEM image in Fig. S14(d),† suggesting the excellent thermal stability of the atomically dispersed Pt/GDC nanocatalysts. Furthermore, prior to electrochemical characterization, the full cell was heat-treated at 800 °C for 2 h to form leak-tight seals using glass-ceramic sealants, and the TEM analysis shown in Fig. S13† indicates that atomic Pt is stable at high temperatures up to 800 °C.
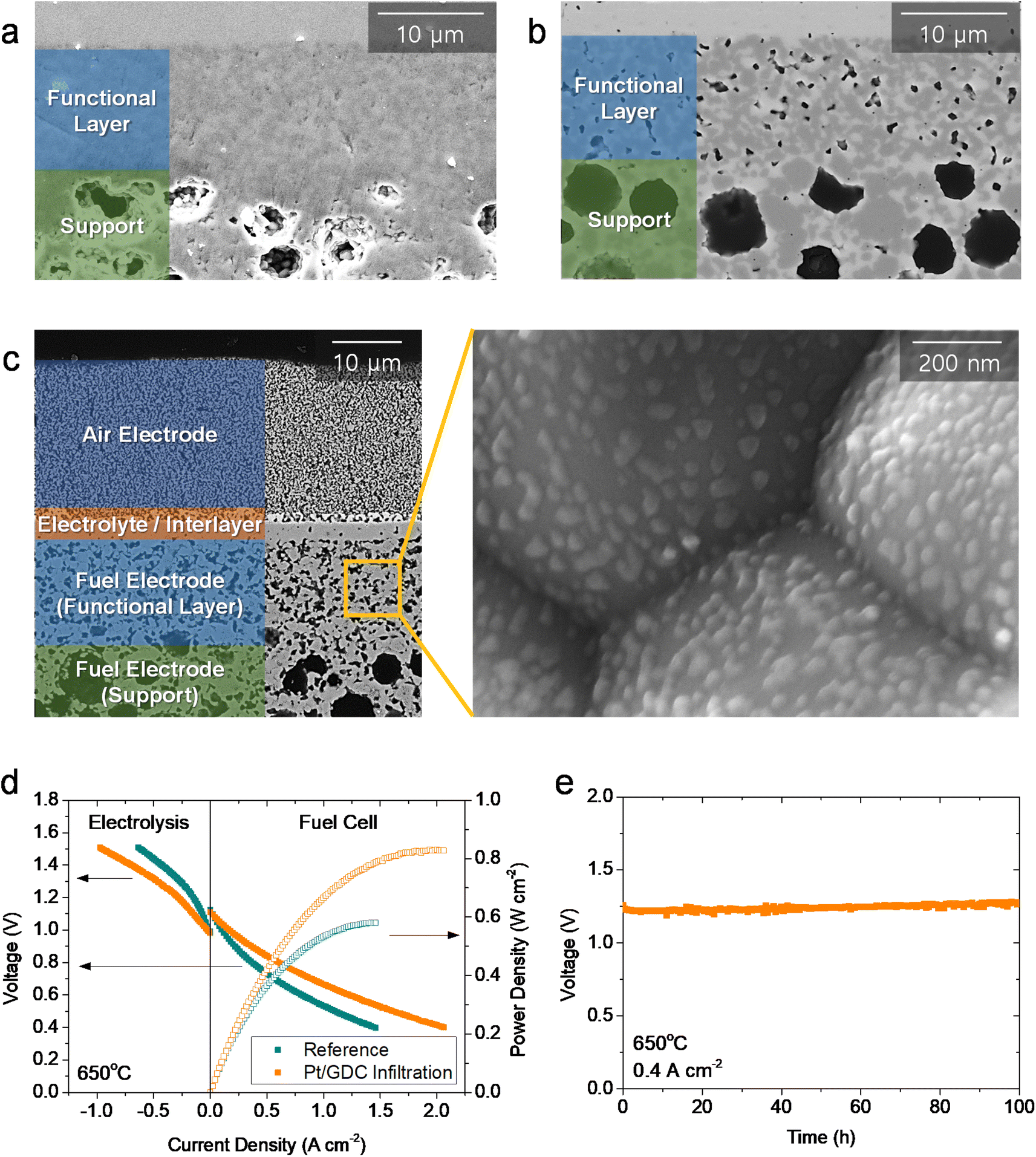 | ||
| Fig. 5 Practical application of atomically dispersed Pt/GDC in fuel-electrode-supported full cells: (a and b) SEM images of fuel electrode functional layer (a) without and (b) with carbon black pore former. (c) Cross-sectional SEM image of the tested Pt/GDC-infiltrated full cell. (d) I–V curves of the cells with and without Pt/GDC in fuel cell and electrolysis modes. (e) Long-term stability test of the cell with Pt/GDC in electrolysis mode. | ||
Conclusions
This study demonstrates that atomically dispersed catalysts could operate stably in high-temperature electrochemical devices at ∼650 °C without thermal degradation. The strong interactions between the atomic metal catalyst and ceramic support prevent aggregation. Atomically dispersed catalysts can be loaded into the porous electrode via an in situ synthetic technique, which remarkably improves the electrochemical performance. Moreover, the thermal stabilization significantly expands the range of potential applications for atomically dispersed catalysts, and the in situ catalyst loading technique enables their practical use in various electrochemical energy devices.Author contributions
