
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging disorder in Gd2(Ti1−xZrx)2O7 pyrochlores matrices for radioactive waste disposal: symmetry lowering versus defect clustering†
Armando
di Biase
 *a,
Carlo
Castellano
*a,
Giorgia
Confalonieri
b,
Patrizia
Fumagalli
c,
Simone
Tumiati
c,
Davide
Ceresoli
d and
Marco
Scavini
*ae
*a,
Carlo
Castellano
*a,
Giorgia
Confalonieri
b,
Patrizia
Fumagalli
c,
Simone
Tumiati
c,
Davide
Ceresoli
d and
Marco
Scavini
*ae
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20131 Milano, Italy. E-mail: armando.dibiase@unimi.it; carlo.castellano@unimi.it; marco.scavini@unimi.it
bESRF – European Synchrotron Research Facility, 71 Avenue des Martyrs, 38043 Grenoble, France
cDipartimento di Scienze della Terra "A. Desio", Università degli Studi di Milano, Via Botticelli 23, 20133 Milano, Italy
dIstituto di Scienze e Tecnologie Chimiche “Giulio Natta” (SCITEC), Consiglio Nazionale delle Ricerche (CNR), Via Golgi 19, Milano 20133, Italy
eConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via Giusti 9, 50121, Firenze, Italy
First published on 26th October 2023
Abstract
Pyrochlore compositions in the Gd2(Ti1−xZrx)2O7 solid solution have gained attention in the field of radioactive waste forms because they are capable of withstanding high doses of ion irradiation without becoming amorphous, as the Zr-content increases. The purpose of this study was to explore the structure of Gd2(Ti1−xZrx)2O7 compounds at various length scales using Synchrotron High-Resolution X-ray Powder Diffraction (HR-XRPD), Pair Distribution Function (PDF) analysis, and Raman spectroscopy. Through Rietveld analysis of HR-XRPD patterns, it was determined that substituting Ti with Zr in the Gd2Ti2O7 compound results in the gradual formation of Anti-Frenkel (AFr) Oxygen defects and, for xZr ≥ 0.75, in slight cation A/B site disordering, while still maintaining the same average pyrochlore structure. Raman spectroscopy shows a marked change in the spectra for xZr ≥ 0.50, with a general broadening of the bands and the emergence of new spectral features, indicating an increase in static disorder and some symmetry breaks at shorter length scales. This outcome was confirmed by PDF modelling in the low r region. Two alternative models were proposed to map the disorder: (i) a Pmma weberite-type structure, which suitable fits the PDFs in the Zr-rich part of the phase diagram only up to r ≈ 8 Å; (ii) a disordered pyrochlore structure containing Anti-Frenkel (AFr) pairs in the form of extended clusters as generated by DFT calculations. The short r range of applicability of the weberite-type model and the computed energy values tip the balance in favor of the AFr clusters.
1. Introduction
Materials with general formula A2B2O7 (A = trivalent rare earth metal, B = tetravalent transition metal) and pyrochlore structure exhibit several interesting properties ranging from high ionic conductivity1 and frustrated magnetic order2 to low thermal conductivity3 and good catalytic activity.4 Moreover, their great chemical durability and radiation tolerance have made them promising candidates for radionuclides disposal in the context of SYNROC (synthetic rock).5–8The irradiation with heavy ions was employed in order to simulate the effect of alpha decay on the crystalline host matrix. In this regard, Gd2(Ti1−xZrx)2O7 pyrochlores exhibit increasing radiation resistance with increasing Zr content. While Gd2Ti2O7 is promptly amorphized under irradiation, Gd2Zr2O7, can effectively dissipate the radiation energy by undergoing a phase transition from the pyrochlore to the defect fluorite structure type, preventing amorphization.9–12 This transformation can be regarded as an order–disorder transition, which occurs through the accumulation of defects within the structure. Nominally, cations are randomized over their crystallographic sites forming cation antisite defects, while anions exchange places with vacancies introducing anion Anti Frenkel (AFr) defects.13,14
In A2B2O7 pyrochlores with complex composition, the chemical substitution alone can drive the structural order–disorder phase transition.15–18 The ratio of the A and B ionic radii rA/rB has been extensively used to predict and explain the formation of different structural type as a function of ionic substitutions.19,20 At atmospheric pressure, a pyrochlore phase is stable if the rA/rB is in the range 1.46–1.78,10,21 whereas an anion deficient fluorite (A0.5B0.5)O1.75 phase appears if rA/rB < 1.46. For rA/rB values exceeding 1.78, a monoclinic perovskite layered structure is favored.22,23 Another fluorite-related structure, known as C-type, is also possible, e.g. in the case of B = Ce, and A = Y,24 Sm,25 Gd,26 Yb.27
The gadolinium titanate Gd2Ti2O7 is inside the pyrochlore stability range (rA/rB ≈ 1.74), whereas the gadolinium zirconate Gd2Zr2O7 lies precisely on the boundary of the pyrochlore/fluorite stability field (rA/rB ≈ 1.46).6,21 Therefore, the Gd2(Ti1−xZrx)2O7 solid solution is a suitable system for studying the disordering mechanism induced by the cationic substitution at the B-site. The progressive disordering of the pyrochlore structure on Ti/Zr-substitution in the Gd2(Ti1−xZrx)2O7 system has been well addressed by several studies using standard diffraction techniques.10,28–33 Despite pure Gd2Zr2O7 still retains the pyrochlore structure on average, EXAFS and Pair Distribution Function (PDF) analysis detected structural deviation at the short-structural scale.34 In the homologous Ho2(Ti1−xZrx)2O7 series, the average structure changes from pyrochlore (xZr = 0) to defect fluorite (xZr = 1), while the evolution of the local structure seems to be best described by a gradual accumulation of weberite-type domains within the pyrochlore matrix.35,36 In recent years, weberite-type domains have been extensively recognized as building blocks of the local structure of several pyrochlore oxides subjected to disordering processes by mechanical milling,37,38 irradiation39–42 and chemical substitution.35,43–45
Aiming to reveal the nature of emergent disorder in Gd2(Ti1−xZrx)2O7 materials, we present here a multiscale study of the structure of Gd2(Ti1−xZrx)2O7 pyrochlores prepared by solid state reaction using synchrotron X-ray diffraction and Raman spectroscopy. The average structure of these materials is examined by analyzing the diffraction patterns via the Rietveld method. Information about the crystallites size and strain is obtained as well. On the other hand, insight into the local structure is given by Raman spectroscopy, DFT calculations and the analysis of the pair distribution function.
Accurately mapping the defect structure of the investigated compounds should pave the way to the comprehension of the mechanism of energy dissipation during α decay events when Gd2(Ti1−xZrx)2O7 materials are adopted to dispose radionuclides.
2. Experimental section
Gd2(Ti1−xZrx)2O7 samples (x = 0.00, 0.15, 0.25, 0.50, 0.75, 0.85 and 1.0) were prepared by the conventional solid-state synthesis. Gd2O3, ZrO2 and TiO2 were selected as starting materials. They were heated at 110 °C Å under vacuum overnight before use in order to remove moisture traces. After that, they were weighed according to the specific stoichiometric ratio and intimately mixed using a mortar and a pestle in an ethanol slurry. The mixture was dried and then pressed into a 2 cm diameter pellet with a 8 ton operating press. The pellet was placed on an alumina crucible and fired in air at 1500 °C in a vertical tube furnace with MoSi2 heaters for 24 hours with heating/cooling rates of 2 °C min−1. Subsequently, the sintered pellet was reground, pressed and subjected to another annealing cycle. The procedure was repeated until a homogeneous sample was produced (typically, 5 and 6 firing cycles). The synthesis temperature (T) was selected in order to obtain a pyrochlore phase at each composition, for it was known that the Gd2Zr2O7 compound crystallizes as defect fluorite at T values higher than 1500 °C.15,46,47The phase homogeneity of each sample during the synthesis step was monitored by laboratory X-ray Powder Diffraction. Powder patterns were collected at room temperature on a Rigaku MiniFlex600 diffractometer with Cu Kα radiation (5° < 2θ < 90°, step 0.02°, 5°/min).
Micro-Raman spectroscopy was carried out using a Horiba LabRam HR evolution at the Dipartimento di Scienze della Terra “A. Desio” of the Università degli Studi di Milano. The spectrometer is equipped with a Nd-YAG 532 nm/100 mW laser with Ultra Low Frequency (ULF) filters. Scattered light was collected by a 100X objective (NA aperture = 0.9) in backscattering geometry; a diffraction grating with 600 lines per mm and the hole set at 100 μm were used. The spectrum has been detected by a Peltier-cooled Charge Couple Detector. To balance signal to noise 2 accumulations for 30 seconds were collected. Instrument calibration was performed before each round of analysis using the peak at 520.70 cm−1 of a silicon wafer. Raman bands were fitted with Gaussian functions after background subtraction using the Origin software.48
Synchrotron High-resolution X-ray Powder Diffraction (HR-XRPD) patterns were collected on all the Gd2(Ti1−xZrx)2O7 samples in transmission geometry during experiment CH-5900 (ref. 49) at the ID22 beamline of the ESRF.50 The powder samples were loaded into 0.5 mm diameter Kapton® capillaries and mounted parallel to the axis of the diffractometer. Two different setups were used to collect data suitable for either Rietveld or Pair Distribution Function (PDF) analysis at 90 K. In the former case the high-resolution setup of ID22 was adopted, using an X-ray wavelength λ = 0.354176(8) Å and a 9-element detector array in the 0° ≤ 2θ ≤ 48° interval (Qmax = 14.4 Å−1), for a total counting time of 60 min per pattern. Data for PDF analysis were collected at 90 K using λ = 0.17704(3) Å and a 2D CCD detector (PerkinElmer XRD 1611CP3) with 100 × 100 μm2 pixel size. An additional data set was collected on an empty Kapton® capillary for background subtraction. Several frames for a total counting time of 40 mn were averaged for each measurement. Wavelength, sample-detector distance (380.4 mm), and azimuthal integration parameters were calibrated on a LaB6 reference. The detector mask was created with the program FIT2D51 calibration and azimuthal integration were all performed using the program pyFAI.52
The Rietveld analysis was applied to the XRPD patterns utilizing the GSAS software suite of programs53 and its graphical interface EXPGUI.54 The background has been fitted through Chebyshev polynomial. Line profile have been fitted using a modification of TCH pseudo-Voigt function as implemented in GSAS. In the refinements, scale factor, cell constants, positional and isotropic atomic mean square displacement parameters have been varied, as well as background, and peak profile parameters.
In addition, diffraction peaks in the 1 ≤ 2θ ≤ 20° interval were fitted using pseudo-Voigt functions using the WinPlotR program.55 After deconvoluting the instrumental contribution to peak broadening calculated on the diffraction pattern of a Si standard, the refined positions 2θi and Integral breaths βi were plotted as β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos(θ) vs. 4sin(θ) and fitted against the Williamson–Hall equation:56
cos(θ) vs. 4sin(θ) and fitted against the Williamson–Hall equation:56
| βcos(θ) = 4εsin(θ)+λ/DV |
For Real space analysis we made use of the reduced PDF, G(r), which is the product of the sine Fourier transform of the experimental total scattering function, S(Q), defined as:57

Atomistic calculation of Gd2Zr2O7 were carried out using the General Lattice Utility Program (GULP)60,61 with polarizable core–shell model62 interatomic potentials to rapidly sample the energetic landscape of defect complexes. Next, the obtained structures were refined with Density Functional Theory (DFT) calculations using the Quantum Espresso code.63,64 We simulated a conventional cubic supercell with 88 atoms and taking the lattice spacing and the volume fixed to the experimental one at room temperature.
3. Results and discussion
3.1. Structures of pyrochlores and pertinent related phases
The structural models of pyrochlore and weberite-type were employed here to describe the long- and short-range order of the crystalline oxide phases under investigation. These two structural types are closely related and can be considered as subtraction derivatives of the fluorite structure type. In the fluorite structure AO2 (s.g. Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, n° 225, Z = 4) the cation A occupies a special position of Wyckoff letter 4c (0, 0, 0) and is 8-fold coordinated in a cubic environment by oxygens placed in 8c sites (1/4, 1/4, 1/4) (see Fig. 1a). The oxygen atoms, conversely, are 4-fold coordinated in a tetrahedral geometry. The only structural degree of freedom is the lattice parameter aF (≈5 Å).
m, n° 225, Z = 4) the cation A occupies a special position of Wyckoff letter 4c (0, 0, 0) and is 8-fold coordinated in a cubic environment by oxygens placed in 8c sites (1/4, 1/4, 1/4) (see Fig. 1a). The oxygen atoms, conversely, are 4-fold coordinated in a tetrahedral geometry. The only structural degree of freedom is the lattice parameter aF (≈5 Å).
