
Quad-metallic MOF-derived carbon-armored pseudo-high entropy alloys as a bifunctional electrocatalyst for alkaline water electrolysis at high current densities†
Duraisamy
Senthil Raja
 ,
Yu-Chieh
Ting
,
Ting-Yu
Lin
,
Chih-Chieh
Cheng
,
Po-Wei
Chen
,
Fan-Yu
Yen
and
Shih-Yuan
Lu
*
,
Yu-Chieh
Ting
,
Ting-Yu
Lin
,
Chih-Chieh
Cheng
,
Po-Wei
Chen
,
Fan-Yu
Yen
and
Shih-Yuan
Lu
*
Department of Chemical Engineering, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: sylu@mx.nthu.edu.tw
First published on 30th October 2023
Abstract
A new catalyst design, pseudo-high entropy alloy (HEA), was proposed and demonstrated for high performance water electrolysis. Pseudo-HEAs, defined as single-phase four-element alloys of molar configurational entropy greater than 1.36R, are in general easier to design and prepare for enhanced electrocatalytic efficiency as compared with traditional HEAs of five or more elements. A carbon armored pseudo-HEA catalyst, FeCoNiMo@C, was synthesized with a simple two-stage thermal conversion of FeCoNiMo-MOF, grown in situ on a nickel foam (NF) substrate, to exhibit outstanding catalytic efficiency and stability toward water electrolysis in alkaline media. It delivered 10 and 500 mA cm−2 at ultra-low overpotentials of 55 and 233 mV, respectively for the hydrogen evolution reaction and 204 and 273 mV, respectively for the oxygen evolution reaction. The FeCoNiMo@C/NF//FeCoNiMo@C/NF couple achieved ultra-low cell voltages of 1.488 V@10 mA cm−2 and 1.725 V@500 mA cm−2 for full cell water splitting and ultra-stability at an industrially applicable high current density of 500 mA cm−2 with a minor decay of 3.4% over 50 h. The success was attributed to the atomic scale synergy between constituent atoms, Fe, Co, Ni and Mo, atomically dispersed in the pseudo-HEA.
1. Introduction
Fossil fuel-free production of green energy carriers is of critical technological importance and challenge to reach the NetZero by 2050 goal. Hydrogen, emitting no CO2 and particulate pollutants when in use, is considered the most promising green energy carrier. Nevertheless, the current major commercial H2 production technology, natural gas steam reforming, uses fossil fuels as the raw material and emits comparable amounts of CO2. A green and clean approach to commercial hydrogen generation primarily includes the electrolytic water splitting process which involves the combination of the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), driven by renewable energies.1,2 Due to the high kinetic energy barrier of the electrochemical reactions involved, especially the OER process, electrolytic water splitting technology is less feasible for commercial hydrogen generation.3,4 Though precious metal (Pt, Ru, and Ir)-based electrocatalysts, such as Pt/C for the HER and RuO2 or IrO2 for the OER, have been used as benchmark electrodes for electrolytic water splitting, the earth scarcity, high price, and insufficient long-term durability of Pt, Ru, and Ir, restrict their large-scale commercial applications.5 It is therefore significantly important to develop non-precious metal-based bifunctional electrocatalysts for both the OER and HER.Metal–organic frameworks (MOFs) are crystalline nano-porous materials with ordered arrangements of metal ions/metal clusters linked together with organic ligands.6,7 They possess many unique features advantageous for catalytic applications, such as tunable pore structure, high specific surface areas, easy tailoring of material compositions, rich material morphologies, and capability to act as precursors for the preparation of a wide range of carbon-based nanocomposites, and have thus gained much research attention in recent years.8–11 In particular, MOF-derived, carbon coated materials have found promising applications as electrocatalysts for water electrolysis.8–13 For instance, Pan et al. demonstrated a hybrid nanosystem, with CoP nanoparticles coated with an N-doped carbon nanotube hollow polyhedron (NCNHP) obtained from a core–shell MOF system, ZIF-8@ZIF-67, as a bifunctional electrocatalyst for water splitting applications. Overpotentials of 140 and 115 mV for the HER in acidic medium (0.5 M H2SO4) and alkaline medium (1 M KOH), respectively to attain the 10 mA cm−2 current density, were achieved and the corresponding overpotential for the OER in 1 M KOH was 310 mV. When employed as both the cathode and anode for an alkaline water electrolyzer, CoP/NCNHP delivered 10 mA cm−2 at a cell voltage of 1.64 V.9 Recently, Sun et al. reported a hierarchical nickel–carbon composite, fabricated through direct growth of sheet-like 2D Ni-MOFs on the surface of nickel foam followed by a high-temperature thermal treatment, as a bifunctional electrocatalyst for water electrolysis. The product catalyst showed overpotentials of 37 and 265 mV for the HER and OER respectively at 10 mA cm−2, and delivered 35.9 mA cm−2 at a cell voltage of 1.60 V for full cell water splitting.12 Zhao et al. reported a bifunctional catalyst (Fe–Ni@NC-CNTs), produced through carbonization of a bimetallic MIL-88-Fe/Ni-MOF, for applications in overall water splitting. This composite catalyst significantly promoted the charge transfer efficiency and restrained the corrosion of the metallic catalysts, leading to high OER and HER activities with overpotentials of 274 and 202 mV, respectively at 10 mA cm−2 in an alkaline solution.13
It is to be noted that electricity consumption accounts for at least 50% of the hydrogen generation cost for water electrolyzers, which can readily be decreased by reducing the involved overpotentials in both the OER and HER. It is thus critical to design and develop low-cost, highly efficient, non-noble metal-based electrocatalysts, offering lower overpotential values, and durable both mechanically and electrochemically, for water splitting to enable prevailing of this green hydrogen generation technology. The key concerns are the overpotentials and durability of the electrodes functioning at large current densities (∼500 mA cm−2), for practical applications in commercial electrolyzers.14,15 However, the catalytic efficiency and long-time durability of most of the reported catalysts for electrocatalytic water splitting were evaluated under lower current density (≤100 mA cm−2) conditions, which is inadequate for commercial applications.
Recently, high entropy alloys (HEAs) have gained rapidly increasing research attention as catalysts for electrocatalytic water splitting because of their favorable characteristics such as tunable compositions, high electrical conductivities,16 excellent mechanical17 and thermal stability,18 versatile surfaces, and anti-corrosion properties.19,20 In addition to electrolytic water-splitting, HEAs also find applications in a wide range of catalytic reactions such as the nitrogen reduction reaction,21 carbon monoxide and carbon dioxide reduction reactions,22 oxygen reduction reaction,23 alcohol oxidation reaction,24 formic acid oxidation reaction,25 and azo dye degradation.26 HEAs were loosely defined as alloys of a crystalline phase composed of five or more major metallic elements with atomic concentrations of constituent elements falling between 5 and 35%.27 HEAs were also recognized as entropy stabilized alloys with their high molar configurational entropies to suppress phase separation. The molar configurational entropy, or called entropy of mixing (ΔSmix), is calculated as ΔSmix = −RΣxi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(xi), with xi denoting molar fractions of the constituent elements and R the molar gas constant.27 The maximum and minimum values of ΔSmix for a five-element HEA can be calculated to be 1.61R and 1.36R, respectively. Hence, single-phase five-element alloys having ΔSmix values of 1.36–1.61R can be called HEAs. Based on this entropy criterion, it is possible to have ΔSmix ≥ 1.36R for four-element alloys with relatively equimolar compositions.28 Note that it is impossible to have ΔSmix ≥ 1.36R for alloys composed of less than four constituent elements. To more clearly describe these special four-element single-phase alloys, having ΔSmix ≥ 1.36R, we introduce a new term “pseudo-HEA” to differentiate them from traditional HEAs composed of five or more elements. The most critical merit of HEAs, particularly for catalytic applications, is the atomic scale synergy between constituent elements. It is however not a trivial task, often difficult, to find five or more elements, possessing all positive mutual synergy toward targeted applications. Furthermore, it is in general more difficult to prepare HEAs composed of a higher number of constituent elements than pseudo-HEAs, considering for example the atomic size differences of constituent elements. In this regard, pseudo-HEAs are promising alternatives to HEAs and deserve intensive and extensive research attention. For applications in electrolytic water splitting, one example has been reported to show that a pseudo-HEA catalyst, CoNiCuMo, exhibited higher catalytic efficiency than a corresponding HEA catalyst, FeCoNiCuMo, for the HER in 1 M KOH.29 The main reason is the relative inertness of Fe toward HER catalysis in alkaline solutions, and the presence of Fe in the FeCoNiCuMo HEA in fact dilutes the synergy between CoNiCuMo.29 With the above observations in mind, this work attempts to develop MOF-derived pseudo-HEAs as highly efficient and stable bifunctional catalysts toward both the HER and OER in alkaline water electrolysis at high current densities, by taking advantage of carbon armor protection from the MOF-derived approach and synergy between HER-active and OER-active elements of the pseudo-HEAs.
ln(xi), with xi denoting molar fractions of the constituent elements and R the molar gas constant.27 The maximum and minimum values of ΔSmix for a five-element HEA can be calculated to be 1.61R and 1.36R, respectively. Hence, single-phase five-element alloys having ΔSmix values of 1.36–1.61R can be called HEAs. Based on this entropy criterion, it is possible to have ΔSmix ≥ 1.36R for four-element alloys with relatively equimolar compositions.28 Note that it is impossible to have ΔSmix ≥ 1.36R for alloys composed of less than four constituent elements. To more clearly describe these special four-element single-phase alloys, having ΔSmix ≥ 1.36R, we introduce a new term “pseudo-HEA” to differentiate them from traditional HEAs composed of five or more elements. The most critical merit of HEAs, particularly for catalytic applications, is the atomic scale synergy between constituent elements. It is however not a trivial task, often difficult, to find five or more elements, possessing all positive mutual synergy toward targeted applications. Furthermore, it is in general more difficult to prepare HEAs composed of a higher number of constituent elements than pseudo-HEAs, considering for example the atomic size differences of constituent elements. In this regard, pseudo-HEAs are promising alternatives to HEAs and deserve intensive and extensive research attention. For applications in electrolytic water splitting, one example has been reported to show that a pseudo-HEA catalyst, CoNiCuMo, exhibited higher catalytic efficiency than a corresponding HEA catalyst, FeCoNiCuMo, for the HER in 1 M KOH.29 The main reason is the relative inertness of Fe toward HER catalysis in alkaline solutions, and the presence of Fe in the FeCoNiCuMo HEA in fact dilutes the synergy between CoNiCuMo.29 With the above observations in mind, this work attempts to develop MOF-derived pseudo-HEAs as highly efficient and stable bifunctional catalysts toward both the HER and OER in alkaline water electrolysis at high current densities, by taking advantage of carbon armor protection from the MOF-derived approach and synergy between HER-active and OER-active elements of the pseudo-HEAs.