J. Min: methodology, investigation, writing – original draft; H. Seo: methodology, investigation, writing – original draft; J. Shin: methodology; M. Y. Park: methodology; S.-Y. Park: investigation; H. Choi: formal analysis, S. Park: formal analysis; S. Yang: formal analysis; H. J. Chang: formal analysis; J. Hong: supervision, funding acquisition; writing – review & editing; K. J. Yoon: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of the Korean Ministry of Science & ICT through the Technology Development Program to Solve Climate Changes (No. 2020M1A2A2080862) and the institutional research program of the Korea Institute of Science and Technology (KIST). The authors thank Lauren Plavisch for English language editing.Notes and references
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed.
- N. Q. Minh, in Hydrogen Science and Engineering: Materials, Processes, Systems and Technology, 2016, pp. 359–390, DOI:10.1002/9783527674268.ch16.
- N. Q. Minh and Y. Shirley Meng, MRS Bull., 2019, 44, 682–683 CrossRef.
- X. I. Pereira-Hernández, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarik, E. J. M. Hensen, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1358 CrossRef PubMed.
- L. Cao, W. Liu, Q. Luo, R. Yin, B. Wang, J. Weissenrieder, M. Soldemo, H. Yan, Y. Lin, Z. Sun, C. Ma, W. Zhang, S. Chen, H. Wang, Q. Guan, T. Yao, S. Wei, J. Yang and J. Lu, Nature, 2019, 565, 631–635 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- Z. Chen, Q. Li, H. Xiang, Y. Wang, P. Yang, C. Dai, H. Zhang, W. Xiao, Z. Wu and L. Wang, Inorg. Chem. Front., 2023, 10, 1493–1500 RSC.
- Z. Wu, Y. Gao, Z. Wang, W. Xiao, X. Wang, B. Li, Z. Li, X. Liu, T. Ma and L. Wang, Chin. J. Catal., 2023, 46, 36–47 CrossRef CAS.
- H. Wang, P. Yang, X. Sun, W. Xiao, X. Wang, M. Tian, G. Xu, Z. Li, Y. Zhang, F. Liu, L. Wang and Z. Wu, J. Energy Chem., 2023, 87, 286–294 CrossRef CAS.
- Q. Li, X. Luan, Z. Xiao, W. Xiao, G. Xu, Z. Li, Z. Wu and L. Wang, Inorg. Chem., 2023, 62, 9687–9694 CrossRef CAS PubMed.