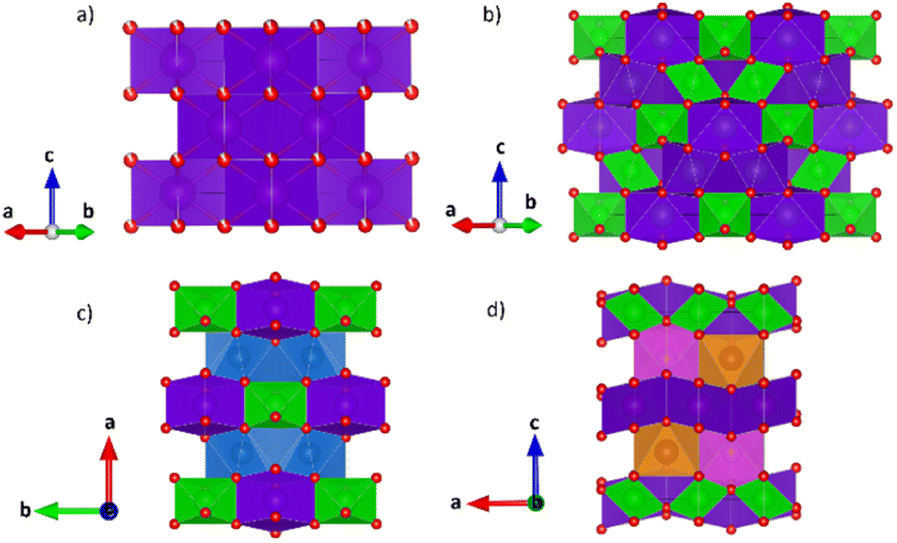 | ||
| Fig. 1 Unit cell of fluorite (a) (s.g. Fmm) and pyrochlore (b) (s.g. Fdm) structure along [110]; unit cell of weberite-type structure in Ccmm space group (c) along [001] and in Pmma space group (d) along [010]. A-site cations 8-fold coordinated are in purple (Wyckoff positions 4a in a, 16d in b, 4b in c and 2b and 2c in d) and B-site cations 6-fold coordinated are in green polyhedra (Wyckoff positions 16c in b, 4a in c and 2a and 2d in d); the 7-fold coordinated site shared by both A- and B-site cations in c (blue polyhedra, Wyckoff position 8g) splits into two separate sites in d (orange and pink polyhedra, Wyckoff position 4k). Oxygen atoms are represented by red spheres. | ||
Expressing the general formula of the fluorite as A4O8−δ (A represents the sum of all metal atoms sites and δ the possible oxygen non stoichiometry) and introducing an anion deficiency δ = 1, one can obtain the general formula of the pyrochlore and weberite-type structures.65
The pyrochlore phase (s.g. Fdm, n° 227, Z = 8, origin at the B site, 16c (0, 0, 0)) belongs to the cubic crystal system but has a doubled cell edge with respect to the fluorite structure (aP = 2aF ≈ 10 Å) and has four crystallographically independent sites.21 The unique cation site of the fluorite is split into two different sites, 16c (0, 0, 0) and 16d (1/2, 1/2, 1/2), occupied by the B and A cation respectively. The anion site of the fluorite, on the other hand, is split into three non-equivalent sites. Those indicated with Wyckoff letters 48f (x, 1/8, 1/8) and 8b (3/8, 3/8, 3/8), hereafter O1 and O2, respectively, host two oxygen atoms, while the third (O3) anion site, 8a (1/8, 1/8, 1/8), is vacant in the ideal pyrochlore structure. In the ESI (ESI), Table ESI1† summarizes the atomic coordinates within the pyrochlore structure. Besides the lattice parameter, the fractional coordinate xO1 is the only structural degree of freedom for the pyrochlore structure. The coordination polyhedra of the two metal ions change with respect to the fluorite structure because of the oxygen deficiency and the variable xO1. In particular, the A cation is 8-fold coordinated and is placed in a scalenohedron, while the B cation is 6-fold coordinated within a trigonal antiprism (Fig. 1b). In contrast, the coordination environment for the oxygen atoms remains fourfold. The 48f oxygen is surrounded by two A and two B neighbors in a A2B2 tetrahedral environment, whereas the 8b oxygen and the 8a vacancy are placed at center of OA4 and OB4 tetrahedra, respectively.
As stated above, the oxygen atoms and the vacancy are ordered over three separate crystallographic sites in the ideal pyrochlore structure. When a phase transition to a defect fluorite structure occurs, the cations randomize as well as the oxygen atoms and the vacancy. In this way, the fractional occupancy of the oxygen site becomes 7/8th in the defect fluorite type, while the A and B cations share the same site with a 50% probability. This can be easily monitored using diffraction techniques. Looking at the transition in reverse, in other words going from a disordered A2B2O7 defect fluorite compound to a perfect pyrochlore phase, one can see that additional reflections appear in the defect fluorite diffraction pattern. In the following discussion, we will refer to them as pyrochlore (superstructure) peaks with respect to the fluorite peaks which appear in both structures. The intensity of the pyrochlore peaks depends on the difference of the scattering power of the A and B cations, the scattering power of oxygen, the distribution of oxygen atoms and vacancies and the fractional coordinate xO1.17,18
As for the pyrochlore, the weberite-type structure is also an anion-deficient fluorite superstructure. It retains the fcc-like packing of the cations and the tetrahedral coordination of the anions. The cell is obtained by a 45° rotation about one of the crystallographic axes of the pyrochlore structure and the resulting lattice parameters are approximately 2aF, aF , aF
, aF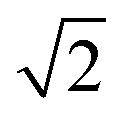 .66,67 Two orthorhombic weberite-type models, Ccmm (a non-standard setting of Cmcm space group, n° 63, Z = 4)36 or C2221 (n° 20, Z = 4)35, have been invoked to interpret the local atomic ordering in the disordered pyrochlore series Ho2(Ti1−xZrx)2O7. The key features of these structural models are an 8-fold coordinated site for the A cation, a 6-fold coordinated site for the B cation and a 7-fold coordinated site that is shared by both cations. Subtle changes in the anion substructure leads to a reduction of symmetry, specifically a splitting of the oxygen ion 16h site into two 8c sites passing from Ccmm to C2221. This, in turn, leads to additional positional degrees of freedom.68 As it will be detailed in Section 3.3.2, in the present work, the same A/B cation ordering of pyrochlore was imposed to the weberite-type models to describe the local structure of Gd2(Ti1−xZrx)2O7 compositions. This further lowers the phase symmetry of Ccmm to Pcmm (a non-standard setting of Pmma space group, n° 51) and of C2221 to P2221 (space group n° 17), causing the splitting of all the sites into two having halved multiplicity, while maintaining the same cell metrics. In particular, the 7-fold coordinated site shared by A and B cations (multiplicity 8) is split into two sets of multiplicity 4 occupied by A and B cations separately (see Fig. 1c and d). The structural models employed in the Reciprocal and Real space analysis will be described in Sections 3.2 and 3.3.2.
.66,67 Two orthorhombic weberite-type models, Ccmm (a non-standard setting of Cmcm space group, n° 63, Z = 4)36 or C2221 (n° 20, Z = 4)35, have been invoked to interpret the local atomic ordering in the disordered pyrochlore series Ho2(Ti1−xZrx)2O7. The key features of these structural models are an 8-fold coordinated site for the A cation, a 6-fold coordinated site for the B cation and a 7-fold coordinated site that is shared by both cations. Subtle changes in the anion substructure leads to a reduction of symmetry, specifically a splitting of the oxygen ion 16h site into two 8c sites passing from Ccmm to C2221. This, in turn, leads to additional positional degrees of freedom.68 As it will be detailed in Section 3.3.2, in the present work, the same A/B cation ordering of pyrochlore was imposed to the weberite-type models to describe the local structure of Gd2(Ti1−xZrx)2O7 compositions. This further lowers the phase symmetry of Ccmm to Pcmm (a non-standard setting of Pmma space group, n° 51) and of C2221 to P2221 (space group n° 17), causing the splitting of all the sites into two having halved multiplicity, while maintaining the same cell metrics. In particular, the 7-fold coordinated site shared by A and B cations (multiplicity 8) is split into two sets of multiplicity 4 occupied by A and B cations separately (see Fig. 1c and d). The structural models employed in the Reciprocal and Real space analysis will be described in Sections 3.2 and 3.3.2.
3.2. Reciprocal space analysis
Panels (a) and (b) of Fig. 2 report as examples the full XRPD patterns collected at 90 K of samples xZr = 0.75 and 0.85 respectively as black crosses, while all the low angle portions of all the patterns are shown in panel (c).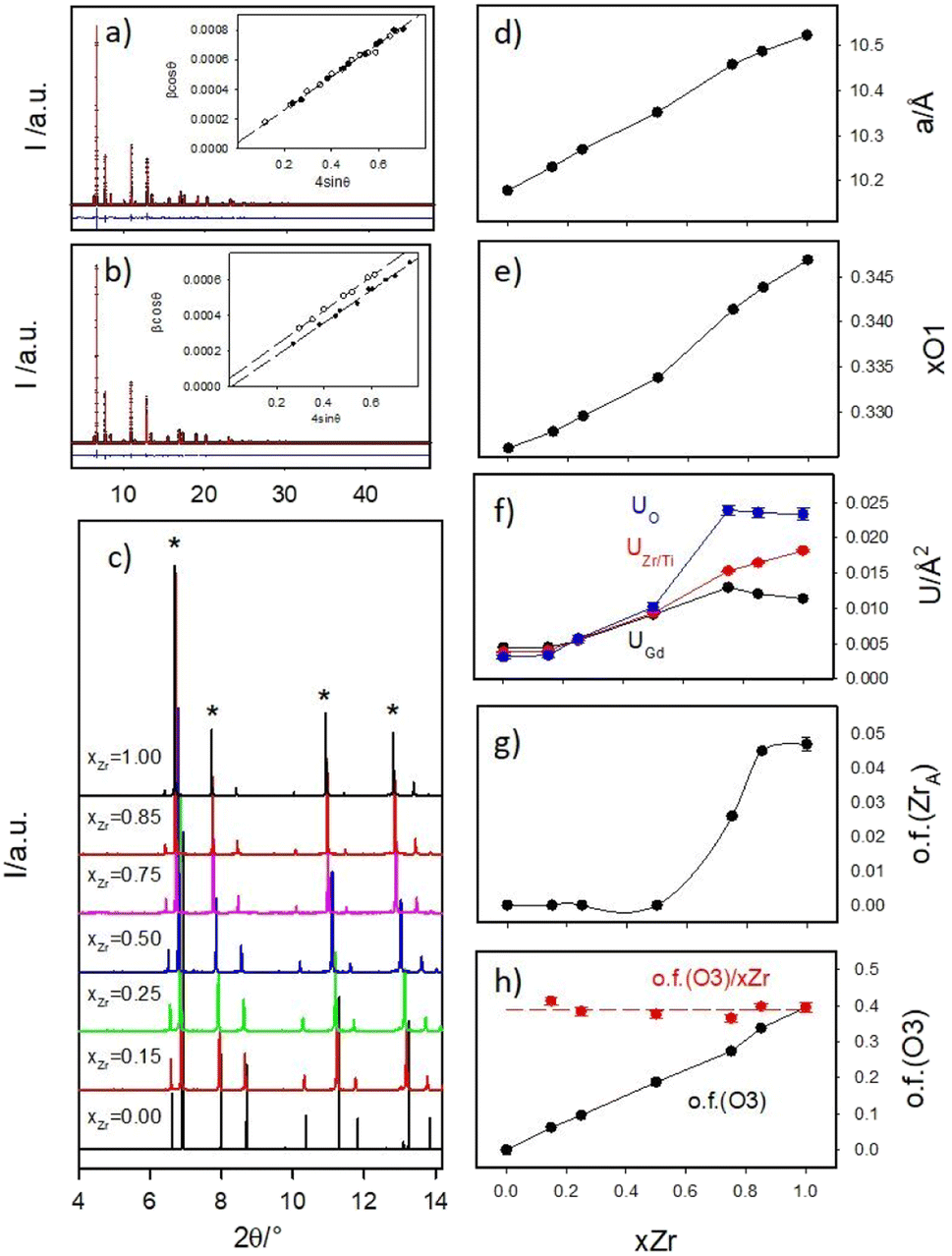 | ||
| Fig. 2 HR-XRPD patterns of Gd2(Ti1−xZrx)2O7 for xZr = 0.75 (a) and 0.85 (b) with corresponding W. H. plots in the insets. HR-XRPD patterns of the whole Gd2(Ti1−xZrx)2O7 series in the low 2θ region (c) with fluorite peaks highlighted by asterisks. Cell constant (d), x coordinate of O1 (e), isotropic displacement parameters (f), occupancy factor of Zr ions at the A site (g) and occupancy factor of oxygen atoms at the O3 site (h) of the pyrochlore structure. | ||
To reveal the compounds microstructure, the experimental peaks up to 2θ = 20° (Qmax ≈ 6 Å−1) were fitted using pseudo-Voigt functions and, after the deconvolution of the instrumental resolution function, their integral breaths were plotted according to the W.-H. equation, to calculate the crystallite size (DV) and strain (ε) contribution to peak broadening. The refined λ/DV and ε values are displayed in Fig. S1 of the ESI† and briefly discussed. The insets of Fig. 2a and b report the results for the xZr = 0.75 and 0.85 samples respectively. The small intercept values of the regression lines testify the large crystallite dimensions of all the samples: DV is always in the order of several μm. In the figures, the experimental βcosθ data for the fluorite peaks are represented by black circles, while data for the pirochlore peaks are depicted as empty circles.
Up to xZr = 0.75, all the experimental points lie on the same straight line. For xZr ≥ 0.85, the empty circles are systematically shift towards higher values pointing to a reduced crystallographic coherence of pyrochlore ordering in respect to the crystallite size. The refined strain value ε for Gd2Ti2O7 is almost 10−4, as expected for a well grown crystalline phase; then it suddenly grows up of an order of magnitude in the xZr = 0.15 sample (1.3 × 10−3) and smoothly decreases raising xZr, reaching ≈8 × 10−4 in the Ti-free sample (see Fig. ESI1B†). This behavior suggests that all the solid solutions and the Gd2Zr2O7 compound are much more strained than the Gd2Ti2O7 one.
An ideal pyrochlore model was initially used in Rietveld refinements to analyze the HR-XRPD patterns. The cell constant was varied along with the displacement parameter xO1 of the O1 site and three isotropic thermal parameters, for Gd, Zr and O.
Some sample dependent peaks asymmetries were detected that could not be ascribed to instrumental aberrations. Similar effects were already detected in patterns collected at synchrotron radiation sources by other authors while investigating the Gd2Zr2O7 composition prepared by solid-state synthesis. The asymmetry was interpreted as residual sample inhomogeneity caused by poorly crystalline defect fluorite material and modelled using an additional cubic fluorite phase in the refinements.69,70 In our case, we decided to refine the patterns using two pyrochlore phases, constraining the structural parameters (xO1 and Uiso) to be the same for the two models. In all samples, either the second phase has a very small weight fraction or the cell constants of the two phases were very close to each other. Small portions of these bi-phasic refined patterns are shown as examples in Fig. ESI2 of the ESI.†
All experimental peaks were included in the model and no indications of symmetry decrease, such as growth of additional reflections, were observed in respect to the calculated patterns. However, Ti/Zr substitution induces a systematic overestimation of the intensities of the pyrochlore peaks by the model. The progressive reduction of the intensity of the pyrochlore peaks in respect to the fluorite ones (labelled with asterisks) is clearly visible Fig. 2c. These findings suggest that the pyrochlore ordering is reduced in Zr-rich samples.