A great number of reports have shown that FeNi,30 FeCo,31 and FeCoNi-based32 materials are excellent electrocatalysts for alkaline OER, whereas NiMo-based33 materials have been repeatedly demonstrated to exhibit remarkable catalytic efficiency toward alkaline HER. Furthermore, it is important to maximize the synergy between constituent elements. With equimolar compositions, the interactions between constituents of a multi-component system are maximum if the constituents are randomly distributed in the system. And the maximum extent of randomness of a multi-component system ensures a maximum configurational entropy of the system. It is thus a rational design to combine all the above-mentioned elements, Fe, Co, Ni, Mo, to form a pseudo-HEA to serve as a bifunctional catalyst for alkaline water splitting. Herein, corresponding quad-metallic FeCoNiMo-MOFs were first grown in situ on nickel foam (NF), FeCoNiMo-MOF/NF, which was subsequently thermally treated to form carbon-armored FeCoNiMo pseudo-HEA, FeCoNoMo@C/NF (Fig. 1). The carbon-coated FeCoNiMo pseudo-HEA catalyst exhibited outstanding OER, HER, and overall water splitting performances in 1 M KOH, achieving ultra-low cell voltages of 1.488 V@10 mA cm−2 and 1.725 V@500 mA cm−2 with remarkable durability at large current densities, a minor increment of 3.4% in the applied cell voltage in a chronopotentiometric durability test carried out at an industrially applicable large current density of 500 mA cm−2 for full water splitting over 50 h. The successful first demonstration of MOF-derived pseudo-HEAs for bifunctionality in electrocatalysis opens up a new route for catalyst design toward electrocatalysis.
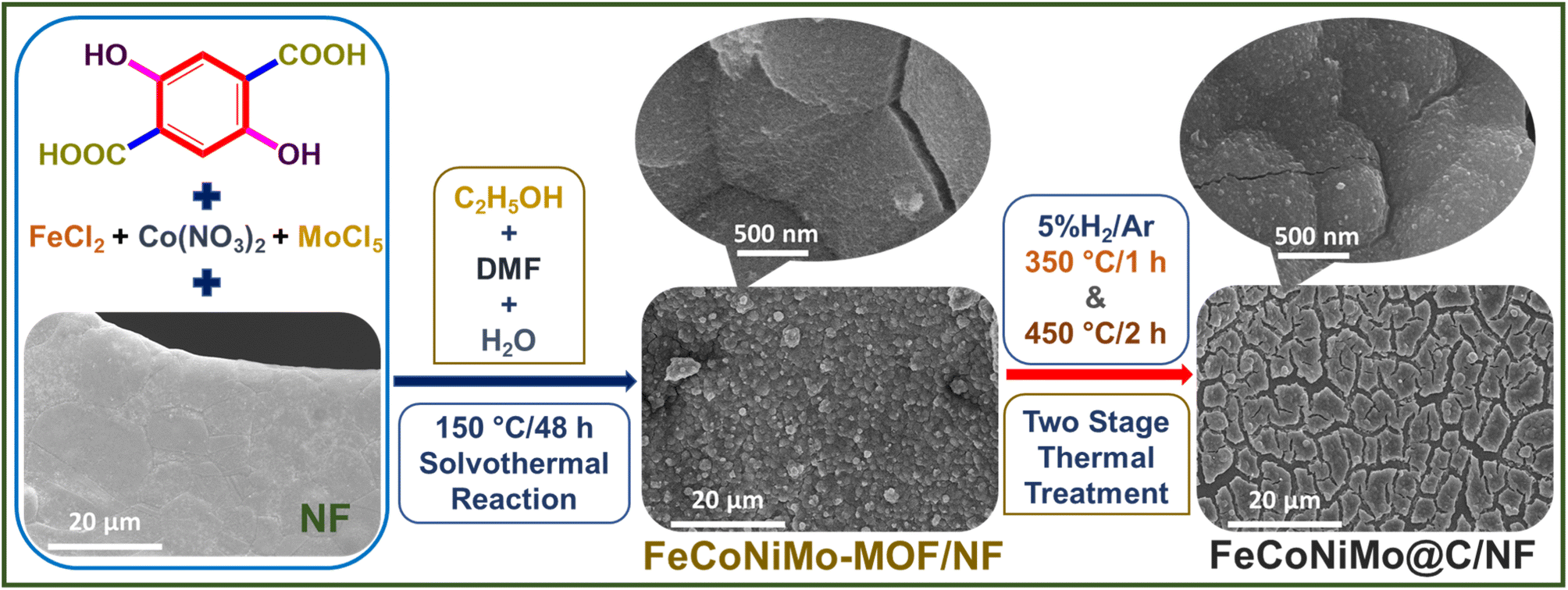 | ||
| Fig. 1 Fabrication processes of FeCoNiMo-MOF on nickel foam (NF) and its pseudo-HEA derivative, FeCoNiMo@C/NF. | ||
2. Experimental section
General experimental details, including chemicals used, instrument descriptions, electrocatalytic measurements, and supporting calculations are presented in the ESI.†2.1. Nickel foam (NF)-based electrode fabrication process
3. Results and discussion
3.1. Structural characterization
Multi-metallic MOFs, precursors for corresponding carbon armored multi-metallic alloys, were first prepared and characterized. In addition to the quad-metallic MOF, FeCoNiMo-MOF, tri-metallic MOFs, including FeCoNi-MOF, FeNiMo-MOF, and CoNiMo-MOF, were also synthesized in situ on NF for comparison. Note that it is impossible to synthesize the Ni absent tri-metallic FeCoMo-MOF with the present synthetic process since Ni ions were generated from the dissolution of NF during the solvothermal reaction to take part in the MOF construction. To avoid interference of the XRD peaks contributed by NF, the synthesized MOF products were scratched off from MOF/NF samples for XRD characterization. Fig. S1a (ESI)† shows the powder XRD patterns of the four scratched-off MOF products. All diffraction patterns match well with that of the theoretically simulated powder XRD pattern of MOF-74,34 clearly suggesting that all synthesized MOFs possess the same crystalline structure as MOF-74. Fig. S1b–i (ESI)† show the SEM images of FeCoNiMo-MOF/NF, FeCoNi-MOF/NF, FeCoMo-MOF/NF, and CoNiMo-MOF/NF, respectively at two magnifications, from which it is clear that the skeleton surfaces of the NF are fully covered by the MOFs. Interestingly, FeCoNiMo-MOF, FeCoMo-MOF, and CoNiMo-MOF exhibit the morphology of aggregated microspheres, whereas FeCoNi-MOF appears as aggregated micro-spindles. Spindle shape is commonly observed for MOF products. Here, the three Mo-containing MOFs, FeCoNiMo-MOF, FeNiMo-MOF, and CoNiMo-MOF, exhibit the morphology of microspheres, which may be caused by the significantly larger size of Mo atoms.35Carbon armored multi-metallic alloy catalysts on NF, namely FeCoNiMo@C/NF, FeCoNi@C/NF, FeNiMo@C/NF, and CoNiMo@C/NF, were produced from thermal treatment of corresponding precursors MOF/NF, FeCoNiMo-MOF/NF, FeCoNi-MOF/NF, FeNiMo-MOF/NF, and CoNiMo-MOF/NF, respectively. Compositions of these multi-metallic alloys were determined with inductively coupled plasma optical emission spectroscopy (ICP-OES), and the corresponding results are summarized in Table 1 for comparison. It is evident that these alloys are relatively composition balanced, with the elemental concentrations of the quad-metallic alloy and the three tri-metallic alloys centering around 25% and 33%, respectively. Also interesting to note is that Ni appears in all samples even though no Ni precursors are used during the synthesis of MOFs. Nickel has originated from the Ni ions generated in situ from slight NF dissolution during the solvothermal process.32 With the composition available, ΔSmix can be readily calculated. Here, a ΔSmix of 1.37R, higher than 1.36R, is obtained for the primary catalyst, FeCoNiMo@C. In general, a solid-solution HEA of a face-centered cubic (FCC) structure commonly possesses: (1) a small atomic size difference (δ) (≤8.5%), (2) a suitable enthalpy of mixing (ΔHmix) (−22 to 7 kJ mol−1), (3) a suitable ΔSmix (11–19.5 J (K mol)−1), and (4) a high valence electron concentration (VEC) (>8).36 The primary catalyst, FeCoNiMo@C, satisfies all four conditions suggested for solid-solution HEAs of an FCC structure, with δ = 3.65%, ΔHmix = −5.54 kJ mol−1, ΔSmix = 11.37 J K−1 mol−1, and VEC = 8.52 (detailed calculations are given in the ESI†). The crystalline structure of all multi-metallic alloys was determined with XRD characterization to be FCC as shown in Fig. 2a.37 Here, again, to avoid the interference of the strong diffraction peaks of NF, these alloys were scratched off from their NF substrates. Evidently, only FCC phase is present in the samples without showing side phases, indicating single-phase products without phase separation. Note that the amount of carbon is negligibly small and no relevant diffraction peaks are observed. With the crystalline structure of FCC confirmed for FeCoNiMo, FeCoNiMo is thus a single-phase alloy having ΔSmix higher than 1.36R and FeCoNiMo@C is a carbon armored pseudo-HEA catalyst.