- Z. Wu, Y. Wang, D. Liu, B. Zhou, P. Yang, R. Liu, W. Xiao, T. Ma, J. Wang and L. Wang, Adv. Funct. Mater., 2023, 2307010 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- W. C. Chueh, Y. Hao, W. Jung and S. M. Haile, Nat. Mater., 2012, 11, 155–161 CrossRef CAS PubMed.
- S. P. Jiang and S. H. Chan, J. Mater. Sci., 2004, 39, 4405–4439 CrossRef CAS.
- C. Wang, F. Feng, J. Du, T. Zheng, Z. Pan and Y. Zhao, ChemCatChem, 2019, 11, 2054–2057 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- T. Theophanides and P. D. Harvey, Coord. Chem. Rev., 1987, 76, 237–264 CrossRef CAS.
- J. Shin, Y. J. Lee, A. Jan, S. M. Choi, M. Y. Park, S. Choi, J. Y. Hwang, S. Hong, S. G. Park, H. J. Chang, M. K. Cho, J. P. Singh, K. H. Chae, S. Yang, H.-I. Ji, H. Kim, J.-W. Son, J.-H. Lee, B.-K. Kim, H.-W. Lee, J. Hong, Y. J. Lee and K. J. Yoon, Energy Environ. Sci., 2020, 13, 4903–4920 RSC.
- T. Shishido, Y. Yamamoto, H. Morioka, K. Takaki and K. Takehira, Appl. Catal., A, 2004, 263, 249–253 CrossRef CAS.
- D. Sordelet and M. Akinc, J. Colloid Interface Sci., 1988, 122, 47–59 CrossRef CAS.
- K. J. Yoon, M. Biswas, H.-J. Kim, M. Park, J. Hong, H. Kim, J.-W. Son, J.-H. Lee, B.-K. Kim and H.-W. Lee, Nano Energy, 2017, 36, 9–20 CrossRef CAS.
- K. J. Yoon, S.-i. Lee, H. An, J. Kim, J.-W. Son, J.-H. Lee, H.-J. Je, H.-W. Lee and B.-K. Kim, Int. J. Hydrogen Energy, 2014, 39, 3868–3878 CrossRef CAS.
- H. Y. Jung, S. H. Choi, H. Kim, J. W. Son, J. Kim, H. W. Lee and J. H. Lee, J. Power Sources, 2006, 159, 478–483 CrossRef CAS.
- A. Uzun, V. Ortalan, Y. Hao, N. D. Browning and B. C. Gates, ACS Nano, 2009, 3, 3691–3695 CrossRef CAS PubMed.
- G. Delahay, D. Valade, A. Guzmán-Vargas and B. Coq, Appl. Catal., B, 2005, 55, 149–155 CrossRef CAS.
- I. Balint, A. Miyazaki and K.-i. Aika, J. Catal., 2003, 220, 74–83 CrossRef CAS.
- A. Singh and K. Miyabayashi, RSC Adv., 2020, 10, 362–366 RSC.
- H. Xu, L.-X. Ding, C.-L. Liang, Y.-X. Tong and G.-R. Li, NPG Asia Mater., 2013, 5, e69 CrossRef CAS.
- L. S. Kibis, D. A. Svintsitskiy, A. I. Stadnichenko, E. M. Slavinskaya, A. V. Romanenko, E. A. Fedorova, O. A. Stonkus, V. A. Svetlichnyi, E. D. Fakhrutdinova, M. Vorokhta, B. Šmíd, D. E. Doronkin, V. Marchuk, J.-D. Grunwaldt and A. I. Boronin, Catal. Sci. Technol., 2021, 11, 250–263 RSC.
- P. Bera, K. R. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayaram and G. N. Subbanna, Chem. Mater., 2003, 15, 2049–2060 CrossRef CAS.
- J. R. McBride, K. C. Hass, B. D. Poindexter and W. H. Weber, J. Appl. Phys., 1994, 76, 2435–2441 CrossRef CAS.