Local probes such as pair distribution function analysis35,71,72 and XAS spectroscopy69,73,74 revealed that in Zirconium based pyrochlores, Zr is preferentially surrounded by seven oxygen ions pointing to the partial occupation of the O3 site. Conversely, the Rietveld refinement of neutron diffraction patterns of Gd2Zr2O7 (using 160Gd to overcome the problem of the high absorption neutron cross section of natural Gd) did not detect significant oxygen positional disorder.75 In addition, cation swapping between the A and B sites should occur when the long-range structure approaches the defect fluorite type.17,35 For these reasons, beside the parameters listed above, the occupancies of oxygen 48f (O1) and 8a (O3) sites were allowed to vary in the final refinements, constraining the oxygen concentration to 7 O/formula. The occupation of 8b (O2) site was fixed to 1 because preliminary tests did not reveal any oxygen depletion on it. At the same time, Zr and Gd ions were allowed to mix in the A, B sites, taking fixed the (full) sites occupations and the Zr:Gd ratio (=1). Panels (d–h) of Fig. 2 display the refinement results, while refined parameters are reported in Table ESI2 of the ESI.†
The cell constant a (Fig. 2d) and the xO1 (Fig. 2e) show the expected trends. The first one is simply related to the different ionic radii of Zr and Ti; the second one is in line with previous studies on compositionally induced disorder in pyrochlore oxides. As the radius ratio rA/rB, decreases, the xO1 parameter approaches the ideal value that it would have in a fluorite structure type (=0.375).17,18,73,76,77
The isotropic mean square displacement parameters U are displayed in Fig. 2f. They all lie around ≈0.005 Å2 in Gd2Ti2O7 compound; then they smoothly raise in Ti-rich solid solutions (xZr ≤ 0.50) and are boosted moving toward the Zr-rich part of the phase diagram; in Gd2Zr2O7 their value span from two (UGd) to five (UO) times their values in Gd2Ti2O7. When complete solid solutions connect end members both exhibiting ordered crystal structures, following the composition coordinate, U values should initially raise (as detected in xZr ≤ 0.50 samples). In fact, cation mixing should promote structural disordering that shifts U values toward larger values. However, they should reach a plateau and start decreasing, dropping to a new minimum for the second end member. This is observed, for example, in the Ce1−xGdxO2−x/2 solid solutions.78 The very high U values detected for Gd2Zr2O7 compound and Zr-rich compounds point again to high structural disorder.
In Fig. 2g, the occupation factor of the A site by Zr (o.f. (ZrA)) is zero up to xZr = 0.50; then, it raises up to ≈0.05 in the Zr-richest samples. However, even in Gd2Zr2O7 endmember, the A site is occupied by ≈0.8 Zr ions per cell. The occupation of the A-site by Zr, and of the B-site by Gd, can be expressed through cation antisite defects as follows:
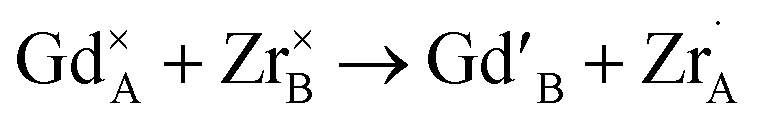
The occupation factor of O3 site (o.f. (O3)) linearly raises with xZr, as shown in Fig. 2h. For Gd2Zr2O7 compound ≈3.2 oxygen ions per cell occupy the O3 interstitial site. When o.f. (O3) is normalized in respect to xZr, o.f. (O3)/xZr function appear to be flat (≈0.4) in the whole compositional range (see red circles in Fig. 2b), enforcing the idea that Zr is systematically surrounded by a larger number of oxygen ions in respect to titanium. In particular, in Gd2Zr2O7 the average Zr coordination number should be ≈6.4.
When one oxygen ion ideally jumps from a O1 site to a O3 one, an anion Anti-Frenkel pair is formed:
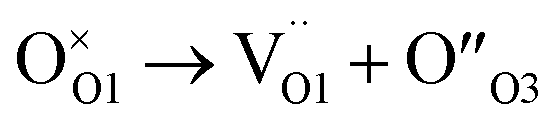
According to the refined pyrochlore average structure, six too short O3–O1 contacts are formed (dO3–O1 ≈ 2.2 Å for xZr = 1.0) that can be reduced to five if the interstitial anion and the vacancy are close to each other. It is worth supposing that some structural relaxation/reconstruction takes place around the formed couple to reduce electrostatic energy, producing the disorder revealed by the large U values in the Zr-rich part of the phase diagram.
The raise of disorder on adding Zr to the structure is not unexpected. Ab initio calculations by Jiang and coworkers79 showed that the disordering energy from pyrochlore to disordered fluorite is doubled passing from Gd2Zr2O7 (≈0.7 eV per formula unit) to Gd2Ti2O7 (≈1.5 eV per formula unit). It is interesting to note that our results point to some hierarchy of disordering: anion Anti-Frenkel defects appear already at the smallest Zr doping value and have larger concentration than cation antisite defects. On the other hand, the pyrochlore peaks are systematically broader than the structure ones almost in the same compositional region were Zr/Gd antisite disorder reaches its maximum value. The coherence length of the pyrochlore ordering seems to be mostly sensitive to cation swapping, at least using X-rays as probes.
3.3. Local probes
The Raman spectra of Gd2(Ti1−xZrx)2O7 samples are presented in Fig. 3a, where the vibrational modes are labelled from M1 to M7.
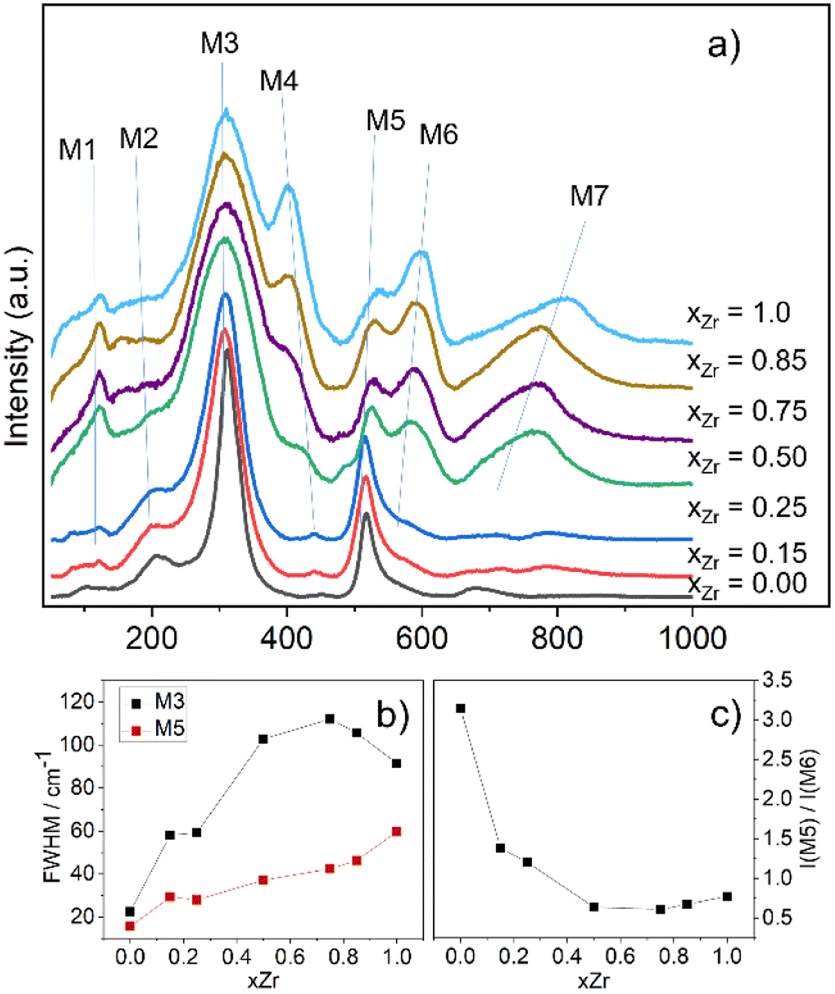 | ||
| Fig. 3 Raman spectra of Gd2(Ti1−xZrx)2O7 series between 50 and 1000 cm−1 (a) offset on the Y axis. After background subtraction, each spectrum was normalized between 0 and 100; FWHM of modes M3 and M5 (b) and intensity ratio between modes M5 and M6 (c) as a function of Zr-content. | ||
The experimental spectra recorded in this study reproduce quite well those already reported in the literature for the Gd2(Ti1−xZrx)2O7 solid solution.10,15,29,31,33,82,83 To identify the relevant bands that change upon Zr-substitution, one may focus on the spectrum of Gd2Ti2O7, which has the sharpest lines among all the spectra. As shown in Fig. 3a, the weak feature at ≈113 cm−1 (M1) in the spectrum of Gd2Ti2O7 sharpens, while the band situated at ≈212 cm−1 (M2) appears to broaden on Zr-substitution. The most intense peak centered at ≈311 cm−1 (M3) experiences an appreciable broadening with increasing xZr as well. A band centered at ≈450 cm−1 (M4) is hardly visible in the spectrum of Gd2Ti2O7. However, as the Zr-content increases, M4 rises and shifts towards lower frequencies (i.e. is red-shifted), until it becomes one of the most prominent features in the Raman spectrum of Gd2Zr2O7 (≈400 cm−1). The lines at ≈516 cm−1 (M5) and ≈550 cm−1 (M6) are both blue-shifted with increasing Zr-content. However, their behavior regarding intensity is different. Specifically, while the intensity of M5 decreases, the intensity of M6 increases. A subtle spectral feature is present in the spectrum of Gd2Ti2O7 around 690 cm−1 (M7). It seems to split into two broad and weak bands for xZr = 0.15, 0.25, and then suddenly grows for xZr = 0.50, while being blue-shifted. The fit of features M1–M7 with Gaussian functions for the Raman spectra of samples xZr = 0, 0.15, 0.50 and 1.0 are reported in Fig. ESI3† as examples, while the trends of their frequency values on varying xZr are displayed in Fig. ESI4.†
The vibrational modes of pyrochlores correspond to spectral bands that lie approximately in the range 200–600 cm−1 of the spectrum.84,85 The assignment of Raman bands to the corresponding lattice modes has been done in previous studies by measuring polarized Raman spectra on single crystals and/or by theoretical calculations.85–90 From the experimental point of view, no spectrum completely fulfills the selection rules under light polarization.90 Nonetheless, there is a general agreement on the assignment of the most intense bands, M3 and M5, which are the fingerprints of the pyrochlore spectrum.91 The peak M3 is actually consisting of two overlapping modes, T2g and Eg, which are placed at 311 and 330 cm−1 respectively and involve O1 at the 48f site through the Ti–O stretching and O–Ti–O bending.85,88–90,92–94 Gupta and coworkers interchanged their frequencies.87,95,96 The band M5 has also been assigned to two vibrational modes, which have adjacent frequencies and symmetry labels A1g and T2g.85,88 The splitting of these two modes is more evident in rare earth zirconate and hafnate pyrochlores.85,97,98 The A1g mode is associated with the modulation of xO1 by the vibration of O1 (O48f) along the [100] axis toward the O3 (8a) vacant site.15,91,99,100 Oueslati and coworkers,82 followed by Mori et al.,90,101,102 assigned the bands at ≈300 and 500 cm−1 to O–Gd–O bending and Gd–O stretching vibrations respectively, on the basis of the evolution of their wavenumbers in the Gd2(Ti1−xZrx)2O7 solid solution with xZr. More controversial is the assignment of the remaining T2g modes, which might be attributed to some of the weaker bands that are visible in the spectrum of Gd2Ti2O7 at ≈113 (M1), 212 (M2), 450 (M4), 550 (M6) and 690 (M7) cm−1. A brief description of these additional bands is provided in the following discussion and in the ESI.†
As can be evidenced from Fig. 3a, the peaks M3 and M5 are clearly affected by line broadening on Zr-substitution. The corresponding values of FWHM as a function of xZr are plotted in Fig. 3b. In particular, the data for M3 (≈311 cm−1) are comparable with those already reported by Hess and coworkers.15 The lines broadening suggests that static disorder is accumulating within the structure via defects formation.82 The partial loss of translational symmetry at the local scale, caused by static disorder, relaxes the k = 0 selection rule and allows phonons from other parts of the Brillouin zone to mix. This, in turn, results in lines broadening in the spectrum.81 What is striking is the evolution of the M5 line with respect to the M6 on Zr-substitution. M6 has been regarded as one of the T2g modes in previous investigations on related pyrochlore systems.15,93,103 However, it might be associated with extra vibrational modes activated by chemical substitution at the B site of the structure.104 It is worth remembering at this point that O3 is surrounded by four B-cations in the pyrochlore structure. The progressive filling of O3 (8a) vacant site through an Anti-Frenkel defect formation would activate a forbidden mode related to B–O vibration. The newly activated mode M6 moves toward higher wavenumbers on increasing xZr and reaches ≈600 cm−1 for the Gd2Zr2O7 compound. The Anti-Frenkel defect leaves the O1 site depleted, affecting the A1g component of band M5 ultimately leading to a decrease in intensity of M5 (ref. 91, 105 and 106) (see Fig. 3c). Both the modes related to M5 and M6 harden on Zr-substitution because of short O1–O3 distances in Zr-rich composition, being M5 particularly sensitive to O–O interactions.91,106 Indeed, as revealed by Rietveld analysis, Zr doping in Gd2(Ti1−xZrx)2O7 brings to progressive O3 site occupation and O1–O3 distances that are much shorter than the O1–O1 (≈2.7 Å) and O1–O2 (≈2.9 Å) ones.
Another interesting spectral feature is the broad band at ≈700 cm−1 (M7), which becomes evident at xZr = 0.50 and is probably a combination of more than just one vibrational mode. This band is commonly assigned to an overtone or second-order scattering process related to the main band at ≈310 cm−1.89,92,107–109 On the other hand, the intensity of such second order line is quite surprising in comparison with the other spectral bands of the first order.31 DFT calculations have shown that a T2g mode is allowed beyond 700 cm−1 in the case of Gd2Ti2O7 (ref. 93) and La2Zr2O7 pyrochlores.110 The high wavenumber suggests that a B–O vibration is involved.111,112 Glerup and coworkers assigned this M7 band to a BO7 cluster in line with a gradual disordering from the pyrochlore to the defect fluorite structure in the Y2Ti2−yZryO7 solid solution and a change of the B-coordination number (CN) from 6 to 7 (ref. 113) Sanjuan and et al.99 argues that the actual coordination of B species is based on preferential CN of each cation involved, causing a disordering of the oxygen atoms in the solid solution based on the following mechanism: an oxygen atom moves from the O1 (48f) site to the vacant O3 (8a), increasing selectively the CN of Zr to 7. Conversely, the CN of Ti should remain unaffected or even decreased to 5. The presence of Ti atoms in a 5-fold or 6-fold coordination environment and a random displacement of the metal from its ideal position in coordination polyhedron would activate breathing modes above 700 cm−1 (M7).99 Moreover, it should be noted that this band is one of the dominant modes associated to B–O stretching in other fluorite-derived superstructure such as weberite,100,114 zirconolite109,115,116 and brannerite.117,118 This observation may account for a local ordering in the Gd2(Ti1−xZrx)2O7 series that is different from the pyrochlore or fluorite structure type. Further insight can be gained by the PDF analysis of the 2D diffraction data.
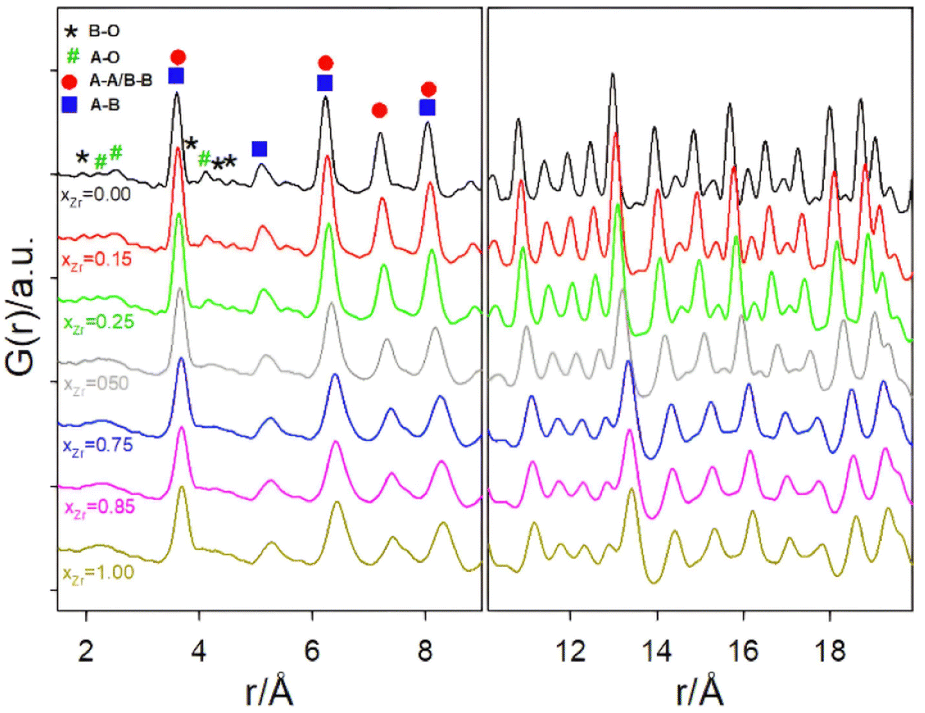 | ||
| Fig. 4 Experimental PDF functions in the 1.5–9 Å (left side panel) and 10–20 Å (right side panel) intervals. Black, green, red, and blue symbols in the left side panels appear in correspondence to the B–O, A–O, A–A/B–B distances, respectively, according to the pyrochlore structure. For sake of clarity, the cation-oxygen distances are shown only up to r ≈ 4.5 Å. | ||
Focusing first on the high r range, the G(r) of Gd2Ti2O7 presents sharp and symmetric peaks. Upon increasing compositional coordinate xZr, the peaks progressively broaden. Being all the G(r) functions collected at the same temperature, the dynamic (vibrational) contribution to peak broadening should be similar for all samples. The observed trend is to be attributed to an increased distribution of the equilibrium interatomic distances and is a fingerprint of static disorder.27,57 All the experimental G(r) functions were fitted using a pyrochlore structure, first. Good fits appear for r distances larger than 8 and 9 Å. The observed peaks broadening is accounted for by increasing atomic mean square displacement parameters values during the fitting process, similarly to the reciprocal space Rietveld refinement findings. In Fig. ESI5† right panel, the fits in the 9–22 Å interval are shown for all the compositions, while in Fig. 5 the Rw values are shown as black squares. Moving on to the short r range (1.8 ≤ r ≤ 8 Å), again the G(r) of Gd2Ti2O7 presents sharp and symmetric peaks; by substituting Ti with Zr, beside broadening, asymmetry in the cation–cation peaks is observed, for example for those at ≈6.5 Å (with A–A, B–B and A–B contributions) and at ≈7.5 Å (with A–A, B–B contributions). The latter peak splits into two in the Zr-richest cases. Applying the pyrochlore model, good agreement between experimental and calculated PDFs is observed for Gd2Ti2O7 (see Fig. 6a). On raising xZr, the fit quality progressively worsens as reveled by the fit residuals Rw, reported in Fig. 5. All the fits in the 2–8 Å range are shown in Fig. ESI5† left panel, while Fig. 6b and c display the cases of xZr = 0.50 and 1.0, respectively. Passing from pure Ti to pure Zr compounds, static disorder, which is related to peak broadening, causes again the increase of the atomic displacement parameters, in particular for UGd (from 0.0034 Å2 to 0.016 Å2) and UO (from 0.021 Å2 to 0.049 Å2). In addition, fit failures appear, especially for the shortest cation-oxygen distances (1.4–2.4 Å interval) and for the peaks centered at ≈3.9 Å, ≈6.5 Å and 7.5 Å, which correspond to the most intense metal–metal correlations. This finding suggests extended relaxation of the cations positions that the pyrochlore structure cannot account for, because no cationic positional degrees of freedom are allowed in it. Accordingly, EXAFS investigations on the Zr–K and Gd-L3 edges revealed that different cation–cation distances exist in Gd2Zr2O7 and that the cations shift from the high symmetrical sites of the pyrochlore structure.15,34 It is worth mentioning that some splitting of the second coordination sphere consisting of cation–cation correlations was observed even in the case of Gd2Ti2O7.15,119 Our PDF results revealed peaks splitting up to ≈8 Å in the G(r) functions of the Zr rich side of the phase diagram, fixing a lower r limit for the coherence length of structural distortions. Two scenarios are possible: on one hand, symmetry could decrease locally, to account for the different characteristic of Zr ions in respect to Ti ones forming (sub)nanodomains that merge into a higher symmetry structure at larger scale. On the other hand, point defects such as, for example, the anion Anti-Frenkel defects detected by the reciprocal space analysis and by Raman spectroscopy could form extended clusters that distort the structure up to almost one nanometer.
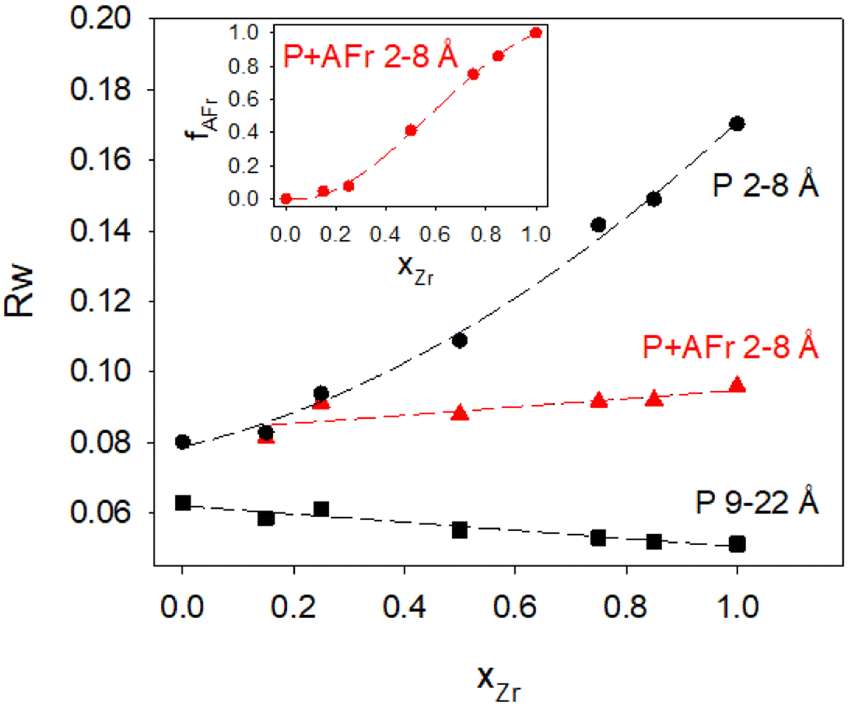 | ||
| Fig. 5 Residuals of G(r) fits as a function of composition. Black circles and squares refer to pyrochlore structural model in the 1.8–8 Å and 9–22 Å intervals, respectively. Red triangles refer to a biphasic fit with defect free Gd2Ti2O7 and Frenkel defective relaxed Gd2Zr2O7 pyrochlore phases. In the inset is reported the fraction fFr of the defective Gd2Zr2O7 pyrochlore phase. Dashed lines are guides for the eyes. See main text for details. | ||
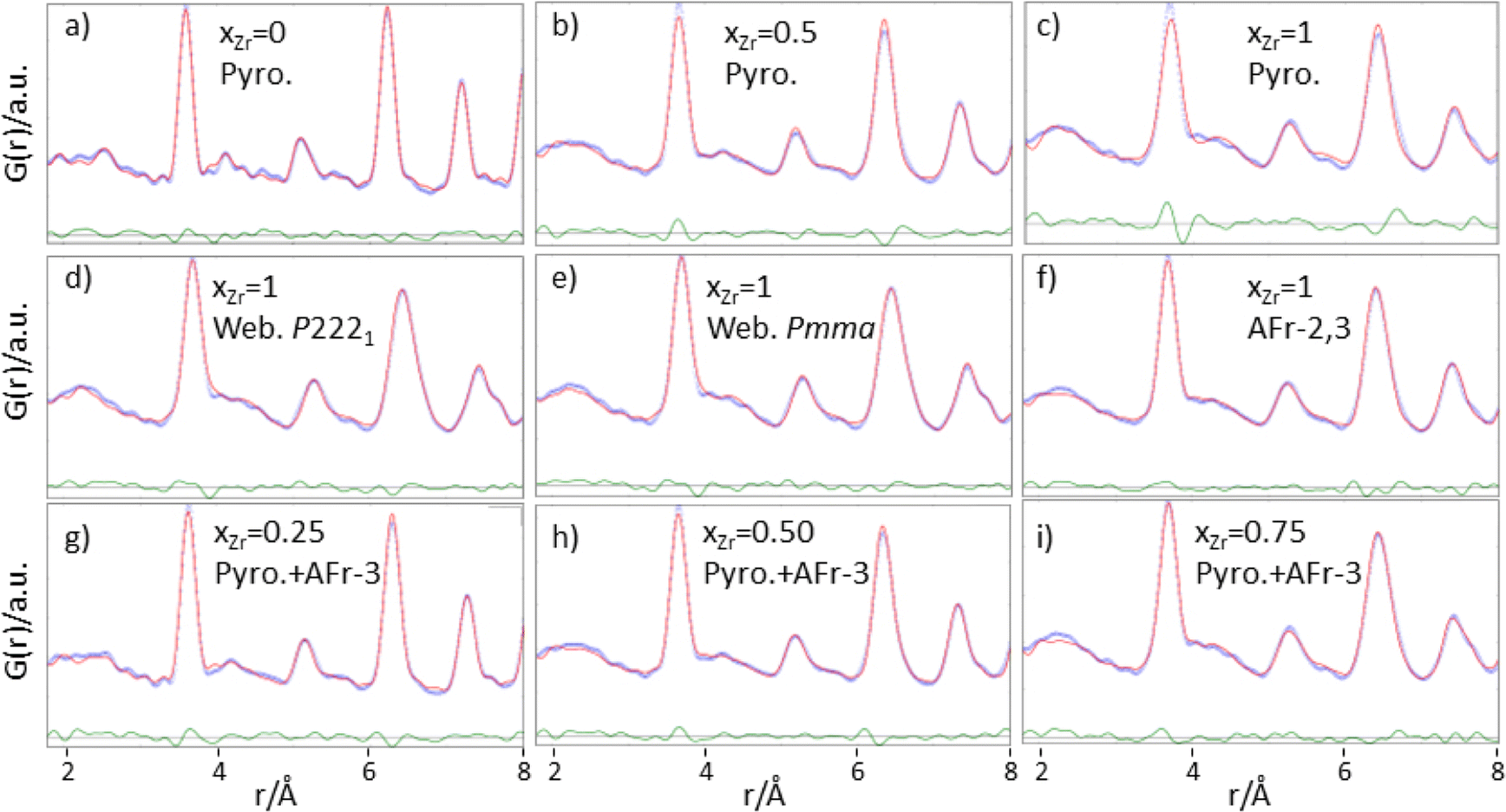 | ||
| Fig. 6 Experimental (light blue circles), calculated (red curves) and difference (green curves) G(r) functions in the 1.8–8 Å. Panels (a–c) report the function relative to xZr = 0, 0.50 and 1.0 samples respectively, fitted using the pyrochlore model. In panels (d–f) the function of the Gd2Zr2O7 sample was fitted using the weberite-type P2221, Pmma models, and with the Frenkel defective relaxed pyrochlore phase, respectively. In panels (g–i) the function relative to xZr = 0.25, 0.50 and 0.75 samples, respectively were fitted using a biphasic fit with defect free Gd2Ti2O7 and Frenkel defective relaxed Gd2Zr2O7 pyrochlore phases. See main text for details. | ||
As to the former scenario, previous investigation on Ho2(Ti1−xZrx)2O7 system highlighted similar progressive structural disordering while substituting Ti with Zr. Taking as an example Ho2Zr2O7, which crystallizes in the cubic defect fluorite structure at the average scale, orthorhombic Ccmm weberite-type (a non-standard setting of e.g. Cmcm space group, n° 63) have been invoked by Shambling and et al., to interpret its local structural arrangement up to 15 Å, as revealed by neutron PDF.36 On the other hand, Drey and co-workers35 revealed compositional and symmetry fluctuations at the local scale in the Ho2(Ti1−xZrx)2O7 system. These fluctuations consisted of pyrochlore-like Ho2Ti2O7 and weberite-like (C2221) Ho2Zr2O7 nanodomains. In both Ccmm and C2221 weberite-type models, cations are located in three crystallographic non-equivalent sites, one 8-fold coordinated for Ho (HoO8 cage), one 6-fold coordinated for Zr (ZrO6 cage), and one 7-fold coordinated shared by both cations ((Ho/Zr)O7 cage). The coordination of this latter site can be regarded as derived from a different ordering of oxygen ions, which results in the occupation of some of the anion sites previously empty in the pyrochlore structure (O3 sites).36 It is worth noting that a group–subgroup relationship exists between the aristotype fluorite and the weberite-type models,120 allowing the structure of Zr-rich Ho2(Ti1−xZrx)2O7 solid solutions and pure Ho2Zr2O7 to converge to the defect fluorite structure at the average scale. The case of Gd2(Ti1−xZrx)2O7 is different from the A = Ho one, because pyrochlore and weberite-type polymorphs belong to different branches of the same group–subgroup tree. The local Gd and Zr mixing of weberite-type is hardly compatible with the (almost) perfect cation ordering detected in Gd2(Ti1−xZrx)2O7 compounds at the average scale. Nonetheless, the weberite-type models described above allow mapping the tendency of Zr to enlarge its coordination number and supply the needed positional degrees of freedom to cations in order to shift their positions in respect to their highly symmetrical sites in pyrochlore. For this reason, we applied two modified weberite-type models to the fitting of the G(r) functions, where the same cation ordering as in pyrochlore is retained causing in both cases the removal of the C face centering. In this way, C2221 and Ccmm models turn into P2221 and Pcmm (a non-standard setting of Pmma space group), respectively. As described in Section 3.1, removing the C face centering increases the number of symmetry independent atomic sites as well as the atomic positional degrees of freedom, so lowering the experimental-points to variable-parameters ratios to unsafe low values. Consequently, the experimental G(r) of Gd2Zr2O7 has been fitted in the 1.8–8.0 Å interval with both weberite-type models P2221 or Pmma preserving the symmetry constrains of C2221 and Cmcm (see Fig. 6d and f, respectively). In order to further reduce the number of degrees of freedom, only the parameters that varied significantly in respect to their special positions in fluorite were allowed to vary. Additionally, three cell parameters, a scale factor, a short range correlated motion parameter (delta2 in PDFGui) and four atomic mean square parameters (one for each of the three cationic sites and one for oxygen) were allowed to vary, for a total number of 16 varied parameters for the former and 15 varied parameters for the latter model. Considering that the number of independent points is n = Qmax (rmax − rmin)/π = 51.3, at least 3 independent data per parameter are present.
Both models significantly improve the fit quality in respect to pyrochlore (Rw = 0.094 and 0.091, respectively). However, in the refined structure derived by the constrained P2221 model, non-physical oxygen–oxygen distances appear; for this reason, the following discussion deals only with the (constrained) Pmma model. Atoms coordinates are listed in Table 1.
| Atom | Wyckoff letter | x | y | z | Occ |
|---|---|---|---|---|---|
| Gd1 | 2b | 0 | 1/2 | 0 | 1 |
| Gd2 | 2c | 0 | 0 | 1/2 | 1 |
| Gd3 | 4k | 1/4 | 0.2385 | 0.75 | 1 |
| Zr1 | 4k | 1/4 | 0.7385 | 0.25 | 1 |
| Zr2 | 2a | 0 | 0 | 0 | 1 |
| Zr3 | 2d | 0 | 1/2 | 1/2 | 1 |
| O1 | 8l | 1/2 | 0.2192 | 0.8788 | 1 |
| O2 | 8l | 1/2 | 0.7192 | 0.3788 | 1 |
| O3 | 2e | 1/4 | 0 | 0.0485 | 1 |
| O4 | 2f | 1/4 | 1/2 | 0.5485 | 1 |
| O5 | 2f | 1/4 | 1/2 | 0.0864 | 1 |
| O6 | 2e | 1/4 | 0 | 0.5864 | 1 |
| O7 | 2f | 1/4 | 1/2 | 0.8348 | 1 |
| O8 | 2e | 1/4 | 0 | 0.3348 | 1 |
As shown in Table 1, only the cation y coordinates of the Gd/Zr 4k sites are shifted significantly from their special position (0.2385 and 0.7385 instead of 0.25 and 0.75). This causes a one-dimensional modulation of the cation–cation distances, allowing resolving the G(r) misfits at ≈3.9 Å, ≈6.5 Å and ≈7.5 Å. In particular, while according to the pyrochlore structure all the nearest cation–cation distances should be at ≈3.71 Å, the applied weberite-type model allows these distances to span in the 3.63–3.76 Å range. As shown in Fig. 6e, the modulation expands to larger interatomic distances allowing the splitting of cation–cation correlations even at ≈7.5 Å. Peak broadening is partially accounted for by the increased positional degrees of freedom, but the mean square parameters U still take large values for the 6-coordinated cation and for the O sites (0.031 Å2 and 0.035 Å2, respectively).
The present results agree with the model proposed by Shambling and coworkers36 on A = Ho compound, with the noticeable difference that significant cation ordering is retained in the A = Gd system. These authors extended positively the same model up to 15 Å, determining the coherence length of the weberite-type nanodomains. Accordingly, we expanded the same model up to at least the length of the c axis. Fig. ESI6† shows the fits in the 1.8–11 Å interval. If the model is expanded as such, without refining any parameter, a very worse fit is obtained (Rw = 0.213, see Fig. ESI6a†). Relaxing the structure improves the residuals (Rw = 0.120, see Fig. ESI6b†) but results in the reappearing of the misfits already observed for the pyrochlore model (Rw = 0.136, see Fig. ESI6c†), despite the much larger structural degrees of freedom of the former model. This suggests that the weberite-type ordering, if present, has a coherence length smaller than one translation vector along its c cell axis. This sows the seed of doubt that a different structural distortion exists mimed by weberite-type structural features, which permits larger coordination numbers for (half of) the Zr ions and modulation of the cation–cation distances.
Despite the shortcomings above described, the modified weberite-type Pmma model has been applied to solid solutions in the 1.8–8 Å range, substituting progressively Zr with Ti on the BO6 site first, and then on the BO7 one. The main finding is that the weberite-type model fits the data better than the pyrochlore one as long as TiO6 polyhedra are formed (down to xZr = 0.50), while for larger Ti concentration, when TiO7 polyhedra are introduced in the model, the pyrochlore model has to be preferred in respect the weberite-type structure (see residuals in Fig. ESI7†).
As a possible alternative, we tested a completely different approach. Instead of looking for some local coherent symmetry break, following the Rietveld refinements results and starting from the average pyrochlore structure, we introduced Anti-Frenkel defects. As described in the next section, oxygen ions were moved from the O1 to the O3 sites and the energies of the derived structures, relaxed by means of force field (FF) and Density Functional Theory (DFT) calculations, were evaluated.
3.4. Atomistic calculations and derived G(r) functions
The weberite-type Pmma, as well as the ideal pyrochlore structure containing anion Anti-Frenkel defects of Gd2Zr2O7 composition, were relaxed in a two steps procedure, first using a simple polarizable force field, then by DFT.To simulate the partial occupations of the O3 site (empty in the pyrochlore structure), we generated 300 atomistic configurations of Gd2ZrO7 by randomly selecting one, two and three oxygen ions sitting in the O1 (48f) sites and placing them in the empty O3 (8a)sites.
Since DFT calculations on the 88-atoms cell are quite expensive, we relaxed the atomic positions in two steps: first using a polarizable core–shell potential.13 Then, we selected the lowest energy configurations and performed a more accurate relaxation at the DFT level with Quantum Espresso 6.8.63,64 We used “f-in-core” ultrasoft pseudopotentials121 with a plane wave cutoff of 45 Ry. The electron interaction was treated with the PBE exchange and correlation functional.
Note that the force field13 yields in rare cases a Zr–O bond of ≈1.8 Å, which is way too short than the Zr–O bonds found in ZrOx polymorphs122 and amorphous zirconia.123 Therefore, we discarded the structures showing too short Zr–O bonds (<1.8 Å).
In agreement with DFT calculations reported in the literature79,124–126 for Gd2Zr2O7, the weberite structure is slightly more stable than the pyrochlore by 0.014 eV per formula unit. Interestingly, the AFr defects have negative formation energies of about 0.5 eV per defect on average. We observed that even when starting from 2 anti-Frenkel defects, the structure relaxation leads to the formation of further AFr defects, up to 5 defects in the 88-atom cell, both with FF and with DFT. This can be due to the tendency of Zr to increase his coordination number. The FF is unable to predict a negative AFr formation energy, while DFT accounts correctly for the relaxation of the electronic structure due to the formation of Zr–O bonds.
The atomistic configurations obtained by minimizing the energy of the partially occupied O3 sites show distinct features. If only one O3 site out of eight is occupied (AFr-1), the oxygen ions tend to stay close to the tetrahedral site, leading to a negligible distortion of the cation and anion sublattices. In Fig. 7b a typical AFr-1 configuration is depicted. Within it (and in the following panels), red spheres indicate oxygen ions close to the ideal O1 and O2 sites, while yellow spheres indicate oxygen ions close to the ideal O3 sites, that should be empty in the pyrochlore structure.
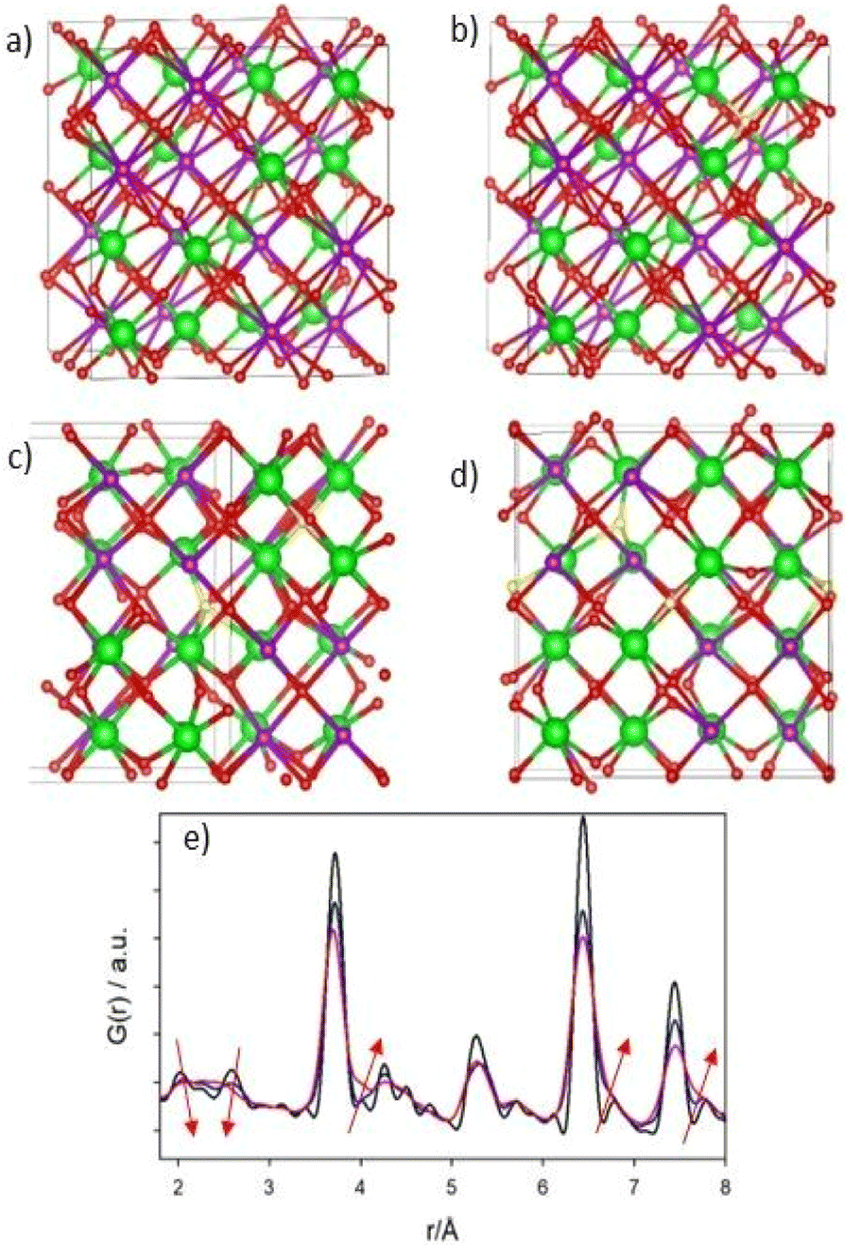 | ||
| Fig. 7 (a–d) Examples of DFT optimized atomistic configuration of the Frenkel pairs defect complexes. Zr and Gd are green and purple in color. (a) ideal pyrochlore; (b–d) configuration with one (b), two (c) and three (d) Frenkel pairs; green and violet spheres represent Gd and Zr ions, respectively. Red spheres indicate oxygen ions close to the ideal O1 and O2 sites. Yellow spheres represent the ones close to the O3 positions. (e) Simulated PDFs of the atomistic simulation. Black curve: G(r) of ideal pyrochlore; blue curve: G(r) calculated from configuration (b); red curve: G(r) calculated from configuration (d). Arrows indicate the direction of the changes of some peaks intensities on increasing the number of Frenkel pairs. | ||
Calculated G(r) functions have been produced starting from the calculated ideal pyrochlore structures and the defective models, using PDFGui, fixing Qmax, and the instrumental parameter Qdamp to the experimental conditions and adopting for all the atoms the same (small) atomic mean square displacement parameter (=0.003 Å2).
As shown in Fig. 7e, the simulated PDF of these configurations (blue curve) is similar to that of the ideal pyrochlore structure (black curve), thus the isolated Anti-Frenkel defect does not provide a good description of the local structure. However, some peak broadening is apparent, due to structural relaxations (see e.g. the shortest M–O distances and the A–B one at r ≈ 5.5 Å). The situation is different when two or more O3 sites are occupied. In most cases, the minimization of the total energy leads to sizable rearrangement of the cation and anion sublattices whose structure mainly depends on the number of anion Anti-Frenkel pairs per cell. Fig. 7c and d depict representative structures when two (AFr-2) or three (AFr-3) defect pairs are present, respectively. Enlarged versions of Fig. 7c and d are reported in the ESI (Fig. ESI8A and 8B†) with selected Zr–O distances.
As shown in Fig. 7c and d, the Anti-Frenkel pair defects tend to cluster around one Zr ions, and to form defect complexes consisting in one oxygen moving towards the edges or the faces of the Zr tetrahedra, accompanied by one or more anion Anti-Frenkel defects. Further stabilization of this defect complex is of electrostatic origin and is provided by the modulation of the cation distances that tends to screen the charge of the oxygen. In fact, while in the ideal pyrochlore the first neighbor cation–cation distance is 3.72 Å, in this defect complex, the first neighbor Zr–Zr distance becomes shorter (3.44–3.69 Å) while the next nearest Zr–Zr distance increases slightly. In addition, the Zr ions have the tendency to increase their coordination number by displacing one oxygen ion from the ideal position to a “bridging” position with a Zr–O distance of ≈1.9 Å. The shortest O–O distance was 2.45 Å.
These complex structural reorganizations deeply affect the calculated G(r) function. The one relative to AFr-3 configuration is depicted as a red curve in Fig. 7e. Beside the additional peak broadening, the double peak structure 2–2.5 Å interval disappears due to the spread on the Zr(Gd)–O nearest neighbor distances. In addition, the modulation of cation–cation distances causes the growth of the shoulder (≈3.8 Å) at the right side of the most intense G(r) peak, whose maximum is shift toward shorter r values. Finally, additional shoulders form in the 5–8 Å range.
The evolution of the calculated G(r) functions on adding Anti-Frenkel pairs to the structure mimes the changes of the experimental ones on progressively substituting Ti with Zr, apart the lattice expansion due to the larger ionic radius of the latter ion in respect to the former one. The appearance of the high-r shoulder to the cation–cation peak around 7.5 Å testifies that the coherent structural distortion related to defect clustering extend up to almost 1 nm. In addition, we note that in all calculated G(r) all the atomic mean square displacement parameters are fixed to the same value (=0.003 Å2). The peak broadening observed in Fig. 7e is totally accounted by structural relaxation.
The relaxed optimized structures exploiting the lowest energies have been adopted to fit the experimental G(r) of Gd2Zr2O7 in the 1–8–8 Å interval, taking fixed the atomic positions and varying a scale factor, correlated motion parameter, the cell a constant of the cubic cell and three atomic means square displacement (one for Gd, Zr and O ions) parameters. In the following they will be referred to as “Anti-Frenkel (AFr) models”. Applying each configuration separately, the AFr-1 model lower the Rw value to 0.151. When two (AFr-2) or three (AF-3) defect couples are formed, the residuals Rw are further reduced to 0.121 and 0.122, respectively.
Low energy Frenkel defect configurations should be simultaneously present in the sample and, therefore, the next step was to use a combination of the AFr-2 and AFr-3 models to fit the experimental G(r) of Gd2Zr2O7 in the 1.8–8 Å interval, as depicted in Fig. 6f. The low residuals (Rw = 0.096) testify the fit quality. It is worth noting that in this fit only 8 parameters are varied, increasing the points/parameter ratio to 6.9. The applied model does not imply a priori any long-range structural correlation, such as the cation–cation distance modulation along the y direction of the weberite-type structure; however, a good fit appears up to 8 Å, suggesting again that clustering of Frenkel defects causes distortions up to this value. Also, atomic mean square displacement values for Gd2Zr2O7 are very similar to the ones fitted for pyrochlore Gd2Ti2O7 being UGd, UB and UO = 0.003 Å2, 0.006 Å2 and 0.021 Å2 respectively for the former and UO = 0.003 Å2, 0.007 Å2 and 0.021 Å2 respectively for the latter case. As a last comment on the present fit, the phase fraction of the AFr-3 configuration (=0.64) is almost two times the phase fraction of the AFr-2 model (=0.36).
The AFr models have been extended to solid solutions. Including Ti to calculations would have brought to an explosion of the number of possible configurations, making quite hard or even impossible the comparison to experiments. For this reason, the AFr models of Gd2Zr2O7 were flanked by a pyrochlore Gd2Ti2O7 phase. For each AFr model, only the cell constant and one scale factor were varied, while for the pyrochlore phase also the xO1 parameter was considered. Three overall U parameters (UGd, UTi/Zr, UO) were refined. Preliminary tests using three phases (AFr-2, AFr-3 and pyrochlore) brought to correlations among the scale factors and, in most cases, the one of AFr-2 was reduced to zero. Consequently, we used a biphasic model in the final refinements, including only the AFr-3 and a pyrochlore model. In Fig. 6g–i the fits for xZr = 0.25, xZr = 0.50 and xZr = 0.75 are shown, respectively.
Increasing Ti concentration, smoothly lowers the Rw residuals (see red triangles in Fig. 5); for xZr ≥ 0.50, the fit quality clearly exceeds the one of pure pyrochlore, while for xZr = 0.25 and 0.15 they are quite similar. In the inset of Fig. 5 is shown the atomic fraction of the AFr (fAFr). For the largest Zr concentration, xZr and fFr values are in suitable accord. For xZr = 0.50, fFr ≈ 0.41 while for xZr = 0.25 and 0.15, fFr is 0.075 and 0.0445, respectively. To introduce in the model the correct Zr/Ti ratio in these last cases, the pyrochlore phase was suitable doped with Zr following an iterative procedure. The small weight of the Frenkel phase suggests that in these samples either the concentration of Frenkel defects is lower than expected in reference to the Zr concentration, or the structural relaxation is somewhat dampened. In fact, the atomic positions in the AFr phase have not been changed in respect to the AFr-3 model computed for Gd2Zr2O7 compound. In both cases, Titanium stiffens the crystal structure.
4. General discussion
The structural disorder induced by chemical substitution in Gd2(Ti1−xZrx)2O7 solid solution has been studied at different length scales. First, High Resolution X-ray Powder Diffraction revealed that the average structure of these materials is pyrochlore-like throughout the whole compositional range and Rietveld analysis shed light on how the disordering mechanism evolves at this length scale. The occupation of the O3 (8a) site with oxygens coming from the O1 (48f) site confirms that the pyrochlore structure can accommodate a certain amount of disorder through Anti-Frenkel defects formation (Fig. 2 panel H). At the same time, the remaining O1 oxygens tend to move away from the newly occupied O3 site, as evidenced by the increasing xO1 coordinate (Fig. 2 panel E). A similar behavior was observed in the case of Y2(Ti1−xZrx)2O7,17 Y2(Sn1−xZrx)2O7 (ref. 18) and Ho2(Ti1−xZrx)2O7 (ref. 35) systems. However, in those cases the Zr-endmember adopts the defect fluorite structure at the average scale and a sudden surge in cation antisite defects with A- and B-cations exchanging places was reported (specifically for 0.6 < xZr < 0.9). As previously stated, even though Gd2Zr2O7 has a radius ratio of rA/rB = 1.46 and is prone to disorder being at the boundary of the stability fields of pyrochlore and defect fluorite,10,21 there is no phase transition at the average scale in the compositions under investigation. Moreover, it is important to remember that the samples were all produced at a temperature of 1500 °C, which is lower than the temperature required for a transition from pyrochlore to defect fluorite in the Gd2Zr2O7 system.15,46,47 Nonetheless, as suggested by Willimson–Hall analysis (Fig. ESI1†), the coherence of pyrochlore ordering diminishes in the Zr-richest part of the solid solution, where moderate A/B swapping is present (Fig. 2, panel G). The pyrochlore model that was employed for Rietveld refinements of HR-XRPD patterns clearly does not account for the complete disorder that exists within the structure. Evidence shows that there is a rise in the Uiso parameters as xZr increases (Fig. 2 panel F), indicating an escalation in static disorder.25,127 Raman spectroscopy supports this idea, as the broadening of spectral bands and the activation of new spectral features indicate an increment of disorder in the anion sub-structure.82 The activation of mode M6 and the concomitant decrease of mode M5 (Fig. 3) can be directly related to the formation of anion Anti-Frenkel defects between the O1 (48f) and O3 (8a). It appears that the oxygen atoms play a major role in the disordering mechanism, while cation antisite defects have only a minor contribution.To explore the source of this static disorder, the short-range structure was investigated through the analysis of pair distribution function. The pyrochlore structure is still the best choice for the description of the structure for interatomic distances r larger than 8 and 9 Å in the whole compositional range of Gd2(Ti1−xZrx)2O7. At these coherence lengths, disordering is identifiable by the progressive broadening of G(r) peaks. At low r values, only the structure of Gd2Ti2O7 and of the Ti-richest solid solutions can be suitably described by using the pyrochlore model. On the other hand, introduction of Zr4+ induces a broadening of all cation–cation peaks and the appearance of shoulders that cannot be modelled simply by introducing oxygen Anti-Frenkel and cation antisite defects in the undistorted pyrochlore structure, because they would not affect the cation–cation distances. As suggested also by Raman measurements, a lowering of the local symmetry with respect to the long-range pyrochlore arrangement is required to properly describe all these correlations. In this regard, following the approach of Shambling and co-workers36 for Ho2Zr2O7, the weberite-type model (s.g. Ccmm) was introduced to describe the local structure of Gd2Zr2O7. This model implies the occupation of one 8-fold coordination site (4b) by the A cation, one 6-fold coordination site (4a) by the B cation and one 7-fold coordination site (8g) equally shared by both cations. It was already stated that an important difference exists between Gd2Zr2O7 in this study and Ho2Zr2O7: when A = Ho, the average structure is defect fluorite, while when A = Gd an average pyrochlore structure is retained. Therefore, it is plausible to assume a local mixing of Ho/Zr in the 7-fold coordination site of the weberite-type model for Ho2Zr2O7. Instead, in the present study a Gd/Zr ordering was preserved at the local scale for Gd2Zr2O7 by reducing the symmetry to Pmma and, at the same time, applying suitable constrains to maintain a reasonable data/parameters ratio. Good fits were obtained at the local scale (2–8 Å) for both Gd2Zr2O7 compound and Zr-rich Gd2(Ti1−xZrx)2O7 solid solutions, progressively substituting Zr with Ti on the ZrO6 site.
This method provides the structural degrees of freedom needed to model the PDFs at low r values and Ti4+ and Zr4+ tend to maintain their preferential CN.99,120 In fact, while the small Ti4+ cation prefers a pseudo-octahedral coordination such that of the B site of the pyrochlore structure, the larger Zr4+ enters the seven-fold coordination site in the weberite-type structure. A CN of 7 is also found for Zr4+ in the stable phase at room temperature of ZrO2, the monoclinic zirconia.128 Obviously, this “rule” can be ideally respected by weberite-type models only for the Gd2(Ti0.5Zr0.5)2O7 composition.
Attempts to extend the weberite-type model to larger r values (e.g. up to ≈11 Å, about one c cell parameter) failed, questioning the presence of a coherent weberite-like local symmetry reduction. As a matter of fact, the application of the weberite-type structure to the description of the short-range ordering in Gd2(Ti1−xZrx)2O7 pyrochlore and related compounds is still debated. For example, Marlton and co-workers studied the long- and short-range structure in the Y2(Sn1−xZrx)2O7 system.72 According to them, the good fits of the X-ray and neutron PDFs with the weberite-type model obtained for the Ho2(Ti1−xZrx)2O7 series by Drey35 and Shambling36 were essentially due to data collected at room temperature. The thermal broadening of the peaks would have hidden subtle structural features, which could not be properly modelled with the weberite-type. Our diffraction data were collected at 90 K to reduce the thermal broadening of G(r) peaks. The use of X-rays allowed us to highlight the cation contributions to the structural distortions that seems to have an important role in the investigated systems. Conversely, the high neutron absorption cross section of natural Gd hinders the use of neutron diffraction that would supply a more accurate picture of the oxygen contributions. This shortcoming and, most important, the very short range of the “weberite-type ordering” have prompted us to take alternative path.
We performed DFT calculations on the Gd2Zr2O7 compound, applying the Pmma weberite-type and the pyrochlore structures. In the latter case, both perfect and Anti-Frenkel pairs containing models were elaborated. Starting from the relaxed structures, we calculated the G(r) functions and fitted them against the experimental one collected on Gd2Zr2O7 taking fixed the atomic positions. A very good agreement was found for AF-2 and AF-3 configurations, which well modelled the G(r) features up to r = 8 Å, despite the small number of variable parameters. We noted that, in contrast with the weberite-type model, the atomic mean square displacement parameters for Gd2Zr2O7 are very similar to those fitted for pyrochlore Gd2Ti2O7, enforcing the idea that the formation of oxygen Anti-Frenkel defects and clustering suitably interpret the local structure of the B = Zr compound and the origin of the structural distortions.
To extend the same approach to the solid solutions, we applied a biphasic model, where the relaxed structure of AF-3 Gd2Zr2O7 phase was flanked by a defect free pyrochlore Gd2Ti2O7 phase. Good fits were obtained in the whole compositional range, especially in the 0.5 ≤ xZr ≤ 1 interval, notably without introducing any atomic degree of freedom in the defective phase. In the Ti-rich part of the solid solution, the phase fraction of the defective structure seems to be smaller than the nominal xZr composition, suggesting that large Ti concentration stiffens the structure, in accord to the trends of the U parameters, as calculated from the Rietveld refinements.
The DFT field calculations supplied also the formation energy for each phase at 0 K, showing that the weberite-type phase of Gd2Zr2O7 is marginally more stable than the pyrochlore structure suggesting the reason of a possible weberite-like local distortion. However, the creation of an isolated Anti Frenkel defect from the ideal pyrochlore is energetically favored due to tendency of Zr towards increasing its coordination number. Next, the coalescence of multiple Anti Frenkel defects is stabilized by the modulation of the cation distances via electrostatic screening of the O2− charge. Moreover, it is conceivable that at finite temperature, the formation of a defect complex is further favored by the configurational entropy SC.
An accurate thermodynamic analysis is behind the scope of this paper. However, we note that in case of Anti-Frenkel defects the configurational entropy SC is the sum of the ones due to O1 vacancies (SV) and interstitial O3 oxygen (SI) formation. Considering the concentration of O3 ions computed by Rietveld refinement for Gd2Zr2O7 (≈3 ions per cell) and the multiplicity of O1 (48) and O3 (8), we can estimate the configurational entropies as:
| SV = −n R [(3/48) log(3/48) + (45/48) log(45/48)] = 0.23379 n R |
| S1 = −n R [(5/8) log(5/8) + (3/8) log(3/8)] = 0.66156 n R, |
At the temperature used for the synthesis (1773 K), the configurational entropy contribution to the Gibbs free energy can be roughly evaluated as −T SC = −T (SV + SI) ≈ −13.20 kJ mol−1 ≈ −1.09 eV for the 88-atoms cell. This lowering of the free energy contributes further to the stabilization of the defective pyrochlore. Please note that in this simple estimate, the vibrational entropy due to structural relaxation around defects has not been considered. In addition, in our model computations we did not consider the contribution of the swapping of Gd/Zr ions (1/16 in pure Zr compound) both to enthalpy and to configurational entropy.
Although the weberite-type model appears to suitably describe the local structure in disordered pyrochlores35,41,42,44 as a coherent symmetry decrease, our study points to an alternative scenario. Our experimental results on the Gd2(Ti1−xZrx)2O7 solid solution can be interpreted in terms of clusters of defects that induce structural relaxations. This outcome reinforces the idea that the weberite-type structure might not be the definitive answer and the question of properly describing the short-range structure in disordered pyrochlore systems requires further research. The development of new structural models will be fundamental in the understanding of pyrochlore materials designed for radionuclides immobilization.
5. Conclusions
In this paper we investigated the crystal structure of the Gd2(Ti1−xZrx)2O7 system in sight of its potential usage in the field of radionuclides disposal. To this purpose, we used synchrotron radiation powder diffraction, analyzing the data both in the reciprocal (Rietveld refinements and Williamson–Hall analysis) and in the real space with Pair Distribution Function analysis, Raman Spectroscopy and DFT calculations.The main findings can be summarized as follows:
(i) Although all samples retain the pyrochlore structure at the average scale, Rietveld refinements of HR-XRPD patterns revealed a gradual increasing of anion Anti-Frenkel defects ( ) with increasing Zr-content. Conversely, a small concentration of cation ZrGd/GdZr antisite defects (
) with increasing Zr-content. Conversely, a small concentration of cation ZrGd/GdZr antisite defects (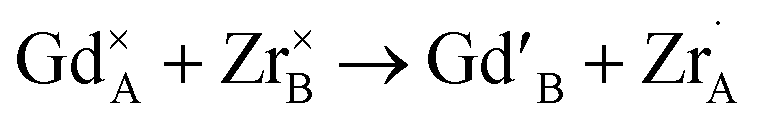 ) appears only at the highest xZr values.
) appears only at the highest xZr values.
(ii) Raman spectroscopy enforces the idea of oxygen anions responsible for the increasing structural disorder in the solid solution upon Zr-substitution. A dramatic change in the spectra occurs when xZr ≥ 0.5; the main pyrochlore bands broaden and new spectral lines suggest that the source of the disorder needs to be sought at the local scale in Zr-rich compositions.
(iii) Analysis of G(r) functions of all the samples shows that with increasing xZr, modulations of the cation–cation distances not compatible with the pyrochlore structure appear up to r ≈ 8 Å. At first, a Pmma weberite-type model providing positional degrees of freedom and increasing average Zr-coordination was tested in the fitting of experimental G(r) of Zr-rich samples. The short coherence length of this structural model (less than c cell constant) has led to another approach. DFT calculations were employed to generate disordered pyrochlore cells in Gd2Zr2O7 composition where variable numbers of AFr defects were introduced and the structures were allowed to relax. The lowest in energy among these structures were suitably fitted against the experimental data without further relaxing the structures, apart cell constants and U parameters. These last models was extended to solid solutions with Ti confined in defect free pyrochlore Gd2Ti2O7 phase.
Author contributions
Armando di Biase conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, funding acquisition. Carlo Castellano conceptualization, methodology, formal analysis, investigation, resources, writing – review & editing, visualization, supervision, funding acquisition. Giorgia Confalonieri methodology, investigation, data curation. Patrizia Fumagalli methodology, formal analysis, investigation, resources. Simone Tumiati methodology, investigation, resources. Davide Ceresoli conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, writing – review & editing. Marco Scavini conceptualization, methodology, formal analysis, investigation, resources, data curation, writing –original draft, writing – review & editing, visualization, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the ESRF for providing beamtime for experiment ch-5900.49 C. C. is thankful for the financial support from the University of Milan (fund PSR2021_DIP_005_PI_CDPIN). M. S. acknowledge partial funding under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 – Call for tender no. 1561 of 11.10.2022 of Ministero dell’ Università e della Ricerca (MUR) funded by the European Union – NextGenerationEU (project code PE0000021, Concession Decree no. 1561 of 11.10.2022 adopted by Ministero dell’Università e della Ricerca (MUR), CUP D43C22003090001, project title “Network 4 Energy Sustainable Transition – NEST”).Notes and references
- A. P. Anantharaman and H. P. Dasari, Ceram. Int., 2021, 47, 4367–4388 CrossRef CAS
.
- J. S. Gardner, M. J. P. Gingras and J. E. Greedan, Rev. Mod. Phys., 2010, 82, 53–107 CrossRef CAS
- E. V. Dudnik, S. N. Lakiza, N. I. Hrechanyuk, A. K. Ruban, V. P. Red’ko, M. S. Hlabay and A. B. Myloserdov, Powder Metall. Met. Ceram., 2018, 57, 301–315 CrossRef CAS
- J. Xu, R. Xi, X. Xu, Y. Zhang, X. Feng, X. Fang and X. Wang, J. Rare Earths, 2020, 38, 840–849 CrossRef
- A. E. Ringwood, S. E. Kesson, N. G. Ware, W. Hibberson and A. Major, Nature, 1979, 278, 219–223 CrossRef CAS
- R. C. Ewing, W. J. Weber and J. Lian, J. Appl. Phys., 2004, 95, 5949–5971 CrossRef CAS
- S. A. McMaster, R. Ram, N. Faris and M. I. Pownceby, J. Hazard. Mater., 2018, 360, 257–269 CrossRef CAS PubMed
- Y. Wang, C. Jing, Z. Ding, Y. Zhang, T. Wei, J. Ouyang, Z. Liu, Y.-J. Wang and Y. Wang, Crystals, 2023, 13, 143 CrossRef CAS
- S. X. Wang, B. D. Begg, L. M. Wang, R. C. Ewing, W. J. Weber and K. V. G. Kutty, J. Mater. Res., 1999, 14, 4470–4473 CrossRef CAS
- B. D. Begg, N. J. Hess, D. E. McCready, S. Thevuthasan and W. J. Weber, J. Nucl. Mater., 2001, 289, 188–193 CrossRef CAS
- M. Lang, F. X. Zhang, R. C. Ewing, J. Lian, C. Trautmann and Z. Wang, J. Mater. Res., 2009, 24, 1322–1334 CrossRef CAS
- M. Lang, J. Lian, J. Zhang, F. Zhang, W. J. Weber, C. Trautmann and R. C. Ewing, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 224105 CrossRef
- L. Minervini, R. W. Grimes and K. E. Sickafus, J. Am. Ceram. Soc., 2000, 83, 1873–1878 CrossRef CAS
- K. E. Sickafus, L. Minervini, R. W. Grimes, J. A. Valdez, M. Ishimaru, F. Li, K. J. McClellan and T. Hartmann, Science, 2000, 289, 748–751 CrossRef CAS PubMed
- N. J. Hess, B. D. Begg, S. D. Conradson, D. E. McCready, P. L. Gassman and W. J. Weber, J. Phys. Chem. B, 2002, 106, 4663–4677 CrossRef CAS
- P. Nachimuthu, S. Thevuthasan, E. M. Adams, W. J. Weber, B. D. Begg, B. S. Mun, D. K. Shun, D. W. Lindle, E. M. Gullikson and R. C. C. Perera, J. Phys. Chem. B, 2005, 109, 1337–1339 CrossRef CAS PubMed
- C. Heremans, B. J. Wuensch, J. K. Stalick and E. Prince, J. Solid State Chem., 1995, 117, 108–121 CrossRef CAS
- B. J. Wuensch, K. W. Eberman, C. Heremans, E. M. Ku, P. Onnerud, E. M. E. Yeo, S. M. Haile, J. K. Stalick and J. D. Jorgensen, Solid State Ionics, 2000, 129, 111–133 CrossRef CAS
- A. F. Fuentes, S. M. Montemayor, M. Maczka, M. Lang, R. C. Ewing and U. Amador, Inorg. Chem., 2018, 57, 12093–12105 CrossRef CAS PubMed
- E. C. O'Quinn, D. L. Drey and M. K. Lang, Front. Chem., 2021, 9, 1–9 Search PubMed
- M. A. Subramanian, G. Aravamudan and G. V. Subba Rao, Prog. Solid State Chem., 1983, 15, 55–143 CrossRef CAS
- E. J. Harvey, S. E. Ashbrook, G. R. Lumpkin and S. A. T. Redfern, J. Mater. Chem., 2006, 16, 4665–4674 RSC
- G. Herrera, J. Jiménez-Mier and E. Chavira, Mater. Charact., 2014, 89, 13–22 CrossRef CAS
- M. Coduri, M. Scavini, M. Allieta, M. Brunelli and C. Ferrero, Chem. Mater., 2013, 25, 4278–4289 CrossRef CAS
- M. Coduri, P. Masala, M. Allieta, I. Peral, M. Brunelli, C. A. Biffi and M. Scavini, Inorg. Chem., 2018, 57, 879–891 CrossRef CAS PubMed
- M. Scavini, M. Coduri, M. Allieta, P. Masala, S. Cappelli, C. Oliva, M. Brunelli, F. Orsini and C. Ferrero, IUCrJ, 2015, 2, 511–522 CrossRef CAS PubMed
- M. Coduri, D. Bozzetti, S. Checchia, M. Brunelli and M. Scavini, Inorganics, 2019, 7, 102 CrossRef CAS
- K. J. Moreno, A. F. Fuentes, M. Maczka, J. Hanuza and U. Amador, J. Solid State Chem., 2006, 179, 3805–3813 CrossRef CAS
- C. Wan, Z. Qu, A. Du and W. Pan, Acta Mater., 2009, 57, 4782–4789 CrossRef CAS
- Y. H. Lee, H. S. Sheu, J. P. Deng and H. C. I. Kao, J. Alloys Compd., 2009, 487, 595–598 CrossRef CAS
- G. Sattonnay, S. Moll, L. Thomé, C. Decorse, C. Legros, P. Simon, J. Jagielski, I. Jozwik and I. Monnet, J. Appl. Phys., 2010, 108, 103512 CrossRef
- A. Kumar, P. K. Kulriya, S. K. Sharma, V. Grover, A. K. Tyagi and V. K. Shukla, J. Nucl. Mater., 2020, 539, 152278 CrossRef CAS
- A. Kumar, S. K. Sharma, V. Grover, Y. Singh, V. Kumar, V. K. Shukla and P. K. Kulriya, J. Nucl. Mater., 2022, 564, 153682 CrossRef CAS
- V. V. Popov, Y. V. Zubavichus, A. P. Menushenkov, A. A. Yaroslavtsev, E. S. Kulik, V. F. Petrunin, S. A. Korovin and N. N. Trofimova, Russ. J. Inorg. Chem., 2014, 59, 279–285 CrossRef CAS
- D. L. Drey, E. C. O'Quinn, T. Subramani, K. Lilova, G. Baldinozzi, I. M. Gussev, A. F. Fuentes, J. C. Neuefeind, M. Everett, D. Sprouster, A. Navrotsky, R. C. Ewing and M. Lang, RSC Adv., 2020, 10, 34632–34650 RSC
- J. Shambling, M. Feygenson, J. Neuefeind, C. L. Tracy, F. Zhang, S. Finkeldei, D. Bosbach, H. Zhou, R. C. Ewing and M. Lang, Nat. Mater., 2016, 15, 507–511 CrossRef PubMed
- E. C. O'Quinn, J. L. Bishop, R. Sherrod, J. Neuefeind, S. M. Montemayor, A. F. Fuentes and M. Lang, J. Mater. Sci., 2018, 53, 13400–13410 CrossRef
- C. K. Chung, E. C. O'Quinn, J. C. Neuefeind, A. F. Fuentes, H. Xu, M. Lang and A. Navrotsky, Acta Mater., 2019, 181, 309–317 CrossRef CAS
- J. Shamblin, C. L. Tracy, R. I. Palomares, E. C. O'Quinn, R. C. Ewing, J. Neuefeind, M. Feygenson, J. Behrens, C. Trautmann and M. Lang, Acta Mater., 2018, 144, 60–67 CrossRef CAS
- C. K. Chung, J. Shamblin, E. C. O'Quinn, A. Shelyug, I. Gussev, M. Lang and A. Navrotsky, Acta Mater., 2018, 145, 227–234 CrossRef CAS
- R. Sherrod, E. C. O'Quinn, I. M. Gussev, C. Overstreet, J. Neuefeind and M. K. Lang, npj Mater. Degrad., 2021, 5, 19 CrossRef CAS
- E. C. O'Quinn, C. L. Tracy, W. F. Cureton, R. Sachan, J. C. Neuefeind, C. Trautmann and M. K. Lang, J. Mater. Chem. A, 2021, 9, 16982–16997 RSC
- D. Drey, E. O'Quinn, S. Finkeldei, J. Neuefeind and M. Lang, Acta Mater., 2022, 225, 117590 CrossRef CAS
- P. R. Jothi, W. Liyanage, B. Jiang, S. Paladugu, D. Olds, D. A. Gilbert and K. Page, Small, 2022, 18, 2101323 CrossRef CAS PubMed
- D. Zhang, Y. Chen, H. Vega, T. Feng, D. Yu, M. Everett, J. Neuefeind, K. An, R. Chen and J. Luo, Adv. Powder Mater., 2023, 2, 100098 CrossRef
- D. Michel, M. Perez y Jorba and R. Collongues, Mater. Res. Bull., 1974, 9, 1457–1468 CrossRef CAS
- J. L. Shi, Z. X. Qu and Q. Wang, Key Eng. Mater., 2016, 697, 386–389 Search PubMed
- OriginLab Corporation Search PubMed.
-
A. di Biase, C. Castellano, M. Scavini and G. Confalonieri, Phase diagram and local structure of Gd2(Zr1-xTix)2O7 and (Gd1-yNdy)2(Zr1-zCez)2O7 pyrochlore matrices for nuclear waste disposal, European Synchrotron Radiation Facility, 2024, https://data.esrf.fr/doi/10.15151/ESRF-ES-404435446 Search PubMed
- A. Fitch, C. Dejoie, E. Covacci, G. Confalonieri, O. Grendal, L. Claustre, P. Guillou, J. Kieffer, W. de Nolf, S. Petitdemange, M. Ruat and Y. Watier, J. Synchrotron Radiat., 2023, 30, 1–10 CrossRef PubMed
- A. P. Hammersley, J. Appl. Crystallogr., 2016, 49, 646–652 CrossRef CAS
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed
- A. C. Larson and R. B. Von Dreele, Los Alamos, NM, 87545, 1994, p. 178.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS
-
T. Roisnel and J. Rodriguez-Carvajal, WinPLOTR: a Windows tool for powder diffraction patterns analysis, Materials Science Forum, Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), 2000, pp. 118–123 Search PubMed
- G. K. Williamson and W. H. Hall, Acta Metall., 1953, 1, 22–31 CrossRef CAS
-
T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Newnes, 2012 Search PubMed
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens.Matter, 2007, 19, 335219 CrossRef CAS PubMed
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291–341 CrossRef CAS
- J. D. Gale, Z. fur Krist.–Cryst. Mater., 2005, 220, 552–554 CrossRef CAS
- M. J. L. Sangster, J. Phys. Chem. Solids, 1974, 35, 195–200 CrossRef CAS
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens.Matter, 2009, 21, 395502 CrossRef PubMed
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens.Matter, 2017, 29, 465901 CrossRef CAS PubMed
- O. Yakubovich, V. Urusov, W. Massa, G. Frenzen and D. Babel, Z. fur Anorg. Allg. Chem., 1993, 619, 1909–1919 CrossRef CAS
- H. P. Rooksby and E. A. D. White, J. Am. Ceram. Soc., 1964, 47, 94–96 CrossRef CAS
- L. Cai and J. C. Nino, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 269–290 CrossRef CAS PubMed
- I. M. Gussev, E. C. O'Quinn, G. Baldinozzi, J. Neuefeind, R. C. Ewing, F. Zhang and M. Lang, Acta Mater., 2020, 196, 704–709 CrossRef CAS
- P. E. R. Blanchard, R. Clements, B. J. Kennedy, C. D. Ling, E. Reynolds, M. Avdeev, A. P. J. Stampfl, Z. Zhang and L.-Y. Jang, Inorg. Chem., 2012, 51, 13237–13244 CrossRef CAS PubMed
- E. Reynolds, P. E. R. Blanchard, B. J. Kennedy, C. D. Ling, S. Liu, M. Avdeev, Z. Zhang, G. J. Cuello, A. Tadich and L.-Y. Jang, Inorg. Chem., 2013, 52, 8409–8415 CrossRef CAS PubMed
- S. T. Norberg, S. Hull, S. G. Eriksson, I. Ahmed, F. Kinyanjui and J. J. Biendicho, Chem. Mater., 2012, 24, 4294–4300 CrossRef CAS
- F. P. Marlton, Z. Zhang, Y. Zhang, T. E. Proffen, C. D. Ling and B. J. Kennedy, Chem. Mater., 2021, 33, 1407–1415 CrossRef CAS
- Z. Zhang, S. C. Middleburgh, M. de los Reyes, G. R. Lumpkin, B. J. Kennedy, P. E. R. Blanchard, E. Reynolds and L.-Y. Jang, J. Phys. Chem. C, 2013, 117, 26740–26749 CrossRef CAS
- B. P. Mandal, K. Bhattacharyya, J. H. Zain, V. Sudarsan, S. Nigam, C. Nayak and A. K. Tyagi, J. Solid State Chem., 2021, 303, 122472 CrossRef CAS
- B. J. Kennedy, Q. Zhou and M. Avdeev, J. Solid State Chem., 2011, 184, 1695–1698 CrossRef CAS
- O. Knop, F. Brisse and L. Castelliz, Can. J. Chem., 1969, 47, 971–990 CrossRef CAS
- L. Minervini, R. W. Grimes, Y. Tabira, R. L. Withers and K. E. Sickafus, Philos. Mag. A, 2002, 82, 123–135 CAS
- M. Scavini, M. Coduri, M. Allieta, M. Brunelli and C. Ferrero, Chem. Mater., 2012, 24, 1338–1345 CrossRef CAS
- C. Jiang, C. R. Stanek, K. E. Sickafus and B. P. Uberuaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 17–21 Search PubMed
- R. A. McCauley, J. Opt. Soc. Am., 1973, 63, 721 CrossRef CAS
- B. E. Scheetz and W. B. White, J. Am. Ceram. Soc., 1979, 62, 468–470 CrossRef CAS
- M. Oueslati, M. Balkanski, P. K. Moon and H. L. Tuller, MRS Proc., 1988, 135, 199 CrossRef
- A. Bhuiyan, V. Wong, J. L. Abraham, R. D. Aughterson, L. Kong, R. Farzana, D. J. Gregg, C. C. Sorrell, Y. Zhang and P. Koshy, Mater. Chem. Phys., 2021, 273, 125058 CrossRef CAS
- N. T. Vandenborre, E. Husson and H. Brusset, Spectrochim. Acta, Part A, 1981, 37, 113–118 CrossRef
- M. T. Vandenborre, E. Husson, J. P. Chatry and D. Michel, J. Raman Spectrosc., 1983, 14, 63–71 CrossRef CAS
- M. T. Vandenborre and E. Husson, J. Solid State Chem., 1983, 50, 362–371 CrossRef CAS
- H. C. Gupta, S. Brown, N. Rani and V. B. Gohel, J. Raman Spectrosc., 2001, 32, 41–44 CrossRef CAS
- S. Saha, D. V. S. Muthu, C. Pascanut, N. Dragoe, R. Suryanarayanan, G. Dhalenne, A. Revcolevschi, S. Karmakar, S. M. Sharma and A. K. Sood, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 064109 CrossRef
- M. Maçzka, J. Hanuza, K. Hermanowicz, A. F. Fuentes, K. Matsuhira and Z. Hiroi, J. Raman Spectrosc., 2008, 39, 537–544 CrossRef
- T. T. A. Lummen, I. P. Handayani, M. C. Donker, D. Fausti, G. Dhalenne, P. Berthet, A. Revcolevschi and P. H. M. Van Loosdrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1–11 Search PubMed
- L. Grima, J. I. Peña and M. L. Sanjuán, J. Alloys Compd., 2022, 923, 166449 CrossRef CAS
- M. Mączka, M. L. Sanjuán, A. F. Fuentes, L. Macalik, J. Hanuza, K. Matsuhira and Z. Hiroi, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 214437 CrossRef
- S. Kumar and H. C. Gupta, Vib. Spectrosc., 2012, 62, 180–187 CrossRef CAS
- V. A. Chernyshev, V. P. Petrov and A. E. Nikiforov, Phys. Solid State, 2015, 57, 996–1002 CrossRef CAS
- H. C. Gupta and S. Brown, J. Phys. Chem. Solids, 2003, 64, 2205–2207 CrossRef CAS
- H. C. Gupta, J. Singh, S. Kumar, Karandeep and N. Rani, J. Mol. Struct., 2009, 937, 136–138 CrossRef CAS
- A. A. Yastrebtsev, V. V. Popov, A. P. Menushenkov, A. I. Beskrovnyi, D. S. Neov, I. V. Shchetinin and K. V. Ponkratov, J. Alloys Compd., 2020, 832, 154863 CrossRef CAS
- A. K. Kushwaha, S. P. Mishra, M. K. Vishwakarma, S. Chauhan, H. R. Jappor, R. Khenata and S. Bin Omran, Inorg. Chem. Commun., 2021, 127, 108495 CrossRef CAS
- M. L. Sanjuan, C. Guglieri, S. Díaz-Moreno, G. Aquilanti, A. F. Fuentes, L. Olivi and J. Chaboy, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 104207 CrossRef
- C. L. Tracy, J. Shamblin, S. Park, F. Zhang, C. Trautmann, M. Lang and R. C. Ewing, Phys. Rev. B, 2016, 94, 064102 CrossRef
- M. Mori, Solid State Ionics, 2003, 158, 79–90 CrossRef CAS
- F. X. Zhang and S. K. Saxena, Chem. Phys. Lett., 2005, 413, 248–251 CrossRef CAS
- B. P. Mandal, A. Banerji, V. Sathe, S. K. Deb and A. K. Tyagi, J. Solid State Chem., 2007, 180, 2643–2648 CrossRef CAS
- F. Poulsen, M. Glerup and P. Holtappels, Solid State Ionics, 2000, 135, 595–602 CrossRef CAS
- P. B. Oliete, A. Orera and M. L. Sanjuán, J. Raman Spectrosc., 2020, 51, 514–527 CrossRef CAS
- M. Suganya, K. Ganesan, P. Vijayakumar, A. S. Gill, S. K. Srivastava, C. K. Singh, R. M. Sarguna, P. K. Ajikumar and S. Ganesamoorthy, Cryst. Res. Technol., 2022, 57, 2100287 CrossRef
- S. Brown, H. C. Gupta, J. A. Alonso and M. J. Martinez-Lope, J. Raman Spectrosc., 2003, 34, 240–243 CrossRef CAS
- F. X. Zhang, B. Manoun and S. K. Saxena, Mater. Lett., 2006, 60, 2773–2776 CrossRef CAS
- M. Jafar, S. N. Achary, N. P. Salke, A. K. Sahu, R. Rao and A. K. Tyagi, J. Nucl. Mater., 2016, 475, 192–199 CrossRef CAS
- V. A. Chernyshev, Opt. Spectrosc., 2019, 127, 825–831 CrossRef CAS
- F. X. Zhang, B. Manoun, S. K. Saxena and C. S. Zha, Appl. Phys. Lett., 2005, 86, 181906 CrossRef
- D. J. Arenas, L. V. Gasparov, W. Qiu, J. C. Nino, C. H. Patterson and D. B. Tanner, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1–8 CrossRef
- M. Glerup, O. F. Nielsen and F. W. Poulsen, J. Solid State Chem., 2001, 160, 25–32 CrossRef CAS
- S. A. Kovyazina, L. A. Perelyaeva, I. A. Leonidov and Y. A. Bakhteeva, J. Struct. Chem., 2003, 44, 975–979 CrossRef CAS
- A. Salamat, P. F. McMillan, S. Firth, K. Woodhead, A. L. Hector, G. Garbarino, M. C. Stennett and N. C. Hyatt, Inorg. Chem., 2013, 52, 1550–1558 CrossRef CAS PubMed
- L. R. Blackburn, S. Sun, L. J. Gardner, E. R. Maddrell, M. C. Stennett and N. C. Hyatt, J. Nucl. Mater., 2020, 535, 1–11 CrossRef
- Y. Zhang, J. Čejka, I. Karatchevtseva, M. Qin, L. Kong, K. Short, S. C. Middleburgh and G. R. Lumpkin, J. Nucl. Mater., 2014, 446, 68–72 CrossRef CAS
- N. Gumber, R. V. Pai, R. Phatak, B. Adiraju, M. Sahu, J. Jagannath and K. Sudarshan, J. Nucl. Mater., 2021, 556, 153191 CrossRef CAS
- V. V. Popov, A. P. Menushenkov, A. A. Ivanov, A. A. Yastrebtsev, B. R. Gaynanov, F. D'Acapito and A. Puri, Radiat. Phys. Chem., 2020, 175, 108469 CrossRef CAS
- E. C. O’Quinn, K. E. Sickafus, R. C. Ewing, G. Baldinozzi, J. C. Neuefeind, M. G. Tucker, A. F. Fuentes, D. Drey and M. K. Lang, Sci. Adv., 2020, 6(35), eabc2758 CrossRef PubMed
- A. Dal Corso, Comput. Mater. Sci., 2014, 95, 337–350 CrossRef CAS
- J. Zhang, A. R. Oganov, X. Li, H. Dong and Q. Zeng, Phys. Chem. Chem. Phys., 2015, 17, 17301–17310 RSC
- X. Zhao, D. Ceresoli and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 085107 CrossRef
- Y. Li, P. M. Kowalski, G. Beridze, A. R. Birnie, S. Finkeldei and D. Bosbach, Scr. Mater., 2015, 107, 18–21 CrossRef CAS
- P. M. Kowalski, Scr. Mater., 2020, 189, 7–10 CrossRef CAS
- P. S. Maram, S. V. Ushakov, R. J. K. Weber, C. J. Benmore and A. Navrotsky, Sci. Rep., 2018, 8, 10658 CrossRef PubMed
- D. N. Argyriou, J. Appl. Crystallogr., 1994, 27, 155–158 CrossRef CAS
- G. Pilania, B. Puchala and B. P. Uberuaga, npj Comput. Mater., 2019, 5, 1–9 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04847k |
| This journal is © The Royal Society of Chemistry 2023 |