| Sample | At% |
|---|---|
| FeCoNiMo@C | Fe = 23.21 |
| Co = 24.59 | |
| Ni = 32.92 | |
| Mo = 19.27 | |
| FeCoNi@C | Fe = 35.21 |
| Co = 32.35 | |
| Ni = 32.44 | |
| FeNiMo@C | Fe = 32.92 |
| Ni = 40.81 | |
| Mo = 26.27 | |
| CoNiMo@C | Co = 40.71 |
| Ni = 39.06 | |
| Mo = 20.23 |
 | ||
| Fig. 2 (a) Powder XRD patterns of scratched-off multi-metallic alloys. SEM images of FeCoNiMo@C/NF (b and c), FeCoNi@C/NF (d and e), FeCoMo@C/NF (f and g), and CoNiMo@C/NF (h and i). | ||
Fig. 2b–i show SEM images of the four carbon armored multi-metallic alloys at high and low magnifications. It is evident that all samples fully cover the NF surface and their primary morphologies resemble those of their parent MOFs, microspheres for FeCoNiMo@C/NF, FeNiMo@C/NF, and CoNiMo@C/NF, and micro-spindles for FeCoNi@C/NF, indicating well-structural preservation during the thermal conversion process. If examined closely, secondary fine structures appear on the primary structures, likely being created from the carbonization of the organic linkers of the MOFs.
Transmission electron microscope (TEM) imaging was used to deeply observe the atomic and nano-structural characteristics of the primary sample, FeCoNiMo@C. The microspheres are actually composed of combined FeCoNiMo@C nanoparticles, having FeCoNiMo nanoparticles of sizes 10–30 nm coated and linked with carbon matrices, as shown in Fig. 3a and b. Interlayer distances of 0.211 and 0.180 nm are determined, in good agreement with the d-spacing of the (111) and (200) crystalline planes, respectively of an FCC structure. Furthermore, a lattice constant of 0.361 nm calculated with Vegard's rule, matches well with 0.356 nm determined based on powder XRD data and 0.363 nm estimated based on high resolution (HR) TEM data. In addition, TEM-energy dispersive X-ray spectrometry (EDX) elemental mapping of FeCoNiMo@C (Fig. 3d) reveals that all constituent elements of FeCoNiMo@C, Fe, Co, Ni, Mo, and C, are uniformly distributed in nanometer domains of the sample, further confirming its solid-solution characteristics. Further, the results of TEM-EDX elemental analysis of the FeCoNiMo@C sample (Fig. S2, ESI†) showed that the atomic composition of Fe, Co, Ni, and Mo is similar to that obtained from ICP-OES analyses (Table 1), further confirming the relatively equimolar composition of FeCoNiMo.
 | ||
| Fig. 3 TEM images of FeCoNiMo@C (a–c) at increasing magnifications, and (d) TEM-EDX elemental mapping of FeCoNiMo@C. | ||
The surface chemical states of FeCoNiMo@C were investigated with X-ray photoelectron spectroscopy (XPS, Fig. 4a–e). The survey spectrum (Fig. 4a) shows the existence of Fe, Co, Ni, Mo, and C, together with a considerable amount of O, which originated from surface oxidation of metallic elements and carbon layers under ambient conditions.38 The HR-XPS spectrum of Fe 2p (Fig. 4b) displays binding energy peaks at 708.4 and 721.1 eV, which correspond to Fe 2p3/2 and Fe 2p1/2 of Fe(0), respectively, whereas the peaks found at 712.3 and 724.8 eV are attributable to Fe 2p3/2 and Fe 2p1/2 of Fe(II), respectively.39 In the HR-XPS spectrum of Co 2p (Fig. 4c), the peaks observed at 778.1 and 793.6 eV are contributed by Co 2p3/2 and Co 2p1/2 of Co(0), respectively, and the peaks at 781.4 and 796.9 eV can be attributed to Co 2p3/2 and Co 2p1/2 of Co(II), respectively.40 The HR-XPS spectrum of Ni 2p (Fig. 4d) exhibits binding energy peaks of Ni 2p3/2 and Ni 2p1/2 of Ni(0) at 852.9 and 866.6 eV, respectively. The peaks located at 856.6 and 872.4 eV can be attributed to Ni 2p3/2 and Ni 2p1/2 of Ni(II), and the peaks observed at 862.2 and 879.2 eV are the corresponding satellite peaks.41 As for the Mo 3d spectrum (Fig. 4e), the two peaks observed at 228.8 and 231.8 eV can be assigned to Mo 3d5/2 and Mo 3d3/2 of Mo(0), respectively, and the peaks found at 230.8 and 234.0 eV correspond to Mo 3d5/2 and Mo 3d3/2 of Mo(IV), respectively.42 Note that the inevitable surface oxidation of metallic samples when exposed to air and the few nanometer sampling depth of XPS lead to dominant showing of positively charged metal ions and minor signals of Fe(0), Co(0), Ni(0), and Mo(0). For comparison purposes, HR-XPS spectra of FeCoNi@C, FeNiMo@C, and CoNiMo@C (Fig. S3, ESI†) were also collected. All spectra of the three trimetallic samples show minor signals of metallic elements as observed in the case of the pseudo-HEA sample, FeCoNiMo@C. Interestingly, if examined closely, the binding energies of Fe, Co, and Ni of the quad-metallic sample, FeCoNiMo@C, are slightly higher than those of Fe, Co, and Ni of the three trimetallic samples, implying changes in the electronic structure induced by the introduction of the fourth metallic element. The upshifts in binding energies favor OER catalysis because of the presence of a higher oxidation state of the active sites.32
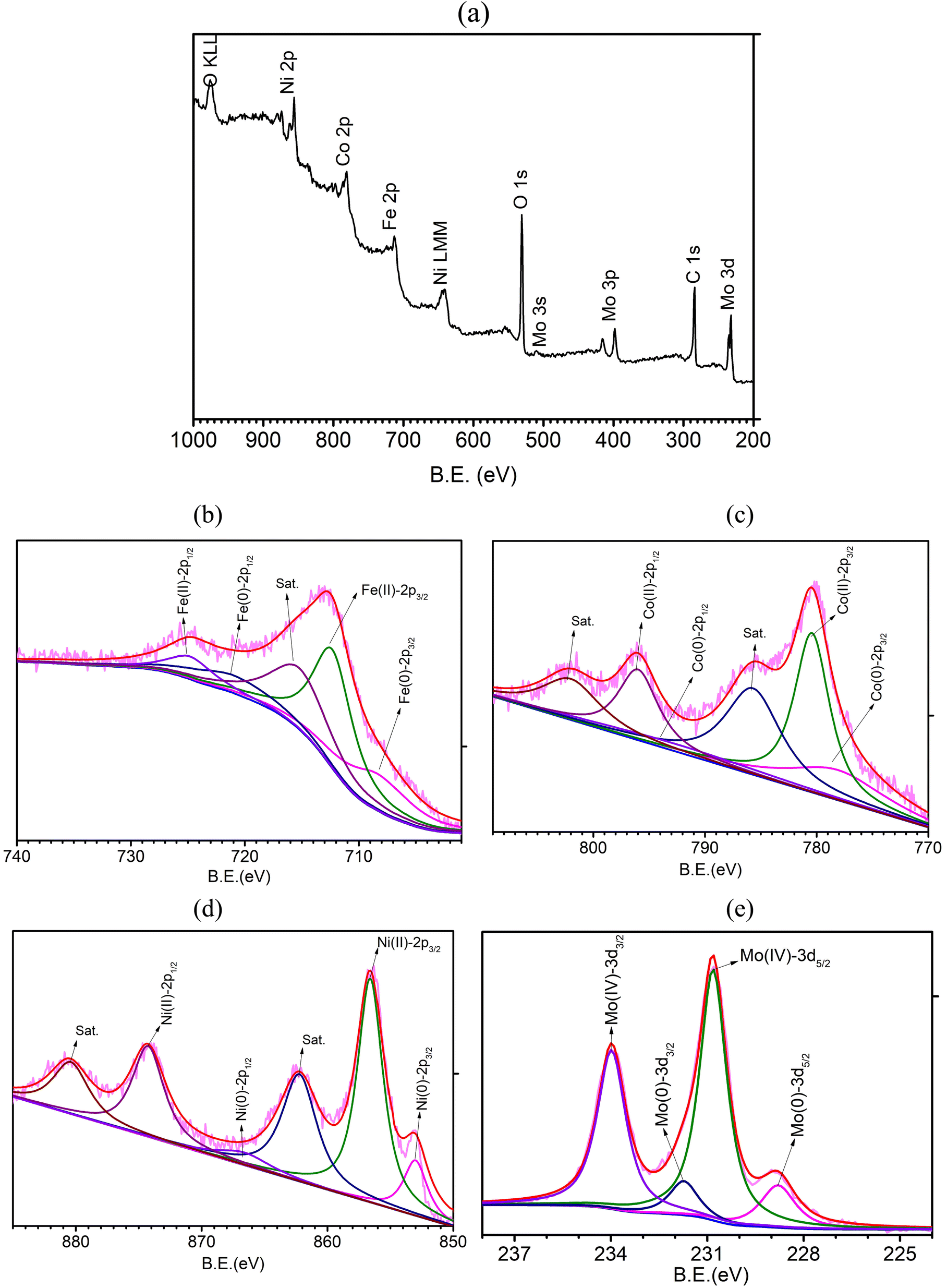 | ||
| Fig. 4 (a) XPS survey spectrum and HR-XPS spectra of (b) Fe 2p, (c) Co 2p, (d) Ni 2p, and (e) Mo 3d of FeCoNiMo@C. | ||
Next, the porous structure of the four carbon armored multi-metallic alloy samples was characterized with the aid of N2 gas adsorption/desorption measurements (Fig. S4, ESI†). The corresponding isotherms are of type IV, suggesting the presence of both micropores and mesopores. The initial fast rise in adsorption in the low pressure region shows the existence of micropores, while the presence of mesopores is confirmed by the observed hysteresis loop in the high pressure region for all the samples. The specific surface areas of FeCoNiMo@C, FeCoNi@C, FeNiMo@C, and CoNiMo@C are determined to be 213, 207, 201, and 221 m2 g−1, respectively. Interestingly, regardless of the morphology, microspheres or micro-spindles, the specific surface area remains roughly constant.
3.2. Electrocatalytic HER performances
The electrocatalytic efficiency of the four MOF-derived samples on NF, including FeCoNiMo@C/NF, FeCoNi@C/NF, FeNiMo@C/NF, and CoNiMo@C/NF, toward HER, OER, and full cell water splitting was examined by recording iR-compensated polarization curves in 1 M KOH solutions. For a fair comparison, the electrocatalytic efficiency of blank NF along with the two benchmark electrodes, Pt–C/NF for the HER and IrO2/NF for the OER, was also examined. The HER electrocatalytic efficiency of sample electrodes is first discussed (Fig. 5). The linear sweep voltammetry (LSV) polarization curves of all the tested electrodes for the HER process are presented in Fig. 5a, and the corresponding overpotentials at low (10 mA cm−2, η10) and high (500 mA cm−2, η500) current densities are given in Table 2. The pseudo-HEA-based FeCoNiMo@C/NF electrode, although not as good as the noble metal-based benchmark electrode, Pt–C/NF (η10 = 21 mV), exhibits the highest HER efficiency among the four multi-metallic alloy-based electrodes, with the lowest η10 of 55 mV, as compared to 65, 78, and 122 mV achieved by the three trimetallic alloy based electrodes, CoNiMo@C/NF, FeNiMo@C/NF, and FeCoNi@C/NF, respectively. All four multi-metallic alloy-based electrodes largely outperform the blank NF electrode (η10 = 245 mV), confirming the dominant contribution of the carbon-armored multi-metallic alloys toward HER catalysis. Among the three trimetallic alloy-based electrodes, the alloy electrodes containing both Ni and Mo show better HER performances, implying the important role played by NiMo in the HER. In addition to η10, catalytic efficiency measured at large current densities, such as η500, is critical for large-scale commercial applications of the catalyst. The overpotentials at 500 mA cm−2 (η500) are also determined and summarized in Table 2 for comparison. Again, FeCoNiMo@C/NF achieves the lowest η500 of 233 mV among the four multi-metallic alloy-based electrodes, with CoNiMo@C/NF (η500 = 276 mV) coming next, followed by FeNiMo@C/NF (η500 = 287 mV) and FeCoNiMo@C/NF (η500 = 343 mV). Interestingly, FeCoNiMo@C/NF outperforms Pt–C/NF (η500 = 245 mV) at high current densities, making it a better electrode for commercial water electrolyzers. The decay in catalytic efficiency of Pt–C toward the HER at high current densities has been commonly observed,43 which also motivates the development of alternative HER catalysts.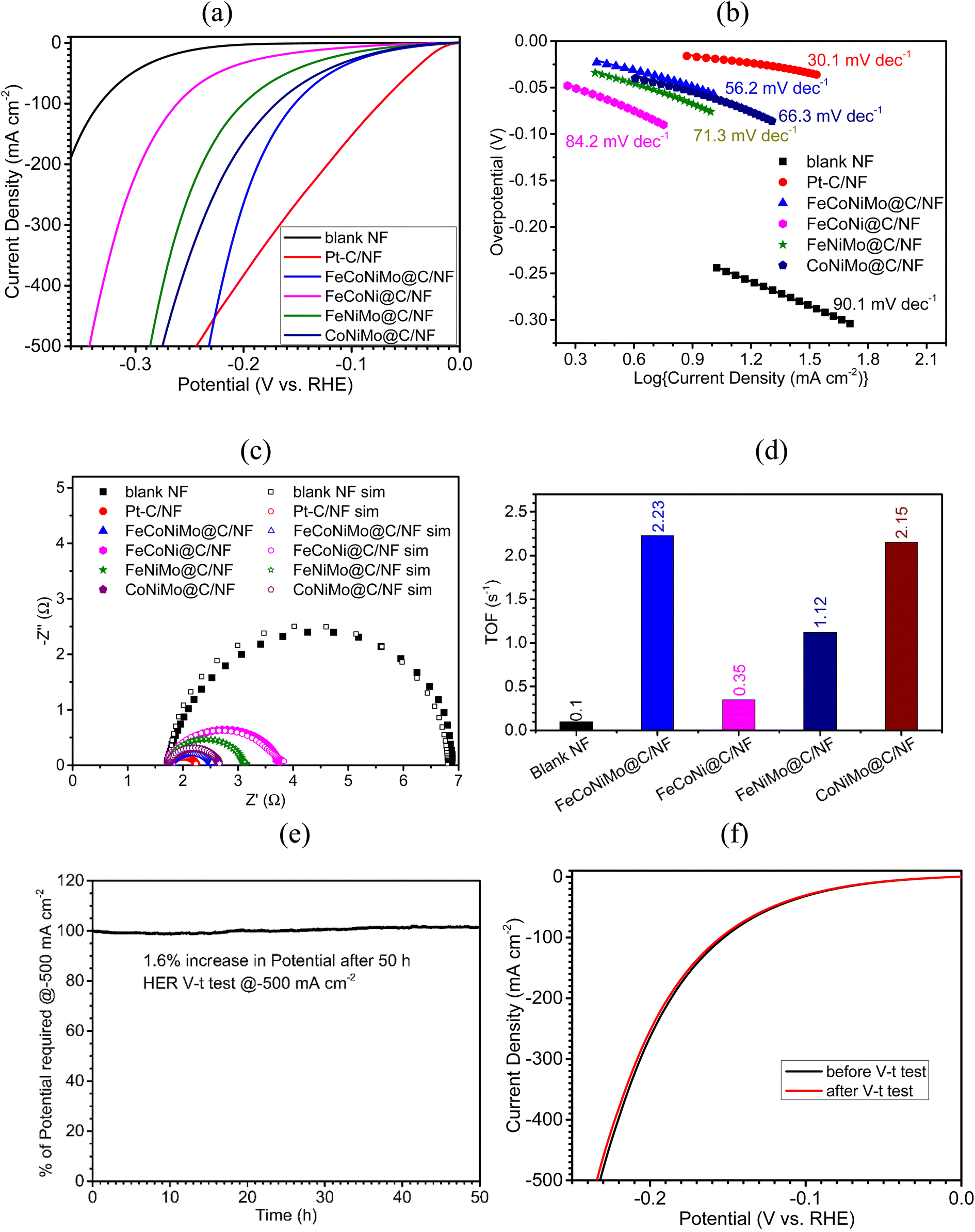 | ||
| Fig. 5 (a) LSV curves recorded in 1 M KOH at scan rate of 1 mV s−1, and corresponding (b) Tafel plots and (c) Nyquist plots of sample electrodes. (d) H2 turnover frequencies (TOFs) of sample catalysts calculated at −0.18 V (vs. RHE). (e) Chronopotentiometric (V–t) curve of FeCoNiMo@C/NF for the HER at −500 mA cm−2 for 50 h. (f) LSV curves of FeCoNiMo@C/NF before and after the V–t stability test for the HER at −500 mA cm−2 for 50 h. | ||
| Sample | Overpotential (η), mV | |
|---|---|---|
| η 10 | η 500 | |
| Blank NF | 245 | — |
| Pt–C/NF | 21 | 245 |
| FeCoNiMo@C/NF | 55 | 233 |
| FeCoNi@C/NF | 122 | 343 |
| FeNiMo@C/NF | 78 | 287 |
| CoNiMo@C/NF | 65 | 276 |
The HER kinetic characteristics of the four multi-metallic alloy-based electrodes, together with blank NF and Pt–C/NF, have next been evaluated with Tafel plots (Fig. 5b). The pseudo-HEA-based electrode, FeCoNiMo@C/NF, displays favorable HER kinetics, observed from the smallest Tafel slope of 56.2 mV dec−1 among the four alloy-based samples (66.3 for CoNiMo@C/NF, 71.3 for FeNiMo@C/NF, and 84.2 for FeCoNi@C/NF). A Tafel slope of around 30 mA dec−1, here 30.1 mV dec−1, is expected for Pt–C/NF, complying with the Volmer–Tafel mechanism having the Tafel step as the rate-determining step. As for FeCoNiMo@C/NF, a Tafel slope of 56.2 mV dec−1 suggests the Volmer–Heyrovsky mechanism with comparable rates of the Volmer and Heyrovsky steps.
Further investigation on the HER kinetics of the fabricated electrodes was carried out with electrochemical impedance spectroscopy (EIS) in the frequency range of 100 kHz to 1 Hz in 1 M KOH. The applied potential was set at −0.20 V (vs. RHE) to ensure the occurrence of the HER for all sample electrodes tested. The recorded EIS data (Fig. 5c) were fitted to an equivalent circuit model (Fig. S5, ESI†) to extract information on the charge transfer resistance (Rct), an important kinetics parameter. The smaller the Rct, the faster the HER kinetics. Evidently, the smallest Rct value of 0.45 Ω was found for FeCoNiMo@C/NF among the four alloy-based samples, which is only slightly higher than that of Pt–C/NF (0.35 Ω), but significantly smaller than those of CoNiMo@C/NF (0.70 Ω), FeNiMo@C/NF (1.01 Ω), FeCoNi@C/NF (1.76 Ω), and blank NF (5.03 Ω), consistent with the performance order observed in η10 and Tafel slope values.
The intrinsic activities of these alloy-based catalysts were quantitatively investigated and compared with turnover frequencies (TOFs). To determine TOFs, the amount of exposed active sites present on the catalyst needs to be measured. A reported reduction peak area quantification method has been adopted to estimate the total number of exposed active sites for the calculation of TOFs.44 Accordingly, the TOFs of all alloy-based samples and blank NF were estimated toward the HER at −0.18 V (vs. RHE) and the results are presented in Fig. 5d for comparison. Evidently, FeCoNiMo@C, the pseudo-HEA-based catalyst, with a TOF of 2.23 s−1, is catalytically more active than the three trimetallic alloy-based ones (2.15 for CoNiMo@C/NF, 1.12 for FeNiMo@C/NF, and 0.35 for FeCoNi@C). The inertness of NF toward HER catalysis is again confirmed with its low TOF of 0.10 s−1. The positive synergy between Fe, Co, Ni, and Mo toward HER catalysis is confirmed, exhibiting the merit of high entropy alloying for catalyst developments.
Among the three trimetallic alloy-based samples, CoNiMo@C and FeNiMo@C, both containing Ni and Mo, possess significantly higher activities than FeCoNi@C, implying the importance of the NiMo alloy toward HER catalysis. It has been well-established, both theoretically and experimentally, that the integration of Mo and Ni in the form of an alloy shows high HER activities, by increasing the rate of water dissociation, decreasing the energy gap of the electrochemical adsorption step, and improving the hydrogen desorption step toward HER catalysis.45 Furthermore, theoretical studies on CoNiMo-based alloys for the HER suggested that incorporation of Co further increases electron-deficient sites and modifies the bond nature, which effectively accelerates the HER process.46 On the other hand, it was reported that the introduction of Fe into NiMo alloys could improve the HER performance by balancing the bond strength of metal–H's by combining stronger Mo–H bonds with weaker Fe–H and Ni–H bonds, subsequently optimizing the free energy for hydrogen adsorption.47 Hence, it is clear that FeCoNiMo@C, having both Fe and Co incorporated in NiMo, exhibited improved HER performances over only Co-incorporated, CoNiMo@C, and only Fe-incorporated, FeNiMo@C, catalysts. And, it is also clear that FeCoNi@C, containing no Mo and thus lacking the strong synergistic effects between Ni and Mo, showed the lowest HER activities among the four alloy-based samples. As for CoNiMo@C and FeNiMo@C, both having the NiMo synergy, CoNiMo@C showed superior HER performances to FeNiMo@C, which may be attributed to the fact that Co is a slightly more effective species than Fe in promoting the formation of coordinated unsaturated sites (CUS) for enhanced hydrogen adsorption.48
Finally, the HER stability of FeCoNiMo@C/NF was tested at −500 mA cm−2 over 50 h in 1 M KOH in a chronopotentiometric mode, and the resulting V–t curve is presented in Fig. 5e. Evidently, the HER stability of FeCoNiMo@C/NF is outstanding, experiencing only a 1.6% increase in applied potentials, even under these harsh operation conditions. The LSV polarization curves recorded before and after the HER V–t test (Fig. 5f) are in good agreement with each other, further indicating the excellent mechanical and electrochemical robustness of FeCoNiMo@C/NF.
3.3. Electrocatalytic OER performances
The catalytic efficiency toward the OER for the four multi-metallic alloy catalysts, together with the blank NF and the OER benchmark catalyst IrO2, was studied in 1 M KOH. The resulting LSV curves and corresponding overpotentials are presented in Fig. 6a and Table 3, respectively. As evident from Table 3, FeCoNiMo@C/NF displays remarkable electrocatalytic OER efficiency with an η10 of only 204 mV, which is significantly lower than those of all tested electrodes, including FeCoNi@C/NF (η10 = 218 mV), FeNiMo@C/NF (η10 = 238 mV), IrO2/NF (η10 = 291 mV), CoNiMo@C/NF (η10 = 303 mV), and blank NF (η10 = 337 mV). Interestingly, the η10 of the present pseudo-HEA catalyst, FeCoNiMo@C, is lower than that of many of the recently reported HEA based OER catalysts in alkaline media, including Al–Ni–Co–Ru–Mo nanowires (η10 = 270 mV),49 HEA-NPs (η10 = 390 mV),50 CoNiCuMnAl@C (η10 = 215 mV),51 and HEAN@NPC/CC-450 (η10 = 263 mV).52 As for high current density overpotentials, FeCoNiMo@C achieves an ultralow η500 of 271 mV, lower than those of FeCoNi@C (η500 = 290 mV), FeNiMo@C (η500 = 325 mV), IrO2 (η500 = 424 mV), and CoNiMo@C (η500 = 447 mV). Evidently, the strong synergy between Fe, Co, Ni, and Mo in FeCoNiMo@C works well, not only for the HER, but also for the OER, making FeCoNiMo@C an outstanding bifunctional catalyst toward water electrolysis.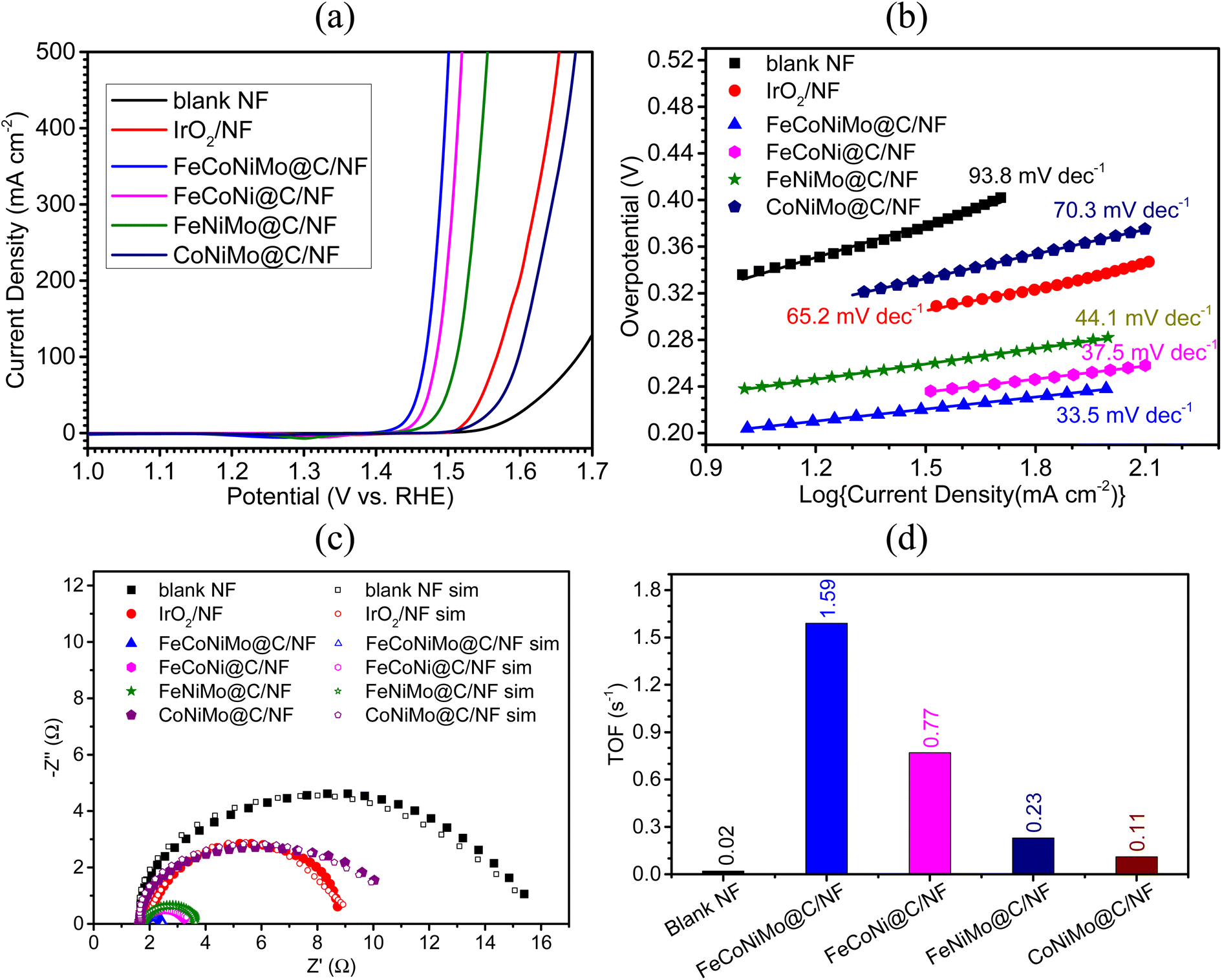 | ||
| Fig. 6 (a) LSV curves recorded in 1 M KOH at scan rate of 1 mV s−1, and corresponding (b) Tafel plots and (c) Nyquist plots of sample electrodes. (d) O2 turnover frequencies (TOFs) of sample catalysts calculated at 1.50 V (vs. RHE). | ||
| Sample | Overpotential (η), mV | |
|---|---|---|
| η 10 | η 500 | |
| Blank NF | 337 | — |
| IrO2/NF | 291 | 424 |
| FeCoNiMo@C/NF | 204 | 271 |
| FeCoNi@C/NF | 218 | 290 |
| FeNiMo@C/NF | 238 | 325 |
| CoNiMo@C/NF | 303 | 447 |
Furthermore, the OER kinetics of all the tested electrodes were examined with Tafel slopes obtained from the corresponding LSV polarization curves. As indicated in Fig. 6b, FeCoNiMo@C/NF exhibits the smallest Tafel slope of 33.5 mV dec−1, which is lower than those of FeCoNi@C/NF (37.5 mV dec−1), FeNiMo@C/NF (44.1 mV dec−1), IrO2/NF (65.2 mV dec−1), CoNiMo@C/NF (70.3 mV dec−1), and blank NF (93.8 mV dec−1). This suggests that FeCoNiMo@C/NF possesses the fastest OER kinetics, the same for the HER process discussed in a previous section.
To further investigate the OER kinetics of the tested electrodes, the EIS experiment was carried out in the frequency range of 100 kHz to 0.1 Hz in 1 M KOH, with the sample electrodes serving as the working electrode operated at 1.57 V (vs. RHE), at which all working electrodes underwent the OER. The obtained EIS data (Fig. 6c) were carefully fitted with a suitable circuit model (Fig. S5, ESI†) to find out the Rct values as in the HER case. Accordingly, the smallest Rct value of 0.71 Ω was found for FeCoNiMo@C/NF, which is relatively lower than that of FeCoNi@C/NF (1.12 Ω) and FeNiMo@C/NF (1.54 Ω) electrodes, and greatly smaller than that of benchmark IrO2/NF (5.64 Ω), CoNiMo@C/NF (8.01 Ω), and blank NF (12.25 Ω), matching well with that of the OER performance order observed in the case of overpotential values (η10 and η500) and Tafel slope values.
To examine the intrinsic activities of the present catalysts toward the OER, TOFs were determined at 1.50 V (vs. RHE) and the corresponding results are displayed in Fig. 5d. As expected, FeCoNiMo@C exhibits the highest TOF value of 1.59 s−1, with FeCoNi@C coming next at 0.77 s−1, followed by FeNiMo@C (0.23 s−1) and CoNiMo@C (0.11 s−1), and the blank NF (0.02 s−1) giving the lowest value as expected. The trend observed in TOF is in excellent agreement with those of the LSV and EIS results.
In order to explore the OER mechanism, in situ Raman spectra for the four multi-metallic alloy catalysts, FeCoNiMo@C (Fig. 7a), FeCoNi@C (Fig. S6, ESI†), FeNiMo@C (Fig. S7, ESI†), and CoNiMo@C (Fig. S8, ESI†), during the OER process in 1 M KOH solution at increasing applied potentials (vs. RHE) were recorded and compared. Evidently, only two broad scattering peaks appear at the open circuit potential (OCP) in the Raman shift range of 1250–1700 cm−1, characteristic of the D-band and G-band of carbon. Interestingly, the intensity ratio of the D band to G band (ID/IG) appears to increase with increasing applied potentials, e.g., 0.80 at 1.20 V to 1 at 1.40 V for FeCoNiMo@C/NF, indicative of the increasing extent of carbon oxidation for generation of the disordered carbon structure during intensifying OER. On the other hand, when the applied potential reaches a critical value, 1.30 V for FeCoNiMo@C/NF, 1.33 V for FeCoNi@C/NF, 1.37 V for FeNiMo@C/NF, and 1.42 V for CoNiMo@C/NF, two new scattering peaks emerge at ∼480 cm−1 and ∼560 cm−1, indicating formation of OER-active species for OER catalysis. It is quite fascinating to note that the onset overpotential required for the peak emergence is the lowest for FeCoNiMo@C/NF, followed by FeCoNi@C/NF, FeCoMo@C/NF, and CoNiMo@C/NF, and the order is consistent with those of η, Tafel slope, Rct, and TOF data. The two new broad Raman peaks, centering at ∼480 cm−1 and ∼560 cm−1, indicate the formation of MOOH (M = Fe, Co, Ni), the well-known active species for OER catalysis in alkaline media.53–57 Nevertheless, considering the single-phase (FCC) of the pristine catalyst, it is possible that the in situ derived MOOH exists in a state of atomic scale mixing of FeOOH, CoOOH, and NiOOH. This clearly suggests that the presence of Fe, Co, and Ni in FeCoNiMo@C/NF accelerates the formation of the active MOOH species and thus gives higher catalytic efficiency toward the OER. Furthermore, theoretical studies clearly suggest that the presence of Mo decreases the OH* adsorption ability of the OER active sites (Fe, Co, Ni) to a certain extent, which facilitates the deprotonation process and greatly improves the OER activity of the catalyst.39 In addition, it has been well-known that the integration of Fe and Ni as electrocatalysts for the OER greatly improves the electrocatalytic performances, because Fe can increase the Ni–O bond length within the oxyhydroxides, which accelerates the decomposition of *OOH intermediates into oxygen molecules.58 In this regard, the lowest OER activity of CoNiMo@C may be attributed to the absence of the FeNi synergy. On the other hand, the OER activity of FeCoNi@C is higher than that of FeNiMo@C, which may be attributed to the favorable formation of Co-based active intermediates (CoOOH) along with Fe and Ni-based oxyhydroxides for FeCoNi@C, whereas the absence of Co-based active intermediates is disadvantageous to the OER activity of FeNiMo@C. Furthermore, the finding obtained from XPS characterizations, upshifts in binding energies of Fe, Co, and Ni upon introduction of Mo favorable for OER catalysis, supports the finding obtained from in situ Raman characterization, formation of Fe, Co, and Ni-based active species, MOOH, during the OER.
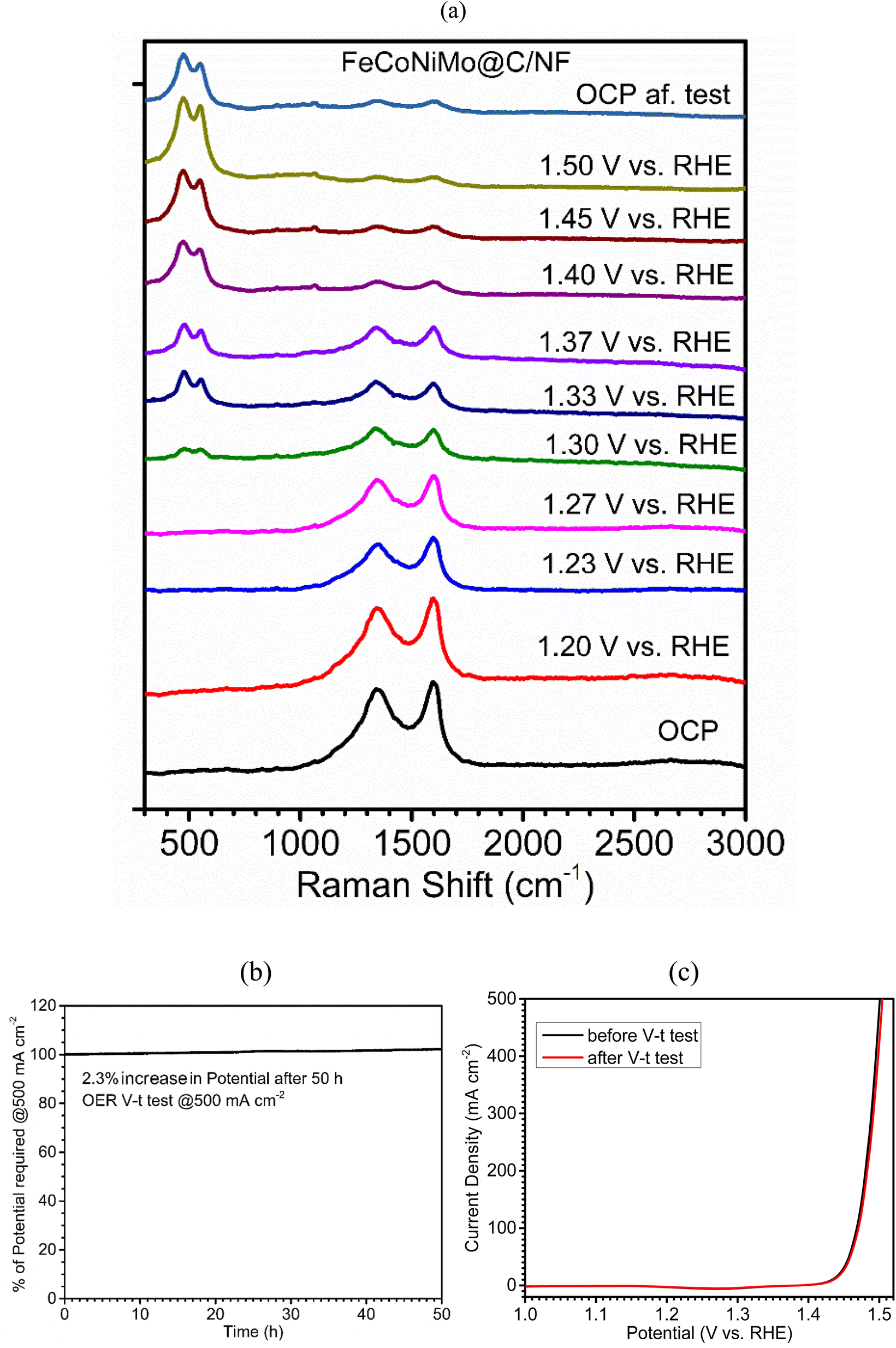 | ||
| Fig. 7 (a) In situ Raman spectra of FeCoNiMo@C/NF at an excitation wavelength of 532 nm and under OER conditions at increasing applied potentials (vs. RHE). (b) Chronopotentiometric (V–t) curve of FeCoNiMo@C/NF for the OER at 500 mA cm−2 for 50 h. (c) LSV curves of FeCoNiMo@C/NF before and after the V–t stability test for the OER at 500 mA cm−2 for 50 h. | ||
Apart from remarkable electrocatalytic efficiency, the long-term stability of the electrocatalyst at high current densities is also a key character for its commercial applications. The OER stability of FeCoNiMo@C/NF was examined under the severe conditions of a high current density operation at 500 mA cm−2 in V–t mode over 50 h. FeCoNiMo@C/NF experienced only a 2.3% increase in applied potentials at the end of the test (Fig. 7b). The excellent OER stability of the FeCoNiMo@C/NF electrode has further been confirmed with the LSV curve recorded after the long-term durability test. It is to be noted that the LSV curve obtained after the OER durability test (Fig. 7c) matched well with its original LSV curve, confirming the excellent mechanical and electrochemical robustness of FeCoNiMo@C/NF.
3.4. Overall water splitting performances
Owing to its outstanding OER and HER performances, the pseudo-HEA-based catalyst, FeCoNiMo@C, was adopted as both the cathode and anode catalysts to fabricate an alkaline water electrolyzer in 1 M KOH. The blank NF//blank NF and Pt–C/NF//IrO2/NF couples were also tested for comparison. An ultra-low cell voltage of 1.488 V was achieved by the FeCoNiMo@C/NF//FeCoNiMo@C/NF couple to deliver a current density of 10 mA cm−2, which is significantly lower than those of the Pt–C/NF//IrO2/NF (1.544 V) and blank NF//blank NF (1.811 V) couples (Fig. 8a). Furthermore, the cell voltage needed to attain the industrially relevant large current density of 500 mA cm−2 is only 1.725 V. In addition, the long-term stability of the FeCoNiMo@C/NF//FeCoNiMo@C/NF couple was tested at a high current density of 500 mA cm−2 in a V–t mode for 50 h (Fig. 8b). The long-term durability of the couple is outstanding, giving a small increase of 3.4% in cell voltages at the end of the test. Moreover, the LSV curve recorded after the V–t test clearly shows only a mild shift to the right when compared to the original one (Fig. 8c), suggesting excellent mechanical and electrochemical robustness of the couple. The stability of the catalyst has further been confirmed by invariant morphologies observed in SEM images (Fig. S9, ESI†) before and after the tests. In addition, TEM images of FeCoNiMo@C scratched off from FeCoNiMo@C/NF as the anode (Fig. S10, ESI†) and cathode (Fig. S11, ESI†) after the V–t test clearly showed the spherical carbon coated pseudo-HEA particles and their FCC (111) crystal lattice fringes, similar to those of original FeCoNiMo@C (Fig. 3a–c). The TEM-EDX elemental mapping of FeCoNiMo@C after the stability test (Fig. S12 for anode and Fig. S13 for cathode, ESI†) clearly suggests the atomically well-dispersed Fe, Co, Ni, Mo, and C elements in the samples, indicating their excellent stability in composition. Next, TEM-EDX elemental analyses (Fig. S14 for the anode and Fig. S15 for the cathode, ESI†) and ICP-OES analyses (Table S2, ESI†) were conducted for the samples after the V–t stability test and the corresponding results clearly supported the composition stability of the samples, with the at% of Fe, Co, Ni, and Mo matching well with those of the original FeCoNiMo@C. Furthermore, the ICP-OES analyses of the electrolyte collected after the stability test indicated that only a small amount (∼1.38%) of catalyst was dissolved during the stability test of 50 h (Table S2, ESI†). The excellent robustness of the catalyst may be partly attributed to the presence of the carbon armor. The carbon layer in FeCoNiMo@C protects and separates the active multi-metallic sites from direct attack of the corrosive alkaline environment, the main advantage of a chainmail catalyst design.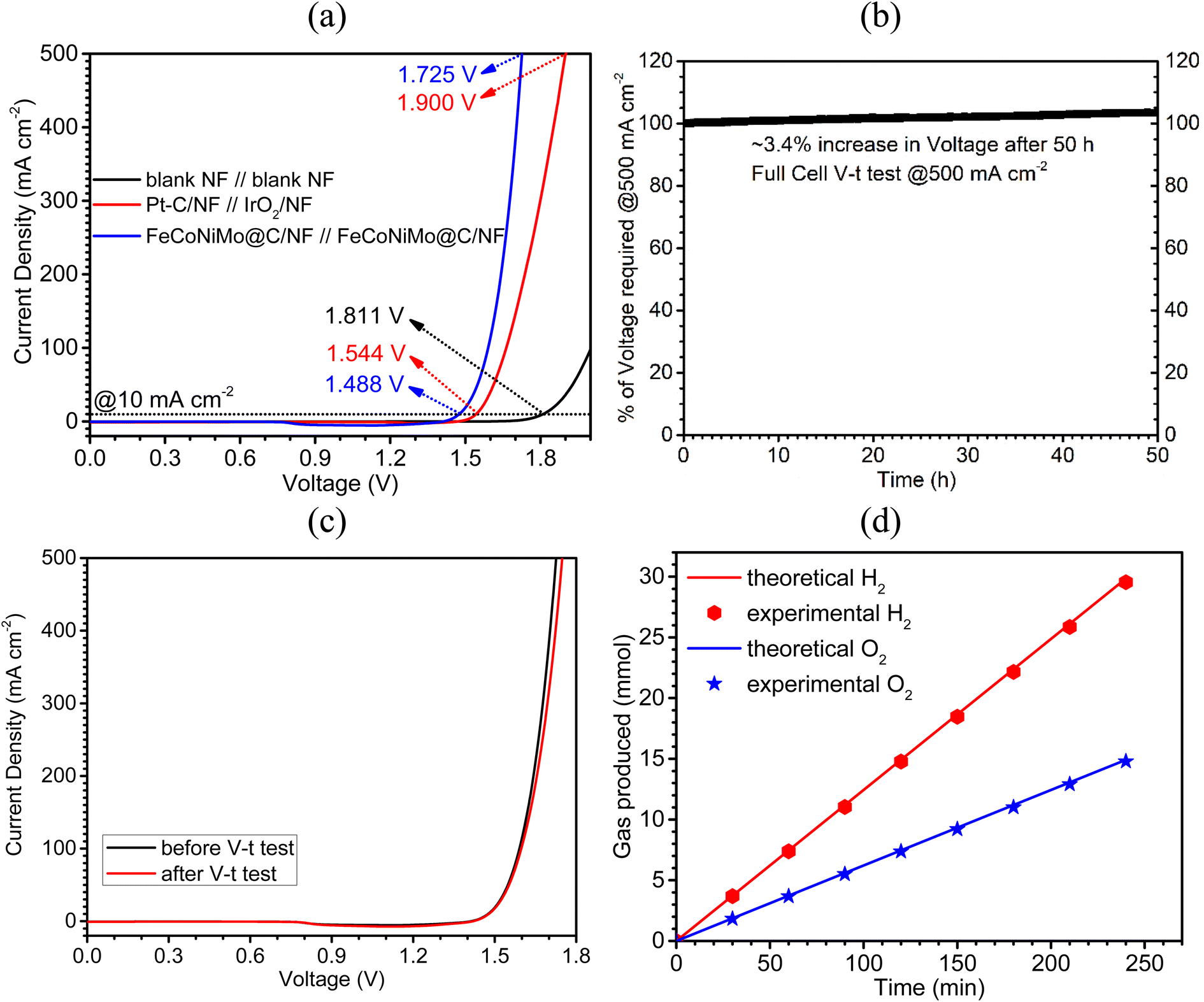 | ||
| Fig. 8 Overall water splitting performances of FeCoNiMo@C/NF//FeCoNiMo@C/NF couple: (a) LSV curve, (b) normalized chronopotentiometric (V–t) curve at 500 mA cm−2 for 50 h, and (c) LSV curves before and after the V–t test. (d) Experimental and theoretical amounts of hydrogen and oxygen produced by the FeCoNiMo@C/NF//FeCoNiMo@C/NF couple during overall water splitting operation at 200 mA cm−2 in 1 M KOH. | ||
Next, the Faradaic efficiency of the present FeCoNiMo@C/NF//FeCoNiMo@C/NF couple was determined to check if side reactions occur to contribute to the currents recorded in LSV curves. The hydrogen and oxygen produced during the electrolytic water splitting process at 200 mA cm−2, were quantified and compared with the values obtained theoretically. The corresponding results (Fig. 8d) show excellent agreements between theoretical and experimental values of hydrogen and oxygen production, confirming near 100% Faradaic efficiency of the present FeCoNiMo@C/NF//FeCoNiMo@C/NF couple and absence of side reactions.
Finally, we compare the HER, OER, and overall water splitting performances of the present carbon armored pseudo-HEA catalyst, FeCoNiMo@C, with recently reported top performing monofunctional (HER or OER) and bifunctional (HER, OER, and full water splitting) MOF-derived non-noble metal based electrocatalysts (in alkaline media) in terms of η10, Tafel slope, and long-term durability in Table S3 (ESI)† (monofunctional HER electrocatalysts), Table S4 (ESI)† (monofunctional OER electrocatalysts), and Table S5 (ESI)† (bifunctional electrocatalysts for overall water splitting). Evidently, on comparing η10 and Tafel slopes, FeCoNiMo@C shows superior/comparable performances to the best MOF-derived noble-metal-free electrocatalysts. Except for very few studies, the stability of all these reported MOF-derived non-noble metal-based electrocatalysts was examined only under lower current density (≤100 mA cm−2) conditions, which is insufficient to assess their potential industrial applicability. In that respect, FeCoNiMo@C/NF, with excellent stability at high current densities, stands out among all recently reported top-tier MOF-derived non-noble metal-based monofunctional (HER or OER) and bifunctional electrocatalysts.
4. Conclusion
A new electrocatalyst design, pseudo-high entropy alloys, was successfully demonstrated for high performance water electrolysis. Pseudo-HEAs, single-phase four-element solid-solution alloys of molar configurational entropy higher than 1.36R, possess the advantages of less complications in acquiring positive constituent synergy and formation of single-phase alloys, as compared with traditional HEAs of five or more elements, and still benefit from atomic scale constituent synergy for enhanced electrocatalytic performances. A pseudo-HEA catalyst, carbon armored FeCoNiMo nanoparticles, FeCoNiMo@C, was successfully developed with a simple two-stage thermal conversion of a quad-metallic MOF, to serve as a highly efficient and stable non-precious metal-based bifunctional electrocatalyst for water electrolysis. The pseudo-HEA catalyst achieved ultralow overpotentials of 204 and 55 mV at 10 mA cm−2 for the OER and HER, respectively, ultralow cell voltages of 1.488 and 1.725 V to reach current densities of 10 and 500 mA cm−2, respectively for overall water splitting, and ultrastability of a minor decay of 3.4% after operations at 500 mA cm−2 for 50 h in alkaline media. The new approach of pseudo-HEA adds a new branch for the development of multi-metallic alloys toward applications in electrocatalysis.Author contributions
Duraisamy Senthil Raja: conceptualization, data curation, formal analysis, investigation, methodology, validation, writing – original draft, and writing – review & editing. Yu-Chieh Ting: data curation, formal analysis, and investigation. Ting-Yu Lin: data curation, formal analysis, and investigation. Chih-Chieh Cheng: data curation and formal analysis. Po-Wei Chen: data curation, and formal analysis. Fan-Yu Yen: data curation, and formal analysis. Shih-Yuan Lu: conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts of interest.Acknowledgements
Financial support received from the National Science and Technology Council of Taiwan under grants MOST 108-2221-E-007-073-MY3 and MOST 110-2811-E-007-515 is gratefully acknowledged. The authors also sincerely acknowledge the use of spherical-aberration corrected filed emission TEM (JEM-ARM200FTH, JEOL Ltd, NSTC 112-2740-M-007-001) and HR-XPS (PHI QuanteraII, ULVAC-PHI Inc.) facilities belonging to the Instrument Center of National Tsing Hua University of Taiwan.References
- R. Eisenberg, Science, 2009, 324, 44–45 CrossRef CAS PubMed.
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- Y.-N. Zhou, F.-L. Wang, S.-Y. Dou, Z.-N. Shi, B. Dong, W.-L. Yu, H.-Y. Zhao, F.-G. Wang, J.-F. Yu and Y.-M. Chai, Chem. Eng. J., 2022, 427, 131643 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- A. Aijaz, J. Masa, C. Rösler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087–4091 CrossRef CAS PubMed.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W.-C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS PubMed.
- B. Weng, C. R. Grice, W. Meng, L. Guan, F. Xu, Y. Yu, C. Wang, D. Zhao and Y. Yan, ACS Energy Lett., 2018, 3, 1434–1442 CrossRef CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- H. Sun, Y. Lian, C. Yang, L. Xiong, P. Qi, Q. Mu, X. Zhao, J. Guo, Z. Deng and Y. Peng, Energy Environ. Sci., 2018, 11, 2363–2371 RSC.
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 8921–8926 CrossRef CAS PubMed.
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2858–2864 RSC.
- H.-J. Liu, R.-N. Luan, L.-Y. Li, R.-Q. Lv, Y.-M. Chai and B. Dong, Chem. Eng. J., 2023, 461, 141714 CrossRef CAS.
- P. Koželj, S. Vrtnik, A. Jelen, S. Jazbec, Z. Jagličić, S. Maiti, M. Feuerbacher, W. Steurer and J. Dolinšek, Phys. Rev. Lett., 2014, 113, 107001 CrossRef PubMed.
- B. Gludovatz, A. Hohenwarter, D. Catoor, E. H. Chang, E. P. George and R. O. Ritchie, Science, 2014, 345, 1153–1158 CrossRef CAS PubMed.
- B. Schuh, F. Mendez-Martin, B. Völker, E. P. George, H. Clemens, R. Pippan and A. Hohenwarter, Acta Mater., 2015, 96, 258–268 CrossRef CAS.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- Y. Zhang, D. Wang and S. Wang, Small, 2022, 18, 2104339 CrossRef CAS PubMed.
- D. Zhang, H. Zhao, X. Wu, Y. Deng, Z. Wang, Y. Han, H. Li, Y. Shi, X. Chen, S. Li, J. Lai, B. Huang and L. Wang, Adv. Funct. Mater., 2021, 31, 2006939 CrossRef CAS.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- T. Löffler, H. Meyer, A. Savan, P. Wilde, A. Garzón Manjón, Y.-T. Chen, E. Ventosa, C. Scheu, A. Ludwig and W. Schuhmann, Adv. Energy Mater., 2018, 8, 1802269 CrossRef.
- H. Li, Y. Han, H. Zhao, W. Qi, D. Zhang, Y. Yu, W. Cai, S. Li, J. Lai, B. Huang and L. Wang, Nat. Commun., 2020, 11, 5437 CrossRef CAS PubMed.
- N. K. Katiyar, S. Nellaiappan, R. Kumar, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, Mater. Today Energy, 2020, 16, 100393 CrossRef.
- Z. Y. Lv, X. J. Liu, B. Jia, H. Wang, Y. Wu and Z. P. Lu, Sci. Rep., 2016, 6, 34213 CrossRef CAS PubMed.
- J. W. Yeh, S. K. Chen, S. J. Lin, J. Y. Gan, T. S. Chin, T. T. Shun, C. H. Tsau and S. Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- Y. Xin, S. Li, Y. Qian, W. Zhu, H. Yuan, P. Jiang, R. Guo and L. Wang, ACS Catal., 2020, 10, 11280–11306 CrossRef CAS.
- C.-L. Huang, Y.-G. Lin, C.-L. Chiang, C.-K. Peng, D. Senthil Raja, C.-T. Hsieh, Y.-A. Chen, S.-Q. Chang, Y.-X. Yeh and S.-Y. Lu, Appl. Catal., B, 2023, 320, 122016 CrossRef CAS.
- X. Gu, Z. Liu, M. Li, J. Tian and L. Feng, Appl. Catal., B, 2021, 297, 120462 CrossRef CAS.
- J.-H. Park, J. C. Ro and S.-J. Suh, Appl. Surf. Sci., 2022, 589, 153041 CrossRef CAS.
- D. Senthil Raja, C.-L. Huang, Y.-A. Chen, Y. Choi and S.-Y. Lu, Appl. Catal., B, 2020, 279, 119375 CrossRef CAS.
- D. Senthil Raja, C.-C. Cheng, Y.-C. Ting and S.-Y. Lu, ACS Appl. Mater. Interfaces, 2023, 15, 20130–20140 CrossRef CAS PubMed.
- M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. Head-Gordon, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 12119–12129 CrossRef CAS PubMed.
- S. Chen, H. Jang, J. Wang, Q. Qin, X. Liu and J. Cho, J. Mater. Chem. A, 2020, 8, 2099–2104 RSC.
- S. Guo and C. T. Liu, Prog. Nat. Sci.: Mater. Int., 2011, 21, 433–446 CrossRef.
- C.-H. Tsau, M.-C. Tsai and W.-L. Wang, Materials, 2022, 15, 888 CrossRef CAS PubMed.
- S. Zhao, J. Huang, Y. Liu, J. Shen, H. Wang, X. Yang, Y. Zhu and C. Li, J. Mater. Chem. A, 2017, 5, 4207–4214 RSC.
- Y. Mei, Y. Feng, C. Zhang, Y. Zhang, Q. Qi and J. Hu, ACS Catal., 2022, 12, 10808–10817 CrossRef CAS.
- W. Luo, Y. Wang, L. Luo, S. Gong, Y. Li and X. Gan, Appl. Surf. Sci., 2022, 606, 154808 CrossRef CAS.
- Z.-Y. Yu, C.-C. Lang, M.-R. Gao, Y. Chen, Q.-Q. Fu, Y. Duan and S.-H. Yu, Energy Environ. Sci., 2018, 11, 1890–1897 RSC.
- Y. Wang, Y. Wang, J. Bai and W.-M. Lau, ChemistrySelect, 2022, 7, e202200468 CrossRef CAS.
- I. K. Mishra, H. Zhou, J. Sun, F. Qin, K. Dahal, J. Bao, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2246–2252 RSC.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- K. Hu, M. Wu, S. Hinokuma, T. Ohto, M. Wakisaka, J.-i. Fujita and Y. Ito, J. Mater. Chem. A, 2019, 7, 2156–2164 RSC.
- F. Qin, Z. Zhao, M. K. Alam, Y. Ni, F. Robles-Hernandez, L. Yu, S. Chen, Z. Ren, Z. Wang and J. Bao, ACS Energy Lett., 2018, 3, 546–554 CrossRef CAS.
- W. Han, S. Wang, X. Li, B. Ma, M. Du, L. Zhou, Y. Yang, Y. Zhang and H. Ge, RSC Adv., 2020, 10, 8055–8065 RSC.
- Z. Jin, J. Lyu, Y.-L. Zhao, H. Li, X. Lin, G. Xie, X. Liu, J.-J. Kai and H.-J. Qiu, ACS Mater. Lett., 2020, 2, 1698–1706 CrossRef CAS.
- H. Li, H. Zhu, Q. Shen, S. Huang, S. Lu, P. Ma, W. Dong and M. Du, Chem. Commun., 2021, 57, 2637–2640 RSC.
- S. Wang, W. Huo, F. Fang, Z. Xie, J. K. Shang and J. Jiang, Chem. Eng. J., 2022, 429, 132410 CrossRef CAS.
- K. Huang, B. Zhang, J. Wu, T. Zhang, D. Peng, X. Cao, Z. Zhang, Z. Li and Y. Huang, J. Mater. Chem. A, 2020, 8, 11938–11947 RSC.
- Z. Qiu, C.-W. Tai, G. A. Niklasson and T. Edvinsson, Energy Environ. Sci., 2019, 12, 572–581 RSC.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. García de Arquer, M. Liu, C. T. Dinh, T. Regier, J. J. Dynes, S. He, H. L. Xin, H. Peng, D. Prendergast, X. Du and E. H. Sargent, Nat. Chem., 2018, 10, 149–154 CrossRef CAS PubMed.
- J. Chen, F. Zheng, S.-J. Zhang, A. Fisher, Y. Zhou, Z. Wang, Y. Li, B.-B. Xu, J.-T. Li and S.-G. Sun, ACS Catal., 2018, 8, 11342–11351 CrossRef CAS.
- X. Zhang, C. Feng, B. Dong, C. Liu and Y. Chai, Adv. Mater., 2023, 35, 2207066 CrossRef CAS PubMed.
- G. Zhang, J. Zeng, J. Yin, C. Zuo, P. Wen, H. Chen and Y. Qiu, Appl. Catal., B, 2021, 286, 119902 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, supporting experimental results, figures, and performance comparison tables. See DOI: https://doi.org/10.1039/d3ta04670b |
| This journal is © The Royal Society of Chemistry 2023 |