- W. Lin, A. A. Herzing, C. J. Kiely and I. E. Wachs, J. Phys. Chem. C, 2008, 112, 5942–5951 CrossRef CAS.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CrossRef CAS.
- H. Inaba and H. Tagawa, Solid State Ionics, 1996, 83, 1–16 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- A. Sin, Y. Dubitsky, A. Zaopo, A. S. Aricò, L. Gullo, D. La Rosa, S. Siracusano, V. Antonucci, C. Oliva and O. Ballabio, Solid State Ionics, 2004, 175, 361–366 CrossRef CAS.
- H. Yahiro, K. Eguchi and H. Arai, Solid State Ionics, 1989, 36, 71–75 CrossRef CAS.
- A. Lekhal, B. J. Glasser and J. G. Khinast, Chem. Eng. Sci., 2001, 56, 4473–4487 CrossRef CAS.
- T. Mahata, S. R. Nair, R. K. Lenka and P. K. Sinha, Int. J. Hydrogen Energy, 2012, 37, 3874–3882 CrossRef CAS.
- B. Mo, J. Rix, U. Pal, S. Basu and S. Gopalan, J. Electrochem. Soc., 2020, 167, 134506 CrossRef CAS.
- I. Jung, D. Lee, S. O. Lee, D. Kim, J. Kim, S.-H. Hyun and J. Moon, Ceram. Int., 2013, 39, 9753–9758 CrossRef CAS.
- D. Marrero-López, J. Peña-Martínez, J. C. Ruiz-Morales, M. Gabás, P. Núñez, M. A. G. Aranda and J. R. Ramos-Barrado, Solid State Ionics, 2010, 180, 1672–1682 CrossRef.
- K. B. Yoo and G. M. Choi, Solid State Ionics, 2009, 180, 867–871 CrossRef CAS.
- A. Ghosh, A. Azad and J. T. Irvine, ECS Trans., 2011, 35, 1337 CrossRef CAS.
- S. Primdahl and M. Mogensen, J. Electrochem. Soc., 1997, 144, 3409–3419 CrossRef CAS.
- S. Gewies, W. G. Bessler, V. Sonn and E. Ivers-Tiffee, ECS Trans., 2007, 7, 1573–1582 CrossRef CAS.
- R. Barfod, M. Mogensen, T. Klemensø, A. Hagen, Y.-L. Liu and P. Vang Hendriksen, J. Electrochem. Soc., 2007, 154, B371–B378 CrossRef CAS.
- M. Vogler, A. Bieberle-Hütter, L. Gauckler, J. Warnatz and W. G. Bessler, J. Electrochem. Soc., 2009, 156, B663–B672 CrossRef CAS.
- P. Holtappels, L. G. J. de Haart and U. Stimming, J. Electrochem. Soc., 1999, 146, 1620–1625 CrossRef CAS.
- S. P. Jiang and S. P. S. Badwal, J. Electrochem. Soc., 1997, 144, 3777–3784 CrossRef CAS.
- S. Raz, K. Sasaki, J. Maier and I. Riess, Solid State Ionics, 2001, 143, 181–204 CrossRef CAS.
- W. Lai and S. M. Haile, J. Am. Ceram. Soc., 2005, 88, 2979–2997 CrossRef CAS.
- C. Lu, W. L. Worrell, J. M. Vohs and R. J. Gorte, J. Electrochem. Soc., 2003, 150, A1357 CrossRef CAS.
- A. Hauch, P. S. Jørgensen, K. Brodersen and M. Mogensen, J. Power Sources, 2011, 196, 8931–8941 CrossRef CAS.
- A. Hauch and M. Mogensen, Solid State Ionics, 2010, 181, 745–753 CrossRef CAS.
- Y. L. Liu and C. Jiao, Solid State Ionics, 2005, 176, 435–442 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05534e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |